

Abstract
Ischemia-reperfusion (I/R) injury is important in the pathogenesis and/or progression of various diseases, including stroke, cardiovascular disease and acute renal injury. Increasing evidence indicates that atorvastatin exerts protective effects in I/R injury-associated diseases; however, the underlying mechanisms remain to be fully elucidated. In the present study, oxygen-glucose deprivation (OGD)/reperfusion-stimulated. RAW264.7 murine macrophages served as a model of I/R injury. The knockdown of peroxisome proliferator activated receptor-γ (PPARγ) expression in these cells increased OGD/reperfusion-induced expression of inducible nitric oxide synthase (iNOS), tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ), and enhanced OGD/reperfusion-induced downregulation of the expression of cluster of differentiation (CD) 206, at the mRNA and protein levels. Conversely, overexpression of PPARγ significantly attenuated OGD/reperfusion-induced alterations in the expression of iNOS, TNF-α, IFN-γ and CD206 at the mRNA and protein levels. Notably, atorvastatin inhibited OGD/reperfusion-induced iNOS expression and reversed OGD/reperfusion-induced downregulation of the expression of CD206 and PPARγ at the mRNA and protein levels. The results of the present study indicate that atorvastatin exhibits significant anti-inflammatory effects in OGD/reperfusion-stimulated RAW264.7 cells, possibly via PPARγ regulation. The findings of the present study may reveal a novel mechanism underlying the protective effects of atorvastatin in I/R injury-associated diseases.
Keywords: atorvastatin, peroxisome proliferator activated receptor-γ, oxygen-glucose deprivation/reperfusion
Introduction
Ischemia-reperfusion (I/R) injury-associated diseases, including stroke, cardiovascular disease and acute renal failure are among the leading causes of mortality or disability worldwide (1,2). I/R injury is a complex pathophysiological process (3) in which a variety of factors are important, including the production of reactive oxygen species (ROS), calcium overload and mitochondrial dysfunction (3,4). Increasing evidence indicates that I/R injury induces the production of numerous cytokines and chemokines, which are key for I/R-induced injury or cell death (3,5).
Renal I/R injury represents a major cause of acute renal injury (ARI) (6). In ARI, macrophages, immune cells that are widely distributed in tissues and organs (7), rapidly enter the site of injury and produce effector molecules, which initiate the inflammatory response and recruit other immune cells (8). Accumulating evidence suggests that inflammatory mediators produced by macrophages are critical for the pathogenesis of I/R injury in ARI (8–10). High levels of pro-inflammatory cytokines, including inducible nitric oxide synthase (iNOS), interleukin (IL)-1β, IL-6 and tumor necrosis factor-α (TNF-α) are present in ARI patients (10,11). Notably, gene polymorphisms and the levels of certain cytokines may have predictive value in clinical ARI (11,12). Furthermore, inhibition of macrophage migration to the site of injury or cytokine production are protective against ARI (9).
Macrophages are polarized into various distinct populations according to the specific microenvironment (7,8). Following stimulation by lipopolysaccharide (LPS) and interferon-γ (IFN-γ), macrophages differentiate into the classically activated pro-inflammatory (M1) subtype, which upregulate chemokine receptor 7 and produce iNOS, TNF-α and other pro-inflammatory cytokines involved in tissue injury (13–16). Conversely, following stimulation with IL-4 and IL-13, macrophages differentiate into the alternatively activated (M2) subtype, which are responsible for anti-inflammatory cytokine production and tissue repair. M2 macrophages are characterized by high expression of cluster of differentiation 206 (CD206; the mannose receptor) and production of chitinase 3-like 3, arginase-1 and transforming growth factor-β (13–16). In ARI, M1 and M2 macrophage phenotypes are significant in kidney injury and repair (17): M1 macrophages contribute to I/R injury, while M2 macrophages alleviate I/R injury and promote tissue repair (17–19). Collectively, these results indicate that regulation of the balance of M1 and M2 macrophages may represent a promising therapeutic strategy for the treatment of ARI.
Currently, no effective treatments for ARI are available. The 3-hydroxy-3-methylglutaryl-coenzyme reductase inhibitors (statins), including atorvastatin, are commonly used to treat high cholesterol, and additionally reduce the risk of stroke and other cardiovascular complications of type 2 diabetes (20). Recent studies have suggested that atorvastatin may exert a beneficial effect in kidney-associated diseases, including ARI (21,22). Although the underlying molecular mechanisms remain to be fully elucidated, atorvastatin has been demonstrated to reduce LPS-induced upregulation of its receptor, Toll-like receptor 4 (23). In addition, our previous study in an experimental rat I/R model suggested that atorvastatin attenuates renal injury partially via its anti-inflammatory effects (24). However, the direct anti-inflammatory effects of atorvastatin in I/R-induced macrophages remain unclear. In the present study, oxygen-glucose deprivation (OGD)/reperfusion-stimulated RAW264.7 murine macrophages were used as a model of I/R injury to investigate the anti-inflammatory effects of atorvastatin, in order to develop novel therapies for treatment of I/R associated disorders.
Materials and methods
Antibodies and chemicals
The primary antibodies raised in rabbits recognizing CD206 (catalog no., sc-48758), PPARγ (catalog no., sc-7196) and GAPDH (catalog no., sc-25778), and the secondary antibodies, CruzFluor™ 488-conjugated goat anti-rabbit IgG (catalog no., sc-362262) and CruzFluor™ 594-conjugated goat anti-rabbit IgG (catalog no., sc-362282) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Rabbit antibodies against IFN-γ (catalog no., ab198801), iNOS (catalog no., ab15323) and TNF-α (catalog no., ab6671) were purchased from Abcam (Cambridge, MA, USA). Chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless indicated otherwise.
Cell culture
RAW264.7 murine macrophages were obtained from the American Type Cell Culture Collection (Manassas, VA, USA) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1% penicillin/streptomycin at 37°C in a humidified 5% CO2 atmosphere.
Overexpression/knockdown of PPARγ
The pBabe-puro-PPARγ expression vector was constructed as described previously (25). Control and PPARγ expression vectors were packaged into retroviruses by transient transfection of ecotropic packaging cells (Phoenix™; Allele Biotechnology, San Diego, CA, USA). RAW264.7 macrophages were infected with equal titers of recombinant retrovirus at 50% confluence and stable cell lines were selected with 2 µg/ml puromycin. For knockdown of PPARγ expression in RAW264.7 cells, the retroviral vector-mediated short hairpin RNA against PPARγ (5′-TAATGTGGAGTAGAAATGCTGTTGATATCCGCAGCATTTCTACTCCACATTA-3′) was cloned into pRNAT-H1.1/adeno vectors (catalog no., SD1213; GenScript, New Brunswick, NJ, USA) and recombinant lentiviruses were produced by cotransfecting 293T cells with the lentivirus expression plasmid and packaging plasmids (psPAX2, catalog no., 12260; pMD2.G, catalog no., 12259, both Addgene, Cambridge, MA, USA) using the calcium phosphate method (26). Cells with stable knockdown of expression were acquired following transfection of the plasmid into cells and selection of stable cells with 1.5 µg/ml G418.
OGD/reperfusion
OGD/reperfusion was conducted as described previously (27). Briefly, to achieve OGD, cells were incubated in a humidified anaerobic chamber (Reming Bioinstruments Co., Redfield, NY, USA) with an atmosphere of 5% CO2 and 95% N2 for 60 min. The culture medium was replaced three times (every 20 min each time) with a glucose-free, balanced salt solution containing 116 mmol/l NaCl, 1.8 mmol/l CaCl2, 0.8 mmol/l MgSO4, 5.4 mmol/l KCl, 1 mmol/l NaH2PO4, 14.7 mmol/l NaHCO3 and 0.1 mol/L4- (2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.4). For reoxygenation, OGD cells were incubated with medium containing 5.5 mg/ml glucose in an atmosphere of 5% CO2 and 95% air at 37°C for 60 min. Control cells were incubated in a normoxic incubator in medium supplemented with 5.5 mmol/l glucose at 37°C for 120 min.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
The mRNA expression levels of iNOS, CD206, PPARγ, TNF-α and IFN-γ were detected by RT-qPCR. Briefly, total RNA was extracted using RNAiso Plus (Takara Bio, Inc., Otsu, Japan) according to the manufacturer's instructions and cDNA was synthesized using a Bio Reverse Transcription kit (Takara Bio, Inc.). RT-qPCR was performed using the SYBR Green kit of the Bio Reaction System according to the manufacturer's instructions. The primer sequences were as follows: Sense, 5′-CCAAGAACGTGTTCACCATG-3′ and anti-sense, 5′-GATGTCCAGGAAGTAGGTGAGG-3′ for iNOS; sense, 5′-CTAAGCCAAGGGGCAACC-3′ and anti-sense, 5′-GAACAGCGACCGGAATCAC-3′ for CD206; sense, 5′-CCTCCCTGATGAATAAAGATGG-3′ and anti-sense, 5′-GCAAACTCAAACTTAGGCTCCA-3′ for PPARγ; sense, 5′-CGTGGAACTGGCAGAAGAGG-3′ and anti-sense, 5′-AGACAGAAGAGCGTGGTGGC-3′ for TNF-α; sense, 5′-AGCAACAACATAAGCGTCAT-3′ and anti-sense, 5′-CCTCAAACTTGGCAATACTCA-3′ for IFN-γ; and sense, 5′-GCCATGTACGTAGCCATCCA-3′ and anti-sense, 5′-GAACCGCTCATTGCCGATAG-3′ for β-actin. Thermal cycling parameters for the amplification were as follows: A denaturation step at 94°C for 2 min, followed by 40 cycles at 95°C for 20 sec, 58°C for 20 sec and 72°C for 20 sec. Relative expression levels of target genes were calculated with Mx3000P (Agilent Technologies, Inc., Santa Clara, CA, USA) according to the expression of 2-∆∆Cq (28).
Administration of atorvastatin and grouping of cells
To evaluate the effect of atorvastatin on the PPARγ associated pathways, PPARγ knockdown RAW264.7 cells were administrated with atorvastin as follows: i) CTRL group, control group, normal RAW264.7 cells; ii) Atorvastatin group, RAW264.7 cells incubated with 10 µM atorvastatin (Haoran Biological Technolgy Co., Ltd., Shanghai, China) for 24 hat room temperature; iii) Model group, RAW264.7 cells underwent OGD/R treatment; iv) Model + atorvastatin group, OGD/R treated RAW264.7 cells incubated with 10 µM atorvastatin for 24 h at room temperature. Subsequently, the expression of PPARγ associated indicators was detected using RT-qPCR, western blotting and immunofluorescence staining.
Western blot analysis
Cellular proteins were extracted using the Total Protein Extraction kit according to the manufacturer's instructions (Wanleibio, Shenyang, China) and analyzed by western blotting. GAPDH was used as internal reference protein. Briefly, 40 µg denatured protein in 20 µl solution samples were loaded onto a 13% SDS-PAGE gel, electrophoresed for 2.5 h at 80 V and transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Following blocking with non-fat milk, membranes were incubated overnight at 4°C with antibodies recognizing iNOS (1:1,000), CD206 (1:200), PPARγ (1:1,000), TNF-α (1:100), IFN-γ (1:1,000) and GAPDH (1:5,000), and incubated with the anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:20,000, diluted in 55 (M/V) skim milk powder). Protein detection was performed using Enhanced Chemilumiscence Advance Western Blotting Detection reagents (GE Healthcare Life Sciences, Chalfont, UK).
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline, permeabilized with 0.5% Triton X-100 and then blocked with 5% normal goat serum. The cells were then incubated with antibodies recognizing iNOS, CD206, PPARγ, TNF-α and IFN-γ in 1% bovine serum albumin (Gibco; Thermo Fisher Scientific, Inc.) at 4°C overnight. The bound antibodies were detected by the secondary antibodies conjugated to CruzFluor 488 or CruzFluor 594. Cell nuclei were indicated by staining with DAPI for 15 min. Images were captured on a Zeiss fluorescence microscope (ICM-405; Carl Zeiss AG, Oberkochen, Germany) at magnification, ×400.
Statistical analysis
Data are presented as the mean ± standard deviation. Statistically significant differences between the groups were identified by one-way analysis of variance followed by paired Student's t-test. P<0.05 was considered to indicate a statistically significant difference. All statistical analyses were performed using SPSS version 19.0 (IBM SPSS, Armonk, NY, USA).
Results
Involvement of PPARγ in OGD/reperfusion-induced alteration of iNOS, CD206, TNF-α and IFN-γ mRNA expression levels
To investigate whether PPARγ is involved in OGD/reperfusion-induced expression of inflammatory mediators in RAW264.7 murine macrophages, knockdown and overexpression of PPARγ in RAW264.7 cells was performed. As presented in Fig. 1A, PPARγ protein expression levels in RAW264.7 cells overexpressing PPARγ were increased compared with cells that received the vehicle control. Conversely, as presented in Fig. 1B, PPARγ protein expression levels in RAW264.7 cells with PPARγ knocked down were decreased compared with cells that received the vehicle control.
Figure 1.

Protein expression levels of PPARγ in PPARγ overexpressed and knockdown RAW264.7 murine macrophages. Western blotting detected (A) the stable overexpression of PPARγ in RAW264.7 cells and (B) knockdown of PPARγ. GAPDH served as a loading control. PPARγ, peroxisome proliferator activated receptor-γ; shRNA, short hairpin RNA; NC, group transfected with control non-targeting shRNA.
OGD/reperfusion-stimulated RAW264.7 murine macrophages were used as a model of I/R injury to examine the effect of OGD on the expression of iNOS, TNF-α, IFN-γ and CD206. As presented in Fig. 2, exposure of RAW264.7 macrophages to 1 h of OGD followed by 1 h of reperfusion induced the upregulation of iNOS, TNF-α and IFN-γ, as well as the downregulation of CD206 at the mRNA level. Overexpression of PPARγ significantly attenuated OGD/reperfusion-induced expression of iNOS, TNF-α and IFN-γ at the mRNA levels (Fig. 2A-C; for iNOS, Model+PPARγ group vs. Model group, P<0.001, Model+PPARγ-shRNA group, P<0.001; for TNF-α, Model+PPARγ group vs. Model group, P<0.001, Model+PPARγ-shRNA group, P<0.001; for IFN-γ, Model+PPARγ group vs. Model group, P<0.001, Model+PPARγ-shRNA group, P<0.001). Furthermore, overexpression of PPARγ significantly increased the expression of CD206, a marker of M2 macrophages (Fig. 2D; Model+PPARγ group vs. Model group, P=0.005, Model+PPARγ-shRNA group, P<0.001).
Figure 2.

Effects of overexpression and knockdown of PPARγ on OGD/reperfusion-induced expression of (A) iNOS, (B) TNF-α, (C) IFN-γ and (D) CD206 at the mRNA level in RAW264.7 murine macrophages. mRNA expression levels were analyzed by reverse transcription-quantitative polymerase chain reaction in cells cultured under normal conditions (control), and in model (OGD/R treated), PPARγ overexpressing and PPARγ knockdown cells that had undergone OGD/reperfusion. The mRNA expression levels of iNOS, TNF-α, IFN-γ and CD206 were determined. PPARγ overexpression significantly decreased the expression of iNOS, TNF-α and IFN-γ, and significantly increased the expression of CD206 at the mRNA level compared with model cells. PPARγ knockdown had significant and opposite effects. Data are presented as the mean ± standard deviation (n=3). **P<0.01 vs. model. PPARγ; peroxisome proliferator activated receptor-γ; OGD, oxygen-glucose deprivation; iNOS, inducible nitric oxide synthase; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ; CD206, cluster of differentiation 206; Ctrl, control; shRNA, short hairpin RNA.
By contrast, knockdown of PPARγ expression significantly increased OGD-induced expression of iNOS, TNF-α and IFN-γ mRNA levels (Fig. 2A-C; P=0.008, P=0.009, P=0.006 for iNOS, TNF-α and IFN-γ, respectively), and significantly decreased OGD-induced downregulation of CD206 mRNA expression levels (Fig. 2D; P=0.009, P=0.007, P=0.007 for iNOS, TNF-α and IFN-γ, respectively). Together, these data demonstrate that PPARγ is important for the regulation of OGD/reperfusion-induced effects on the mRNA expression levels of iNOS, TNF-α, IFN-γ and CD206.
Involvement of PPARγ in OGD/reperfusion-induced alterations of iNOS, CD206, TNF-α and IFN-γ protein expression levels
To evaluate whether PPARγ is essential for OGD/reperfusion-induced alterations of iNOS, CD206, TNF-α and IFN-γ protein expression levels, western blotting was performed. OGD/reperfusion induced the upregulation of iNOS, TNF-α and IFN-γ, and the downregulation of CD206, at the protein level (Fig. 3). Notably, overexpression of PPARγ in RAW264.7 cells abrogated the OGD/reperfusion-induced expression of iNOS, TNF-α and IFN-γ. In addition, western blotting indicated that overexpression of PPARγ reversed the OGD/reperfusion-induced downregulation of CD206 expression (Fig. 3). By contrast, knockdown of PPARγ expression increased OGD-induced expression of iNOS, TNF-α and IFN-γ protein, and decreased the OGD-induced downregulated expression of CD206 protein (Fig. 3).
Figure 3.
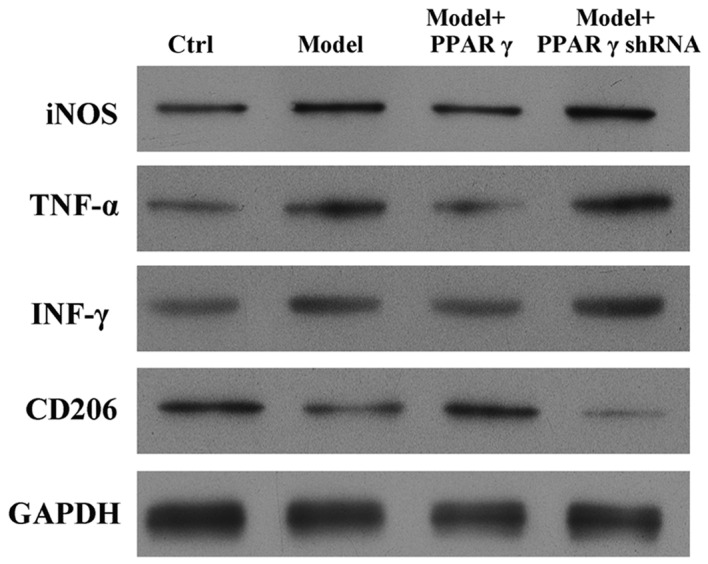
Effects of overexpression and knockdown of PPARγ on OGD/reperfusion-induced expression of iNOS, TNF-α, IFN-γ and CD206 at the protein level in RAW264.7 murine macrophages. Protein expression was detected by western blotting in cells cultured under normal conditions (control), and in model (OGD/R treated), PPARγ overexpressing and PPARγ knockdown cells that had undergone OGD/reperfusion. PPARγ overexpression decreased the expression of iNOS, TNF-α and IFN-γ, and increased the expression of CD206 at the protein level compared with model cells. PPARγ knockdown had the opposite effects. GAPDH served as a loading control. PPARγ; peroxisome proliferator activated receptor-γ; OGD, oxygen-glucose deprivation; iNOS, inducible nitric oxide synthase; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ; CD206, cluster of differentiation 206; Ctrl, control; shRNA, short hairpin RNA.
Furthermore, immunofluorescence was performed to evaluate the effect of PPARγ on OGD/reperfusion-induced alterations in iNOS, CD206, TNF-α and IFN-γ expression levels. As presented in Fig. 4, overexpression of PPARγ reversed OGD/reperfusion-induced expression of iNOS, TNF-α and IFN-γ, and downregulation of CD206. By contrast, knockdown of PPARγ expression enhanced OGD/reperfusion-induced upregulation of iNOS, TNF-α and IFN-γ, and downregulated the expression of CD206. Collectively, these data suggest that overexpression of PPARγ reversed OGD/reperfusion-induced effects on iNOS, CD206, TNF-α and IFN-γ. Conversely, knockdown of PPARγ expression enhanced the OGD/reperfusion-induced effects on iNOS, CD206, TNF-α and IFN-γ.
Figure 4.
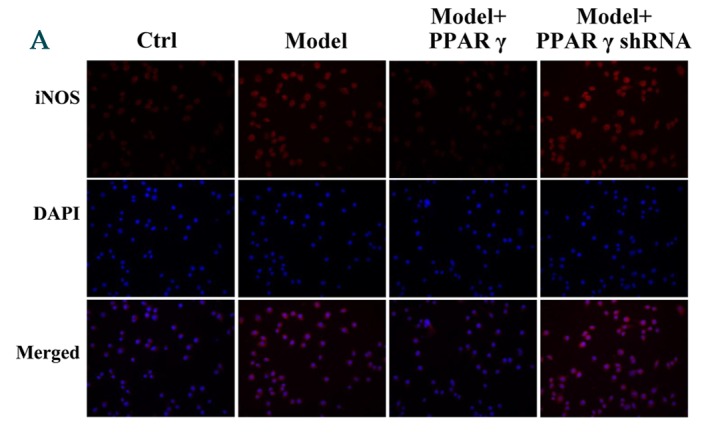
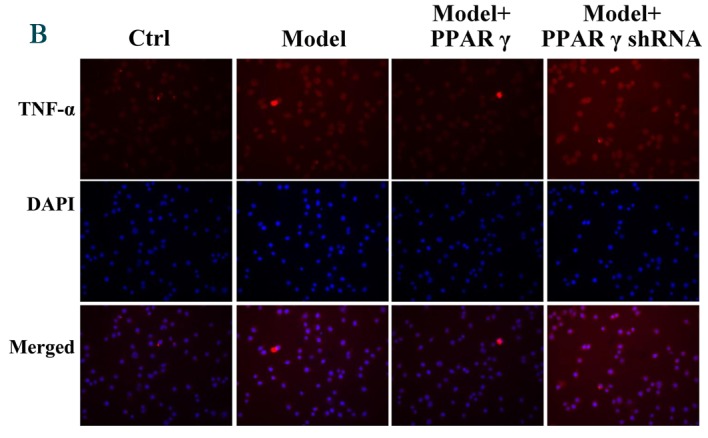
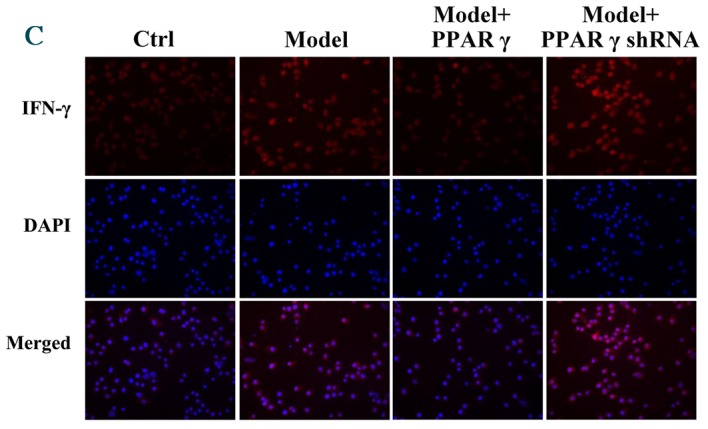
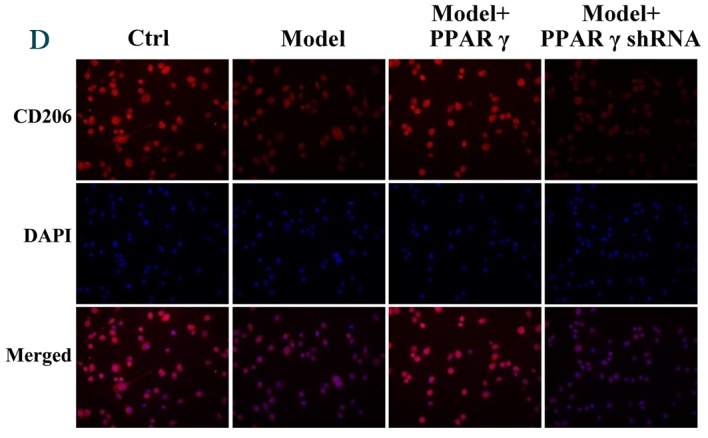
Immunofluorescence staining detected effects of overexpression and knockdown of PPARγ on OGD/reperfusion-induced expression of (A) iNOS, (B) TNF-α, (C) IFN-γ and (D) CD206 in RAW264.7 murine macrophages. Cells were cultured under normal conditions (control), and in model (OGD/R treated), PPARγ overexpressing and PPARγ knockdown cells that had undergone OGD/reperfusion. Cells were stained with antibodies recognizing iNOS, TNF-α, IFN-γ and CD206 and nuclei were stained with DAPI. Representative images from at least three independent experiments are presented (magnification, ×400). PPARγ; peroxisome proliferator activated receptor-γ; OGD, oxygen-glucose deprivation; iNOS, inducible nitric oxide synthase; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ; CD206, cluster of differentiation 206; Ctrl, control; shRNA, short hairpin RNA.
Effects of atorvastatin on the mRNA expression levels of iNOS and CD206 in cells that had undergone OGD/reperfusion
A previous study suggested that PPARγ is a target of atorvastatin (29). In the present study, PPARγ was demonstrated to be important for OGD/reperfusion-induced alterations in the expression of iNOS, TNF-α, IFN-γ and CD206. Thus, it was investigated whether atorvastatin may regulate the OGD/reperfusion-induced alterations in PPARγ, iNOS and CD206 expression. OGD/reperfusion was demonstrated to downregulate PPARγ mRNA expression levels; atorvastatin reversed this effect (Fig. 5A), and the difference between the model group and the model + atorvastatin group was significant (P<0.001). In addition, atorvastatin significantly inhibited OGD/reperfusion-induced upregulation of the mRNA expression levels of iNOS (Fig. 5B; P<0.001) and reversed OGD/reperfusion-induced downregulation of the mRNA expression levels of CD206 (Fig. 5C; P=0.007).
Figure 5.
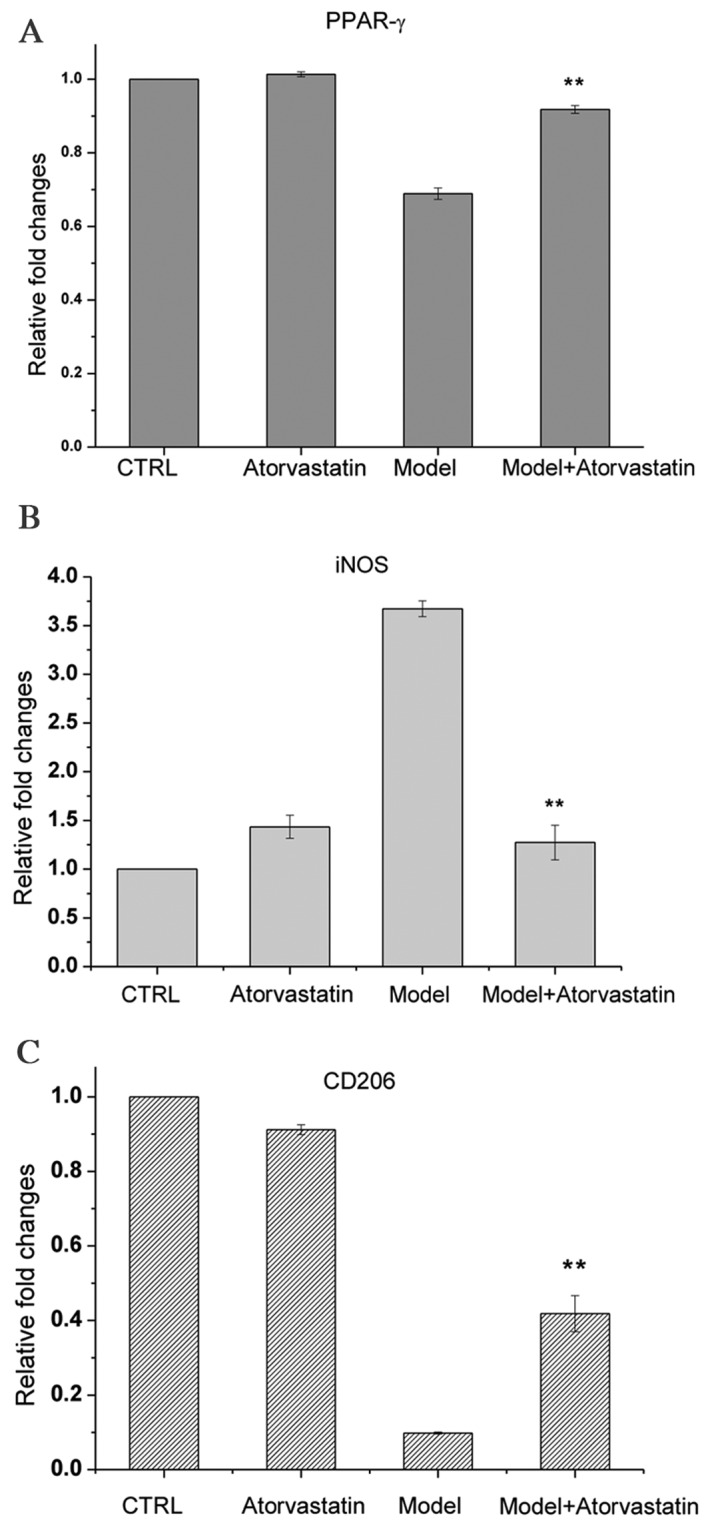
Effects of atorvastatin on the mRNA expression levels of (A) PPARγ, (B) iNOS and (C) CD206 in RAW264.7 murine macrophages that had undergone OGD/reperfusion. mRNA expression levels were analyzed by reverse transcription-quantitative polymerase chain reaction in cells cultured under normal conditions in the absence (control) or presence (atorvastatin) of atorvastatin, and in the model (OGD/R treated) and atorvastatin-treated (model+atorvastatin) cells that had undergone OGD/reperfusion. The mRNA expression levels of PPARγ, iNOS and CD206 were determined. Atorvastatin treatment significantly abrogated the OGD/reperfusion-induced increase in iNOS and significantly reversed the OGD/reperfusion-induced decrease in CD206 and PPARγ. Data are presented as the mean ± standard deviation (n=3). **P<0.01 vs. model. PPARγ; peroxisome proliferator activated receptor-γ; OGD, oxygen-glucose deprivation; iNOS, inducible nitric oxide synthase; CD206, cluster of differentiation 206; Ctrl, control.
Effects of atorvastatin on the protein expression of iNOS and CD206 in cells that had undergone OGD/reperfusion
It was investigated whether atorvastatin may regulate the OGD/reperfusion-induced alterations in iNOS and CD206 protein expression. Consistent with the results of the RT-qPCR, western blotting indicated that atorvastatin reversed OGD/reperfusion-induced downregulation of PPARγ expression at the protein level, blocked OGD/reperfusion-induced upregulation of iNOS protein expression and reversed OGD/reperfusion-induced downregulation of CD206 protein expression (Fig. 6). These results were confirmed by immunofluorescence staining (Fig. 7). Taken together, these results suggest that atorvastatin is an important regulator of OGD/reperfusion-induced alterations in iNOS and CD206 expression at the mRNA and protein levels, and that the underlying mechanism may involve PPARγ.
Figure 6.
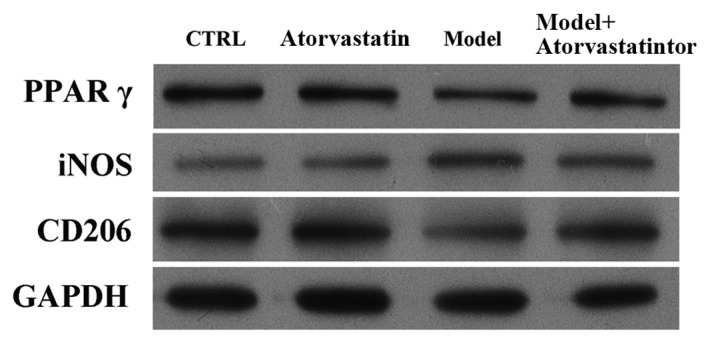
Effects of atorvastatin on the protein expression levels of PPARγ, iNOS and CD206 in RAW264.7 murine macrophages that had undergone OGD/reperfusion. Protein expression was detected by western blotting in cells cultured under normal conditions in the absence (control) or presence (atorvastatin) of atorvastatin, and in model (OGD/R treated) and atorvastatin-treated (model+atorvastatin) cells that had undergone OGD/reperfusion. Atorvastatin treatment significantly abrogated the OGD/reperfusion-induced increase in iNOS and significantly reversed the OGD/reperfusion-induced decrease in CD206 and PPARγ. GAPDH served as a loading control. PPARγ; peroxisome proliferator activated receptor-γ; OGD, oxygen-glucose deprivation; iNOS, inducible nitric oxide synthase; CD206, cluster of differentiation 206; Ctrl, control.
Figure 7.

Immunofluorescencestaining detected the effects of atorvastatin on the protein expression levels of (A) PPARγ, (B) iNOS and (C) CD206 in RAW264.7 murine macrophages that had undergone OGD/reperfusion. Cells were cultured under normal conditions in the absence (control) or presence (atorvastatin) of atorvastatin, and in model (OGD/R treated), and atorvastatin-treated (model + atorvastatin) cells that had undergone OGD/reperfusion. Cells were then stained with antibodies recognizing PPARγ, iNOS and CD206 and nuclei were stained with DAPI. Representative images from at least three independent experiments are presented (magnification, ×400). PPARγ; peroxisome proliferator activated receptor-γ; OGD, oxygen-glucose deprivation; iNOS, inducible nitric oxide synthase; CD206, cluster of differentiation 206; Ctrl, control.
Discussion
The results of the present study demonstrate that overexpression of PPARγ attenuates the OGD/reperfusion-induced upregulation of iNOS, TNF-α and IFN-γ expression, and the downregulation of CD206 expression in RAW264.7 murine macrophages. Conversely, knockdown of PPARγ expression enhanced OGD/reperfusion-induced alterations in iNOS, TNF-α, IFN-γ and CD206 expression. Notably, atorvastatin reversed OGD/reperfusion-induced alterations in iNOS and CD206 expression at the mRNA and protein levels; the underlying mechanism may be via the regulation of PPARγ expression.
Macrophages are dynamically regulated by their microenvironment, and may differentiate into the pro-inflammatory M1 subtype or the anti-inflammatory M2 subtype depending on the stimuli (16). These two types play important roles in the production of cytokines and chemokines and are key for the regulation of I/R injury in various types of disease, including stroke (30), ARI (17) and cardiovascular disease (31). PPARγ belongs to the nuclear receptor superfamily, and is central to the regulation of lipid and glucose metabolism, adipogenesis, glucose homeostasis, cellular differentiation, apoptosis and inflammation (32,33). Notably, PPARγ is important for regulating macrophage phenotype and the production of cytokines (34,35). However, the roles of I/R injury in the induction of cytokine production by macrophages and PPARγ in regulating I/R-induced cytokine production and macrophage phenotype remain to be fully elucidated. In the present study, OGD/reperfusion-stimulated RAW264.7 murine macrophages were used as a model of I/R injury. OGD/reperfusion upregulated the expression of iNOS, TNF-α, IFN-γ and downregulated the expression of CD206, a typical M2-type macrophage marker. Overexpression of PPARγ abrogated the OGD/reperfusion-induced alterations of iNOS, TNF-α, IFN-γ and CD206 expression levels. Conversely, knockdown of PPARγ expression enhanced the OGD/reperfusion-induced alterations of iNOS, TNF-α, IFN-γ and CD206 expression levels. These results suggest that PPARγ may be essential for regulating OGD/reperfusion-induced release of pro-inflammatory cytokines in RAW264.7 cells. Notably, PPARγ regulates the OGD/reperfusion-induced alterations in the expression of the M1 macrophage marker, iNOS and the M2 macrophage marker, CD206. However, whether OGD/reperfusion induced the phenotype changes of macrophages and whether PPARγ has a role in macrophage phenotype regulation requires further investigation.
Recent studies have suggested that atorvastatin may have a beneficial effect in kidney-associated diseases, including ARI (21,22). However, the protective effects of atorvastatin in I/R injury-associated diseases and the underlying mechanisms remain to be fully elucidated. Our previous study in an experimental rat I/R model indicated that atorvastatin attenuates renal injury partially via its anti-inflammatory effects (24). However, the effect of atorvastatin on the OGD/reperfusion-induced production of inflammatory cytokines, and the underlying mechanism, remain unclear. It is well-established that atorvastatin activates PPARγ in various cell types. Grip et al (36) observed that atorvastatin activates PPAR-γ and attenuates the inflammatory response in human monocytes. Additionally, Planavila et al (37) demonstrated that atorvastatin improved peroxisome proliferator-activated receptor signaling in cardiac hypertrophy. In the present study, PPARγ was demonstrated to regulate OGD/reperfusion-induced alterations in the expression levels of iNOS, CD206, TNFα and IFNγ. Thus, the roles and underlying molecular mechanisms of atorvastatin in the regulation of OGD/reperfusion-induced alterations in iNOS and CD206 expression were investigated. OGD/reperfusion was revealed to significantly downregulate PPARγ expression at the mRNA and protein levels, an effect that was reversed by atorvastatin. Notably, atorvastatin significantly inhibited the OGD/reperfusion-induced upregulation of iNOS expression and downregulation of CD206 expression. Together these results demonstrated that atorvastatin regulates OGD/reperfusion-induced inflammation, and that the underlying mechanisms may involve PPARγ. In conclusion, the findings of the present study may reveal a novel mechanism underlying the protective effects of atorvastatin in I/R injury-associated diseases by the targeting of OGD/reperfusion-induced stimulation of macrophages. The present study is a supplement to the understanding of the mechanism driving OGD/R injuries and also provides potential for the development of therapies for the treatment of OGD/reperfusion-induced inflammation in the clinic.
Acknowledgements
The present study was supported by Guangzhou Medical Key Subject Construction Project (grant no., XM203190, 2013–2015).
References
- 1.Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng ZJ, Flegal K, O'Donnell C, Kittner S, et al. Heart disease and stroke statistics-2006 update: A report from the American heart association statistics committee and stroke statistics subcommittee. Circulation. 2006;113:e85–e151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 2.Ricci Z, Cruz D, Ronco C. The RIFLE criteria and mortality in acute kidney injury: A systematic review. Kidney Int. 2008;73:538–546. doi: 10.1038/sj.ki.5002743. [DOI] [PubMed] [Google Scholar]
- 3.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. doi: 10.1016/B978-0-12-394309-5.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Groot H, Rauen U. Ischemia-reperfusion injury: Processes in pathogenetic networks: A review. Transplant Proc. 2007;39:481–484. doi: 10.1016/j.transproceed.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 5.Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol. 2006;17:1503–1520. doi: 10.1681/ASN.2006010017. [DOI] [PubMed] [Google Scholar]
- 6.Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380:756–766. doi: 10.1016/S0140-6736(11)61454-2. [DOI] [PubMed] [Google Scholar]
- 7.Varol C, Mildner A, Jung S. Macrophages: Development and tissue specialization. Annu Rev Immunol. 2015;33:643–675. doi: 10.1146/annurev-immunol-032414-112220. [DOI] [PubMed] [Google Scholar]
- 8.Cao Q, Harris DC, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 2015;30:183–194. doi: 10.1152/physiol.00046.2014. [DOI] [PubMed] [Google Scholar]
- 9.Bonventre JV, Zuk A. Ischemic acute renal failure: An inflammatory disease? Kidney Int. 2004;66:480–485. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 10.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simmons EM, Himmelfarb J, Sezer MT, Chertow GM, Mehta RL, Paganini EP, Soroko S, Freedman S, Becker K, Spratt D, et al. Plasma cytokine levels predict mortality in patients with acute renal failure. Kidney Int. 2004;65:1357–1365. doi: 10.1111/j.1523-1755.2004.00512.x. [DOI] [PubMed] [Google Scholar]
- 12.Jaber BL, Rao M, Guo D, Balakrishnan VS, Perianayagam MC, Freeman RB, Pereira BJ. Cytokine gene promoter polymorphisms and mortality in acute renal failure. Cytokine. 2004;25:212–219. doi: 10.1016/j.cyto.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 14.Kawanishi N, Yano H, Yokogawa Y, Suzuki K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc Immunol Rev. 2010;16:105–118. [PubMed] [Google Scholar]
- 15.Mantovani A, Sica A, Locati M. New vistas on macrophage differentiation and activation. Eur J Immunol. 2007;37:14–16. doi: 10.1002/eji.200636910. [DOI] [PubMed] [Google Scholar]
- 16.Goerdt S, Politz O, Schledzewski K, Birk R, Gratchev A, Guillot P, Hakiy N, Klemke CD, Dippel E, Kodelja V, Orfanos CE. Alternative versus classical activation of macrophages. Pathobiology. 1999;67:222–226. doi: 10.1159/000028096. [DOI] [PubMed] [Google Scholar]
- 17.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22:317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Jiang ZP, Su N, Fan JJ, Ruan YP, Peng WX, Li YF, Yu XQ. The role of peritoneal alternatively activated macrophages in the process of peritoneal fibrosis related to peritoneal dialysis. Int J Mol Sci. 2013;14:10369–10382. doi: 10.3390/ijms140510369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jo SK, Sung SA, Cho WY, Go KJ, Kim HK. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol Dial Transplant. 2006;21:1231–1239. doi: 10.1093/ndt/gfk047. [DOI] [PubMed] [Google Scholar]
- 20.Meier CR, Schlienger RG, Kraenzlin ME, Schlegel B, Jick H. HMG-CoA reductase inhibitors and the risk of fractures. JAMA. 2000;283:3205–3210. doi: 10.1001/jama.283.24.3205. [DOI] [PubMed] [Google Scholar]
- 21.Lewicki M, Ng I, Schneider AG. HMG CoA reductase inhibitors (statins) for preventing acute kidney injury after surgical procedures requiring cardiac bypass. Cochrane Database Syst Rev. 2015:CD010480. doi: 10.1002/14651858.CD010480.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zamorskiĭ II, Zeleniuk VG. Renoprotective effects of statins under the conditions of acute renal failure, caused by rhabdomyolysis. Biofizika. 2014;59:1027–1030. (In Russian) [PubMed] [Google Scholar]
- 23.Wang Y, Zhang MX, Meng X, Liu FQ, Yu GS, Zhang C, Sun T, Wang XP, Li L, Wang YY, et al. Atorvastatin suppresses LPS-induced rapid upregulation of Toll-like receptor 4 and its signaling pathway in endothelial cells. Am J Physiol Heart Circ Physiol. 2011;300:H1743–H1752. doi: 10.1152/ajpheart.01335.2008. [DOI] [PubMed] [Google Scholar]
- 24.Wu K, Lei W, Tian J, Li H. Atorvastatin treatment attenuates renal injury in an experimental model of ischemia-reperfusion in rats. BMC Nephrol. 2014;15:14. doi: 10.1186/1471-2369-15-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-X. [DOI] [PubMed] [Google Scholar]
- 26.Chen CA, Okayama H. Calcium phosphate-mediated gene transfer: A highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- 27.Guo J, Krause DN, Horne J, Weiss JH, Li X, Duckles SP. Estrogen-receptor-mediated protection of cerebral endothelial cell viability and mitochondrial function after ischemic insult in vitro. J Cereb Blood Flow Metab. 2010;30:545–554. doi: 10.1038/jcbfm.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Brault M, Ray J, Gomez YH, Mantzoros CS, Daskalopoulou SS. Statin treatment and new-onset diabetes: A review of proposed mechanisms. Metabolism. 2014;63:735–745. doi: 10.1016/j.metabol.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 30.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 31.Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:1120–1126. doi: 10.1161/ATVBAHA.112.300173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tontonoz P, Spiegelman BM. Fat and beyond: The diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 33.Lamers C, Schubert-Zsilavecz M, Merk D. Therapeutic modulators of peroxisome proliferator-activated receptors (PPAR): A patent review (2008-present) Expert Opin Ther Pat. 2012;22:803–841. doi: 10.1517/13543776.2012.699042. [DOI] [PubMed] [Google Scholar]
- 34.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 35.Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23:351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 36.Grip O, Janciauskiene S, Lindgren S. Atorvastatin activates PPAR-γ and attenuates the inflammatory response in human monocytes. Inflammation Research. 2002;51:58–62. doi: 10.1007/BF02684000. [DOI] [PubMed] [Google Scholar]
- 37.Planavila A, Laguna JC, Vázquez-Carrera M. Atorvastatin improves peroxisome proliferator-activated receptor signaling in cardiac hypertrophy by preventing nuclear factor-κB activation. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2005;1687:76–83. doi: 10.1016/j.bbalip.2004.11.004. [DOI] [PubMed] [Google Scholar]