

Abstract
Rab5a, a key member of the Rab family of GTPases, was determined to be a regulator of vascular smooth muscle cell (VSMC) proliferation and migration. However, the exact regulatory mechanism remains unclear. As Rab5a has been shown to be associated with autophagy, which is essential for the conversion of VSMCs from a contractile to a synthetic phenotype in order to prevent cell death due to oxidative stress. The present study hypothesized that autophagy may be responsible for the proliferation and migration of VSMCs via the Rab5a protein. The aim of the present study was to evaluate the effect of Rab5a on autophagy in VSMCs. The human aorta vascular smooth muscle cell line, T/G HA-VSMCs, was treated with small interfering (si)RNA against Rab5a and/or platelet-derived growth factor (PDGF). Following treatment, the phenotype transition of the VSMCs was evaluated by detecting the mRNA and protien expression levels of VSMC molecular markers using reverse transcription-quantitative polymerase chain reaction and western blotting, respectively. In addition, autophagy in VSMCs was evaluated by western blotting for autophagy-associated proteins, flow cytometry of acidic vesicular organelles, punctate fluorescence of microtubule associated protein light chain 3 and transmission electron microscopy of typical scattered double-membrane vacuolar structures. Additionally, the proliferation, migration, cell cycle and apoptotic response of VSMCs were detected by sulforhodamine B assay, transwell assay and flow cytometry, respectively. The results revealed that transfection with siRNA against Rab5a led to a significant decrease in Rab5a protein expression, while the reduced expression trend of Rab5a was rescued by intervention with PDGF. Furthermore, cells transfected with siRNA against Rab5a inhibited the autophagy of VSMCs. Downregulated Rab5a inhibited the phenotype transition of VSMCs. Additionally, downregulated Rab5a led to slowed cell growth, decreased numbers of migrated cells, decreased numbers of cells at the G0-G1 phase and a higher apoptosis rate. However, PDGF significantly rescued these phenomena caused by siRNA against Rab5a. These results indicated that Rab5a-mediated autophagy may regulate the phenotype transition and cell behavior of VSMCs through the activation of the extracellular-regulated kinase 1/2 signaling pathway.
Keywords: Rab5a, vascular smooth muscle cells, autophagy, phenotype transition, ERK1/2 signaling pathway
Introduction
Vascular smooth muscle cells (VSMCs), as the primary cellular components of the normal blood vessel wall, are associated with the integrity of vascular structure, regulation of vascular function and stability of vascular lesions (1). Notably, VSMCs are considered to be vital to the initiation and progression of atherosclerotic lesions (2). In response to trauma and altered hemodynamics of vascular reconstruction, the phenotypic modulation of VSMCs from a contractile to a synthetic phenotype is observed during atherogenesis, which promotes not only the migratory and proliferative capacity of VSMCs, but also the synthesis of extracellular matrix proteins (3,4). Therefore, it is essential to understand the molecular mechanism underlying the phenotype transition and cell behavior of VSMCs.
Rab proteins are Ras-related small GTPases, which may regulate exocytic and endocytic membrane trafficking by vesicle docking and fusion (5,6). As a key member of the Ras GTPase superfamily, Rab5a has been shown to be associated with not only the heterozygous fusion of early endosomes and endocytic vesicles, but also the homomorphous fusion between early endosomes (7). Ravikumar et al (8) suggested that Rab5a can promote autophagosome formation, indicating that Rab5a is associated with autophagy. In addition, Rab5a may influence the morphogenesis and metastasis of various cancer types, including breast cancer, cervical cancer, ovarian cancer and hepatocellular carcinoma (9–12). As the pathogenesis of intimal hyperplasia is somewhat similar to neoplasia, Rab5a may also be involved in the intimal hyperplasia and arterial restenosis. A previous study indicated that Rab5a is involved in VSMC proliferation and migration (13), while autophagy induced by platelet-derived growth factor (PDGF) serves an essential role in the conversion of VSMCs from the contractile to synthetic phenotype in order to prevent cell death due to oxidative stress (14). Therefore, the present study hypothesized that autophagy may be responsible for the proliferation and migration of VSMCs, and that Rab5a was essential in this process.
In the present study, a human aorta vasuclar smooth muscle cell line, referred to as T/G HA-VSMCs, was treated with small interfering (si)RNA against Rab5a and/or PDGF, and the phenotype transition and cell behaviors, including proliferation, cell cycle, migration, apoptosis and autophagy, were assessed. The present study aimed to reveal the effects of Rab5a on autophagy in VSMCs, and whether the phenotype transition and cell behaviors of VSMCs are accompanied by autophagy.
Materials and methods
Cell culture and treatment
T/G HA-VSMCs were obtained from American Type Culture Collection (Rockefeller, MD, USA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), penicillin (100 U/ml) and streptomycin (100 mg/ml) at 37°C with 5% CO2. The cells were transfected with control siRNA (siC), Rab5a siRNA (siR; a pool of four siRNAs; Dharmacon Research, Lafayette, CO, USA), siC combined with PDGF (siC + P; 20 ng/ml; R&D Biosystems, Minneapolis, MN, USA) and siR combined with PDGF (siR + P; 20 ng/ml) prior to experiments. Transfection was performed using DharmaFECT transfection reagent in serum-free medium (GE Healthcare Life Sciences, Chalfont, UK) following manufacturer's protocol.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Following treatment with siRNA and/or PDGF for 24 h, the total RNA from cells was obtained using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. The RNA (25 nM) was subsequently reverse transcribed using the RevertAid First-Strand cDNA Synthesis kit (Fermentas, Vilnius, Lithuania). PCR amplification was performed with the SYBR Green Premix Ex Taq kit (Takara Bio., Inc., Dalian, China). Primer sequences are listed in Table I. The PCR program included denaturation at 95°C for 10 sec, amplification with 40 cycles at 95°C for 5 sec and 60°C for 31 sec, and a final 2 min extension at 72°C. Finally, the 2−ΔΔCq method (15) was used to calculate the expression levels of genes, normalized against the levels of GAPDH.
Table I.
Primer sequences.
| Gene | Primer sequence (5′→3′) |
|---|---|
| Rab5a | |
| Forward | CAGTTCAAACTAGTACTTCTGG |
| Reverse | GCTAGGCTATGGTATCGTTCTTG |
| α-SMA | |
| Forward | GCGTGGCTATTCCTTCGTTA |
| Reverse | ATGAAGGATGGCTGGAACAG |
| Calponin | |
| Forward | AGCTAAGAGAAGGGCGGAAC |
| Reverse | CATCTGCAGGCTGACATTGA |
| Vimentin | |
| Forward | AAAACACCCTGCAATCTTTCAGA |
| Reverse | CACTTTGCGTTCAAGGTCAAGAC |
| Osteopontin | |
| Forward | GGACAGCCGTGGGAAGG |
| Reverse | TCAATCACATCGGAATGCTCA |
| GAPDH | |
| Forward | GCACCGTCAAGGCTGAGAAC |
| Reverse | TGGTGAAGACGCCAGTGGA |
α-SMA, α-smooth muscle actin.
Western blotting analysis
Following treatment for 48 h with siRNA and/or PDGF, the cells were lysed in radioimmunoprecipitation buffer (Beyotime Institute of Biotechnology, Inc., Jiangsu, China) containing 1 mM phenylmethanesulfonyl fluoride. The supernatants were acquired by centrifugation at 13,440 × g for 15 min at 4°C. The proteins (30 µg/lane) were separated and subsequently transferred onto a polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat milk at room temperature for 2 h. Following blocking, the membranes were incubated with specific primary antibodies overnight at 4°C and then incubated with horserdish peroxidase-conjugated secondary antibodies (cat. nos. A0181; A0216; A0208; 1:1,000; Beyotime Institute of Biotechnology, Inc.) at room temperature for 2 h. The antibodies used were as follows: anti-Rab5a (cat. no. 2143; 1:1,000), anti-survivin (cat. no. 2803), anti-caspase-3 (cat. no. 9662; 1:1,000), anti-Beclin1 (cat. no. 3738; 1:1,000), anti-Light chain (LC)3-I/II (cat. no. 12741; 1:1,000), anti-phosphorylated (p-) (cat. no. 4060; 1:1,000) and total (t-)protein kinase B (Akt) (cat. no. 4691; 1:1,000), anti-p-mammalian target of rapamycin (mTOR; cat. no 5536; 1:1,000) and anti-t-mTOR (cat. no. 2972; 1:1,000), and p-extracellular signal-regulated kinase (ERK cat. no. 4370; 1:1,000) and t-ERK (cat. no. 4695; 1:1,000; all from Cell Signaling Technology, Inc., Danvers, MA, USA. Anti-α-smooth muscle actin (SMA; cat. no. SAB2500963; 0.5 µg/ml) and anti-calponin (cat. no. C2687; 1:10,000) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Anti-osteopontin (cat. no. sc-10591; 1:200) was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA), and anti-GAPDH (cat. no. AF0006; 1:1,000) was purchased from Beyotime Institute of Biotechnology, Inc. Following antibody incubations, an enhanced chemiluminescence assay (PerkinElmer, Waltham, MA, USA) was used to detect the proteins. GAPDH was used as a loading control.
Cell proliferation assay
The proliferation of VSMCs was detected using a sulforhodamine B (SRB) assay. A total of 3.0×103 T/G HA-VSMCs were seeded into 96-well plates. The next day, the cells were assigned into the following four groups: i) siC group, cells treated with control siRNA; ii) siC + P group, cells treated with control siRNA and PDGF; iii) siR group, cell treated with Rab5a siRNA; and iv) siR+P group, cells treated with Rab5a siRNA and PDGF. Following treatment for 24, 48 and 72 h, the cells were fixed in 30% (w/v) trichloroacetic acid and were subsequently stained with 0.4% SRB (Sigma-Aldrich) diluted with 1% acetic acid for 30 min at 25°C. The excess SRB was rinsed off using 1% (v/v) acetic acid and the proteins were dissolved in 10 mM Tris solution. Finally, a microplate reader (iMark; Bio-Rad, Hercules, CA, USA) was used to measure the value of SRB at a wavelength of 570 nm.
Migration analysis
The migration of T/G HA-VSMCs was detected using a Transwell system with 24-well inserts and 8 µm pores (Corning Costar, Lowell, MA, USA). Matrigel (30 µl; BD Biosciences, San Jose, CA, USA) was used to coat the inserts and 600 µl complete medium was added into the lower chamber. Following treatment for 48 h, trypsin was used to detach the cells and the cells were resuspended in fresh medium (0.2 ml) without FBS at a concentration of 1×104 cells. These cell suspensions were seeded into the upper chamber. Following incubation for 24 h, the cells in the upper chamber were removed with cotton swabs, while non-migrating cells were gently removed and rinsed out with phosphate-buffered saline (PBS) three times. Cells in the lower chamber were fixed with methanol at room temperature for 30 min and were subsequently stained using hematoxylin at room temperature for 5 min. Migrating cells were counted in six randomly selected representative fields of each insert by light microscopy (BX51; Olympus, Tokyo, Japan).
Cell cycle and apoptosis analysis
Flow cytometry was performed to detect the cell cycle and apoptosis in each group. Following treatment for 48 h, the cells were trypsinized, collected and fixed with pre-cooled 70% ethanol overnight at 4°C. The cells were subsequently centrifuged at 1,120 × g for 10 min at 4°C and the ethanol was washed off with PBS. The cells were trypsinized for 30 min at 37°C. Propidium iodide (PI; 50 µg/ml; Sigma-Aldrich) was used to stain the cells for 30 min at 25°C for cell cycle analysis.
Cell apoptosis analysis was performed using a Vybrant® Apoptosis assay kit (Invitrogen; Thermo Fisher Scientific, Inc.). Following treatment for 48 h, the cells were co-stained with annexin V and PI, according to the manufacturer's protocol. The results were detected using flow cytometry (FACS Calibur; BD Biosciences, Franklin Lakes, NJ, USA). The data regarding cell cycle and apoptosis were acquired using CellQuest 7.0 software (Beckman Coulter, Brea, CA, USA) and subsequently analyzed using ModFit3.0 software (Verity Software House, Maine, ME, USA).
Acridine orange staining assay
To observe the characteristics of autophagic cell death, acidic vesicle organelles (AVOs) were stained with acridine orange (Sigma-Aldrich). Following treatment for 48 h, the cells in different groups were stained with acridine orange (1 µg/ml) for 15 min at 37°C in the dark. The cells were subsequently rinsed with PBS, collected with trypsin and then resuspended in PBS, supplemented with 1% FBS. Finally, the fluorescence emission was detected using CellQuest 7.0 software.
Detection of autophagy with LC3 localization
To further reveal the autophagy of cells in each group, the cells were co-transfected with pEGFP-LC3 (National Institute for Basic Biology, Okazaki, Japan) using Lipofectamine2000 (Invitrogen; Thermo Fisher Scientific, Inc.). Following transfection for 48 h, the cells were fixed in 95% ethanol in the dark and fluorescence was detected by fluorescent microscopy (BX51; Olympus).
Transmission electron microscopy (TEM)
TEM was performed to observe the formation of autophagosomes. Following treatment for 48 h, the cells in each group were fixed in 3% glutaraldehyde at 25°C for 2 h. The cells were subsequently fixed in 1% osmium tetroxide, and were sectioned and embedded in LX112 plastic. Finally, uranyl acetate combined with lead citrate was used to stain the ultrathin sections, and electron micrographs were obtained by TEM (Philips Medical Systems, BG Eindhoven, The Netherlands).
Statistical analysis
Statistical analysis was performed using SPSS version 15.0 software (SPSS Inc., Chicago, IL, USA). All data were expressed as the mean ± standard deviation. Comparisons among different groups were analyzed by one-way analysis of variance, while comparisons between two groups were analyzed by Student's t-test. P≤0.05 was considered to indicate a statistically significantly difference.
Results
Effect of Rab5a siRNA transfection and PDGF on the expression of Rab5a in T/G HA-VSMCs
The knockdown efficiency of Rab5a siRNA transfection and the effects of PDGF on T/G HA-VSMCs were determined using RT-qPCR and western blotting. As a result, transfection with siRNA against Rab5a led to a significant decrease in the mRNA and protein expression levels of Rab5a (P<0.01); however, the expression of Rab5a in the siR + P group was relatively close to the siC group (Fig. 1). These data revealed that Rab5a was effectively downregulated by Rab5a siRNA transfection and the reduced expression trend of Rab5a can be rescued by the intervention of PDGF in T/G HA-VSMCs.
Figure 1.
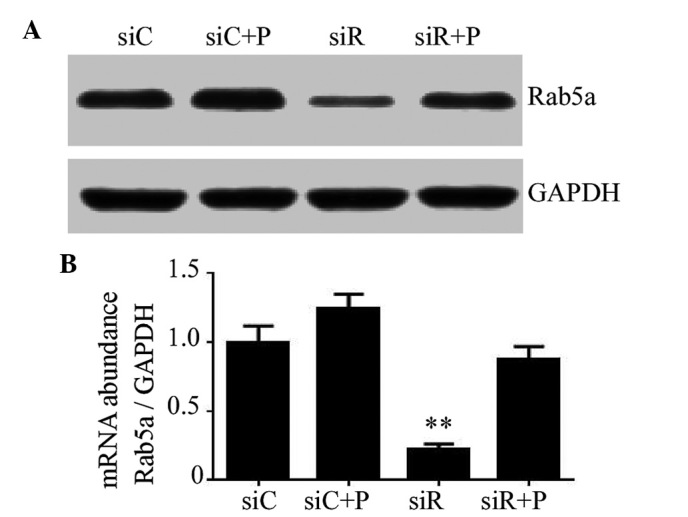
Expression of Rab5a in T/G HA-VSMCs treated in various manners. (A) The protein expression levels of Rab5a were determined by western blotting analysis following various treatments. GAPDH was used as a loading control. (B) The mRNA expression levels of Rab5a were determined by reverse transcription-quantitative polymerase chain reaction analysis following various treatments. The data were quantified and mRNA expression levels were determined relative to the expression of GAPDH. The data are presented as the mean ± standard deviation (**P<0.01 compared with the siC group.) si, siRNA; C, control; R, Rab5a; P, PDGF; PDGF, platelet-derived growth factor; T/G HA-VSMC, human aorta-vascular smooth muscle cell line.
Effect of Rab5a siRNA transfection and PDGF on the phenotype transition of T/G HA-VSMCs
The expression of molecular markers of the T/G HA-VSMC phenotype, including α-SMA, calponin, osteopontin and vimentin, were detected to evaluate the phenotypic transition of T/G HA-VSMCs in different groups. As a result, the mRNA expression levels of α-SMA and calponin were significantly increased, while osteopontin and vimentin were significantly reduced in the siR group compared with the siC group (P<0.01). However, the expression of these proteins was restored to normal levels by the intervention of PDGF, which was relatively close to siC (Fig. 2A). Similarly, the changes in the protein expression levels of α-SMA, calponin and osteopontin were consistent with that of the mRNA level (Fig. 2B). These results indicated that Rab5a may promote the phenotypic transition of T/G HA-VSMCs.
Figure 2.

Expression of molecular markers of the VSMC phenotype following various treatments. (A) The mRNA expression levels of α-sm-actin, Calponin, Osteopontin and Vimentin were determined by reverse transcription-quantitative polymerase chain reaction in T/G HA-VSMCs treated with siC, siR, siC + P, and siR + P. The mRNA expression levels were normalized against that of GAPDH. The data are presented as the mean ± standard deviation (*P<0.05 and **P<0.01 compared with the siC group.) (B) The protein expression levels of α-sm-actin, Calponin and Osteopontin were determined by western blotting analysis. The protein and mRNA expression changes were confirmed to be the same. GAPDH was used as a loading control. si, siRNA; C, control; R, Rab5a; P, PDGF; PDGF, platelet-derived growth factor; T/G HA-VSMC, human aorta-vascular smooth muscle cell line; α-sm-actin, α-smooth muscle actin.
Effect of Rab5a siRNA transfection and PDGF on the proliferation, cell cycle and migration of T/G HA-VSMCs
In addition to the phenotypic transition of T/G HA-VSMCs, the proliferation, cell cycle and migration of T/G HA-VSMCs were investigated in the different groups. Compared with the siC group, the growth of the cells was significantly reduced in the siR group at 24 (P<0.05), 48 (P<0.01) and 72 h (P<0.01). This growth trend was rescued by the intervention of PDGF in the siR + P group (Fig. 3A). According to the cell cycle analysis, the percentage of cells in the siR group was significantly decreased in the G0/G1 phase and increased at the G2/M and S phases; however, the percentage of cells at the G0/G1, G2/M and S phases were not significantly changed in the siR + P group compared with the siC group (Fig. 3B). These data indicated that Rab5a modulated T/G HA-VSMC proliferation at different phases of the cell cycle. In addition, compared with the siC group, the average number of migrated cells in the siR group was revealed to be significantly decreased (P<0.01). However, the percentage of migrated cells was not markedly changed in the siR + P group. Additionally, more migrated cells were observed in the siC + P compared with the siC group (P<0.05; Fig. 3C). These results indicated that the knockdown of Rab5a can inhibit T/G HA-VSMCs migration and that PDGF treatment can reverse the decreased migration level in Rab5a siRNA transfected cells.
Figure 3.

Proliferation, cell cycle and migration in T/G HA-VSMCs following various treatments. (A) A sulforhodamine B assay was used to demonstrate the growth of the cells following treatment that compared with siC, siR, siC + P and siR + P at 24, 48, and 72 h,. (B) Cell cycle analysis was performed by flow cytometry following treatment with siC, siR, siC + P and siR + P. The percentage of apoptotic, G0-G1, G2-M or S phase cells were calculated. (C) Microscopy observation (magnification, ×20) of the cells was performed following migration assay. The percentage of cells migrating was calculated. Quantified data are presented as the mean ± standard deviation (**P<0.01 and *P<0.05 compared with the siC group.) si, siRNA; C, control; R, Rab5a; P, PDGF; PDGF, platelet-derived growth factor; T/G HA-VSMC, human aorta-vascular smooth muscle cell line.
Effect of Rab5a siRNA transfection and PDGF on apoptosis in T/G HA-VSMCs
The early apoptotic rate was quantitatively analyzed by flow cytometry and the results demonstrated that cells transfected with siRNA against Rab5a exhibit a significantly higher apoptotic rate compared with the siC group (P<0.01), while the percentage of apoptotic cells was significantly reduced by the intervention of PDGF (Fig. 4A). In addition, the expression of the apoptotic proteins survivin and caspase-3 was detected by western blotting. These results revealed reduced expression of survivin and increased expression of cleaved caspase-3 in the siR group compared with the siC group, while a combination treatment with PDGF and siRNA (siCon or siRab) transfection did not alter the expression levels of survivin and caspase-3 (Fig. 4B).
Figure 4.
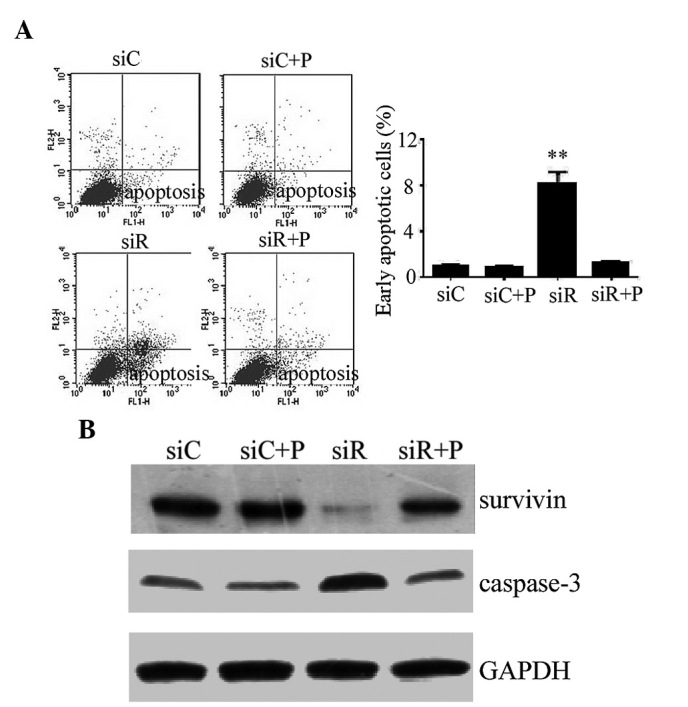
Apoptotic response in T/G HA-VSMCs following various treatments. (A) Flow cytometry was performed to detemin the percentage of cells undergoing apoptosis following treatment with siC, siR, siC + P and siR + P. The data are presented as the mean ± standard deviation (**P<0.01 compared with the siC group.) (B) The protein expression levels of apoptosis-associated proteins, survivin and caspase-3, were determined by western blotting analysis. GAPDH was used as a loading control. si, siRNA; C, control; R, Rab5a; P, PDGF; PDGF, platelet-derived growth factor; T/G HA-VSMC, human aorta-vascular smooth muscle cell line.
Effect of Rab5a siRNA transfection and PDGF on autophagy in T/G HA-VSMCs
The autophagy in T/G HA-VSMCs from each group was first detected by flow cytometry. As a result, the percentage of cells with AVOs were significantly decreased (P<0.01), while autophagy returned to a normal degree following treatment with PDGF (Fig. 5A). In addition, the autophagy in these cells was also analyzed by fluorescence localization of LC3. As a result, the percentage of cells with punctate fluorescence was significantly reduced in the siR group (P<0.05), while no difference was observed in the other three groups (Fig. 5B). Additionally, VSMCs were visualized by TEM to examine the formation of autophagosomes. It was revealed that Rab5a siRNA transfection caused a significantly reduced number of autophagosomes with typical scattered double-membrane vacuolar structures (P<0.05), while the number of phagosomes in cells treated with PDGF remained unchanged compared with the siC group (Fig. 5C). Furthermore, the expression levels of the autophagic proteins Beclin1, LC3-I and LC3-II were also analyzed by western blotting. As an autophagy-related protein, Beclin1 is required for the initiation of autophagy. Additionally, LC3-II formation is also a confirmed marker of autophagy. As the formation of LC3-II is transient and the protein can be rapidly degraded in the lysosome, a conversion of LC3-I to LC3-II may also indicate autophagy in cells. Compared with the siC group, the protein expression of Beclin1 and the conversion of LC3-I to LC3-II was significantly reduced in the siR group, whereas it was elevated to a normal level following treatment with PDGF (Fig. 5D).
Figure 5.

Autophagy in T/G HA-VSMCs following various treatments. (A) Flow cytometry was performed to determine the percentage of cells with AVOs as a sign of autophagy following treatment with siC, siR, siC + P and siR + P. (B) Representative microscopy images (magnification, ×40) of fluorescence localization of LC3 were captured and the percentage of cells with punctate fluorescence was calculated. (C) Transmission electron microscopy (magnification, 1,000x) was performed to determine the number of autophagosomes with typical scattered double-membrane vacuolar structures (arrows). (D) The protein expression levels of Beclin1, LC3 I and LC3 II were determined by western blotting analysis. Quantified data are presented as the mean ± standard deviation (**P<0.01 and *P<0.05 compared with the siC group.) AVOs, acidic vesicular organelles; LC3, light chain 3; si, siRNA; C, control; R, Rab5a; P, PDGF; PDGF, platelet-derived growth factor; T/G HA-VSMC, human aorta-vascular smooth muscle cell line.
Effect of Rab5a siRNA transfection and PDGF on the PI3K/AKT/mTOR and ERK1/2 signaling pathways in T/G HA-VSMCs
To assess the mechanism of Rab5a siRNA transfection-related autophagy, the PI3K/AKT/mTOR and ERK1/2 signaling pathways in T/G HA-VSMCs were assessed by western blotting. The result revealed that compared with the siC group, the expression levels of p-AKT, p-mTOR and p-ERK were markedly reduced in the siR group, while PDGF recovered the normal expression of these proteins. In addition, no differences were observed in the expression levels of t-AKT, t-mTOR and t-ERK among these four groups (Fig. 6). The results suggested that the MAPK/ERK pathway may be involved in Rab5a-related autophagy, which did not occur via the inhibition of the classical PI3K/AKT/mTOR pathway.
Figure 6.
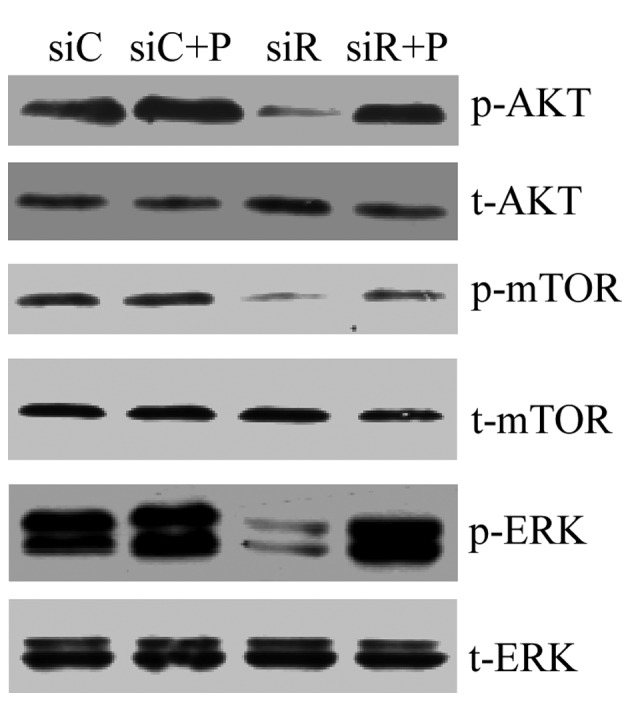
PI3K/AKT/mTOR and ERK1/2 signaling pathways in T/G HA-VSMCs following various treatments. The mRNA expression levels of p-AKT, t-AKT, p-mTOR, t-mTOR, p-ERK and t-ERK were determined by western blotting analysis following treatment with siC, siR, siC + P and siR + P. p-, phosphorylated; t-, total; Akt, protein kinase B; mTOR, mammalian target of rapamycin; ERK, extracellular signal-regulated kinase; si, siRNA; C, control; R, Rab5a; P, PDGF; PDGF, platelet-derived growth factor; T/G HA-VSMC, human aorta-vascular smooth muscle cell line.
Discussion
It is generally accepted that accelerated proliferation and migration of VSMCs are essential for the development of neointimal hyperplasia, which is also a key pathological feature of atherosclerosis and vascular reconstruction with restenosis (16). The present study revealed that downregulated expression of Rab5a inhibited the phenotypic transition, proliferation, migration and autophagy of T/G HA-VSMCs, while it increased apoptosis in T/G HA-VSMCs. However, PDGF treatment reversed these changes. Furthermore, Rab5a-related autophagy may be induced via the ERK pathway, however, not the PI3K/AKT/mTOR pathway.
Previous studies have demonstrated that phenotype transition in VSMCs is regulated by numerous factors, including contractile agonists and extracellular matrix components (17–19). Notably, the present study found that downregulation of Rab5a resulted in increased expression levels of α-SMA and calponin, as well as decreased expression levels of osteopontin and vimentin. These results indicated that Rab5a may be associated with the phenotype transition from contractile to synthetic in VSMCs; however, the underlying mechanism remains unclear. Su et al (20) suggested that Rab5a was associated with autophagy induced by hepatitis C virus non-structural protein 4B, while autophagy was an essential regulator for the phenotype transition of VSMCs (14). Therefore, it was speculated that autophagy may serve a vital role in the effect of Rab5a on the phenotype transition of VSMCs. In addition, it was revealed that PDGF treatment can significantly reverse the phenotype switching of VSMCs caused by Rab5a siRNA transfection. Notably, PDGF is usually overexpressed in atherosclerotic lesions and can promote VSMC proliferation (21,22), which may induce autophagy and phenotype switching in VSMCs (14). These results further indicated that the phenotypic transition of VSMCs induced by Rab5a may be regulated by autophagy.
Furthermore, the present study demonstrated that Rab5a promoted the proliferation and migration of T/G HA-VSMCs. Similarly, our previous study also suggested a critical role for Rab5a in rat VSMC proliferation and migration (13). However, the exact mechanism remains to be elucidated. Autophagy was considered to be the basic catabolic mechanism that is essential for cell survival, differentiation, development and the cellular response to stress via the regulation of the actions of lysosomes (23). Du et al (24) suggested that autophagy can promote angiogenesis by AKT activation in aortic endothelial cells, while increased angiogenesis may promote tube formation and endothelial cell migration (25). In addition, in lung cancer cells, autophagy also facilitated cell proliferation, migration and invasion by increasing the expression levels of various cytokines, including interleukin 6, vascular endothelial growth factor A and matrix metallopeptidase 2 (26). Therefore, the present study speculated that the proliferation and migration of T/G HA-VSMCs induced by Rab5a may also be regulated by autophagy.
The crosstalk between apoptosis and autophagy is very complex. Autophagy is a physiological process accompanied by the degradation of disordered cellular components, while apoptosis is programmed cell death. Under stress circumstances, cells exhibit a stress adaptation by inducing low levels of autophagy, and the intrinsic and extrinsic pathways of apoptosis are inhibited in order to avoid cell death (27). However, in certain circumstances, autophagy acts as a key regulator to activate the programmed cell death pathway, causing massive cell death and eventually leading to functional impairment in vivo (28). In the present study, Rab5a was revealed to inhibit the apoptosis of VSMCs, which exhibited reduced expression of survivin, high expression levels of caspase-3 and a higher apoptosis rate. However, Rab5a was demonstrated to induce autophagy in VSMCs that exhibited significantly reduced expression of Beclin1, to downregulated the conversion of LC3-I to LC3-II, reduce AVOs, LC3 punctate fluorescence and autophagosomes with typical scattered double-membrane vacuolar structures in Rab5a siRNA transfected cells. This may be explained by the fact that autophagy confers cytoprotection to VSMCs. In addition, Shen et al (29) observed that high autophagy levels induced cell death in tumor cells receiving anticancer treatments, while low levels of autophagy may promote cell survival by removing disordered intracellular proteins and organelles (30). These two previous studies indicated that the cell survival or death was associated with the level of autophagy, which may also explain the present results. Therefore, Rab5a-induced autophagy may protect against the onset of apoptosis, and the inhibition of autophagy by Rab5a siRNA transfection can trigger the apoptosis of T/G HA-VSMCs, and subsequently regulate the proliferation and migration of T/G HA-VSMCs.
The present study further examined various signaling pathways involved in autophagy. Previously, the PI3K/AKT/mTOR signaling pathway has been considered to be crucial in the regulation of cellular survival, differentiation, proliferation, metabolism, migration and angiogenesis (31). Additionally, it was also a well-known pathway in the regulation of autophagy in mammalian cells, which can activate autophagy by the inhibition of AKT and mTOR (32,33). However, the present results demonstrated that the expression of phosphorylated AKT and mTOR was reduced by the knockdown of Rab5a, demonstrating that Rab5a-related autophagy was independent of the PI3K/AKT/mTOR pathway. Notably, a reduced expression trend of Rab5a was also observed, to be consistent with the phosphorylation of ERK1/2. The MAPK/ERK pathway served a primary role in the promotion of cellular proliferation in response to growth factors (34) and the induction of autophagy by the activation of the ERK1/2 pathway (35,36). These results indicated that Rab5a-related autophagy correlated with the ERK1/2 pathway; however, not the PI3K/AKT/mTOR pathway.
Rab5a is pivotal for autophagy in VSMCs, and the phenotype transition, proliferation, cell cycle, migration and apoptosis induced by Rab5a may be regulated by autophagy via the activation of the ERK1/2 pathway. The present results supported a critical role for Rab5a in atherogenesis. However, further studies are required to detail the underlying molecular mechanisms of Rab5a-induced autophagy.
Acknowledgements
The authors would like to thank the Electron Microscopy Laboratory of Fudan University for technical assistance in the TEM analysis. This work was supported by the Outstanding Leaders Training Program of Pudong Health Bureau of Shanghai (PWRI2011-02), the Project of Science and Technology Commission of Shanghai Municipality (12jc1408101) and the Shanghai Pudong District Science Project (PKJ2012-Y68).
References
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 2.Pyle AL, Young PP. Atheromas feel the pressure: Biomechanical stress and atherosclerosis. Am J Pathol. 2010;177:4–9. doi: 10.2353/ajpath.2010.090615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones. 2007;39:86–93. [PubMed] [Google Scholar]
- 4.Mack CP. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler Thromb Vasc Biol. 2011;31:1495–1505. doi: 10.1161/ATVBAHA.110.221135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jordens I, Marsman M, Kuijl C, Neefjes J. Rab proteins, connecting transport and vesicle fusion. Traffic. 2005;6:1070–1077. doi: 10.1111/j.1600-0854.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 6.Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: Achieving specificity in membrane traffic. Proc Natl Acad Sci USA. 2006;103:11821–11827. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91:119–149. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ravikumar B, Imarisio S, Sarkar S, O'Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci. 2008;121:1649–1660. doi: 10.1242/jcs.025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang PS, Yin PH, Tseng LM, Yang CH, Hsu CY, Lee MY, Horng CF, Chi CW. Rab5A is associated with axillary lymph node metastasis in breast cancer patients. Cancer Sci. 2011;102:2172–2178. doi: 10.1111/j.1349-7006.2011.02089.x. [DOI] [PubMed] [Google Scholar]
- 10.Liu SS, Chen XM, Zheng HX, Shi SL, Li Y. Knockdown of Rab5a expression decreases cancer cell motility and invasion through integrin-mediated signaling pathway. J Biomed Sci. 2011;18:58. doi: 10.1186/1423-0127-18-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Z, Liu XF, Wu HC, Zou SB, Wang JY, Ni PH, Chen XH, Fan QS. Rab5a overexpression promoting ovarian cancer cell proliferation may be associated with APPL1-related epidermal growth factor signaling pathway. Cancer Sci. 2010;101:1454–1462. doi: 10.1111/j.1349-7006.2010.01558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukui K, Tamura S, Wada A, Kamada Y, Igura T, Kiso S, Hayashi N. Expression of Rab5a in hepatocellular carcinoma: Possible involvement in epidermal growth factor signaling. Hepatol Res. 2007;37:957–965. doi: 10.1111/j.1872-034X.2007.00143.x. [DOI] [PubMed] [Google Scholar]
- 13.Ma Z, Wang H, Wu L, Zhui L, Shi W, Ma D, Chen Z, Yu B. RNAi-mediated Rab5a suppression inhibits proliferation and migration of vascular smooth muscle cells. Acta Cardiol. 2010;65:507–514. doi: 10.1080/ac.65.5.2056236. [DOI] [PubMed] [Google Scholar]
- 14.Salabei JK, Cummins TD, Singh M, Jones SP, Bhatnagar A, Hill BG. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem J. 2013;451:375–388. doi: 10.1042/BJ20121344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Butoi E, Gan AM, Manduteanu I. Molecular and functional interactions among monocytes/macrophages and smooth muscle cells and their relevance for atherosclerosis. Crit Rev Eukaryot Gene Expr. 2014;24:341–355. doi: 10.1615/CritRevEukaryotGeneExpr.2014012157. [DOI] [PubMed] [Google Scholar]
- 17.Garat C, Van Putten V, Refaat ZA, Dessev C, Han SY, Nemenoff RA. Induction of smooth muscle alpha-actin in vascular smooth muscle cells by arginine vasopressin is mediated by c-Jun amino-terminal kinases and p38 mitogen-activated protein kinase. J Biol Chem. 2000;275:22537–22543. doi: 10.1074/jbc.M003000200. [DOI] [PubMed] [Google Scholar]
- 18.Hautmann MB, Thompson MM, Swartz EA, Olson EN, Owens GK. Angiotensin II-induced stimulation of smooth muscle alpha-actin expression by serum response factor and the homeodomain transcription factor MHox. Circ Res. 1997;81:600–610. doi: 10.1161/01.RES.81.4.600. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Zheng J, Du Y, Huang Y, Li J, Liu B, Liu CJ, Zhu Y, Gao Y, Xu Q, et al. Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin. Circ Res. 2010;106:514–525. doi: 10.1161/CIRCRESAHA.109.202762. [DOI] [PubMed] [Google Scholar]
- 20.Su WC, Chao TC, Huang YL, Weng SC, Jeng KS, Lai MM. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J Virol. 2011;85:10561–10571. doi: 10.1128/JVI.00173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myllärniemi M, Calderon L, Lemström K, Buchdunger E, Häyry P. Inhibition of platelet-derived growth factor receptor tyrosine kinase inhibits vascular smooth muscle cell migration and proliferation. FASEB J. 1997;11:1119–1126. doi: 10.1096/fasebj.11.13.9367346. [DOI] [PubMed] [Google Scholar]
- 22.Yu JC, Lokker NA, Hollenbach S, Apatira M, Li J, Betz A, Sedlock D, Oda S, Nomoto Y, Matsuno K, et al. Efficacy of the novel selective platelet-derived growth factor receptor antagonist CT52923 on cellular proliferation, migration, and suppression of neointima following vascular injury. J Pharmacol Exp Ther. 2001;298:1172–1178. [PubMed] [Google Scholar]
- 23.Levine B, Klionsky DJ. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 24.Du J, Teng RJ, Guan T, Eis A, Kaul S, Konduri GG, Shi Y. Role of autophagy in angiogenesis in aortic endothelial cells. Am J Physiol Cell Physiol. 2012;302:C383–C391. doi: 10.1152/ajpcell.00164.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slevin M, Krupinski J, Rovira N, Turu M, Luque A, Baldellou M, Sanfeliu C, de Vera N, Badimon L. Identification of pro-angiogenic markers in blood vessels from stroked-affected brain tissue using laser-capture microdissection. BMC Genomics. 2009;10:113. doi: 10.1186/1471-2164-10-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhan Z, Xie X, Cao H, Zhou X, Zhang XD, Fan H, Liu Z. Autophagy facilitates TLR4-and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2014;10:257–268. doi: 10.4161/auto.27162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: Cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 28.Platini F, Pérez-Tomás R, Ambrosio S, Tessitore L. Understanding autophagy in cell death control. Curr Pharm Des. 2010;16:101–113. doi: 10.2174/138161210789941810. [DOI] [PubMed] [Google Scholar]
- 29.Shen S, Kepp O, Michaud M, Martins I, Minoux H, Métivier D, Maiuri MC, Kroemer RT, Kroemer G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene. 2011;30:4544–4556. doi: 10.1038/onc.2011.168. [DOI] [PubMed] [Google Scholar]
- 30.Amaravadi RK. Autophagy-induced tumor dormancy in ovarian cancer. J Clin Invest. 2008;118:3837–3840. doi: 10.1172/JCI37667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K, et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2011;7:176–187. doi: 10.4161/auto.7.2.14074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956–959. doi: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaul YD, Seger R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem. 2009;284:21412–21424. doi: 10.1074/jbc.M109.026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cagnol S, Chambard JC. ERK and cell death: Mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]