

Abstract
Background
Although resurrecting ancestral proteins is a powerful tool for understanding the molecular-functional evolution of gene families, nearly all studies have examined proteins functioning in relatively stable biological processes. The extent to which more dynamic systems obey the same ‘rules’ governing stable processes is unclear. Here we present the first detailed investigation of the functional evolution of the RIG-like receptors (RLRs), a family of innate immune receptors that detect viral RNA in the cytoplasm.
Results
Using kinetic binding assays and molecular dynamics simulations of ancestral proteins, we demonstrate how a small number of adaptive protein-coding changes repeatedly shifted the RNA preference of RLRs throughout animal evolution by reorganizing the shape and electrostatic distribution across the RNA binding pocket, altering the hydrogen bond network between the RLR and its RNA target. In contrast to observations of proteins involved in metabolism and development, we find that RLR-RNA preference ‘flip flopped’ between two functional states, and shifts in RNA preference were not always coupled to gene duplications or speciation events. We demonstrate at least one reversion of RLR-RNA preference from a derived to an ancestral function through a novel structural mechanism, indicating multiple structural implementations of similar functions.
Conclusions
Our results suggest a model in which frequent shifts in selection pressures imposed by an evolutionary arms race preclude the long-term functional optimization observed in stable biological systems. As a result, the evolutionary dynamics of immune receptors may be less constrained by structural epistasis and historical contingency.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-016-0818-6) contains supplementary material, which is available to authorized users.
Keywords: RIG-I, RIG-like receptors, Ancestral reconstruction, Antiviral immunity, Evolution of immunity, Molecular evolution, Innate immunity
Background
Resurrection and biochemical analysis of ancestral proteins is a powerful technique for understanding the molecular-structural basis of functional evolution [1, 2]. Example studies have elucidated the precise details by which evolutionary changes in sequence produce changes in protein structure and how structural changes alter molecular function, generating a wealth of potentially generalizable results about how structural properties may affect the evolution of molecular function [3–18].
One of the major limitations of current ancestral sequence resurrection (ASR) studies is that they have—almost without exception—examined molecular systems that function in basic cellular processes and organism development, systems that are expected to be relatively slow-evolving [19–24]. In contrast, proteins comprising the immune systems of multicellular eukaryotes are often involved in evolutionary arms races with pathogens, leading to rapid and highly variable evolutionary trajectories [25–27]. Proteins directly interacting with pathogen factors are expected to evolve particularly rapidly, and genome-wide comparisons have generally supported this expectation [28–30]. No detailed ASR studies have yet been conducted on primary immune receptors, so we have little experimental information about how the evolution of structure-function occurs in these systems, particularly across large evolutionary timescales.
Another major limitation of existing ASR studies is that they have focused almost exclusively on proteins that function by binding small ligands such as sugars, hormones or other metabolites [8, 13, 16–18, 31, 32] or by light activation [12, 15]. Short-term evolution of macromolecular interactions has been examined through experimental evolution and studies of protein affinity maturation [33–36], and a few ASR studies have begun investigating the evolution of protein interactions with larger macromolecules [37–40]. However, the long-term molecular evolution of macromolecular interactions remains under-studied. Structural properties important for the evolution of macromolecular interactions may be different from those driving protein-small ligand interactions, and the extent to which results obtained in one case can be generalized to the other is not clear [41, 42].
The RIG-like receptors, RIG-I (DDx58) MDA5 (IFIH1) and LGP2 (DHx58), form a complement of cytoplasmic RNA-binding proteins contributing to innate antiviral immunity in a wide variety of vertebrates, including all mammals [43–46]. RIG-like receptors (RLRs) bind viral-derived RNAs and signal cellular immune responses, primarily through direct interactions with the mitochondrion-anchored signal transducer, IPS1 [47, 48] (Additional file 1: Figure S1).
In order to function as effective antiviral receptors, RLRs must collectively recognize a variety of viral-associated RNA types without binding the specific motifs associated with cytoplasmic host RNAs. Although the precise ligand complement of human and other vertebrate RLRs has not been determined, RLRs have been shown to recognize specific end structures of RNA molecules via a C-terminal RNA recognition domain (RD), providing a structural mechanism capable of differentiating viral-derived from host RNA [49–51]. An upstream helicase and intervening pincer domain also contribute to RLR-RNA binding, primarily through interactions with the RNA backbone [52].
RIG-I—by far the best understood of the RLRs—recognizes single- and double-stranded RNA molecules with and without 5′ triphosphate (5′ppp) moieties but does not recognize the 7-methylguanylate cap typical of eukaryote mRNA [52–54] Additionally, RIG-I exhibits severely reduced immune signaling activity from dsRNA with 3′ or 5′ overhangs, which are typical of mature host tRNAs, rRNAs and microRNAs [55–57]. Less is known about the specific RNA ligands bound by MDA5 and LGP2. LGP2 appears to bind RNA moieties similar to those of RIG-I as well as additional RNA end structures [58, 59]. The natural MDA5 ligands have remained the most mysterious, although evidence suggests that MDA5 may cooperatively bind long—and possibly short—blunt-ended dsRNA molecules or other specific virus-produced RNAs [59–61].
We have recently found that RLRs were present in the earliest multicellular animals and functionally diversified through a series of gene duplication events [46]. Our analysis uncovered evidence for recurrent protein-coding adaptation in RLRs throughout the mammalian lineage and in the human population, particularly targeting the RNA recognition domain, consistent with an ‘arms race’ model in which RLRs adapt their RNA-binding repertoire in response to rapidly-evolving viral threats.
In our view, the RLRs provide an excellent model with which to begin extending current ASR results obtained from slow-evolving small-ligand-binding proteins to fast-evolving immune receptors interacting with larger macromolecules. Better understanding the evolutionary trajectories leading to RLR functional diversity is expected to shed light on how pathogen-driven arms races can shape the molecular functions of primary immune receptors and how structural features of the receptors affect potential evolutionary trajectories.
Results and discussion
The aim of this study is to characterize how RNA preference changed during early RIG-like receptor (RLR) evolution. We employed an approach beginning with phylogenetic analysis to establish the RLR protein family tree, followed by analysis of protein-coding adaptation to identify potential functional shifts in RNA binding. Functional shifts were confirmed using ancestral sequence reconstruction and kinetic analyses of ancestral proteins. We used molecular dynamics simulations of ancestral proteins to characterize the likely mechanisms driving observed shifts in RNA preference and confirmed these by measuring the RNA-binding kinetics of mutant ancestral proteins incorporating historical amino-acid substitutions. This strategy represents a general approach for examining the mechanistic basis for molecular-functional evolution in protein families [62] (see Methods for details).
RIG-like receptors (RLRs) arose in early animals and diversified by gene duplication and protein-coding adaptation targeting the RNA recognition domain (RD) in deuterostomes and jawed vertebrates
In order to establish a robust framework within which to examine RIG-like receptor (RLR) functional evolution, we reconstructed the phylogeny of full-length RLR protein sequences using maximum likelihood, assuming two different alignments in order to incorporate alignment uncertainty (see Methods; Additional file 1: Table S1). Consistent with our previous study [46], the consensus tree across different alignments suggested the RLRs originated in the earliest multicellular animals and diversified by two major gene duplication events, one in bilateria and the other early in the jawed vertebrate lineage (Fig. 1). All bilaterian RLRs formed a monophyletic clade separated from Amphimedon and Nematostella RLRs with ≥0.94 SH-like aLRT, depending on the alignment. Our analysis confidently grouped deuterostome and protostome RIG-Is (support ≥0.92), suggesting that the earliest RLR gene duplication occurred before the protostome-deuterostome split. Vertebrate MDA5 and LGP2 sequences—along with Saccoglossus and Branchiostoma MDA5/LGP2—were monophyletic with maximal support, placing the MDA5-LGP2 duplication very early in the jawed vertebrate lineage, consistent with our previous findings.
Fig. 1.
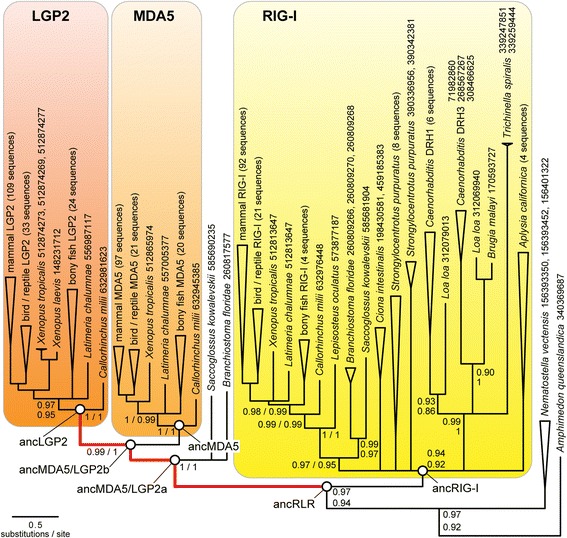
RIG-like receptors (RLRs) arose in early animals and diversified by gene duplications in bilateria and jawed vertebrates. We reconstructed the RLR gene phylogeny by maximum likelihood using two different alignments of all available RLR protein sequences (see Methods). Branch lengths are scaled to the average number of substitutions/site. We plot SH-like aLRT clade support from PROBALIGN (top) and MAFFT (bottom) alignments; clades with <0.8 support are collapsed to polytomies. Sequences from major monophyletic taxonomic groups are collapsed, and genbank IDs are provided for sequences from individual species. Bold red branches indicate significant support for protein-coding adaptation specific to the RNA-recognition (RD) domain (p < 0.01 after correction for multiple tests; see Methods). White circles indicate ancestral proteins resurrected in this study
The phylogeny reconstructed using sequence data implies a loss of MDA5/LGP2 in all protostomes and a second loss of RIG-I in arthropods (Additional file 1: Figure S2). The alternative phylogeny, in which the first RLR gene duplication occurred in deuterostomes after the protostome-deuterostome split, is more parsimonious in gene loss events—requiring only a single loss of the ancestral RLR in arthropods—but is consistently rejected by phylogenetic analysis (SH-test p < 0.023; see also [46]). Although it is impossible to completely rule out phylogenetic error, we have not observed any evidence for long-branch attraction or other systematic artifacts; in our previous analysis, the same phylogeny was consistently recovered using various alignments, inference methods, inclusion/exclusion of fast-evolving species and inclusion/exclusion of outgroup sequences [46]. The resolution of our previous phylogeny using additional sequence data and new alignments further supports its general robustness.
Although the evolution of a protein family’s molecular function can occur in the absence of adaptation or positive selection, reliable identification of protein-coding adaptation is a strong predictor of changes in molecular function, as adaptive processes can only occur when there is some phenotypic change visible to selection. We investigated the evolutionary forces driving RLR functional divergence using a branch-sites test to identify protein-coding adaptation in specific functional domains across specific branches on the phylogeny (see Methods). After correcting for multiple tests, we found strong support for protein-coding adaptation in one lineage following each of the major RLR duplication events (Fig. 1). In all cases, adaptation was specific to the C-terminal RNA recognition domain (RD) and was not found affecting any other functional domain (Additional file 1: Figure S3). These results are generally consistent with our previous findings of recurrent protein-coding adaptation in mammalian and human RLR RDs [46].
Following the earliest duplication of the ancestral RLR (ancRLR), protein-coding adaptation specific to the RD was detected along the branch leading to the first ancestral MDA5/LGP2 (ancMDA5/LGP2a) but not along that leading to ancRIG-I. We observed evidence for continued adaptation of the RD along the ancestral MDA5/LGP2 branch before the MDA5-LGP2 duplication and again in the LGP2 lineage after the MDA5-LGP2 split (Fig. 1). We did observe evidence for protein-coding adaptation in the helicase, pincer, CARD signaling domains and other regions of the RLR protein sequence following the initial diversification of RIG-I, MDA5 and LGP2 lineages (Additional file 1: Figure S3). However, only the RD was inferred to have evolved adaptively along the earliest branches of the RLR tree prior to the establishment of these three major RLR lineages.
Although the branch-sites test is generally considered robust [63–66], concerns have been raised about potentially high false-positive rates under some conditions [67, 68]. However, false-positive rates were always <0.05 when we simulated codon sequences along the maximum-likelihood RLR phylogeny using a variety of empirically-derived neutral scenarios (Additional file 1: Figure S4). Although it is impossible to completely rule out false-positive detection of protein-coding adaptation, we observed no evidence suggesting inflated false-positive rates in this case.
Consistent with evidence from analyses of protein-coding adaptation, we found that the branch lengths—measuring the expected number of protein substitutions/site—were generally longer on the earliest branches of the RLR phylogeny when optimized using only RD sequences, whereas later branches had longer lengths when optimized using the helicase + pincer domains, compared to the RD (Additional file 1: Figure S5). Together, these findings suggest that the C-terminal RNA recognition domain (RD) tended to evolve faster during the earlier history of RLR evolution, whereas the helicase and pincer domains exhibited faster protein-coding evolution only after the major RIG-I, MDA5 and LGP2 lineages were established.
The helicase, pincer and RD domains cooperatively bind viral RNA ligands, with the RD specifically recognizing the end structure of the RNA and the helicase + pincer interacting primarily with the RNA backbone [52]. Our results suggest that early functional evolution of RLR-RNA recognition may have occurred primarily through adaptively-driven changes in the RD, whereas later changes in RLR-RNA binding may have involved evolution of the helicase + pincer domains.
To examine the early functional evolution of RLR-RNA recognition in more detail, we reconstructed ancestral protein sequences at key early nodes on the phylogeny (Fig. 1), inferred structural models of ancestral sequences (see Methods) and mapped putatively-adaptive substitutions to specific locations on the protein sequence (Fig. 2). In general, we found that inferred adaptive substitutions tended to cluster around the canonical “RNA-binding loop,” a flexible region of the RD that anchors the RNA ligand and makes key polar contacts with 5′ppp moieties in human RIG-I [53, 69]. Consistent with a potential functional role for adaptive changes in the RNA-binding loop, we observed a shift in the electrostatic distribution of the RNA-binding loop from strongly basic in ancRLR, ancRIG-I and human RIG-I to strongly acidic in ancMDA5/LGP2a and b (Fig. 3). The evolution of an acidic ‘RNA-binding loop’ in ancMDA5/LGP2a is expected to alter key electrostatic interactions likely to be important for RNA binding [46, 69, 70].
Fig. 2.
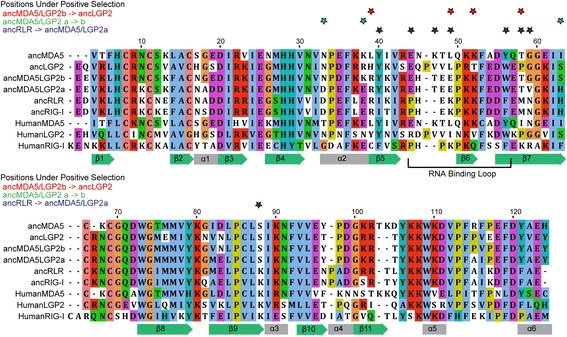
Adaptive protein-coding substitutions clustered near the RNA binding loop in the C-terminal RNA recognition domain (RD) along the lineages leading from ancRLR to ancLGP2 (see Fig. 1). We show the consensus sequence alignment of the three human RLR RDs and the ancestral RLRs resurrected in this study, with residues colored by biochemical classification and sequence conservation. Stars above the alignment indicate significant support for protein-coding adaptation at specific residues along the branches separating ancLGP2 from ancMDA5/LGP2b (red), ancMDA5/LGP2b from ancMDA5/LGP2a (green) and ancMDA5/LGP2a from ancRLR (blue), respectively (see Fig. 1). Adaptive substitutions were inferred by Bayes-empirical-Bayes posterior probability >0.95 using the branch-sites test for positive selection (see Methods). The location of the RNA binding loop is indicated, and approximate secondary structural elements are shown below the alignment
Fig. 3.
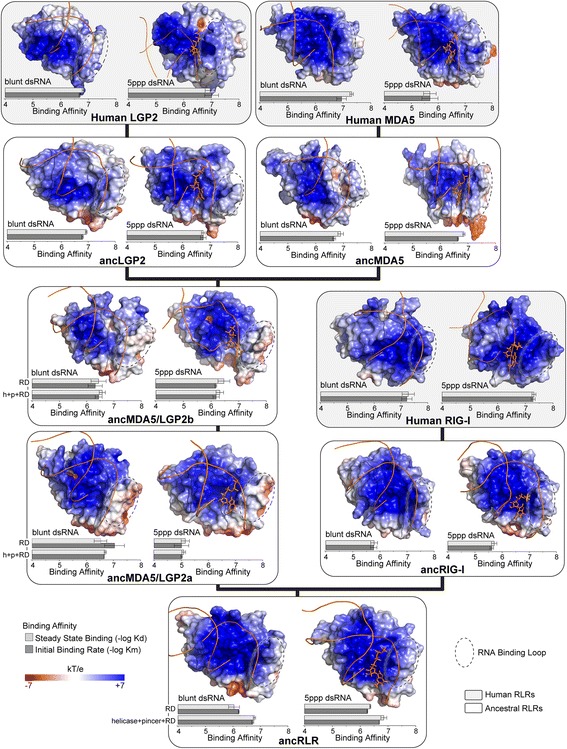
Adaptive substitutions alter RNA recognition domain (RD) structure and RNA-binding preference throughout RIG-like receptor (RLR) evolution. We inferred structural models of human and ancestral RLR RDs (see Figs. 1 and 2) bound to blunt-ended double-stranded RNA and dsRNA having a 5′ triphosphate (5ppp). We show the central structures of each RD-RNA from replicate molecular dynamics simulations, with electrostatic potential (kT/e) displayed on the molecular surface (see Methods). Dotted ovals indicate the location of the canonical RNA binding loop on each structural model. We resurrected ancestral RLR RDs and measured steady-state (Kd) and initial (Km) RNA binding affinities (see Methods). We plot –log-transformed binding affinities, with longer bars indicating higher affinity. Standard errors over three replicates are indicated. For ancRLR, ancMDA5/LGP2a and ancMDA5/LGP2b, we compare RNA binding affinities measured for the RD to affinities measured using the combined helicase + pincer + RD domains
Also consistent with a potential role in altering RD-RNA interactions, we observed changes in the stability of the RNA-binding loop during molecular dynamics simulations of ancestral RDs bound to RNA (Fig. 4). Specifically, residues comprising the RNA-binding loop fluctuated very little over the course of molecular dynamics simulations of ancRLR, ancRIG-I and human RIG-I RDs bound to blunt-ended or 5′ppp double-stranded RNA ligands, suggesting that these residues are likely stabilized by strong interactions with the RNA. Other ancestral RLRs in the MDA5/LGP2 lineage exhibited much larger root mean square fluctuation (RMSF) of residues in the RNA-binding loop, suggesting a general loss of stabilizing RNA interactions along this lineage. We observed a similar increase in the fluctuation of the RNA-binding loop in ancMDA5/LGP2a and ancMDA5/LGP2b during molecular dynamics simulations of the combined helicase + pincer + RD domains, suggesting that this observation is likely relevant to general RLR-RNA interactions and not an artifact of simulating only the RD bound to RNA (Additional file 1: Figure S6). Our results are generally consistent with recent structural studies of chicken LGP2 and MDA5, which also found the ‘RNA-binding loop’ failed to make strong contacts with RNA ligands [59]. Overall, these results suggest that the RNA-binding loop lost the capacity to bind blunt-ended and 5′ppp dsRNA ligands early in the MDA5/LGP2 lineage following the first RLR gene duplication in bilateria, and this functional shift was likely retained throughout MDA5/LGP2 evolutionary history.
Fig. 4.
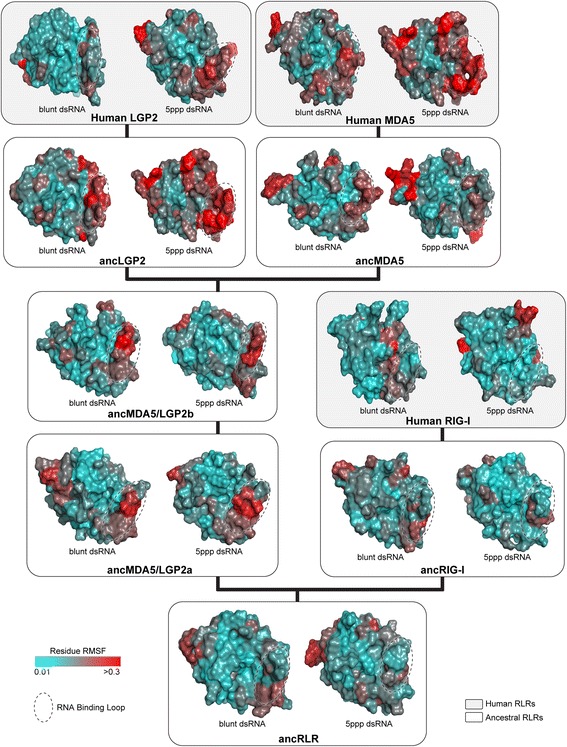
The RNA-binding loop of ancRLR and the RIG-I lineage fluctuates less over the course of molecular dynamics simulations, compared to the RNA-binding loop of other ancestral and human RLRs. We plot the root mean square fluctuation (RMSF) of each residue on the molecular surface of ancestral (white boxes) and human (gray boxes) RLR RNA-recognition domains (RD), averaged over replicate molecular dynamics simulations (see Methods). Higher values of RMSF indicate that the residue moved more over the course of the dynamics simulations. Dotted oval indicates location of the RNA-binding loop on each structure. See Figs. 1 and 2 for locations of each ancestral RLR on the phylogeny and ancestral RD sequences, respectively
Consistent with this general model, we found that the “RNA-binding loop” of ancMDA5/LGP2a and ancMDA5/LGP2b exhibited reduced capacity to form hydrogen bonds with RNA ligands over the course of molecular dynamics simulations, compared to ancRIG-I (Fig. 5; Additional file 1: Figure S7). Specifically, MDA5/LGP2a lost a key polar contact conserved in ancRLR and the RIG-I lineage (K49, see Fig. 2), which appears to stabilize both blunt-ended and 5′ppp dsRNA (Fig. 5). Additionally, K88 appears to form a strong hydrogen bond with the 5′ppp moiety in ancRLR and ancRIG-I, whereas the corresponding Ser in the ancMDA5/LGP2 lineage does not stabilize the 5′ppp dsRNA ligand (see Figs. 2 and 5). These losses of key polar contacts with the RNA ligand along the MDA5/LGP2 lineage were observed whether we simulated only the RD bound to RNA (Fig. 5) or the complete helicase + pincer + RD (Additional file 1: Figure S7), suggesting that adaptively-driven changes in the RLR RD may have altered the hydrogen bond network stabilizing RLR-RNA interactions, particularly early in the establishment of the MDA5/LGP2 lineage.
Fig. 5.
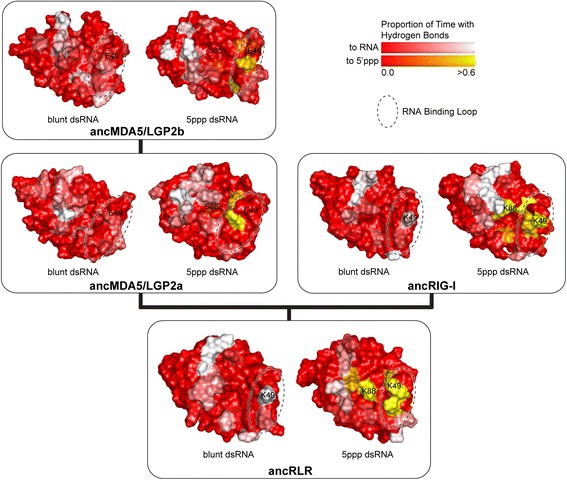
The RNA-binding loop loses hydrogen bonding to RNA ligands in the MDA5/LGP2 lineage after the first RLR gene duplication. We plot the proportion of molecular dynamics time points during which each residue was observed to form hydrogen bonds with the RNA ligand (red-white gradient) or the 5′ppp moiety in particular (red-yellow gradient), averaged over replicate simulations of ancestral RLR RDs (see Methods). The RNA-binding loop and specific residues exhibiting reduced hydrogen bonding to 5′ppp dsRNA in ancMDA5/LGP2a-b are indicated
Maximum likelihood reconstructions of ancestral RD sequences were highly similar to those found in our previous study [46], and support for ancestral sequences was generally improved (Additional file 1: Figure S8). For each of the ancestral sequences reconstructed, > 47.5 % of ancestral states were reconstructed with >0.95 posterior probability; > 50.8 % of states were reconstructed with >0.9 posterior probability, and >65 % of states had >0.8 posterior probability. Only two sites in each of ancRLR, ancMDA5/LGP2a and ancMDA5/LGP2b had alternative reconstructions with posterior probability >0.3, and ancRIG-I had three such sites. All alternative reconstructions were considered biochemically conservative, and positions with plausible alternative reconstructions generally occurred in parts of the RD structure unlikely to strongly impact RNA binding (Additional file 1: Figure S8).
When we simulated sequence data using the maximum-likelihood tree and best-fit evolutionary model, error rates for ancRLR and ancRIG-I RD reconstructions were <0.03, with remaining ancestral sequences having error rates <0.02 (Additional file 1: Figure S8). Considering residues of the same general biochemical class as equivalent reduced all error rates to < 0.01. Ancestral reconstructions of the helicase + pincer domains were even more strongly supported than those of the RD (Additional file 1: Figure S9). The number of ancestral states across the helicase + pincer reconstructed with >0.9 posterior probability was always >53 %; fewer than 0.06 % of states had alternative reconstructions with posterior probability >0.3, and estimated error rates were always <0.03.
Ancestral sequences were reconstructed using full-length RLRs aligned by PROBALIGN, which produced slightly stronger support for the consensus phylogeny, compared to the MAFFT alignment (see Fig. 1). Reconstructing ancestral RLR RDs at key nodes on the phylogeny using the MAFFT alignment produced sequences >73 % identical (>79 % identical when considering biochemically similar residues as equivalent) to those produced from the PROBALIGN alignment, and the important sequence differences among ancestral RLR RDs outlined above were present in the alternative ancestral reconstructions (Additional file 1: Figure S10A).
We found ample sequence similarity between all ancestral RLR RDs and human RIG-I RD to support accurate structural homology modeling of ancestral proteins (48 % identity for ancRLR, 43 % for ancMDA5/LGP2a, 41 % for ancMDA5/LGP2b, 37 % for ancMDA5, 36 % for ancLGP2 and 49 % for ancRIG-I) [71–73]. Sequence identity between ancestral RLR helicase + pincer domains and human RIG-I helicase + pincer were similarly high (46 % for ancRLR, 37 % for ancMDA5/LGP2a and 36 % for ancMDA5/LGP2b). Consistent with expectation based on high sequence similarity of ancestral RLRs to the crystalized human RIG-I, objective measurements of structural modeling quality were generally high (Additional file 1: Table S2). Conducting replicate molecular dynamics simulations and examining the resulting ‘central’ structures (see Methods) further improved the quality of inferred structural models (Additional file 1: Table S2).
We ran molecular dynamics simulations for 11 ns, excluding the first nanosecond as ‘burnin’ (see Methods). All simulations of RDs bound to RNA appear to have reached stationarity following burnin (Additional file 1: Figures S11–S13). Energy, temperature and pressure parameter values fluctuated randomly around a relatively stable average (Additional file 1: Figure S11), as did the volume and density of the simulated solvent box (Additional file 1: Figure S12). The radius of gyration, root mean squared deviations and minimum atom-atom distances also exhibited little directional trend following burnin—although some simulations did exhibit directional trends during the burnin period—and the minimum atom-atom distance never fell below 2 nm (Additional file 1: Figure S13). Molecular dynamics simulations of combined helicase + pincer + RD domains bound to RNA exhibited similar evidence for stationarity (Additional file 1: Figures S14–S16). Best-fit linear regressions to each simulation time course series exhibited little deviation from zero slope. The average absolute-value of best-fit slope was 4.4e−3, the median was 2.1e−5 and the largest inferred slope was 7.6e−2 in absolute value.
Although it is not possible to completely rule out potential errors in ancestral sequence reconstruction, structural modeling or dynamics simulations, we did not observe any compelling evidence to suggest high potential for errors in these inferences. As a whole, our results suggest that the RLRs functionally diversified via gene duplications in bilateria and jawed vertebrates, associated with protein-coding adaptation specifically targeting residues in the RD likely affecting RNA binding by altering hydrogen bond networks.
RIG-like receptors (RLRs) repeatedly lost and gained affinity for 5′-triphosphate (5′ppp) double-stranded RNA (dsRNA)
To examine the evolution of RNA preference across the early RLR phylogeny experimentally, we measured the affinity with which each ancestral RLR RD bound double-stranded RNAs (dsRNAs) having either a blunt end or a 5′-triphosphate (5′ppp) moiety (see Methods). These particular RNA types were chosen due to their association with viral infections [56, 74–79], observed differences in blunt-ended vs. 5′ppp dsRNA preference among human and ancestral RLRs [46, 52, 53, 80–83] and availability of crystal structures of human RIG-I bound to both RNAs to support structural modeling and molecular dynamics [51, 52, 84]. While we are aware that these two RNA types may not represent the primary drivers of historical RLR evolution, they do provide an important starting point from which to begin examining the evolution of RLR-RNA interactions (see Conclusions).
We observed a dramatic shift in RNA preference between the ancestral RLR (ancRLR) and the first ancestral MDA5/LGP2 after the bilaterian gene duplication (ancMDA5/LGP2a), followed by an immediate reversion back to the ancestral binding preference along the ancestral MDA5/LGP2 lineage (ancMDA5/LGP2b; Fig. 3; Additional file 1: Figures S10B, S17–S19). Consistent with our previous results [46], the ancestral RLR RD had equal preference for blunt-ended and 5′ppp dsRNA (p = 0.09 for steady-state affinities; p = 0.33 for initial binding rates), whereas ancMDA5/LGP2a RD evolved ~20-fold reduced affinity for 5′ppp dsRNA (25.8-fold decrease in steady-state affinity, p = 0.04; 18-fold decrease in initial binding rate, p = 0.014). The ~3-4-fold increase in affinity for blunt-ended dsRNA we observed along this branch was not significant (p > 0.15). In contrast, the RNA binding affinities of ancRIG-I RD were much more similar to those of the ancestral RLR and human RIG-I RD (p > 0.03; Additional file 1: Figures S17 and S18).
Surprisingly, we found that the RD of ancMDA5/LGP2b bound blunt-ended and 5′ppp dsRNAs with the same affinity, similar to what we observed for ancRLR RD and RDs in the RIG-I lineage (p > 0.64). This was caused by an observed ~20-fold increase in ancMDA5/LGP2b RD’s affinity for 5′ppp dsRNA, compared to the RD of ancMDA5/LGP2a (21-fold increase in steady state affinity, p = 0.04; 18.5-fold increase in initial binding rate, p = 0.01). Binding to both 5′ppp and blunt-ended RNA types by ancMDA5/LGP2b RD were equivalent to that of ancRLR RD, suggesting an evolutionary reversion to an ancestral function (p > 0.08 and p > 0.73, respectively). Ancestral RDs resurrected immediately following the MDA5-LGP2 duplication also bound blunt-ended and 5′ppp dsRNAs with equivalent affinities (p > 0.7), arguing against sequence reconstruction errors in ancMDA5/LGP2b as a major explanation of our results (Fig. 3; Additional file 1: Figures S17 and S18). Ancestral sequences reconstructed using an alternative sequence alignment exhibited the same evolutionary shifts in RNA preference, further supporting the robustness of our findings to ancestral reconstruction uncertainty (Additional file 1: Figure S10B).
Human LGP2 RD also bound both RNA types with equal affinity (p > 0.24), whereas human MDA5 RD appears to have re-evolved a preference for blunt-ended over 5′ppp dsRNA (24.7-fold higher steady-state preference for blunt-ended dsRNA, p = 0.03; 45.3-fold higher initial binding rate, p = 0.006), similar to what we observed for the RD of ancMDA5/LGP2a (Additional file 1: Figures S17 and S18). Previous studies have established that human LGP2 RD binds RNA molecules using structural mechanisms distinct from those of human RIG-I RD, despite having similar RNA preference [51, 61, 69, 85]. Our results suggest that the similarity in RNA preference between human LGP2 and RIG-I RDs originated prior to the MDA5-LGP2 duplication (in ancMDA5/LGP2b), although it did evolve from a (functionally) MDA5-like ancestor (ancMDA5/LGP2a). Our finding that human MDA5 prefers blunt-ended dsRNA over 5′ppp dsRNA is also consistent with recent analyses of chicken MDA5 [59].
Previous studies have shown that—in addition to the C-terminal RD—RLR helicase + pincer domains contribute to RNA binding and may affect RNA preference [52, 60, 61, 85]. To determine whether changes in ancestral RLR helicase + pincer contributed to the evolution of RNA preference in early animal RLRs, we compared the RNA preferences of ancestral RLR constructs encoding the complete helicase + pincer + RD domains to those of ancestral RDs, alone. We found that ancestral helicase + pincer + RDs exhibited the same loss of 5′ppp dsRNA affinity along the branch leading from ancRLR to ancMDA5/LGP2a, as well as the same reversion back to equal blunt-vs-5′ppp dsRNA affinities in ancMDA5/LGP2b (Fig. 3; Additional file 1: Figures S17–S19). Specifically, we observed a ~50-fold decrease in ancMDA5/LGP2a helicase + pincer + RD’s affinity for 5′ppp dsRNA, compared to ancRLR (p < 0.0002). AncMDA5/LGP2b helicase + pincer + RD displayed equal preference for 5′ppp and blunt-ended dsRNA (p > 0.58), similar to ancRLR (p > 0.42).
These results indicate that the binding shift we observed for ancestral RDs is not an artifact of measuring RD-RNA binding in the absence of other protein functional domains and suggest that changes in the helicase + pincer domain likely contributed little to the early evolution of RLR-RNA preference, at least for these two RNA types. However, later changes in the helicase + pincer may have impacted RLR-RNA binding of more recently-derived receptors. We did not examine the impact of ancestral RLR CARD domains on RNA binding, as RLR CARD sequences were too divergent to reliably reconstruct ancestral CARDs on these deep nodes of the phylogeny, and CARDs are not considered to strongly impact RNA binding in extant receptors [54, 86, 87].
Our examination of RNA preference across the early RLR phylogeny paints a general picture in which RLRs repeatedly gain and lose affinity for 5′ppp dsRNA, first by losing ancestral high-affinity after the first RLR duplication in deuterostomes, then by immediately re-gaining 5′ppp dsRNA affinity along the MDA5/LGP2 lineage and finally losing it again sometime after MDA5 diverged from LGP2 in jawed vertebrates. In contrast to what has been observed for proteins involved in metabolic and developmental processes, in which major changes in ligand preference generally occur following gene duplication or speciation events and are fairly conserved across large evolutionary distances [8, 13, 16, 23, 31, 88, 89], our results suggest that the evolution of ligand preference in immune receptors can occur much more rapidly, independently of gene duplications, and may repeatedly ‘flip flop’ between two or more functional states. In general, we would expect the strong and variable selection pressures exerted on immune receptors by fast-evolving pathogens to result in more rapid and variable evolution of receptor function, and our results are consistent with this expectation.
A complex substitution in the RNA binding loop reduced affinity for 5′ppp dsRNA
Integrating results from analysis of protein-coding adaptation (Figs. 1 and 2) and molecular dynamics (Figs. 3, 4 and 5), we hypothesized that two historical substitutions were primarily responsible for the observed loss of 5′ppp dsRNA binding between ancRLR and ancMDA5/LGP2a: a complex ΔEK47TEE substitution within the canonical RNA-binding loop and a second K88S substitution within the β9-α3 transition (Figs. 2 and 5). When we introduced these mutations into the ancRLR background (ancRLR∆EK47TEE,K88S), binding to 5′ppp dsRNA was reduced, similar to what we observed in ancMDA5/LGP2a (7.8-fold reduction in steady-state binding, p = 0.0008; 6.7-fold reduction in initial binding rate, p = 0.003; Fig. 6; Additional file 1: Figures S17 and S18), but binding to blunt-ended dsRNA was retained (p > 0.18). These results indicate that the combined ΔEK47TEE and K88S substitutions are sufficient to recapitulate the observed functional shift between ancRLR and ancMDA5/LGP2a. Introducing other historical substitutions around the RNA-binding pocket into the ancRLR background did not shift ancRLR’s RNA preference (p > 0.17; Additional file 1: Figure S17), suggesting that ΔEK47TEE and K88S played a specific role in reducing 5′ppp dsRNA binding during early RLR evolution.
Fig. 6.
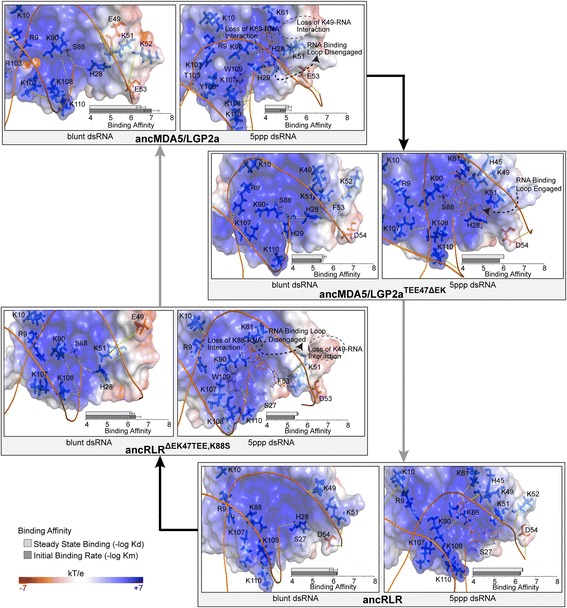
Two complex substitutions are sufficient to recapitulate the observed shift in RNA-binding preference following the earliest RLR gene duplication. We reconstructed ancestral RD protein sequences before and after the first RLR gene duplication (see Fig. 1), constructed structural models of RDs bound to blunt-ended and 5′ppp dsRNAs and performed replicate molecular dynamics simulations to characterize the structural basis for RNA binding (see Methods). We introduced the historical ΔEK47TEE and K88S substitutions into the ancRLR background as well as the ‘reverse’ TEE47ΔEK substitution into the ancMDA5/LGP2a background. We show the central structures of each RD-RNA interaction from replicate molecular dynamics simulations, with electrostatic potential (kT/e) displayed on the molecular surface. Residues forming hydrogen bonds to the RNA molecule in at least 50 % of sampled time points from molecular dynamics simulations (see Additional file 1: Figures S19 and S20) are shown as sticks, with dashed yellow lines indicating hydrogen bonds. We measured steady-state (Kd) and initial (Km) RNA-binding affinities of ancestral and mutant RDs bound to each RNA type (see Methods). We plot –log-transformed binding affinities, with longer bars indicating tighter affinity. Standard errors over three replicates are indicated
Hydrogen bond networks are expected to be important for stabilizing protein-RNA interactions. Consistent with this expectation, we found that all ionizable residues potentially contacting RNA ligands were heavily protonated during molecular dynamics simulations (Additional file 1: Figure S20). Molecular dynamics simulations support the general conclusion that the ΔEK47TEE and K88S substitutions disrupted hydrogen bonding between the ancestral RLR RD and 5′ppp dsRNA (Additional file 1: Figure S21). Hydrogen bonding was reduced in the ancRLR∆EK47TEE,K88S mutant, compared to the ancestral RLR, both to the 5′ppp dsRNA as a whole (p = 0.03) and to the 5′ppp moiety in particular (p = 0.02). We observed no differences in hydrogen bonding to 5′ppp dsRNA between the ancRLR∆EK47TEE,K88S mutant and ancMDA5/LGP2a (p > 0.13), and no overall differences in hydrogen bonding to blunt-ended dsRNA were observed among any of the ancestral or mutant RDs (p > 0.23). These results suggest that the ancRLR∆EK47TEE,K88S mutant is sufficient to recapitulate not only the observed functional shift in RNA preference but also the changes in hydrogen bond networks expected to be responsible for this functional shift.
Structural modeling and molecular dynamics suggest that the ΔEK47TEE substitution introduced an electrostatic clash between the canonical ‘RNA-binding loop’ and the large 5′ppp moiety, disrupting a number of favorable protein-5′pppRNA contacts but having minimal effect on interactions between the protein and blunt-ended dsRNA (Fig. 6). The K49E substitution directly replaces a strong polar contact between the protein and the 5′ppp with a repulsive acidic residue; K49 formed hydrogen bonds with the 5′ppp moiety in 56 % of the sampled time points during molecular dynamics simulations of ancRLR, whereas the corresponding 49E residue did not form any hydrogen bonds in either ancMDA5/LGP2a or the mutant ancRLR∆EK47TEE,K88S (p < 0.002; Fig. 5; Additional file 1: Figure S22). The Δ47T insertion is expected to exert some force to reposition the acidic E48 closer to the bulky 5′ppp, further interfering with its ability to engage the ‘RNA-binding loop’ (Fig. 6).
The ancestral K88 formed hydrogen bonds with the 5′ppp moiety in 47 % of the molecular dynamics samples from ancRLR, whereas hydrogen bonds were formed between K88 and blunt-ended dsRNA in only 2 % of ancRLR dynamics simulation samples (p = 0.02; Additional file 1: Figure S20). In the case of ancMDA5/LGP2a, the corresponding 88S residue formed hydrogen bonds with 5′ppp 13 % of the time and hydrogen bonds with blunt-ended dsRNA 33 % of the time (p = 0.17; Fig. 5; Additional file 1: Figure S22). The ancRLR∆EK47TEE,K88S mutant recapitulated this loss of 5′ppp-specific hydrogen bonding (p = 7.1e−5; Additional file 1: Figure S22), suggesting that the K88S substitution specifically stabilizes blunt dsRNA.
The combined effect of these functional substitutions was to shift the orientation of the 5′ppp dsRNA in the ligand-binding pocket away from its ancestral orientation with the 5′ppp moiety engaged in the RNA-binding loop (Fig. 6) and into an orientation more similar to that of the blunt-ended dsRNA (Fig. 6). In this new orientation, the bulky 5′ppp moiety clashes with the ‘acidified RNA-binding loop’ and is not stabilized by 88S (Fig. 6). The blunt-ended dsRNA lacks a bulky 5′ppp, reducing its clash with the ‘acidified RNA-binding loop’ and allowing 88S to stabilize the sugar backbone of the 5′ terminal base (Fig. 6).
Previous studies of slow-evolving receptors have revealed instances in which, after a functional shift, subsequent “restrictive” substitutions can occur that prevent the direct reversal of receptor preference [9, 90–92]. However, when we introduced the “reverse” TEE47∆EK substitution into the derived ancMDA5/LGP2a (ancMDA5/LGP2aTEE47∆EK), we observed a complete reversion to the ancestral ancRLR function, in which blunt-ended and 5′ppp dsRNAs are bound with equal affinity (p > 0.13; Fig. 6; Additional file 1: Figures S17 and S18). The “reverse” ancMDA5/LGP2aTEE47∆EK mutant was also sufficient to revert overall hydrogen bonding to an ‘ancestral’ condition (Fig. 6; Additional file 1: Figures S21 and S22). The number of hydrogen bonds formed over molecular dynamics simulations was indistinguishable between ancRLR and ancMDA5/LGP2aTEE47∆EK, both to the RNA as a whole (p > 0.21) and to the 5′ppp moiety in particular (p = 0.13). Additionally, the 5′ppp dsRNA adopted the ancestral RLR conformation, with the 5′ppp moiety engaged in the RNA-binding loop (Fig. 6). These results suggest that the TEE47∆EK substitution may be primarily responsible for the differences in 5′ppp binding between ancRLR and ancMDA5/LGP2a, and that ‘restrictive’ substitutions required to optimize shifts in receptor function may be less likely in fast-evolving immune receptors than in more slowly-evolving systems.
In addition to changes in the hydrogen bond network, we also observed some significant differences in sidechain and RD backbone flexibility (Additional file 1: Figure S23), the distance between the RD and its bound ligand (Additional file 1: Figure S24), and protein secondary structure (Additional file 1: Figure S25) across molecular dynamics simulations of different ancestral and mutant RDs. These changes in other molecular-dynamics properties were largely consistent with inferred changes in hydrogen bonding networks, which appear sufficient to explain the observed differences in molecular binding kinetics.
A single amino-acid substitution restored affinity for 5′ppp dsRNA
Between the origin of ancMDA5/LGP2a and the MDA5-LGP2 duplication in early vertebrates, we observed a functional reversion back to ancRLR’s equal preference for blunt-ended and 5′ppp dsRNAs, even though the ‘RNA binding loop’ remained largely acidic (ancMDA5/LGP2b; Fig. 3). Combining information about protein-coding adaptation (Figs. 1 and 2) and molecular dynamics (Figs. 3, 4 and 5) suggested that a single H63S substitution may have contributed to the observed reversion in binding preference (see Fig. 2). To test this hypothesis, we measured changes in RNA preference caused by the single historical H63S substitution in the ancMDA5/LGP2a background (ancMDA5/LGP2aH63S). We found that this single substitution was sufficient to revert ancMDA5/LGP2a’s function to the ancestral-like pattern of equal binding to blunt-ended and 5′ppp dsRNA observed in ancMDA5/LGP2b (Fig. 7; Additional file 1: Figures S17 and S18). The H63S substitution increased affinity for 5′ppp dsRNA by ~20-fold (p < 0.02), while affinity for blunt-ended dsRNA remained unchanged (p > 0.06). RNA binding by ancMDA5/LGP2aH63S was indistinguishable from that of ancMDA5/LGP2b (p > 0.28), suggesting that the single substitution was sufficient to recapitulate the observed functional shift from ancMDA5/LGP2a to ancMDA5/LGP2b.
Fig. 7.
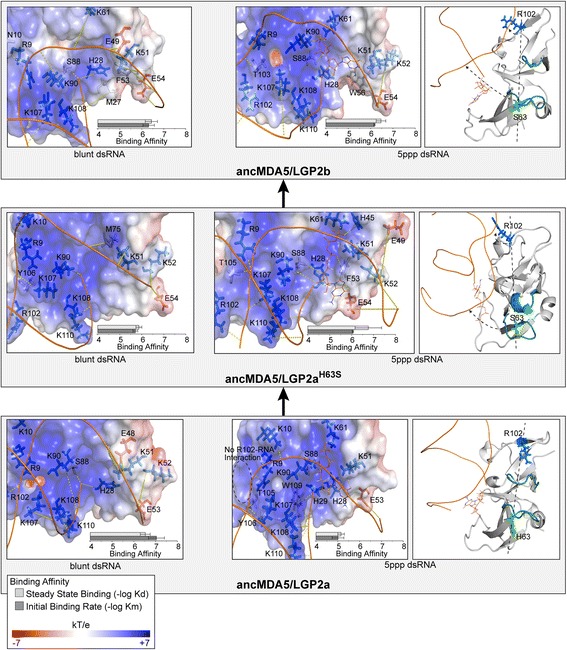
A single amino acid substitution is sufficient to recapitulate the observed re-evolution of high-affinity 5′ppp dsRNA binding between ancMDA5/LGP2a and ancMDA5/LGP2b. We reconstructed RD protein sequences of the first (ancMDA5/LGP2a) and last (ancMDA5/LGP2b) ancestral RLRs between the first and second major RLR gene duplications (see Fig. 1). We additionally introduced a single historical H63S substitution into the ancMDA5/LGP2a background. We show the central structures from replicate molecular dynamics simulations of each RD bound to blunt-ended and 5′ppp dsRNA (see Methods). Electrostatic potential (kT/e) is displayed across the molecular surface of each RNA-binding pocket (left panels). Residues forming hydrogen bonds to the RNA in at least 50 % of molecular dynamics time points (see Additional file 1: Figures S24 and S25) are shown as sticks, with dashed yellow lines indicating hydrogen bonds. We plot –log-transformed steady-state (Kd) and initial (Km) RD-RNA binding rates, with bars indicating standard errors. Right panels show each RD bound to 5′ppp dsRNA. A dotted line connects the H/S63 (green) and R102 (blue) residues and the zinc-binding pocket (teal)
Indroducing the H63S mutation into the ancMDA5/LGP2a background produced a modest increase in the average number of hydrogen bonds observed over molecular dynamics simulations between the protein and the 5′ppp dsRNA ligand as a whole (p = 0.04) as well as to the 5′ppp moiety in particular (p = 0.005; Additional file 1: Figure S26). No differences in overall protein-RNA hydrogen bonding were observed between the ancMDA5/LGP2aH63S mutant and ancMDA5/LGP2b (p > 0.09), suggesting that the single H63S substitution is sufficient to shift the overall hydrogen bond network from the ancestral ancMDA5/LGP2a to the derived ancMDA5/LGP2b conformation.
We observed a single residue, R102, which dramatically increased its capacity to form hydrogen bonds with the RNA ligand in molecular dynamics simulations, from forming no observed hydrogen bonds in ancMDA5/LGP2a to forming favorable protein-RNA contacts in over 50 % of sampled timepoints in ancMDA5/LGP2b and the ancMDA5/LGP2aH63S mutant (p < 0.02; Additional file 1: Figure S27). R102 is located at the end of β11 in ancMDA5/LGP2a, which is disordered in ancMDA5/LGP2b and the ancMDA5/LGP2aH63S mutant and is far away from the H63S substitution (Fig. 7).
Examination of the central structures from molecular dynamics simulations revealed that the substitution of the small serine residue for the bulky histidine at position 63 introduced space into the region of the RNA-binding pocket between the ‘acidified RNA-binding loop’ and the Zn-binding finger, allowing the Zn finger to shift away from the RNA ligand. This allowed the RNA to move closer to the protein and rotate in the binding pocket to engage R102, which is flipped up in both ancMDA5/LGP2b and the ancMDA5/LGP2aH63S mutant, compared to the ancestral MDA5/LGP2a (Fig. 7). As in our analysis of the ancRLR—ancMDA5/LGP2a transition, changes in a number of molecular-dynamics properties were consistent with a primary role for alteration of hydrogen bonding networks in explaining the differences in RNA preference between ancMDA5/LGP2a and ancMDA5/LGP2b (Additional file 1: Figures S28–S30).
Overall, these results reveal how the introduction of a single ‘destabilizing’ H63S substitution had a ‘ripple effect’ through the RD structure, changing the way existing residues far away from the substitution interact with the large RNA ligand. They also reveal that RLR RD evolution has exploited multiple, structurally-distinct evolutionary trajectories to implement and re-implement similar RNA-binding functions.
Conclusions
Ancestral sequence resurrection (ASR) has allowed researchers to experimentally evaluate hypotheses about how the structures and functions of biological molecules evolve, deepening our understanding of important biological systems and informing molecular-evolutionary theory [5, 23, 37, 93]. To date, ASR studies have highlighted potentially strong epistasis among substitutions and historical contingency as major features of protein evolution [9, 10, 17, 94, 95] and have supported a model in which ancestral ‘promiscuity’ gives rise to increasing specialization as proteins ‘optimize’ specific functions over evolutionary time [39, 96, 97]. Major changes in protein function are generally observed to occur following gene duplication events [98–100] or speciation events [96, 101–103] and appear to be preserved over long periods of time. However, all such studies have examined molecular systems involved in conserved metabolic or developmental processes that we expect to be under relatively stable selection pressures. It is unclear whether the results of these studies generalize to fast-evolving immune receptors, which are expected to experience strong and frequent shifts in selection pressures.
Our study is the first to examine the structural mechanisms underlying long-term functional evolution in a fast-evolving primary immune receptor. In contrast to previous studies of slow-evolving proteins, we found no evidence for strong epistasis among substitutions, historical contingency or long-term ‘functional optimization.’ Rather, the repeated functional ‘flip flopping’ we observed was primarily caused by small numbers of substitutions that clustered in and around the ligand-binding pocket and exerted functional effects by reorganizing local structural dynamics. We also observed little evidence for strong structural constraints on RLR evolution, with similar functions evolving and re-evolving through novel structural mechanisms.
This study focused on two model RNA ligands that have been associated with viral infections [56, 74–79] and exhibit differential binding among RLRs [53, 80–83]. We observed changes in RLR binding to these model RNAs across evolutionary history, and the observed functional changes were associated with adaptive protein-coding substitutions. However, the functional shifts we observed could also be side-effects of adaptation primarily targeting other functions not examined in our study, including binding to other ligands or viral antagonization.
Unlike studies of ligand-binding proteins involved in more stable processes, the exact ligands recognized by modern human RLRs are still unclear [83, 104]. The dynamic nature of host-pathogen molecular interactions may result in potentially radical changes over time in the specific pathogen-associated molecules recognized by host immune receptors. Viral RNA sequences can change extremely rapidly, and although structural analyses and early functional studies have suggested that RLR-RNA interactions are not strongly affected by sequence variation [52, 84], changes in viral RNA sequences might impact RLR immune signaling under some circumstances [105–107]. Many viruses biochemically ‘hide’ their RNAs to avoid host detection [108–110], and viruses are known to antagonize various aspects of the RLR system [111–113]. Unfortunately, we know almost nothing about the ecologically important viruses that may have plagued ancient animals, so determining the precise native ligands that may have driven the evolution of RLRs is not possible. At this point, we must content ourselves with characterizing how interactions between RLRs and model biochemical ligands evolved. Although these studies can shed light on the structural and molecular-functional evolution of immune receptors, it is not possible to confidently infer specific properties of ancient viruses based on limited analyses of receptor-RNA interactions.
While our study has focused on the evolution of receptor-ligand binding affinity, factors other than ligand affinity affect how receptors ultimately function in immune signaling, including how receptors interact with themselves [114–117] with cofactors [48, 118, 119] and with signaling adaptors [47, 48, 120]. Making sense of the functional evolution of RLRs and other immune receptors will ultimately require considering the full breadth of molecular interactions determining a receptor’s cellular function.
While speculative, we feel our results may reflect general differences in the evolutionary dynamics of immune receptors, compared to those of proteins involved in more stable biological processes. Because the functional requirements of pathogen recognition are likely to change rapidly, immune receptors may exist in constant non-equilibrium, unable to optimize a specific functional repertoire. In contrast, proteins evolving toward a more stable evolutionary goal have more time to optimize specific functions. This general model would predict that immune receptors should exhibit less epistasis among substitutions overall, and extant receptors should be less optimally tuned to their functions, compared to slow-evolving counterparts. Determining the validity of this general model will require characterizing the molecular-functional evolution of RLRs across a broader range of ligands and over a larger slice of evolutionary history. Testing generalizability will also require mechanistic investigations of the evolution of other immune receptors. Characterizing a large number of receptor-ligand systems is expected to provide important clues about the specific aspects of pathogen-receptor interactions of primary importance in evolutionary history, advancing our understanding of host-pathogen interactions in particular as well as molecular-evolutionary theory in general.
Methods
Sequence data and gene family phylogeny
RIG-like receptor (RLR) protein sequences were retrieved from the NCBI Reference Sequence database [121] using DELTA-BLAST [122] with an e-value cutoff of 1.0e−5. We used the RNA recognition domains (RDs) from human RIG-I, MDA5, LGP2 and our previously reconstructed ancestral RLR RD [46] as BLAST queries, and merged the results into a single RLR dataset. We confirmed the presence of a DEAD/Helicase domain in each sequence by RPS-BLAST against the NCBI Conserved Domain Database [123], using an e-value cutoff of 0.01.
Sequences were aligned using PROBALIGN v1.4 (default parameters [124]) and MAFFT v7.158b (einsi parameters [125]). Initial maximum likelihood trees were built from each alignment using FastTree v2.1.7 (default parameters [126]) and further refined using the “BEST” topology search heuristic in PhyML v20140606 [127], with the best-fit evolutionary model inferred by AIC using ProtTest v3.4 [128]. We inferred SH-like aLRT clade support using PhyML [129]. Ancestral sequences were inferred by marginal maximum likelihood reconstruction using RAxML v8.0.25 [130], with insertions and deletions reconstructed by maximum likelihood from the presence-absence alignment matrix. Support for each ancestral state was assessed by calculating posterior probabilities [131], and ancestral sequences were compared to our previously reconstructed ancestral RLRs [46].
Protein-coding adaptation was assessed using the branch-sites model implemented in PAML v4.7 [132, 133], which uses a mixture distribution to model a combination of negatively-selected, neutral and positively-selected positions in the protein sequence [66]. For each branch on the phylogeny, we tested the hypothesis that some sites experienced adaptive protein-coding substitutions against the null hypothesis of neutral evolution using a likelihood ratio test. P values were calculated using the χ 2 distribution [66], and we used a Bonferroni correction for multiple testing.
Structural modeling and molecular dynamics
We constructed structural models of RLR RDs using MODELLER v9.13 [134]. The structures of human RIG1 RD bound to blunt-ended double-stranded RNA (PDB ID: 3OG8 [51]) and 5′-triphosphate dsRNA (PDB ID: 3LRN [84]) were used as templates. We constructed 100 preliminary models of each RD-RNA and scored each model using the MODELLER objective function (molpdf), DOPE score and DOPEHR [135]. Each score was re-scaled to units of standard-deviation across the 100 models [136], and we selected the best structural model as that with the best average of re-scaled molpdf, DOPE and DOPEHR scores. Spatial and thermodynamic quality scores were assigned to each structural model using QMean [137], DFIRE [138] and Procheck [139]. Structural models of ancestral RLR helicase + pincer + RD domains bound to blunt dsRNA (PDB ID: 5F9F) and 5′ppp dsRNA (PDB ID: 5F9H) were constructed using the same approach [52].
Structural models were processed for electrostatic surface visualization using PROPKA v3.1 and PDB2PQR v1.7 to determine residue side-chain pKas, optimize the structure for favorable hydrogen bonding and calculate charge and radius parameters from amber electrostatic force fields [140, 141]. These calculations were performed using protein-RNA complexes to ensure proper side-chain orientation and protonation in the presence of ligand. For visualization, we estimated electrostatic surface potentials in the absence of RNA ligands using APBS v1.4.1 [142] and projected them onto the protein’s molecular surface.
For each RLR structure bound to each RNA type, we ran 4 replicate molecular dynamics simulations using GROMACS v4.6.5 [143]. Dynamics simulations of RLR RDs bound to RNA contained between 22,056 and 64,408 water molecules, 43–146 sodium and 44–120 chloride ions. Simulations of helicase + pincer + RD domains bound to RNA contained 32,906–35,871 waters, 67–83 sodium ions and 68–73 chloride ions. We used the amber99sb-ildn force field [144] and the tip3p water model. Initial dynamics topologies were generated for each RLR-RNA complex using the GROMACS pdb2gmx algorithm with default parameters. Topologies were relaxed into simulated solvent at pH = 7 using a 50,000-step steepest-descent energy minimization. The system was then brought to 300K using a 50-ps dynamics simulation under positional restraints, followed by pressure stabilization for an additional 50 ps. Unconstrained molecular dynamics were run for 11 ns using a 0.002-ps integration time step, with the system sampled every 5 ps. Simulations were run using Particle-Mesh Ewald electrostatics with cubic interpolation and grid spacing of 0.12 nm. Van der Waals forces were calculated using a cutoff of 1.0 nm. We used Nose-Hoover temperature coupling, with protein, RNA and solvent systems coupled separately and the period of temperature fluctuations set to 0.1 ps. Pressure coupling was applied using the Parrinello-Rahman approach, with a fluctuation period of 2.0 ps. Non-bonded cutoffs were treated using buffered Verlet lists. We discarded the first 1 ns of each simulation.
From each dynamics simulation, we inferred the central structure by calculating pairwise root mean square deviations (RMSDs) between every pair of simulation samples and identifying the sampled structure most equidistant to the others, using the g_cluster function in GROMACS. We measured the root mean square fluctuation (RMSF) of each residue’s side-chain and backbone over the dynamics simulation. We additionally calculated a number of biochemical properties from each sample taken from each molecular dynamics simulation. Secondary structure was calculated using DSSP v2.2.1 [145], and we report the proportion of samples from which each residue was a member of a helix, strand or loop. We estimated a consensus secondary structure over the course of each simulation by assigning each residue to helix or strand if it was assigned to that secondary structure by DSSP in >50 % of samples; we required at least 3 consecutive residues to infer consensus helices and strands. We inferred hydrogen bonds using a radius cutoff of 0.3 nm and an angle cutoff of 20 ° and report, for each residue, the proportion of simulation samples from which that residue forms a hydrogen bond with the RNA molecule or the 5′ppp moiety. We calculated the minimum distance between each residue and the RNA molecule over the course of each dynamics simulation. We considered a residue as potentially contributing to RNA contact if its average minimum distance across the entire dynamics simulation was significantly <4 angstroms.
Significance of differences in each biochemical property inferred by structural dynamics was assessed using the 2-tailed, 2-sample independent t-test, assuming unequal variances [146]. We corrected for multiple testing using a false discovery rate correction [147].
Molecular binding kinetics
We generated GC-rich 29-base-pair RNA molecules in vitro using T7 RNA reverse transcriptase and synthetic dsDNA as template (3′-GAAAGAGGUGCGGAAAGAGGUAGAGGAGG-5′). Complementary purified single-stranded RNAs were annealed to produce double-stranded RNA by combining at 1:1 ratio, heating to 95 °C for 5 min and then cooling to 25 °C. Blunt-ended dsRNA was produced from 5′-triphosphate RNA by exposure to alkaline phosphatase or synthesized de novo by IDT (Iowa, USA). The 3′ end of one RNA strand was biotinylated to facilitate kinetics assays using the Pierce™ 3′ End RNA Biotinylation Kit (Thermo).
RLR RDs were expressed in E. coli Rosetta™ 2(DE3)pLysS cells using pET-22b(+) constructs, which were verified by Sanger sequencing. Proteins were purified by His-affinity purification and visualized by SDS-page stained with 1 % coomassie. Protein concentrations were measured using a linear-transformed Bradford assay [148].
We measured RD-RNA binding using a label-free in vitro kinetics assay at pH = 7 [149]. Biotinylated RNA molecules were bound to a series of 8 streptavidin probes for 15 min, until saturation was observed. Probes were washed and then exposed to 25 μg/ml biocytin to bind any remaining free streptavidin. The probes were then exposed in parallel to RDs at various concentrations in 1 × Kinetics Buffer (ForteBio) for 30 min, followed by dissociation in Kinetics Buffer for an additional 30 min. Molecular binding at each concentration over time was measured as the change in laser wavelength when reflected through the probe in solution, sampled every 3 ms.
For each replicate experiment, we estimated the RD concentration at which ½-maximal steady-state RNA binding was achieved (Kd) by fitting a one-site binding curve to the steady-state laser wavelengths measured across RD concentrations at saturation (60 min), using nonlinear regression. We additionally fit 1-site association/dissociation curves to the full time-course data in order to estimate the initial rates of RNA binding across RD concentrations and used these rates to calculate the RD concentration at which the ½-maximal RNA-binding rate was achieved (Km). Kds and Kms were negative-log transformed to facilitate visualization, and standard errors across 3 experimental replicates were calculated. We calculated the statistical significance of differences between Kds and Kms using the 2-tailed, 2-sample independent t-test, assuming unequal variances [146].
Acknowledgments
This research was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (grant number R21AI101571), the United States Department of Agriculture, National Institute of Food and Agriculture (grant number FLA-MCS-005262) and by the University of Florida. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the US Department of Agriculture or the University of Florida.
Availability of data and materials
All data not obtained from public databases or presented in the main text is available in Additional file 1.
Authors’ contributions
CP synthesized RNA, carried out binding kinetics assays, performed sequence alignments and phylogenetic analyses, conducted molecular dynamics simulations and drafted the manuscript. OK constructed and verified expression plasmids, purified protein and performed kinetics assays. AM performed structural modeling and molecular dynamics simulations. BKorithoski participated in the design of the study and conducted binding kinetics assays. BKolaczkowski conceived of the study, participated in its design, conducted ancestral sequence reconstructions and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
This study does not involve human subject data and therefore does not require informed consent to publish.
Ethics approval and consent to participate
This study was approved by the University of Florida Institutional Review Board. This study does not involve human subjects or data, beyond publicly available gene sequence data.
Abbreviations
- 5′ppp
Five-prime triphosphate
- ASR
Ancestral sequence resurrection
- dsRNA
Double-stranded RNA
- RD
RNA recognition domain
- RLR
RIG-like receptor
Additional file
Supplementary figures and tables. (PDF 9731 kb)
Contributor Information
Charles Pugh, Email: cpugh1355@ufl.edu.
Oralia Kolaczkowski, Email: oraliak@ufl.edu.
Austin Manny, Email: austin.manny@ufl.edu.
Bryan Korithoski, Email: bkorithoski@ufl.edu.
Bryan Kolaczkowski, Email: bryank@ufl.edu.
References
- 1.Dean AM, Thornton JW. Mechanistic approaches to the study of evolution: the functional synthesis. Nat Rev Genet. 2007;8(9):675–688. doi: 10.1038/nrg2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harms MJ, Thornton JW. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat Rev Genet. 2013;14(8):559–571. doi: 10.1038/nrg3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harms MJ, Thornton JW. Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature. 2014;512(7513):203–207. doi: 10.1038/nature13410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bridgham JT, Keay J, Ortlund EA, Thornton JW. Vestigialization of an allosteric switch: genetic and structural mechanisms for the evolution of constitutive activity in a steroid hormone receptor. PLoS Genet. 2014;10(1):e1004058. doi: 10.1371/journal.pgen.1004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harms MJ, Eick GN, Goswami D, Colucci JK, Griffin PR, Ortlund EA, Thornton JW. Biophysical mechanisms for large-effect mutations in the evolution of steroid hormone receptors. Proc Natl Acad Sci U S A. 2013;110(28):11475–11480. doi: 10.1073/pnas.1303930110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eick GN, Colucci JK, Harms MJ, Ortlund EA, Thornton JW. Evolution of minimal specificity and promiscuity in steroid hormone receptors. PLoS Genet. 2012;8(11):e1003072. doi: 10.1371/journal.pgen.1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll SM, Ortlund EA, Thornton JW. Mechanisms for the evolution of a derived function in the ancestral glucocorticoid receptor. PLoS Genet. 2011;7(6):e1002117. doi: 10.1371/journal.pgen.1002117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bridgham JT, Eick GN, Larroux C, Deshpande K, Harms MJ, Gauthier ME, Ortlund EA, Degnan BM, Thornton JW. Protein evolution by molecular tinkering: diversification of the nuclear receptor superfamily from a ligand-dependent ancestor. PLoS Biol. 2010;8(10). doi: 10.1371/journal.pbio.1000497. [DOI] [PMC free article] [PubMed]
- 9.Bridgham JT, Ortlund EA, Thornton JW. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature. 2009;461(7263):515–519. doi: 10.1038/nature08249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. Crystal structure of an ancient protein: evolution by conformational epistasis. Science. 2007;317(5844):1544–1548. doi: 10.1126/science.1142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bridgham JT, Carroll SM, Thornton JW. Evolution of hormone-receptor complexity by molecular exploitation. Science. 2006;312(5770):97–101. doi: 10.1126/science.1123348. [DOI] [PubMed] [Google Scholar]
- 12.Bloch NI, Morrow JM, Chang BS, Price TD. SWS2 visual pigment evolution as a test of historically contingent patterns of plumage color evolution in warblers. Evolution. 2015;69(2):341–356. doi: 10.1111/evo.12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voordeckers K, Brown CA, Vanneste K, van der Zande E, Voet A, Maere S, Verstrepen KJ. Reconstruction of ancestral metabolic enzymes reveals molecular mechanisms underlying evolutionary innovation through gene duplication. PLoS Biol. 2012;10(12):e1001446. doi: 10.1371/journal.pbio.1001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Hazel I, Sabouhanian A, Day L, Endler JA, Chang BS. Functional characterization of spectral tuning mechanisms in the great bowerbird short-wavelength sensitive visual pigment (SWS1), and the origins of UV/violet vision in passerines and parrots. BMC Evol Biol. 2013;13:250. doi: 10.1186/1471-2148-13-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H, Grunkemeyer TJ, Modi C, Chen L, Fromme R, Matz MV, Wachter RM. Acid-base catalysis and crystal structures of a least evolved ancestral GFP-like protein undergoing green-to-red photoconversion. Biochemistry. 2013;52(45):8048–8059. doi: 10.1021/bi401000e. [DOI] [PubMed] [Google Scholar]
- 16.Kratzer JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, Ortlund EA, Johnson RJ, Gaucher EA. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc Natl Acad Sci U S A. 2014;111(10):3763–3768. doi: 10.1073/pnas.1320393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Risso VA, Gavira JA, Mejia-Carmona DF, Gaucher EA, Sanchez-Ruiz JM. Hyperstability and substrate promiscuity in laboratory resurrections of Precambrian beta-lactamases. J Am Chem Soc. 2013;135(8):2899–2902. doi: 10.1021/ja311630a. [DOI] [PubMed] [Google Scholar]
- 18.Lunzer M, Miller SP, Felsheim R, Dean AM. The biochemical architecture of an ancient adaptive landscape. Science. 2005;310(5747):499–501. doi: 10.1126/science.1115649. [DOI] [PubMed] [Google Scholar]
- 19.Thomson JM, Gaucher EA, Burgan MF, De Kee DW, Li T, Aris JP, Benner SA. Resurrecting ancestral alcohol dehydrogenases from yeast. Nat Genet. 2005;37(6):630–635. doi: 10.1038/ng1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams SG, Harms MJ, Hall KB. Resurrection of an Urbilaterian U1A/U2B″/SNF protein. J Mol Biol. 2013;425(20):3846–3862. doi: 10.1016/j.jmb.2013.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez C, Rest JS, Kim AR, Ludwig M, Kreitman M, White K, Reinitz J. Ancestral resurrection of the Drosophila S2E enhancer reveals accessible evolutionary paths through compensatory change. Mol Biol Evol. 2014;31(4):903–916. doi: 10.1093/molbev/msu042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alderson RG, Barker D, Mitchell JB. One origin for metallo-β-lactamase activity, or two? An investigation assessing a diverse set of reconstructed ancestral sequences based on a sample of phylogenetic trees. J Mol Evol. 2014;79(3–4):117–129. doi: 10.1007/s00239-014-9639-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howard CJ, Hanson-Smith V, Kennedy KJ, Miller CJ, Lou HJ, Johnson AD, Turk BE, Holt LJ: Ancestral resurrection reveals evolutionary mechanisms of kinase plasticity. Elife. 2014;3. doi: 10.7554/eLife.04126. [DOI] [PMC free article] [PubMed]
- 24.Hobbs JK, Prentice EJ, Groussin M, Arcus VL. Reconstructed Ancestral Enzymes Impose a Fitness Cost upon Modern Bacteria Despite Exhibiting Favourable Biochemical Properties. J Mol Evol. 2015;81:110–120. doi: 10.1007/s00239-015-9697-5. [DOI] [PubMed] [Google Scholar]
- 25.Chuang C, Prasanth KR, Nagy PD. Coordinated function of cellular DEAD-box helicases in suppression of viral RNA recombination and maintenance of viral genome integrity. PLoS Pathog. 2015;11(2):e1004680. doi: 10.1371/journal.ppat.1004680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffmann HH, Schneider WM, Rice CM. Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol. 2015;36(3):124–138. doi: 10.1016/j.it.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duggal NK, Emerman M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol. 2012;12(10):687–695. doi: 10.1038/nri3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Areal H, Abrantes J, Esteves PJ. Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evol Biol. 2011;11:368. doi: 10.1186/1471-2148-11-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemos de Matos A, McFadden G, Esteves PJ. Evolution of viral sensing RIG-I-like receptor genes in Leporidae genera Oryctolagus, Sylvilagus, and Lepus. Immunogenetics. 2014;66(1):43–52. doi: 10.1007/s00251-013-0740-7. [DOI] [PubMed] [Google Scholar]
- 30.Meyerson NR, Rowley PA, Swan CH, Le DT, Wilkerson GK, Sawyer SL. Positive selection of primate genes that promote HIV-1 replication. Virology. 2014;454–455:291–298. doi: 10.1016/j.virol.2014.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuang D, Yao Y, Maclean D, Wang M, Hampson DR, Chang BS. Ancestral reconstruction of the ligand-binding pocket of Family C G protein-coupled receptors. Proc Natl Acad Sci U S A. 2006;103(38):14050–14055. doi: 10.1073/pnas.0604717103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tufts DM, Natarajan C, Revsbech IG, Projecto-Garcia J, Hoffmann FG, Weber RE, Fago A, Moriyama H, Storz JF. Epistasis constrains mutational pathways of hemoglobin adaptation in high-altitude pikas. Mol Biol Evol. 2015;32(2):287–298. doi: 10.1093/molbev/msu311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lorenzo-Redondo R, Borderia AV, Lopez-Galindez C. Dynamics of in vitro fitness recovery of HIV-1. J Virol. 2011;85(4):1861–1870. doi: 10.1128/JVI.01254-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanczyc MM, Dorit RL. Experimental evolution of complexity: in vitro emergence of intermolecular ribozyme interactions. RNA. 1998;4(3):268–275. [PMC free article] [PubMed] [Google Scholar]
- 35.Cho S, Swaminathan CP, Yang J, Kerzic MC, Guan R, Kieke MC, Kranz DM, Mariuzza RA, Sundberg EJ. Structural basis of affinity maturation and intramolecular cooperativity in a protein-protein interaction. Structure. 2005;13(12):1775–1787. doi: 10.1016/j.str.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, Depontieu FR, Sidney J, Salay TM, Engelhard VH, Hunt DF, Sette A, Topalian SL, Mariuzza RA. Structural basis for the presentation of tumor-associated MHC class II-restricted phosphopeptides to CD4+ T cells. J Mol Biol. 2010;399(4):596–603. doi: 10.1016/j.jmb.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKeown AN, Bridgham JT, Anderson DW, Murphy MN, Ortlund EA, Thornton JW. Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell. 2014;159(1):58–68. doi: 10.1016/j.cell.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finnigan GC, Hanson-Smith V, Stevens TH, Thornton JW. Evolution of increased complexity in a molecular machine. Nature. 2012;481(7381):360–364. doi: 10.1038/nature10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pougach K, Voet A, Kondrashov FA, Voordeckers K, Christiaens JF, Baying B, Benes V, Sakai R, Aerts J, Zhu B, et al. Duplication of a promiscuous transcription factor drives the emergence of a new regulatory network. Nat Commun. 2014;5:4868. doi: 10.1038/ncomms5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korithoski B, Kolaczkowski O, Mukherjee K, Kola R, Earl C, Kolaczkowski B. Evolution of a Novel Antiviral Immune-Signaling Interaction by Partial-Gene Duplication. PLoS One. 2015;10(9):e0137276. doi: 10.1371/journal.pone.0137276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lynch M, Hagner K. Evolutionary meandering of intermolecular interactions along the drift barrier. Proc Natl Acad Sci U S A. 2015;112(1):E30–E38. doi: 10.1073/pnas.1421641112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dias R, Kolazckowski B. Different combinations of atomic interactions predict protein-small molecule and protein-DNA/RNA affinities with similar accuracy. Proteins. 2015;83(11):2100–2114. doi: 10.1002/prot.24928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A. 2002;99(2):637–642. doi: 10.1073/pnas.022637199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175(8):5260–5268. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Wang C, Schook LB, Hawken RJ, Rutherford MS. An RNA helicase, RHIV −1, induced by porcine reproductive and respiratory syndrome virus (PRRSV) is mapped on porcine chromosome 10q13. Microb Pathog. 2000;28(5):267–278. doi: 10.1006/mpat.1999.0349. [DOI] [PubMed] [Google Scholar]
- 46.Mukherjee K, Korithoski B, Kolaczkowski B. Ancient origins of vertebrate-specific innate antiviral immunity. Mol Biol Evol. 2014;31(1):140–153. doi: 10.1093/molbev/mst184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 48.Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, Chen ZJ. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36(6):959–973. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marques JT, Devosse T, Wang D, Zamanian-Daryoush M, Serbinowski P, Hartmann R, Fujita T, Behlke MA, Williams BR. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat Biotechnol. 2006;24(5):559–565. doi: 10.1038/nbt1205. [DOI] [PubMed] [Google Scholar]
- 50.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23(1):19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 51.Lu C, Ranjith-Kumar CT, Hao L, Kao CC, Li P. Crystal structure of RIG-I C-terminal domain bound to blunt-ended double-strand RNA without 5′ triphosphate. Nucleic Acids Res. 2011;39(4):1565–1575. doi: 10.1093/nar/gkq974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devarkar SC, Wang C, Miller MT, Ramanathan A, Jiang F, Khan AG, Patel SS, Marcotrigiano J. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc Natl Acad Sci U S A. 2016;113(3):596–601. doi: 10.1073/pnas.1515152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 54.Jiang F, Ramanathan A, Miller MT, Tang GQ, Gale M, Patel SS, Marcotrigiano J. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479(7373):423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baum A, Sachidanandam R, García-Sastre A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A. 2010;107(37):16303–16308. doi: 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marq JB, Hausmann S, Veillard N, Kolakofsky D, Garcin D. Short double-stranded RNAs with an overhanging 5′ ppp-nucleotide, as found in arenavirus genomes, act as RIG-I decoys. J Biol Chem. 2011;286(8):6108–6116. doi: 10.1074/jbc.M110.186262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eigenbrod T, Keller P, Kaiser S, Rimbach K, Dalpke AH, Helm M. Recognition of Specified RNA Modifications by the Innate Immune System. Methods Enzymol. 2015;560:73–89. doi: 10.1016/bs.mie.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 58.Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M., Jr Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007;104(2):582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uchikawa E, Lethier M, Malet H, Brunel J, Gerlier D, Cusack S. Structural Analysis of dsRNA Binding to Anti-viral Pattern Recognition Receptors LGP2 and MDA5. Mol Cell. 2016;62(4):586–602. doi: 10.1016/j.molcel.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li X, Ranjith-Kumar CT, Brooks MT, Dharmaiah S, Herr AB, Kao C, Li P. The RIG-I-like receptor LGP2 recognizes the termini of double-stranded RNA. J Biol Chem. 2009;284(20):13881–13891. doi: 10.1074/jbc.M900818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, Lu C, Stewart M, Xu H, Strong RK, Igumenova T, Li P. Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch Biochem Biophys. 2009;488(1):23–33. doi: 10.1016/j.abb.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 62.Liberles DA, Teichmann SA, Bahar I, Bastolla U, Bloom J, Bornberg-Bauer E, Colwell LJ, de Koning AP, Dokholyan NV, Echave J, et al. The interface of protein structure, protein biophysics, and molecular evolution. Protein Sci. 2012;21(6):769–785. doi: 10.1002/pro.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gharib WH, Robinson-Rechavi M. The branch-site test of positive selection is surprisingly robust but lacks power under synonymous substitution saturation and variation in GC. Mol Biol Evol. 2013;30(7):1675–1686. doi: 10.1093/molbev/mst062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu A, Guindon S. Performance of standard and stochastic branch-site models for detecting positive selection among coding sequences. Mol Biol Evol. 2014;31(2):484–495. doi: 10.1093/molbev/mst198. [DOI] [PubMed] [Google Scholar]
- 65.Yang Z, dos Reis M. Statistical properties of the branch-site test of positive selection. Mol Biol Evol. 2011;28(3):1217–1228. doi: 10.1093/molbev/msq303. [DOI] [PubMed] [Google Scholar]
- 66.Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22(12):2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 67.Nozawa M, Suzuki Y, Nei M. Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc Natl Acad Sci U S A. 2009;106(16):6700–6705. doi: 10.1073/pnas.0901855106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suzuki Y. False-positive results obtained from the branch-site test of positive selection. Genes Genet Syst. 2008;83(4):331–338. doi: 10.1266/ggs.83.331. [DOI] [PubMed] [Google Scholar]
- 69.Takahasi K, Kumeta H, Tsuduki N, Narita R, Shigemoto T, Hirai R, Yoneyama M, Horiuchi M, Ogura K, Fujita T, et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: identification of the RNA recognition loop in RIG-I-like receptors. J Biol Chem. 2009;284(26):17465–17474. doi: 10.1074/jbc.M109.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iwakiri J, Tateishi H, Chakraborty A, Patil P, Kenmochi N. Dissecting the protein-RNA interface: the role of protein surface shapes and RNA secondary structures in protein-RNA recognition. Nucleic Acids Res. 2012;40(8):3299–3306. doi: 10.1093/nar/gkr1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Srinivasan N, Blundell TL. An evaluation of the performance of an automated procedure for comparative modelling of protein tertiary structure. Protein Eng. 1993;6(5):501–512. doi: 10.1093/protein/6.5.501. [DOI] [PubMed] [Google Scholar]
- 72.Sánchez R, Sali A. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins. 1997;29(1):50–58. [DOI] [PubMed]
- 73.Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2014;47:5.6.1–5.6.32. doi: 10.1002/0471250953.bi0506s47. [DOI] [PubMed] [Google Scholar]
- 74.Feng Q, Langereis MA, Olagnier D, Chiang C, van de Winkel R, van Essen P, Zoll J, Hiscott J, van Kuppeveld FJ. Coxsackievirus cloverleaf RNA containing a 5′ triphosphate triggers an antiviral response via RIG-I activation. PLoS One. 2014;9(4):e95927. doi: 10.1371/journal.pone.0095927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rehwinkel J, Reis e Sousa C. Targeting the viral Achilles’ heel: recognition of 5′-triphosphate RNA in innate anti-viral defence. Curr Opin Microbiol. 2013;16(4):485–492. doi: 10.1016/j.mib.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weber M, Gawanbacht A, Habjan M, Rang A, Borner C, Schmidt AM, Veitinger S, Jacob R, Devignot S, Kochs G, et al. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe. 2013;13(3):336–346. doi: 10.1016/j.chom.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abbas YM, Pichlmair A, Górna MW, Superti-Furga G, Nagar B. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature. 2013;494(7435):60–64. doi: 10.1038/nature11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nallagatla SR, Toroney R, Bevilacqua PC. A brilliant disguise for self RNA: 5′-end and internal modifications of primary transcripts suppress elements of innate immunity. RNA Biol. 2008;5(3):140–144. doi: 10.4161/rna.5.3.6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31(1):25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M, Akira S, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175(5):2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 81.Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, Jung JU, Hopfner KP, Ha T. Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science. 2009;323(5917):1070–1074. doi: 10.1126/science.1168352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Luo D, Kohlway A, Vela A, Pyle AM. Visualizing the determinants of viral RNA recognition by innate immune sensor RIG-I. Structure. 2012;20(11):1983–1988. doi: 10.1016/j.str.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bruns AM, Horvath CM. Antiviral RNA recognition and assembly by RLR family innate immune sensors. Cytokine Growth Factor Rev. 2014;25(5):507–512. doi: 10.1016/j.cytogfr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu C, Xu H, Ranjith-Kumar CT, Brooks MT, Hou TY, Hu F, Herr AB, Strong RK, Kao CC, Li P. The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure. 2010;18(8):1032–1043. doi: 10.1016/j.str.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Luo D. Toward a crystal-clear view of the viral RNA sensing and response by RIG-I-like receptors. RNA Biol. 2014;11(1):25–32. doi: 10.4161/rna.27717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147(2):423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 87.Berke IC, Modis Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012;31(7):1714–1726. doi: 10.1038/emboj.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 89.He X, Zhang J. Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics. 2005;169(2):1157–1164. doi: 10.1534/genetics.104.037051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anderson DW, McKeown AN, Thornton JW. Intermolecular epistasis shaped the function and evolution of an ancient transcription factor and its DNA binding sites. Elife. 2015;4:e07864. doi: 10.7554/eLife.07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaltenbach M, Jackson CJ, Campbell EC, Hollfelder F, Tokuriki N: Reverse evolution leads to genotypic incompatibility despite functional and active site convergence. Elife. 2015;4. doi: 10.7554/eLife.06492. [DOI] [PMC free article] [PubMed]
- 92.Breen MS, Kemena C, Vlasov PK, Notredame C, Kondrashov FA. Epistasis as the primary factor in molecular evolution. Nature. 2012;490(7421):535–538. doi: 10.1038/nature11510. [DOI] [PubMed] [Google Scholar]
- 93.Boucher JI, Jacobowitz JR, Beckett BC, Classen S, Theobald DL: An atomic-resolution view of neofunctionalization in the evolution of apicomplexan lactate dehydrogenases. Elife. 2014;3. doi: 10.7554/eLife.02304. [DOI] [PMC free article] [PubMed]
- 94.Soylemez O, Kondrashov FA. Estimating the rate of irreversibility in protein evolution. Genome Biol Evol. 2012;4(12):1213–1222. doi: 10.1093/gbe/evs096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gong LI, Suchard MA, Bloom JD. Stability-mediated epistasis constrains the evolution of an influenza protein. Elife. 2013;2:e00631. doi: 10.7554/eLife.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carroll SM, Bridgham JT, Thornton JW. Evolution of hormone signaling in elasmobranchs by exploitation of promiscuous receptors. Mol Biol Evol. 2008;25(12):2643–2652. doi: 10.1093/molbev/msn204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zou T, Risso VA, Gavira JA, Sanchez-Ruiz JM, Ozkan SB. Evolution of conformational dynamics determines the conversion of a promiscuous generalist into a specialist enzyme. Mol Biol Evol. 2015;32(1):132–143. doi: 10.1093/molbev/msu281. [DOI] [PubMed] [Google Scholar]
- 98.Furumizu C, Alvarez JP, Sakakibara K, Bowman JL. Antagonistic roles for KNOX1 and KNOX2 genes in patterning the land plant body plan following an ancient gene duplication. PLoS Genet. 2015;11(2):e1004980. doi: 10.1371/journal.pgen.1004980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kondrashov FA. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc Biol Sci. 2012;279(1749):5048–5057. doi: 10.1098/rspb.2012.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roselló PO, Kondrashov FA. Long-term asymmetrical acceleration of protein evolution after gene duplication. Genome Biol Evol. 2014;6(8):1949–1955. doi: 10.1093/gbe/evu159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Keay J, Thornton JW. Hormone-activated estrogen receptors in annelid invertebrates: implications for evolution and endocrine disruption. Endocrinology. 2009;150(4):1731–1738. doi: 10.1210/en.2008-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bridgham JT, Brown JE, Rodriguez-Mari A, Catchen JM, Thornton JW. Evolution of a new function by degenerative mutation in cephalochordate steroid receptors. PLoS Genet. 2008;4(9):e1000191. doi: 10.1371/journal.pgen.1000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Keay J, Bridgham JT, Thornton JW. The Octopus vulgaris estrogen receptor is a constitutive transcriptional activator: evolutionary and functional implications. Endocrinology. 2006;147(8):3861–3869. doi: 10.1210/en.2006-0363. [DOI] [PubMed] [Google Scholar]
- 104.Dixit E, Kagan JC. Intracellular pathogen detection by RIG-I-like receptors. Adv Immunol. 2013;117:99–125. doi: 10.1016/B978-0-12-410524-9.00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sanchez David RY, Combredet C, Sismeiro O, Dillies MA, Jagla B, Coppee JY, Mura M, Guerbois Galla M, Despres P, Tangy F, et al. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. Elife. 2016;5:e11275. doi: 10.7554/eLife.11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Uzri D, Gehrke L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J Virol. 2009;83(9):4174–4184. doi: 10.1128/JVI.02449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chiang C, Beljanski V, Yin K, Olagnier D, Ben Yebdri F, Steel C, Goulet ML, DeFilippis VR, Streblow DN, Haddad EK, et al. Sequence-Specific Modifications Enhance the Broad-Spectrum Antiviral Response Activated by RIG-I Agonists. J Virol. 2015;89(15):8011–8025. doi: 10.1128/JVI.00845-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Garcin D, Lezzi M, Dobbs M, Elliott RM, Schmaljohn C, Kang CY, Kolakofsky D. The 5′ ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J Virol. 1995;69(9):5754–5762. doi: 10.1128/jvi.69.9.5754-5762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Habjan M, Andersson I, Klingström J, Schümann M, Martin A, Zimmermann P, Wagner V, Pichlmair A, Schneider U, Mühlberger E, et al. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS One. 2008;3(4):e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Girardi E, Chane-Woon-Ming B, Messmer M, Kaukinen P, Pfeffer S. Identification of RNase L-dependent, 3′-end-modified, viral small RNAs in Sindbis virus-infected mammalian cells. MBio. 2013;4(6):e00698–00613. doi: 10.1128/mBio.00698-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rodriguez KR, Horvath CM. Paramyxovirus V protein interaction with the antiviral sensor LGP2 disrupts MDA5 signaling enhancement but is not relevant to LGP2-mediated RLR signaling inhibition. J Virol. 2014;88(14):8180–8188. doi: 10.1128/JVI.00737-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nan Y, Nan G, Zhang YJ. Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses. 2014;6(12):4999–5027. doi: 10.3390/v6124999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Davis ME, Wang MK, Rennick LJ, Full F, Gableske S, Mesman AW, Gringhuis SI, Geijtenbeek TB, Duprex WP, Gack MU. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe. 2014;16(1):19–30. doi: 10.1016/j.chom.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, Hur S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci U S A. 2011;108(52):21010–21015. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, Hur S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152(1–2):276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 116.Peisley A, Wu B, Xu H, Chen ZJ, Hur S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature. 2014;509(7498):110–114. doi: 10.1038/nature13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bruns AM, Horvath CM. LGP2 synergy with MDA5 in RLR-mediated RNA recognition and antiviral signaling. Cytokine. 2015;74(2):198–206. doi: 10.1016/j.cyto.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141(2):315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Castanier C, Zemirli N, Portier A, Garcin D, Bidère N, Vazquez A, Arnoult D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012;10:44. doi: 10.1186/1741-7007-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschlager N, Schlee M, Rothenfusser S, Barchet W, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11(1):63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 121.Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42(Database issue):D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boratyn GM, Schäffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL. Domain enhanced lookup time accelerated BLAST. Biol Direct. 2012;7:12. doi: 10.1186/1745-6150-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43(Database issue):D222–D226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Roshan U, Livesay DR. Probalign: multiple sequence alignment using partition function posterior probabilities. Bioinformatics. 2006;22(22):2715–2721. doi: 10.1093/bioinformatics/btl472. [DOI] [PubMed] [Google Scholar]
- 125.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26(7):1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 128.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rannala B, Yang Z. Probability distribution of molecular evolutionary trees: a new method of phylogenetic inference. J Mol Evol. 1996;43(3):304–311. doi: 10.1007/BF02338839. [DOI] [PubMed] [Google Scholar]
- 132.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13(5):555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 133.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 134.Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 135.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15(11):2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Eramian D, Eswar N, Shen MY, Sali A. How well can the accuracy of comparative protein structure models be predicted? Protein Sci. 2008;17(11):1881–1893. doi: 10.1110/ps.036061.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27(3):343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhao H, Yang Y, Zhou Y. Structure-based prediction of DNA-binding proteins by structural alignment and a volume-fraction corrected DFIRE-based energy function. Bioinformatics. 2010;26(15):1857–1863. doi: 10.1093/bioinformatics/btq295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Laskowski R, MacArthur M, Moss D, Thornton J. PROCHECK - a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 140.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35(Web Server issue):W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61(4):704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 142.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98(18):10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 144.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78(8):1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Touw WG, Baakman C, Black J, te Beek TA, Krieger E, Joosten RP, Vriend G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015;43(Database issue):D364–D368. doi: 10.1093/nar/gku1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.WELCH BL. The generalisation of student’s problems when several different population variances are involved. Biometrika. 1947;34(1–2):28–35. doi: 10.1093/biomet/34.1-2.28. [DOI] [PubMed] [Google Scholar]
- 147.Storey JD. A direct approach to false discovery rates. J R Stat Soc B. 2002;64:479–498. doi: 10.1111/1467-9868.00346. [DOI] [Google Scholar]
- 148.Zor T, Selinger Z. Linearization of the Bradford protein assay increases its sensitivity: theoretical and experimental studies. Anal Biochem. 1996;236(2):302–308. doi: 10.1006/abio.1996.0171. [DOI] [PubMed] [Google Scholar]
- 149.Abdiche Y, Malashock D, Pinkerton A, Pons J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal Biochem. 2008;377(2):209–217. doi: 10.1016/j.ab.2008.03.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data not obtained from public databases or presented in the main text is available in Additional file 1.