

SUMMARY
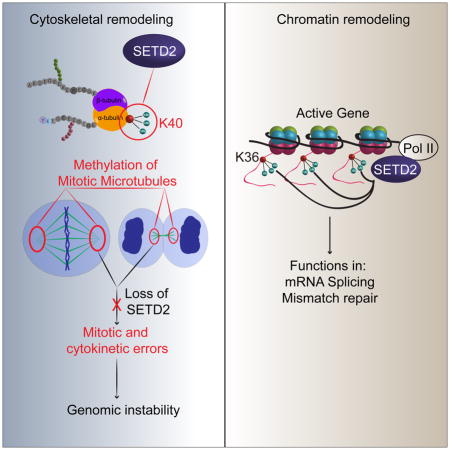
Posttranslational modifications (PTMs) of tubulin specify microtubules for specialized cellular functions and comprise what is termed a “tubulin code”. PTMs of histones comprise an analogous “histone code”, although the “readers, writers and erasers” of the cytoskeleton and epigenome have heretofore been distinct. We show that methylation is a PTM of dynamic microtubules, and that the histone methytransferase, SETD2, which is responsible for H3 lysine 36 trimethylation (H3K36me3) of histones, also methylates α-tubulin at lysine 40, the same lysine that is marked by acetylation on microtubules. Methylation of microtubules occurs during mitosis and cytokinesis, and can be ablated by SETD2 deletion, which causes mitotic spindle and cytokinesis defects, micronuclei and polyploidy. These data now identify SETD2 as a dual function methyltransferase for both chromatin and the cytoskeleton, and show a requirement for methylation in maintenance of genomic stability and the integrity of both the tubulin and histone codes.
In Brief
Tubulin and histones share a methyltransferase, suggesting a new basis for known tumorigenic mutations in the enzyme.
INTRODUCTION
SET-domain containing 2 (SETD2), also known as HYPB and KMT3A, is a histone methyltransferase responsible for histone H3 lysine 36 trimethylation (H3K36me3) of chromatin, an epigenetic mark associated with gene transcription (Edmunds et al., 2008; Hu et al., 2010). It is also one of the many “readers, writers and erasers” of the histone code (Jenuwein and Allis, 2001; Strahl and Allis, 2000), which is comprised of post-translational modifications (PTMs) including acetylation, phosphorylation, ubiquitination and methylation of multiple sites on histone tails, which together constitute a complex language for transcriptional regulation (Lee et al., 2010). SETD2 is able to add a methyl group to a di-methylated lysine to generate a trimethyl mark, as well as de novo mono- and di-methylation to generate a trimethyl mark on histone H3 (Wagner and Carpenter, 2012; Yuan et al., 2009). H3K36me3 of chromatin is a non-redundant SETD2 function (Edmunds et al., 2008), and loss of SETD2 is embryonic lethal (Hu et al., 2010).
Recently, the concept of the “histone code” has been parlayed into a “tubulin code” hypothesis to describe how PTMs distinctly mark subsets of microtubules in the cytoskeleton (Verhey and Gaertig, 2007). The cytoskeleton is a network of fibers that maintains cell shape, allows cells to move and divide, and forms specialized structures such as cilia and microvilli. Although the name implies a stable structure, many parts of the cytoskeleton are dynamic and constantly remodeled, with some parts assembled while others are dismantled. An important component of the cytoskeleton is microtubules, which are built from heterodimers of α- and β-tubulin, and are required for many diverse functions such as mitosis, where they form the mitotic spindle and participate in chromosome segregation and cytokinesis (Walczak and Heald, 2008). The “tubulin code” hypothesis posits that PTMs of specific tubulin subunits within the polymer direct microtubule-based functions at that location (Verhey and Gaertig, 2007). Indeed, PTMs including phosphorylation, detyrosination, polyglutamylation, polyglycylation and acetylation are enriched on specialized microtubule structures such as centrioles and basal bodies, neuronal axons, and primary cilia (Janke, 2014; Song and Brady, 2015).
Most microtubule PTMs have been discovered serendipitously, usually as the result of generation of antibodies later found to react with specific modified residues of α- or β-tubulin (Magiera and Janke, 2013), and as a result, the complete repertoire of microtubule PTMs has yet to be fully elucidated. PTMs of microtubules serve many functions, for example recognition by microtubule-associated proteins (MAPs), which can regulate microtubule dynamics and function. These modifications play important roles during mitosis, where for example, detyrosination guides chromosome congression (Barisic et al., 2015). However for many PTMs, the underlying functionality is still undetermined (Janke, 2014; Song and Brady, 2015). Compared to PTMs that specify stable cytoplasmic microtubules, which are easily detected by purification or immunoreactivity with PTM-specific antibodies, little is known about PTMs associated with mitotic spindle and midbody microtubules that direct their dynamic polymerization and depolymerization during mitosis (Kwok and Kapoor, 2007; Walczak and Heald, 2008).
Importantly, the “readers, writers and erasers” of the histone and tubulin codes identified to date have been distinct, even for PTMs such as acetylation that occur on both histones and microtubules. As a result, in settings such as cancer, where defects in chromatin remodelers such as SETD2 occur with a high frequency, research to understand how loss of “readers, writers and erasers” such as SETD2 contribute to disease pathogenesis has focused on chromatin and the impact of loss of H3K36me3 on the epigenome.
We have now found that SETD2 is required for the integrity of both the histone and tubulin codes, providing evidence for cross-talk between the epigenome and cytoskeleton. We show that methylation is a PTM of mitotic microtubules, and that SETD2 binds to and methylates α-tubulin. Methylation occurs at lysine 40 (α-TubK40me3), the same residue that is acetylated on microtubules (α-TubK40ac). Methylation occurs on the spindle during mitosis and the midbody during cytokinesis, and SETD2-null cells exhibit loss of spindle and midbody methylation, and numerous mitotic and cytokinesis defects, identifying a role for microtubule methylation by SETD2 in genomic stability.
RESULTS
SETD2 binds α-tubulin
We found that endogenous SETD2 could be co-immunoprecipitated with α-tubulin in SETD2-proficient human 786-0 cells (Figure 1A). A direct interaction between SETD2 and α-tubulin was demonstrated by incubating SETD2 protein immunopurified using a SETD2-specific antibody with human recombinant α-tubulin protein (TUBA1A), followed by co-immunoprecipitation (co-IP) of α-tubulin (Figure 1B).
Figure 1. SETD2 binds α-tubulin.

(A) Co-immunoprecipitation of endogenous α-tubulin and SETD2 from 786-0 cells with two different SETD2 antibodies (Sigma-Aldrich and Abcam).
(B) Co-immunoprecipitation of immunopurified SETD2 from MEFs using SETD2 antibody and recombinant α-tubulin. Input - recombinant α-tubulin, 10ng.
(C) Co-immunoprecipitation of exogenous SETD2 and α-tubulin from HEK-293T cells co-expressing SETD2, and EGFP-α-tubulin or EGFP-K40R-α-tubulin.
(D) Schematic of GST-SETD2 fusion protein constructs.
(E) GST pull down assays of α-tubulin with the GST-SETD2 fusion constructs incubated with 1μg of recombinant α-tubulin (TUBA1A) protein and immunoblotted using α-tubulin antibody (upper panel). GST-SETD2 construct expression assessed using Coomassie blue staining (lower panel).
(F) Peptide pull down using biotin-labeled K40 peptide of α-tubulin with the G12 or GST only constructs. Input 50ng.
SETD2 binding and H3K36 methylation of histone tails coincides with histone acetylation at active genes (Edmunds et al., 2008), leading us to ask if acetylation, which is known to occur on microtubules, might play a role in SETD2 binding to α-tubulin. Treatment of cells with trichostatin A (TSA), an HDAC inhibitor that selectively inhibits class I and II mammalian HDACs including the α-tubulin deacetylase HDAC6 (Matsuyama et al., 2002) had no effect on SETD2 binding to α-tubulin (data not shown), and co-expression of wild-type EGFP-α-tubulin and mutant EGFP-K40R α-tubulin (which cannot be acetylated) with full length SETD2 revealed that SETD2 could co-IP with both wild-type and K40R mutant α-tubulins, indicating that the interaction of SETD2 with α-tubulin did not require acetylation of K40 (Figure 1C).
To determine the α-tubulin binding domain of SETD2, we constructed a series of GST fusion proteins containing human SETD2 fragments purified from E. coli (Figures 1D and 1E). While SETD2 protein levels expressed from various constructs differed, we consistently observed that the presence of the SET (Drosophila Su(var)3–9 and ‘Enhancer of zeste’ proteins) domain was required for binding to α-tubulin. As shown in Figure 1E, GST pull down assays revealed that the SET domain was sufficient for binding to α-tubulin, consistent with an enzyme-substrate relationship.
To identify potential sites for methylation on lysine residue(s) of tubulin, we analyzed the amino acid sequence of α-tubulin for motifs that fit the structural requirements for substrates of SETD2 and its corresponding demethylase, JMJD2A (also known as KDM4A, JHDM3A, and TDRD14A). The catalytic domain of both SETD2 and JMJD2A requires a curved or flexible structure containing Pro at +2 or Gly-Gly at +3 and +4 after the substrate lysine residue (Chen et al., 2007; Nelson et al., 2006). We found only one candidate lysine within α-tubulin that met these structural requirements, K40 where the sequence ‘KTIGG’, is a good fit as a structural motif for a SETD2 or JMJD2A substrate. A biotin-tagged peptide around this region “IQPDGGMPSDKTIGGGDDSF-biotin” could co-immunoprecipitate a GST-tagged SET domain (Figure 1F). An antibody raised against the H3K36me3 epitope also could immunopecipitate α-tubulin (Figure S1A), supporting this as a potential recognition site for SETD2 methylation.
To further confirm these data, we generated a polyclonal antibody against trimethylated K40 peptide of α-tubulin (α-TubK40me3). This antibody had minimal cross reactivity with a di-methylated K40 peptide and no detectable cross reactivity with mono- or un-methylated K40 peptides (Figure S1B). We found that similar to the commercially available H3K36me3 antibody, the α-TubK40me3-specific antibody generated to the K40 peptide also detected and immunoprecipitated native α-tubulin (Figure S1A), indicating similarity between the trimethyl SETD2-epitope of chromatin and the methyl-specific epitope recognized by the α-TubK40me3-specific antibody. The specificity of the α-TubK40me3 antibody was demonstrated using a peptide competition assay where an α-TubK40me3 peptide strongly, and α-TubK40me2 peptide modestly, competed for binding of the α-TubK40me3-specific antibody to α-tubulin, with little loss of immunoreactivity seen with mono-methylated, acetylated or unmethylated α-TubK40 peptides (Figure S1C).
Microtubule methylation occurs during mitosis
Because binding of the SET domain to α-tubulin and recognition by both H3K36me3 and α-TubK40me3-specific antibodies suggested that SETD2 could be methylating microtubules, we immunostained mouse embryonic fibroblasts (MEFs) using either the α-TubK40me3-specific or H3K36me3 antibodies together with either α-tubulin (Figures 2A–2B) or acetylated α-tubulin (Figure S2A–S2B). In mitotic cells, in addition to the expected immunostaining of chromatin, we observed distinct immunoreactivity with both the α-TubK40me3 and H3K36me3 antibodies on spindle microtubules but not astral microtubules (Figures 2A–2B and S2A–S2B). During metaphase, methylation of spindle microtubules was concentrated near the spindle poles as shown in Figures 2A–2B and S2A–S2B and illustrated with a line profile through a representative metaphase spindle stained with α-TubK40me3 and α-tubulin antibodies (Figure S2C). Methylation at the minus ends of bundled microtubules becomes especially evident when the spindle midzone compacts into the midbody during cytokinesis (Figures 2A–2C and S2A–S2B).
Figure 2. Microtubule methylation during mitosis and cytokinesis in MEFs.

(A) Representative images of cells stained using α-tubulin (red) and α-TubK40me3 (green) antibodies, counterstained with DAPI (blue). The far right panel (Zoom) indicates higher magnification images for better visualization. Scale bar =10μm.
(B) Representative images of cells stained using α-tubulin (red) and H3K36me3 (green) antibodies, counterstained with DAPI (blue). The far right panel (Zoom) indicates higher magnification images for better visualization. Scale bar =10μm.
(C) Quantitative analysis of average fluorescent intensities of midbodies immunostained with H3K36me3 (green curve) and acetylated α-tubulin (red curve) antibodies (N = 66 midbodies, from 3 independent experiments). Representative image showing H3K36me3 (green), acetylated α-tubulin (red) and DAPI (blue). Scale bar =10μm.
See also Figure S2 and S3.
To further investigate the methylation state of spindle microtubules, we stained cells with a series of pan mono-, di-, and trimethyl lysine antibodies (Guo et al., 2014). In addition to the expected mono-, di- and tri-methylation of chromatin (Figures 2, S2 and S3), we also observed immunoreactivity of spindle and midbody microtubules using a pan tri-methyl lysine (Kme3) antibody (Figure S3A), but not however, with the pan mono-methyl (Kme1) or di-methyl (Kme2) lysine antibodies (Figures S3B and S3C). Antibodies to methyl lysine marks made by other histone methyltransferases, H3K4me3 (COMPASS complex) or H3K27me3 (EZH2) showed no immunoreactivity for these microtubule structures (Figure S3D). Together, these data identify a methylated epitope on mitotic microtubules recognized by the α-TubK40me3, H3K36me3 and pan-Kme3 antibodies, rather than non-specific association of histones/chromatin with this structure.
A striking feature of microtubule trimethylation was the apparently exclusive relationship between trimethylation and the well-known α-tubulin K40ac mark, particularly during the later stages of mitosis. Co-staining with α-TubK40me3 and H3K36me3 and α-tubulin antibodies showed trimethylation on the distal, minus ends of midbodies distinct from chromatin H3K36me3 staining (Figures 2A–2C), while co-staining with α-TubK40me3, H3K36me3 and acetylated α-tubulin showed the methyl mark occurred more distally on the midbody than the acetyl mark (Figures and S2A–S2B). As shown in Figure 2C, quantification (N= 66) of the fluorescence intensity of trimethyl- and acetyl-specific antibody staining along the length of the midbody revealed an inverse relationship between acetylation and trimethylation. This suggested the possibility that microtubule acetylation and methylation were reciprocal marks, and focused our attention on the site for microtubule acetylation, lysine 40 of α-tubulin (Janke, 2014; Verhey and Gaertig, 2007).
SETD2 methylates lysine 40 of α-tubulin
To examine whether methylation at K40 of α-tubulin occurs in vivo, we used mass spectrometry to analyze human myc-tagged α-tubulin (TUBA1B-myc) proteins purified from HEK293T cells. Trimethylation at K40 of α-tubulin was recognized by peaks corresponding to calculated molecular weights from the N- and C-terminus of the peptide (Figure 3A). We next determined if α-tubulin could be directly methylated by SETD2. SETD2 is able to add a methyl group to di-methylated lysine to generate H3K36me3, but is also capable of de novo mono- and di-methylation to generate a trimethyl mark (Wagner and Carpenter, 2012; Yuan et al., 2009), indicating SETD2 substrate recognition is not confined to di-methylated lysine residues. Using purified α-tubulin, bovine microtubules or histones (control) as substrates, in vitro methylation assays revealed that recombinant SETD2 was able to methylate both tubulin proteins and histones (Figures 3B and S4A). Polymerization of microtubules was not required for methylation by SETD2 since SETD2 methylated both soluble tubulin and polymerized microtubules, as well as recombinant α-tubulin protein (Figures 3B and S4A). Together, these data demonstrate that trimethylation of α-tubulin is a new PTM of microtubules, and identify SETD2 as a dual function methyltransferase that can directly methylate both histones and α-tubulin.
Figure 3. SETD2 methylates lysine 40 of α-tubulin.

(A) Mass spectrometry analysis showing trimethylation at K40 on α-tubulin from HEK-293T cells. Expected molecular weights of trimethylated peptides from the N-terminus and C-terminus are shown as peaks in red and blue, respectively.
(B) In vitro methylation of bovine microtubule proteins using recombinant SETD2 and S-[methyl-3H]-Adenosyl-L-Methionine (SAM) as a methyl donor.
See also Figure S4.
SETD2 is a mitotic microtubule methyltransferase
To definitively determine if SETD2 methylates mitotic microtubules, we generated a line of Setd2flox/flox mice, isolated Setd2flox/flox MEFs, transfected these cells with a tamoxifen-inducible ER-Cre expression vector and isolated stable Cre-expressing cells after selection with blasticidin. Setd2flox/flox;ER-Cre MEFs were treated with 4-hydroxytamoxifen or vehicle, and loss of SETD2 activity observed by the absence of SETD2 protein (Figure S4B), loss of α-TubK40me3-immunoreactivity of α-tubulin (Figure S4B), and loss of nuclear H3K36me3 immunoreactivity (Figure 4A). Immunostaining with H3K36me3 and α-TubK40me3 antibodies confirmed that methylation of microtubules was lost in Setd2 knockout MEFs (Figure 4A).
Figure 4. Loss of methylation on midbody and mitosis delay caused by Setd2 ablation.

(A) Representative cell images of Setd2+/+ and Setd2−/− cells using H3K36me3 or α-TubK40me3 (green), and acetylated α-tubulin (red) antibodies, counterstained with DAPI (blue). Scale bar =10μm. Note that pre-warming the PFA to 37°C significantly reduced cross reactivity of the α-TubK40me3 antibody with chromatin while retaining recognition of the α-TubK40me3 epitope on microtubules, as described in Experimental Procedures.
(B) Live cell images of Setd2+/+ and Setd2−/− cells undergoing mitosis and cytokinesis. Red and green arrowheads denote corresponding cells tracked during cell division.
See also Figure S4.
SETD2 is required for genomic stability
To examine the impact of loss of Setd2 during mitosis, we clonally selected stable Setd2flox/flox;ER-Cre MEFs and carried out live cell imaging. In control cells, the average time in mitosis was 1.4 hours (S.D. = 0.57, N = 63 and illustrated using time-lapse images in Figure 4B). However, after 4-hydroxytamoxifen treatment and excision of Setd2, the average time in mitosis was increased to 2.0 hours (S.D. = 0.68, N = 37 and illustrated using time-lapse images in Figure 4B), with Setd2-null cells taking significantly longer to complete mitosis (p ≤ 0.0001 unpaired Welch t-test), and often failing to undergo cytokinesis, resulting in retained cytoplasmic bridges (Figure 4B) and binucleation (Supplemental Movie 1).
This phenotype suggested that acute loss of Setd2 function caused mitotic and/or cytokinesis defects. Consistent with induction of the tetraploid checkpoint (Ganem and Pellman, 2007), tamoxifen-induced excision of Setd2 caused dramatic increase in the sub-G1 population, and death of Setd2-null cells (Figure S5A). When we analyzed the DNA content of clonally-derived Setd2flox/flox MEFs stably expressing tamoxifen-inducible ER-Cre (S4 cells) by flow cytometry after exclusion of the majority of the sub-G1 population (Figures S5B and S5C), we observed increased ploidy, with ~30% of S4 cells becoming polyploid (>4N complement of DNA) upon loss of Setd2 compared to ~14% of controls (Figure 5A). To eliminate the possibility that the observed increase in >4N DNA was due to cell aggregation, we used automated confocal microscopy to capture images from 3,000 cells 72 hours after Cre-mediated excision of Setd2, and quantitated DAPI fluorescence intensities and DAPI staining area by dot plot using algorithms developed in Pipeline Pilot (Accelrys) (Figure 5B). Consistent with the flow cytometry data, polyploidy in Setd2 knockout cells (~41%) was significantly higher than controls (~10% and 17% in pharmacologic and genetic controls). To rule out artifacts associated with clonal selection, we also assessed polynucleation in a pool of stably expressing Setd2flox/flox;ER-Cre MEFs after tamoxifen-induced Cre-mediated excision of Setd2. In three biological replicates counting 500 cells each (total 1,500 cells), polynucleation in Setd2 knockout cells was significantly higher than pharmacological and genetic controls (Figures 5C and 5D). Together, these data demonstrated that loss of Setd2 led to an increase in ploidy and polynucleation.
Figure 5. Loss of Setd2 induces polyploidy.

(A) FACS analysis of Setd2−/− cells (red) and Setd2+/+ controls (blue). Error bars represent the mean and standard deviations of 4 biological replicates. *p<0.0001, n.s. = not significant.
(B) Scatterplot of DNA content in Setd2 knockout (KO) MEFs with two control cells; Setd2flox/flox MEFs treated with 4-hydroxytamoxifen (genetic control) and ER-Cre transfected Setd2flox/flox MEFs treated with vehicle (ethanol) (pharmacologic control). Statistics comparing Setd2 KO to pharmacological control, *p<0.05. Scale bar=25μm, N=3.
(C) Number of nuclei in Setd2 KO and with control MEFs. Cells were stained with DAPI and the nuclear content of ~1500 cells (~500 cells in three independent experiments) was counted manually from multiple confocal images. Scale bar=50μm. N=3.
(D) Percentage of cells (from Figure 5C) with more than 1 nucleus. **p=0.0012, ***p=0.0009. Error bars represent standard error of the mean.
See also Figure S5.
SETD2 is required for normal mitosis and cytokinesis
To examine the mitotic defects caused by loss of SETD2 activity in more detail, we used automated confocal microscopy to examine Setd2flox/flox;ER-Cre MEF clones 72 hours following Cre-mediated excision of Setd2. Images of mitotic cells were obtained and abnormal mitotic events were identified (Figure 6A) and quantified for N=191 Setd2+/+ cells and N=216 Setd2−/− cells (Figure 6B). A significant increase in mitotic and cytokinesis defects was observed following Setd2 loss, including a failure of chromosomes to congress and an increase in multipolar spindles at prometaphase, lagging chromosomes at anaphase, and chromosomal bridges at cytokinesis. An increase in micronuclei was also observed in Setd2−/− cells. This suggested that loss of the α-TubK40me3 mark was responsible for these defects, as other known microtubule PTMs were still present in the spindles of Setd2−/− cells (Figure S6).
Figure 6. Mitotic and cytokinesis defects induced by loss of Setd2.

(A) Representative images of mitotic and cytokinesis defects in Setd2 KO MEFs using an acetylated α-tubulin (green) antibody and counterstained with DAPI (blue). Scale bar=10μm.
(B) Quantitation of abnormal mitotic events in Setd2+/+ control and Setd2−/− cells. N=number of dividing cells counted. ****p<0.0001.
See also Figure S6.
Loss of SETD2 α-tubulin methylation causes mitotic and cytokinesis defects
As a non-redundant histone methyltransferase, loss of SETD2 results in an absence of H3K36me3, making it at least a formal possibility that mitotic defects observed in in SETD2-null cells occurred secondary to loss of H3K36me3 and disrupted epigenomic programming. To explore this possibility, RNA-seq studies were performed on SETD2-proficient and SETD2-disrupted (via TALEN) 786-0 and HKC (human kidney proximal tubule epithelial) cells (Figure S7). In two biological replicates each for SETD2 disrupted 786-0 and HKC cells, as compared with wild type cells, analysis of differentially expressed genes meeting p<0.05 threshold for significance showed no pathways were consistently observed to be enriched upon loss of SETD2 in these cells (Figure S7).
Next, a series of rescue experiments were performed in 786-0 cells in which SETD2 had been deleted, and then clones selected to re-express either an N-terminal truncated (but functional) wild-type (tSETD2-WT), a pathogenic catalytically-dead SET-domain mutation (tSETD2-R1625C) or a pathogenic SRI-domain mutation (tSETD2-R2510H). The SRI domain of SETD2 is thought to interact with Pol II during transcription and “marking” of histones by SETD2 (Kizer et al., 2005; Rebehmed et al., 2014). Many pathogenic mutations in this domain have been noted in RCC, suggesting that in addition to the SET-domain, this domain plays an important, albeit not completely characterized, role in SETD2 function. As shown in Figure 7A, H3K36me3 histone methylation disappeared in 786-0 SETD2-null cells. Expression of tSETD2-WT restored histone methylation, while the tSETD2-R1625C SET-domain mutant (as expected) failed to restore H3K36me3. Mitotic defects followed a similar pattern: the incidence of mitotic defects was higher in SETD2-null 786-0 cells relative to the parental cells (p<0.0001), and tSETD2-WT, but not tSETD2-R1625C SET-domain mutant (p<0.0001), rescued this increase in mitotic defects (Figures 7B and 7C). Importantly, while the tSETD2-R2510H SRI-domain mutant fully rescued H3K36me3 to levels equivalent to tSETD2-WT, it was not able to rescue mitotic defects in 786-0 SETD2-null cells (Figures 7B and 7C). The tSETD2-R2510H SRI-domain mutant was also unable to rescue K40me3 of α-tubulin (Figure 7D), even when expressed at an equivalent level with tSETD2-WT (Figure 7D), consistent with microtubule methylation defects driving genomic instability in these cells. Together, these data separating loss of histone methylation from genomic instability point to SETD2 methylation of microtubules as being critical for proper mitosis and cytokinesis.
Figure 7. SETD2 SRI mutant rescues histone methylation but not tubulin methylation or mitotic defects.

(A) Western blot analyses using histone extracts from parental 786-0 cells, SETD2-null 786-0 cells or SETD2-null 786-0 cells expressing tSETD2 (truncated SETD2 with intact methylation activity), R1625C mutant (SET domain mutant), or R2510H (SRI domain mutant).
(B) Mitotic defects including chromosome bridges (white) and lagging chromosomes (black) quantitated for cells from three technical replicates, in each of two biologically independent experiments for a total of 200 cells for each condition.*p<0.0001.
(C) Representative images of cells stained using α-tubulin (grey), centromere (CREST, red) antibodies and DAPI (blue). Scale bar =10μm.
(D) Immunoprecipitation of cytoplasmic fraction using lysates from (A) with the α-TubK40me3 antibody and immunoblotted using a α-tubulin antibody. In the immunoblot using SETD2 antibody, the tSETD2-2A-GFP fusion protein was autocleaved to tSETD2 and GFP by self-cleaving 2A peptide after translation. The amounts of α-tubulin K40 trimethylation were quantitated by analyzing band intensities in five independent experiments (N=5). *p<0.05, error bars denote standard error of the mean.
See also Figure S7.
DISCUSSION
Given the differential expression of tubulin isotypes in specific cells and tissues and the identification of several PTMs of tubulin subunits within the microtubule polymer, the concept of a ‘tubulin code’ has been suggested (Verhey and Gaertig, 2007), parlaying the ‘histone code’ hypothesis that chromatin structure and gene expression are regulated by a constellation of PTMs (Jenuwein and Allis, 2001; Strahl and Allis, 2000). Although identifying the “readers, writers and erasers” of both the tubulin and histone codes is a focus of much research, they have heretofore appeared to be distinct. We report here the identification of methylation as a modification of α-tubulin within microtubules, thus identifying methylation as critical component of the tubulin code. Furthermore, we identified SETD2 as an α-tubulin methyltransferase, defining SETD2 as a dual function methyltransferase and “writer” required for the integrity of both the histone and tubulin codes. Although dual function chromatin and cytoskeleton remodelers have not been previously identified, it is possible that other “readers, writers and erasers” of the histone code may also have functions associated with the cytoskeleton. Indeed, while we find acute loss of SETD2 leads to loss of microtubule methylation, often accompanied by catastrophic microtubule defects, with prolonged selection, we have been able to isolate SETD2-null cell lines, suggesting the possibility that other methyltransferase(s) may be able to substitute for SETD2 to provide a similar function.
The finding that microtubules are methylated at K40 of α-tubulin, the same site used for acetylation, suggests that methylation and acetylation have opposing functionality for microtubules. There is precedent for acetylation and methylation at a single histone lysine residue to provide mutually exclusive regulatory events. Acetylation of histone H3 at lysine 9 (H3K9ac) activates transcription while di-and trimethylation of this same lysine repress transcription (Latham and Dent, 2007). Similarly, mutually exclusive acetylation/methylation occurs on H3K14, H3K23, H3K27, H3K36, H4K12, H4K20 and H2BK5 (Latham and Dent, 2007).
α-Tubulin K40 acetylation is associated with certain extraordinarily stable microtubules, including some cytoplasmic microtubules, the axoneme of cilia, and microtubules in neuronal axons. It is thus tempting to speculate that methylation may serve an opposing function, regulating microtubule depolymerization or destruction during mitosis. However, α-TubK40ac is not directly responsible for microtubule stability (Perdiz et al., 2011); some highly acetylated spindle microtubules are also highly dynamic. We thus propose that α-TubK40ac and α-TubK40me3 may recruit different “readers” to the microtubule shaft and minus ends, respectively, in order to regulate different microtubule-based activities. For example, it was recently shown that detyrosination of spindle microtubules provides an epigenetic mark that is read by the kinesin-7 family motor CENP-E during chromosome congression to the equator (Barisic et al., 2015). As cells proceed toward cytokinesis, α-TubK40me3 likely also recruits ‘reader’ proteins to the spindle midzone and the midbody. No proteins are known to localize specifically to the minus ends of these microtubules (Green et al., 2012), but the defects seen upon loss of Setd2 suggest that microtubule methylation maybe critical for activation of the NoCut checkpoint (Agromayor and Martin-Serrano, 2013) and recognition by “readers” to complete cytokinesis.
What might be the consequences in disease settings such as cancer for cells that lose SETD2? Loss of SETD2 function occurs in approximately 20% of human renal cell carcinomas (Gerlinger et al., 2012). In some tumors, loss of SETD2 occurs as a subclonal alteration, with tumors exhibiting loss of this methyltransferase having a more aggressive phenotype and worse prognosis (Bi et al., 2016; Cancer Genome Atlas Research, 2013; Hakimi et al., 2013; Ho et al., 2016; Ho et al., 2015; Kovac et al., 2015; Liao et al., 2015; Liu et al., 2015). A high rate of SETD2 mutation has also been observed in bladder and lung cancer (Cerami et al., 2012; Gao et al., 2013) and SETD2 is also mutated in T-cell acute lymphoblastic leukemia (Zhang et al., 2012), and acute leukemia (6 %) (Zhu et al., 2014). Similarly, SETD2 loss has been associated with more aggressive GI stromal tumors (GIST)(Huang Gut 2015) and high-grade gliomas (Fontebasso et al., 2013; Huether et al., 2014). Recurrent loss-of-function SETD2 mutations are also observed in leukemia (Zhu et al., 2014), which interestingly, are most commonly truncating mutations leading to loss of the SRI domain. SETD2 mutations are also enriched in relapsed pediatric leukemias (Mar et al., 2014), suggesting therapy supplies a selective pressure for mutation due to loss of this, and other chromatin remodelers, and leading these authors to suggest that SETD2 inactivation may confer a mutator phenotype that increases mutational diversity, adaptability, and clonal survival, foreshadowing the genomic instability we see with loss of SETD2.
In this regard, H3K36me3 has recently been shown to play important roles in DNA repair (Aymard et al., 2014; Li et al., 2013), and the absence of H3K36me3 is associated with chromatin organizational changes (Simon et al., 2014). These studies suggested a protective role for SETD2 in maintaining genomic stability, and identified a requirement for H3K36me3 in double strand break resection and homologous recombination repair (Carvalho et al., 2014; Pfister et al., 2014), as well as nucleosome stabilization and suppression of replication stress (Kanu et al., 2015). However, a role for mitotic defects resulting from loss of SETD2 functioning as a driver of genomic instability is a previously unappreciated mechanism by which loss of this methyltransferase may contribute to cancer development and progression. In this regard, clear cell and papillary RCC have relatively low mutational loads (Alexandrov et al., 2013; Lawrence et al., 2013), suggesting an alternative mechanism may exist by which SETD2 maintains genomic stability. Our data point to an additional, and heretofore unknown mechanism by which SETD2 participates in genomic stability via its function as a microtubule methyltransferase, and implicate the SRI domain of SETD2 in its function as an α-tubulin methyltransferase. The SRI domain of SETD2 binds the phosphorylated c-terminal domain of Pol II (Kizer et al., 2005; Rebehmed et al., 2014), and pathogenic mutations in the SRI domain of SETD2 have been seen in cancers such as RCC and acute lymphoblastic leukemia (Cancer Genome Atlas Research, 2013, Mar et al., 2014). While deletion of the SRI domain abrogates SETD2 histone methyltransferase activity, less is known regarding the impact of SRI missense mutations. Data that a pathogenic SRI mutation causes loss of microtubule, but not histone methylation, is potentially significant, as it suggests that loss of microtubule methylation may participate in tumorigenesis even in cells that retain H3K36me3 of chromatin.
In conclusion, the discovery that SETD2 is a dual-function methyltransferase for histones and microtubules, sets the stage for future studies identifying the ‘readers and erasers’ of K40me3 on microtubules, and determining whether other epigenetic ‘readers, writers and erasers’ also impinge on the cytoskeleton. These studies now open the door to understanding regulation and cross-talk between the cytoskeleton and epigenome, and the role of SETD2 in both normal and pathophysiological conditions such as cancer.
EXPERIMENTAL PROCEDURES
Cell culture and generation of Setd2-null Setd2flox/flox mouse embryonic fibroblasts (MEFs)
Setd2flox/flox MEFs were generated from 13.5 days postcoitum (d.p.c.) embryos of Setd2flox/flox mice. The cells were spontaneously immortalized via serial passaging. The immortalized cells were transfected with an ER-Cre vector expressing Cre recombinase fused with a mutated ligand-binding domain for the human estrogen receptor (ER-Cre). Cells expressing this ER-Cre were cultured in phenol-red free media (DMEM, high glucose, HEPES, no phenol red) supplemented with Sodium Pyruvate and GlutaMAX (ThermoFisher Scientific), to prevent any spontaneous activation of the Cre recombinase by estrogenic compounds found in phenol red containing media. Stable cell lines expressing ER-Cre were generated by selection using 5μg/ml blasticidin (Thermo Fisher Scientific). Parental Setd2flox/flox MEFs treated with 4-hydroxytamoxifen (the active metabolite of tamoxifen, Sigma-Aldrich) and Setd2flox/flox MEFs transfected with ER-Cre, treated with vehicle (0.01% ethanol) were used to controls. Setd2flox/flox MEFs expressing ER-Cre were treated with 3μM 4-hydroxytamoxifen for three to five days for efficient Setd2 knockout.
Generation of anti-α-TubK40me3 antibody
The α-TubK40me3 antibody was generated in rabbit using trimethylated K40 peptide (Ac-GQMPSD(KMe3)TIGGGDC-amide) conjugated to KLH as an immunogen (Covance). α-TubK40me3 specific antibody was purified using serial columns coupled with unmethylated and trimethylated K40 peptide.
Immunocytochemistry
Cells were cultured on coverslips and immediately fixed using 4% paraformaldehyde solution in PEM/PEG buffer (80mM PIPES (pH7.0), 1mM EGTA, 1mM MgCl2, 4% w/v PEG 8000) at room temperature for 30 mins (we failed to observe a robust signal for methylation of microtubules using a shorter, more conventional 15 mins fixation), followed by permeabilization using a 0.5% Triton X-100 solution in PEM/PEG buffer for 30 mins. We found nonspecific chromatin immunostaining with α-TubK40me3 antibody was reduced by fixation at 37°C using pre-warmed 4% paraformaldehyde solution in PEM/PEG buffer. See Supplemental Experimental Procedures for details.
Statistics
Statistical significance was determined as indicated in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Dynamic microtubules are methylated during mitosis and cytokinesis
SETD2 methylates α-tubulin at lysine 40, the same lysine that is acetylated
Loss of microtubule methylation causes genomic instability
Acknowledgments
We thank Dr. Claire Walczak (U Indiana) for valuable discussions concerning mitotic defects, Tia Berry and Xuefei Tong for technical assistance, Austin Hepperla (UNC-Chapel Hill) for designing the pipeline used to align the RNAseq data, and Dr. Sung Yun Jung (Baylor College of Medicine) for mass spectrometry analysis. This work was supported in part by the Robert E. Welch Foundation (BE-0023), the National Institutes of Health (RC2ES018789, R01ES008263, R01ES023206 and P30ES023512) and the Cancer Prevention and Research Institute of Texas (CPRIT) (DP150086) grants to CLW; R01GM070862 to KV; R01 CA198482-01 to WKR & IJD and a V Foundation for Cancer Research award (T2012-008) to WKR; R01CA166447 to IJD; T32 GM008719 and NRSA award F30 CA192643-02 to CCF; CPRIT RP110471 to MTB; K12CA90628 and Gerstner Family Career Development Award to THH; and NIH/NCI grant P30CA016672 to EJ. The authors have no conflicting financial interests.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one movie and can be found with this article online at.
AUTHOR CONTRIBUTIONS
Conceptualization, C.L.W. and I.Y.P.; Methodology, C.L.W., I.Y.P., R.T.P.; Validation, D.N.T., K.E.H., Y-C.C.; Formal Analysis, R.T.P.; Investigation, I.Y.P., R.T.P., D.N.T., I.J.D., C.C.F., K.E.H., T.L.B., L.B., Y-C.C.; Resources, T.H.H., W.K.R., K.E.H., Y-C.C., Writing-Original Draft, C.L.W. and I.Y.P.; Writing-Review and Editing, R.D., C.L.W., I.Y.P., R.T.P.; Visualization, R.T.P. and I.Y.P.; Supervision, C.L.W., R.D., K.V., W.K.R., M.T.B., E.J.; Project Administration, C.L.W., I.Y.P., R.T.P., R.D.; Funding Acquisition, C.L.W., T.H.H., K.V., W.K.R., M.T.B. and E.J.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agromayor M, Martin-Serrano J. Knowing when to cut and run: mechanisms that control cytokinetic abscission. Trends in cell biology. 2013;23:433–441. doi: 10.1016/j.tcb.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nature structural & molecular biology. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barisic M, Silva e Sousa R, Tripathy SK, Magiera MM, Zaytsev AV, Pereira AL, Janke C, Grishchuk EL, Maiato H. Mitosis. Microtubule detyrosination guides chromosomes during mitosis. Science. 2015;348:799–803. doi: 10.1126/science.aaa5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi M, Zhao S, Said JW, Merino MJ, Adeniran AJ, Xie Z, Nawaf CB, Choi J, Belldegrun AS, Pantuck AJ, et al. Genomic characterization of sarcomatoid transformation in clear cell renal cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:2170–2175. doi: 10.1073/pnas.1525735113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho S, Vitor AC, Sridhara SC, Martins FB, Raposo AC, Desterro JM, Ferreira J, de Almeida SF. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. eLife. 2014;3:e02482. doi: 10.7554/eLife.02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zang J, Kappler J, Hong X, Crawford F, Wang Q, Lan F, Jiang C, Whetstine J, Dai S, et al. Structural basis of the recognition of a methylated histone tail by JMJD2A. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:10818–10823. doi: 10.1073/pnas.0704525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. The EMBO journal. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, Liu XY, Sturm D, Korshunov A, Jones DT, Witt H, Kool M, Albrecht S, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta neuropathologica. 2013;125:659–669. doi: 10.1007/s00401-013-1095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D. Limiting the proliferation of polyploid cells. Cell. 2007;131:437–440. doi: 10.1016/j.cell.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RA, Paluch E, Oegema K. Cytokinesis in animal cells. Annual review of cell and developmental biology. 2012;28:29–58. doi: 10.1146/annurev-cellbio-101011-155718. [DOI] [PubMed] [Google Scholar]
- Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee KA, Yang V, Aguiar M, Kornhauser J, Jia X, et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Molecular & cellular proteomics: MCP. 2014;13:372–387. doi: 10.1074/mcp.O113.027870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK, et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:3259–3267. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TH, Kapur P, Joseph RW, Serie DJ, Eckel-Passow JE, Tong P, Wang J, Castle EP, Stanton ML, Cheville JC, et al. Loss of histone H3 lysine 36 trimethylation is associated with an increased risk of renal cell carcinoma-specific death. Mod Pathol. 2016;29:34–42. doi: 10.1038/modpathol.2015.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TH, Park IY, Zhao H, Tong P, Champion MD, Yan H, Monzon FA, Hoang A, Tamboli P, Parker AS, et al. High-resolution profiling of histone h3 lysine 36 trimethylation in metastatic renal cell carcinoma. Oncogene. 2015;35:1565–74. doi: 10.1038/onc.2015.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Sun XJ, Zhang YL, Kuang Y, Hu CQ, Wu WL, Shen SH, Du TT, Li H, He F, et al. Histone H3 lysine 36 methyltransferase Hypb/Setd2 is required for embryonic vascular remodeling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:2956–2961. doi: 10.1073/pnas.0915033107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nature communications. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C. The tubulin code: molecular components, readout mechanisms, and functions. The Journal of cell biology. 2014;206:461–472. doi: 10.1083/jcb.201406055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kanu N, Gronroos E, Martinez P, Burrell RA, Yi Goh X, Bartkova J, Maya-Mendoza A, Mistrik M, Rowan AJ, Patel H, et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene. 2015;34:5699–708. doi: 10.1038/onc.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac M, Navas C, Horswell S, Salm M, Bardella C, Rowan A, Stares M, Castro-Giner F, Fisher R, de Bruin EC, et al. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nature communications. 2015;6:6336. doi: 10.1038/ncomms7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok BH, Kapoor TM. Microtubule flux: drivers wanted. Current opinion in cell biology. 2007;19:36–42. doi: 10.1016/j.ceb.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Latham JA, Dent SY. Cross-regulation of histone modifications. Nature structural & molecular biology. 2007;14:1017–1024. doi: 10.1038/nsmb1307. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010;142:682–685. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Mao G, Tong D, Huang J, Gu L, Yang W, Li GM. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, Testa JR, Yang H. The roles of chromatin-remodelers and epigenetic modifiers in kidney cancer. Cancer Genet. 2015;208:206–214. doi: 10.1016/j.cancergen.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Fu Q, An H, Chang Y, Zhang W, Zhu Y, Xu L, Xu J. Decreased Expression of SETD2 Predicts Unfavorable Prognosis in Patients With Nonmetastatic Clear-Cell Renal Cell Carcinoma. Medicine (Baltimore) 2015;94:e2004. doi: 10.1097/MD.0000000000002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiera MM, Janke C. Investigating tubulin posttranslational modifications with specific antibodies. Methods in cell biology. 2013;115:247–267. doi: 10.1016/B978-0-12-407757-7.00016-5. [DOI] [PubMed] [Google Scholar]
- Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, Neuberg DS, Sinha AU, Sallan SE, Silverman LB, et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nature communications. 2014;5:3469. doi: 10.1038/ncomms4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. The EMBO journal. 2002;21:6820–6831. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CJ, Santos-Rosa H, Kouzarides T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell. 2006;126:905–916. doi: 10.1016/j.cell.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Perdiz D, Mackeh R, Pous C, Baillet A. The ins and outs of tubulin acetylation: more than just a post-translational modification? Cellular signalling. 2011;23:763–771. doi: 10.1016/j.cellsig.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ, Helleday T, Legube G, La Thangue NB, Porter AC, et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell reports. 2014;7:2006–2018. doi: 10.1016/j.celrep.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebehmed J, Revy P, Faure G, de Villartay JP, Callebaut I. Expanding the SRI domain family: a common scaffold for binding the phosphorylated C-terminal domain of RNA polymerase II. FEBS Lett. 2014;588:4431–4437. doi: 10.1016/j.febslet.2014.10.014. [DOI] [PubMed] [Google Scholar]
- Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M, Ho TH, Kuan PF, Jonasch E, Furey TS, et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome research. 2014;24:241–250. doi: 10.1101/gr.158253.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Brady ST. Post-translational modifications of tubulin: pathways to functional diversity of microtubules. Trends in cell biology. 2015;25:125–136. doi: 10.1016/j.tcb.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Gaertig J. The tubulin code. Cell cycle. 2007;6:2152–2160. doi: 10.4161/cc.6.17.4633. [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nature reviews Molecular cell biology. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak CE, Heald R. Mechanisms of mitotic spindle assembly and function. International review of cytology. 2008;265:111–158. doi: 10.1016/S0074-7696(07)65003-7. [DOI] [PubMed] [Google Scholar]
- Yuan W, Xie J, Long C, Erdjument-Bromage H, Ding X, Zheng Y, Tempst P, Chen S, Zhu B, Reinberg D. Heterogeneous nuclear ribonucleoprotein L Is a subunit of human KMT3a/Set2 complex required for H3 Lys-36 trimethylation activity in vivo. The Journal of biological chemistry. 2009;284:15701–15707. doi: 10.1074/jbc.M808431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, Yan X, Wei W, Pang Y, Cheng H, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nature genetics. 2014;46:287–293. doi: 10.1038/ng.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.