

Abstract
Orai1 is a transmembrane protein that forms homomeric, calcium-selective channels activated by stromal interaction molecule 1 (STIM1) after depletion of intracellular calcium stores. In adult skeletal muscle, depletion of sarcoplasmic reticulum calcium activates STIM1/Orai1-dependent store-operated calcium entry. Here, we used constitutive and inducible muscle-specific Orai1-knockout (KO) mice to determine the acute and long-term developmental effects of Orai1 ablation on muscle structure and function. Skeletal muscles from constitutive, muscle-specific Orai-KO mice exhibited normal postnatal growth and fiber type differentiation. However, a significant reduction in fiber cross-sectional area occurred by 3 mo of age, with the most profound reduction observed in oxidative, fatigue-resistant fiber types. Soleus muscles of constitutive Orai-KO mice exhibited a reduction in unique type I fibers, concomitant with an increase in hybrid fibers expressing both type I and type IIA myosins. Additionally, ex vivo force measurements showed reduced maximal specific force and in vivo exercise assays revealed reduced endurance in constitutive muscle-specific Orai-KO mice. Using tamoxifen-inducible, muscle-specific Orai-KO mice, these functional deficits were found to be the result of the delayed fiber changes resulting from an early developmental loss of Orai1 and not the result of an acute loss of Orai1-dependent store-operated calcium entry.—Carrell, E. M., Coppola, A. R., McBride, H. J., Dirksen, R. T. Orai1 enhances muscle endurance by promoting fatigue-resistant type I fiber content but not through acute store-operated Ca2+ entry.
Keywords: development, exercise, myosin, skeletal muscle
Mammalian skeletal muscle is a highly adaptable tissue that changes in response to input from innervating motor nerves. Each muscle is composed of a heterogeneous population of fibers with differing myosin isoforms and metabolic profiles. Typically, fast-twitch muscles contain a high proportion of fibers that express fast type IIX and type IIB myosins, whereas slow-twitch muscles contain a high proportion of fast type IIA and slow type I myosins. Muscle fibers expressing type IIB and IIX myosins primarily utilize glycolytic metabolism, exhibit lower mitochondrial density, and are more fatigable, while fibers expressing type IIA and type I myosin exhibit primarily oxidative metabolism and higher mitochondrial content, and are more fatigue resistant (1). Under chronic stimulation paradigms, such as during athletic endurance training, relative myosin expression transitions along the following axis: IIB→IIX→IIA→I. In contrast, inactivity results in a reverse pattern of myosin switching from I→IIA→IIX→IIB.
It is well established that in adult muscle calcineurin, a calcium (Ca2+)-dependent serine/threonine protein phosphatase regulates the expression of slow fiber genes in response to chronic, low-frequency motor neuron activity (1). This pattern of activity leads to sustained elevations in cytoplasmic Ca2+ ions that bind calmodulin needed to activate calcineurin (2). Under these conditions, activated calcineurin dephosphorylates the nuclear factor of activated T cell (NFAT) transcription factor, resulting in translocation of NFAT from the myoplasm into the nucleus, ultimately leading to the transcription of genes in the slow fiber program including type I myosin, myoglobin, and troponin I slow (3, 4). However, the precise molecular mechanisms by which chronic, low-frequency motor neuron activity promotes calcineurin-dependent NFAT nuclear localization and activation of the slow fiber muscle program are unclear.
Orai1 is a highly selective Ca2+ entry channel composed of 4 or 6 subunits, each containing 4 transmembrane segments and cytoplasmic N and C termini. As a homomeric channel, Orai1 functions as the calcium release-activated calcium (CRAC) channel that is activated when intracellular Ca2+ stores are depleted. In nonexcitable cells, CRAC channels are activated by direct interaction with the Ca2+ sensor stromal interaction molecule 1 (STIM1) present in the endoplasmic reticulum (ER) membrane. Upon intracellular store depletion, Ca2+ dissociates from the luminal Ca2+ binding EF hand of STIM1, triggering the protein to unfold and oligomerize. STIM1 oligomers then mobilize to regions of the ER membrane closely apposed to the plasma membrane where they interact directly with the N and C termini of Orai1 to drive CRAC channel opening (5). In the presence of extracellular Ca2+, open CRAC channels exhibit a sustained inwardly rectifying current, consistent with high Ca2+-selectivity and the absence of intrinsic voltage gating (6).
Orai1 is also expressed in immature skeletal muscle myotubes and in fully differentiated adult skeletal muscle (7–11). Similar to nonexcitable cells, depletion of the specialized intracellular Ca2+ storage compartment in skeletal muscle, termed the sarcoplasmic reticulum (SR), results in STIM1-dependent activation of a highly Ca2+-selective and inwardly rectifying Orai1 current (12, 13). The physiologic importance of functional CRAC channels in skeletal muscle is underlined by the presence of myopathy in patients with loss-of-function mutations in either STIM1 or ORAI1, while gain-of-function mutations in STIM1 and ORAI1 cause tubular aggregate myopathy (14). Additionally, numerous mouse models have implicated CRAC channels in proper SR Ca2+ store refilling, muscle differentiation, and hypertrophy, as well as in limiting fatigue during sustained muscle activity (8, 15, 16). However, because of the efficient recycling and extremely high buffering capacity of skeletal muscle Ca2+ stores, controversy remains over the role of active CRAC channels in adult muscle (8, 17, 18).
We generated inducible and constitutive muscle-specific Orai1-knockout (KO) mouse models to isolate the acute and long-term developmental effects of Orai1 ablation. Using these models, we found that Orai1 channel activation during muscle use is not required for maximal performance and that profound endurance deficits that occur in the constitutive model can be explained by changes in myosin expression and reduced fatigue-resistant fiber size.
MATERIALS AND METHODS
Generation of muscle-specific Orai-KO mice
The generation of floxed Orai1 mice (Orai1fl/fl; Orai1tm1.2Hjm) has been described previously (19). These mice were crossed to muscle creatine kinase (MCK)-Cre mice on the FVB background (Jackson Laboratory [JAX] strain 006475; The Jackson Laboratory, Bar Harbor, ME, USA) and then back-crossed to the same background as the original Orai1fl/fl mice, Taconic C57BL6/N. All control Orai1fl/fl (wild-type [WT]) and Orai1fl/fl:MCK-Cre [referred to as constitutive Orai1-KO (cOrai1-KO)] mice used in this study were over 98% C57BL6/N. Inducible Orai-KO mice were generated by crossing Orai1fl/lfl mice with muscle-specific, inducible human skeletal actin (HSA) mutated estrogen receptor (Mer)-Cre-Mer (MCM) mice (20). Original HSA-MCM mice were on a C57BL6/J background, and the line was maintained as a mixed C57BL6/N and J mix after breeding with Orai1fl/fl animals. Genotypes were verified using primers: Cre Recombinase (650 bp): (forward) 5′–GCCTGCATTACCGGTCGATGCAACGA–3′, (reverse) 5′–GTGGCAGATGGCGCGGCAACACCATT–3′; flox Orai1 (WT 131 bp, floxed 257 bp, KO 207 bp): (forward) 5′–TATGGTAAGGCTGGGAGACACT–3′, (forward) 5′–GGGACAAAACACTAACCTGTCAT–3′, (reverse) 5′–GGAGTAGAATTCAGTGGGAGAGT–3′.
Tamoxifen preparation and administration
Tamoxifen (Tam); Sigma-Aldrich, St. Louis, MO, USA; T5648) was dissolved in pure corn oil (CO) (Sigma-Alrich; C8267) at 20 mg/ml by shaking overnight in a 37°C dry shaker. Dissolved Tam was stored, protected from light, at 4°C. Four-month-old animals (Orai1fl/fl:HSA-MCM) were given interperitoneal injections on 5 consecutive days with 40 mg/kg/d Tam or an equivalent volume of CO (vehicle) as control.
Animal housing and care
Sex-matched male and female mice were used between 4 and 52 wk of age and were cared for in accordance with the Guide for the Care and Use of Laboratory Animals [National Institutes of Health (NIH), Bethesda, MD, USA]. Mice 4 to 6 wk old were considered young, and mice 3 to 12 mo were categorized as adult. WT/CO matched littermates were used for all experiments. Mice were group housed in sterile ventilated microisolator cages on corncob bedding in an Association for Assessment and Accreditation of Laboratory Animal Care International–accredited facility. Animals were provided ad libitum access to pelleted feed (LabDiet 5010) and water (standard drinking water of Rochester, NY, USA; pH 7.8) via Hydropac. Animals were maintained on a 12:12 h light:dark cycle in rooms at 64–79°F with 30–70% humidity under pathogen-free conditions (Supplemental Table 1).
Immunohistochemistry and histology
Tibialis anterior (TA), extensor digitorum longus (EDL), and soleus (Sol) muscles were mounted in optimal cutting temperature medium and snap frozen in liquid nitrogen–cooled 2-methylbutane. Muscles were cut into 10-µm-thick sections and mounted on SuperFrost Plus slides (Thermo Fisher Scientific, Waltham, MA, USA; Cat. 12-550-15). Sections were incubated with a rabbit anti-dystrophin and mouse anti-type IIb myosin antibodies, followed by goat anti-rabbit Alexa Fluor 488 and goat anti-mouse IgM rhodamine secondary antibodies (Supplemental Table 2). The same sections were then probed with mouse anti-type IIa myosin, followed by goat anti-mouse IgG (H+L) Pacific Blue antibodies. Each incubation was for 1 h at room temperature. Adjacent sections were probed using rabbit antidystrophin and mouse anti-type I myosin antibodies, followed by goat anti-rabbit Alexa Fluor 488 and goat anti-mouse IgG1 secondary antibodies. Fiber boundaries were defined by the dystrophin signal, and cross-sectional area (CSA) was calculated using ImageJ software as described previously (Image Processing and Analysis in Java; NIH) (8). Myosin contributions were analyzed in order of type I > type IIA > type IIB, with any remaining unlabeled fibers classified as IIX. Any fibers labeled with IIA but already classified as type I were considered hybrid I/IIA fibers. Scale bars shown in images represent 100 µm. Similar sections were stained with hematoxylin and eosin to examine gross muscle structure and the presence of centrally nucleated fibers.
Silver stain and Western blot analyses
TA, EDL, and Sol muscles were excised and snap frozen in liquid nitrogen. Silver-stained samples and gels were prepared according to Talmadge and Roy (21). Myosin bands were exposed using the Bio-Rad Silver Stain Plus kit (1610449; Bio-Rad, Hercules, CA, USA). For Western blot analysis, muscles were homogenized in ice-cold RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Igepal CA630, 0.25% sodium deoxycholate, pH 7.4). Samples were centrifuged at 13,000 g for 20 min to remove large debris, and supernatants were retained for analysis. Protein concentrations were determined using the Bio-Rad DC assay (500-0116), and 9 µg whole muscle lysate was loaded onto the gel. All antibodies used (Supplemental Table 2) were diluted in 5% nonfat milk (1706404; Bio-Rad) in TBS-T (20 mM Tris, 150 mM NaCl, 0.1% Tween 20, pH 7.6).
RNA isolation and cDNA preparation
TA, EDL, Sol, and flexor digitorum brevis (FDB) muscles, as well as heart, lung, and spleen, were excised and snap frozen in liquid nitrogen. RNA was isolated using Trizol reagent according to the manufacturer’s protocol (15596-018; Thermo Fisher Scientific). A total of 1 µg RNA was then DNase treated according to manufacturer’s protocol (EN0525; Thermo Fisher Scientific). cDNA libraries were created using the Super Script III First-Strand Synthesis System primed with oligo(dT) (18080-051; Thermo Fisher Scientific).
Transcript quantification
Semiquantitative PCR was performed using 5′ end fluorescein (6-FAM)-labeled forward primers (Integrated DNA Technologies, Coralville, IA, USA) on 10 ng cDNA prepared as described above. Reactions were quantified every 2 cycles from 24 to 34 cycles to verify amplification of both controls [calcium channel, voltage-dependent, L type, α 1S subunit (Cacna1s) for muscle or glyceraldehyde-3-phosphate dehydrogenase (Gapdh) for total] and Orai1 cDNA were within the linear range. Cacna1s (855 bp): (forward) 5′–ATCATCTTCACCCTGGAGATG–3′, (reverse) 5′–TACCCTGTGTGGCAGAACTT–3′; Gapdh (921 bp): (forward) 5′–AGGCCG GTGCTGAGTATGTC–3′, (reverse) 5′–GGGTGCAGCGAACTTTATTGATGG–3′; Orai1 (307 bp): (forward) 5′–TTTAGTGCCTGCACCACAGTGCTA–3′, (reverse) 5′–TGTGGTTGGCGACGATGACTGATT–3′.
Mn2+ quench
Individual FDB fibers were isolated by trituration after a 45 min incubation in 0.1% collagenase A in regular rodent Ringer solution (in mM: 146 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, and 10 HEPES, pH 7.4) at 37°C while shaking at 40 rpm. Store content was passively depleted by incubation in a Ca2+ depletion cocktail (consisting of 15 μM cyclopiazonic acid, 1 μM thapsigargin, and 30 μM N-benzyl-p-toluene sulfonamide) in 0 Ca2+ Ringer (in mM: 145 NaCl, 5 KCl, 3 MgCl2, 10 HEPES, 0.2 EGTA, pH 7.4) for 1 h at 37°C while simultaneously being loaded with 5 µM fura-2 acetoxymethyl ester (AM) (F1221; Thermo Fisher Scientific) (8). Immediately before fura-2 measurements, the depletion cocktail was replaced with 0 Ca2+ Ringer. During the experiment, fibers were excited at 362 nm, and emission was collected at 510 nm using a DeltaRam illumination system (Photon Technology, Birmingham, NJ, USA).The maximum rate of store-operated calcium entry (SOCE) was quantified from the maximum quench rate of quench of fura-2 fluorescence upon the addition of Mn2+ as described previously (8) and was expressed in units of photon counts per second.
Total Ca2+ store content
FDB fibers were loaded with 4 µM fura-FF AM (Tef Labs, Austin, TX, USA; 0136) in Ringer solution at room temperature for 30 min, followed by a 30 min wash in dye-free Ringer. Total Ca2+ store content was calculated from the peak differential of the fura-FF emission trace during application of a rapid Ca2+ release cocktail (1 µM ionomycin, 30 µM cyclopiazonic acid, 0 Ca2+/1 mM EGTA; ICE) as previously described (8).
Ex vivo muscle contraction
Muscle strength and fatigability were assessed in excised EDL and Sol muscles using an ASI muscle contraction system (Aurora Scientific, Aurora, ON, Canada) equipped with a 300C-LR dual mode force transducer and a 701C stimulator as described previously (8). Excised EDL and Sol muscles were mounted between 2 platinum electrodes in oxygenated Tyrode solution (in mM: 1.2 NaH2PO4, 1 MgSO4, 4.83 KCl, 137 NaCl, 2 CaCl2, 10 glucose, 24 NaHCO3, pH 7.4) as described by Hakim et al. (22). Muscle optimal length (Lo) was determined using a series of 1 Hz stimulations. Stimulus output was set at 120% of the voltage required to elicit maximal force. All muscles were equilibrated using three 500 ms, 150 Hz tetani at 1 min intervals and then subjected to a force frequency (EDL 500 ms, Sol 600 ms tetani, 0.04 duty cycle) or a repetitive moderate-frequency stimulation fatigue protocol (60 successive 50 Hz tetani: Sol 1 s duration, 0.4 duty cycle; EDL 500 ms duration, 0.2 duty cycle). Muscle force was recorded using the Dynamic Muscle Control software (Aurora Scientific) and analyzed using Clampfit 10.3 software (Molecular Devices, Sunnyvale, CA, USA). Muscle CSA and specific force were calculated as previously described (22).
Treadmill endurance running assay
Adult mice were acclimated to treadmill running on an Exer 3/6 Treadmill (Columbus Instruments, Columbus, OH, USA) for 3 d by running at a speed of 5 m/min for 5 min at a 15° incline. On the fourth day, mice underwent a 1 h endurance run, described previously (8). Exhaustion was defined as an inability of the mouse to reengage the treadmill after 3 consecutive, <1 s sprits of air using a WhooshDuster (Control Company, Webster, TX, USA), and 3 gentle taps by hand on the animal’s backside.
Rotarod coordination and fatigue assays
Before the coordination experiment, mice were trained at 24 rpm on an Economex rotarod fit with a 3-cm-diameter rod (Columbus Instruments), and the latency to fall (maximum 60 s) was measured during 4 trials on 3 consecutive days. On the fourth day, each mouse underwent 2 trials at 5, 8, 15, 20, 24, 31, 33, and 37 rpm, and average latency to fall was recorded for each speed. After 3 d, mice underwent a rotarod fatigue assay. Rotation speed began at 15 rpm for 15 min, followed by an increase in speed by 1 rpm every 5 min up to 21 rpm; then a maximum speed of 22 rpm was used for the final 15 min of the 1 h protocol. In the event of a fall, the fall was recorded and the mouse returned to the rod. The number of cumulative falls, binned every 5 min, was determined for each animal.
Wire hang test
Mice were placed on a wire grid and gently waved to encourage gripping. The grid was then inverted approximately 10 to 12 inches above a padded surface, and the latency to fall was measured in 3 consecutive trials, with a 5 min rest between trials, on 3 consecutive days. Average latency per trial per day was used as a single data point for each animal.
Statistical analyses
Student’s t test with significance reported at P < 0.05 was used for all data sets. All results were reported as means ± sem.
RESULTS
MCK-Cre efficiently reduces Orai1 expression and function in skeletal muscle of cOrai-KO animals
In order to define the long-term effects of Orai1 ablation in skeletal muscle, we generated a constitutive muscle-specific KO of Orai1 in cOrai1-KO mice, using the cre-loxP system. In this model, previously described floxed Orai1 mice (Orai1fl/fl) were crossed with mice expressing Cre recombinase driven by the MCK promoter (19) (Fig. 1A). Unlike previously described global Orai1-KO animals, these mice were viable and fertile, and lived a normal life span. In addition, WT and cOrai-KO littermates exhibited similar postnatal growth curves. No differences in total body mass were observed up to 16 wk of age, and after 16 wk, only modest reductions in body mass were measured, with slightly larger effects found in male than in female mice (Fig. 1B). To assess the efficacy and specificity of Orai1 ablation in this model, we measured Orai1 transcript levels using semiquantitative PCR. As shown in Fig.1C, Orai1 transcript was markedly reduced in EDL, TA, and Sol muscles from young (4–6 wk) cOrai-KO animals. No significant reduction was observed in the FDB muscle. In muscles isolated from adult (3–12 mo) animals, Orai1 transcript was significantly reduced in all muscles analyzed (Fig. 1D), while Orai1 transcript levels in nonskeletal muscle tissues, including heart, lung, and spleen, were not significantly reduced (Fig. 1E). To ensure the reduction in transcript translated to a reduction in protein, we measured Orai1 function in SOCE using a Mn2+ quench assay in FDB fibers from control and cOrai-KO animals (Fig. 1F, G). Despite significant levels of Orai1 transcript in FDB muscle, the magnitude of SOCE, assessed from the maximum rate of Mn2+ quench of fura-2 fluorescence after store depletion, was reduced >90% in fibers isolated from young cOrai-KO mice, similar to that observed in fibers from adult muscle (Fig. 1G). In addition, total releasable SR Ca2+ content was significantly reduced in FDB fibers from adult cOrai-KO mice (Fig. 1H).
Figure 1.

Specificity and efficiency of muscle-specific cOrai-KO in mice. A) Diagram of muscle-specific Orai-KO breeding strategy. Mice containing 2 floxed Orai1 alleles (WT) were crossed with mice that contained 2 floxed alleles and Cre recombinase cassette driven by MCK promoter (cOrai-KO mice). B) Six-month weekly growth curves for male and female WT and Orai-KO mice (n = 5–10 animals/group). C, D) Orai1 transcript levels normalized to Cacna1s transcript levels in EDL, FDB, TA, and Sol muscles from young (C) and adult (D) WT and cOrai-KO mice, measured using semiquantitative PCR with 6-FAM-labeled primers (n = 3–4 animals). E) Orai1 transcript levels normalized to Gapdh transcript levels in nonskeletal muscle tissues measured as in C (n = 3–4 animals). F) Representative Mn2+ quench traces. Scale bars: x = 100 s, y = 4 × 105 counts. G) Maximal rate of Mn2+ quench in store-depleted FDB fibers isolated from young and adult WT and cOrai-KO mice (n = 5–7 animals). H) Total releasable Ca2+ content measured using fura-FF in single isolated FDB fibers. ΔRatio = 340/380max –340/380 baseline (n = 4–5 animals).
Muscle-specific Orai1 ablation results in a reduction in muscle fiber CSA
Gross analysis of muscle cross sections from cOrai-KO mice revealed no signs of muscle necrosis, fibrosis, or increased incidence of fibers with centrally localized nuclei (Supplemental Fig. 1). Previous studies have reported reduced muscle fiber CSA in muscle-specific STIM1-KO mice (18), muscle-specific dominant negative (dn) Orai1 transgenic mice (8), and human patients with loss-of-function ORAI1 mutations (23). Thus, we quantified fiber CSA in EDL, TA, and Sol muscles. Despite little to no change in overall body mass in adult cOrai-KO mice, a significant reduction in fiber CSA was observed in muscles isolated from these animals (Fig. 2B). Interestingly, the magnitude of the reduction in CSA, consistent with reduced muscle mass (Fig. 2A), was most profound in the Sol, a characteristically slow, fatigue-resistant oxidative muscle.
Figure 2.

Sol muscles from Orai-KO mice develop normally and subsequently undergo fiber type-specific atrophy. A) Average weights of EDL and Sol muscles from WT and cOrai-KO mice (n = 25–36 muscles). B) Average fiber CSA in TA, EDL, and Sol muscle sections from WT and cOrai-KO mice (n = 3–6 animals). C, F) Top: sections of Sol muscles taken from young (C) and adult (F) WT animals. Bottom: sections of Sol muscles taken from young (C) and adult (F) cOrai-KO animals. Sections were stained for type I (magenta), type IIB (blue), and type IIA (red) myosin isotypes. Dystrophin (green) was used as marker of sarcolemma. Scale bar, 100 μm (white line) shown in applies to all images. D, G) Fractional contribution of fibers containing only type I, only type IIA, or both type I and type IIA myosin in Sol muscles from young (D) or adult (G) WT and cOrai-KO mice (n = 4–7 animals). E, H) Average CSA of different fiber types in Sol muscles from young (E) or adult (H) WT and cOrai-KO mice (n = 4–7 animals).
Skeletal muscle from cOrai-KO mice develops normally and subsequently undergoes oxidative fiber specific atrophy
This finding suggested that growth and maintenance of slow oxidative fibers may be more dependent on Orai1-mediated signaling than the fast glycolytic fibers that predominate in TA and EDL muscles. To test this hypothesis, we used myosin-specific antibodies to label individual fiber types and measured the corresponding fiber-type CSA in Sol muscles from young (Fig. 2C–E) and adult (Fig. 2F–H) WT and cOrai-KO mice. In Sol muscles from young mice, no difference was observed in the fractional contribution of type I, type IIA, and hybrid type I/IIA fibers (Fig. 2D) or in the average CSA of each fiber type (Fig. 2E). In contrast, Sol muscles from adult cOrai-KO mice exhibited a >2-fold reduction in the fraction of unique type I fibers compared to WT controls. This occurred in parallel with an increase in the fraction of hybrid type I/IIA fibers, thus leaving the total percentage population of fibers expressing type I myosin unchanged (mean ± sem, WT 0.51 ± 0.04, and cOrai-KO 0.53 ± 0.09) (Fig. 2G). No differences in the fractional contribution of unique type IIA fibers were observed (Fig. 2G). In addition, Sol muscles from adult cOrai-KO mice exhibited a >80% reduction in type I fiber CSA compared to those from adult WT mice. The reduction in type IIA fiber CSA was considerably less, at only ∼30%, while the CSA of hybrid fibers was intermediate, at ∼60% (Fig. 2H). Together, the combined reduction in unique type I fiber fractional contribution and CSA of fibers expressing type I myosin resulted in a reduction in relative type I myosin content in Sol of cOrai-KO mice. While type I myosin accounted for 40% of the total myosin expression in Sol muscles of WT mice, this was reduced to only 20% in muscles from cOrai-KO mice (Supplemental Fig. 2).
Quantification of fiber type content and CSA in EDL muscles from adult WT and cOrai-KO mice revealed a similar overall pattern (Supplemental Fig. 3A–C). Specifically, fiber CSA was significantly reduced in oxidative type IIA fibers and in type IIX fibers, while no change in CSA was observed in fast glycolytic type IIB fibers (Supplemental Fig. 3C). No change in the relative contribution of type IIA, IIX, and IIB fibers was observed in EDL muscles from adult WT and cOrai-KO mice (Supplemental Fig. 3B). This was consistent with no measurable difference in myosin isoform expression in whole muscle lysates (Supplemental Fig. 3D).
cOrai-KO muscles exhibit reduced ex vivo muscle force generation and maintenance
To determine the effect of a constitutive loss of Orai1 protein on whole muscle contractile behavior, we performed ex vivo muscle contractility measurements in Sol (Fig. 3A–D) and EDL (Figs. 3E–H) muscles from adult animals. In Sol, maximal specific force and contractile kinetics (i.e., maximum rate of force generation and relaxation) during single twitch stimulations were not different between muscles from WT and cOrai-KO mice (Fig. 3A, D). Additionally, no changes were observed in the expression of various proteins involved in excitation–contraction coupling (Supplemental Fig. 4), including the type 1 ryanodine receptor, dihydropyridine receptor, and SR/ER Ca2+-ATPase, consistent with proper functioning of the excitation–contraction coupling machinery. Similar to twitch contractions, maximal specific force in Sol muscles from WT and cOrai-KO mice were not different when stimulated with low nonsummating frequencies (5 and 10 Hz). However, a significant reduction in peak specific force was observed when Sol muscles from cOrai-KO mice were stimulated with frequencies that result in a summating contraction (20–200 Hz) (Figs. 3B–D). Sol muscles were also exposed to repetitive, moderate-frequency stimulations in order to assess susceptibility to fatigue. The magnitude of force decay during the initial several trains was significantly greater in Sol muscles from cOrai-KO mice (Fig. 3I).
Figure 3.

Maximal and sustained contractile force during high-frequency stimulation is reduced in muscles from cOrai-KO mice. A–C) Representative ex vivo specific force traces in Sol muscles from adult WT and cOrai-KO mice stimulated at 1 (A), 20 (B), or 100 Hz (C). Timescale shown in B also applies to panels C, F, and G. Total duration for twitch traces in panels A and E is 250 ms. D) Force-frequency summary for Sol muscles from adult WT and Orai-KO mice (n = 12–13 muscles). E–G) Representative ex vivo specific force traces in EDL muscles from adult WT and cOrai-KO mice stimulated at 1 (E), 20 (F), or 100 Hz (G). H) Force-frequency summary of EDL muscles from adult WT and cOrai-KO mice (n = 14–15 muscles). I) Normalized specific force during repetitive, high-frequency stimulation (1 s tetani, 50 Hz, 0.4 duty cycle) for Sol muscles from adult WT and cOrai-KO mice (n = 6–9 muscles). J) Normalized specific force decay during repetitive, high-frequency stimulation (500 ms, 50 Hz, 0.2 duty cycle) in EDL muscles from WT and cOrai-KO mice (n = 6–10 muscles).
Similar to Sol, peak specific force and contractile kinetics during twitch and low-frequency stimulations were not different in EDL muscles from adult WT and cOrai-KO mice (Fig. 3E, F, H). Furthermore, maximal specific force at high frequencies (75–250 Hz) was significantly reduced in EDL from cOrai-KO mice (Fig. 3G, H). During sustained high-frequency stimulation (50–250 Hz for 500 ms), EDL muscles from cOrai-KO mice exhibited a force decay of ∼20% that was not observed in EDL muscles from WT mice (Supplemental Fig. 5D). Finally, a repetitive, moderate-frequency stimulation paradigm was used to assess the susceptibility of EDL muscles to fatigue. EDL muscles from cOrai-KO mice exhibited a more pronounced force decrement after the first 2 stimulus trains compared to that observed for EDL muscles from age-matched WT mice (Fig. 3J).
Interestingly, muscles isolated from young cOrai-KO mice produced maximal specific force that was not different than WT controls (Supplemental Fig 5A, B) and only a slight, yet statistically significant, decay in force was observed in EDL from cOrai-KO mice during high-frequency tetani (Supplemental Fig. 5C).
Consistent with the observed reduction in maximal specific force generation by EDL and Sol muscles from cOrai-KO mice, total releasable SR Ca2+ was reduced in FDB fibers isolated from adult but not young animals (Fig. 1H) (24). We hypothesized that store content in Sol muscles from adult cOrai-KO mice is reduced to an even greater extent than in FDB as a result of a severe reduction the primary SR Ca2+ buffer, calsequestrin 1 (Casq), with no compensatory up-regulation of the minor calsequestrin 2 (Casq2) isoform (Supplemental Fig. 4).
Constitutive reduction in Orai1 results in reduced in vivo sustained grip strength and treadmill endurance
Ex vivo contractile measurements revealed a deficit in maximal force generation and enhanced force decline during prolonged high-frequency tetanic stimulation in muscles from adult cOrai-KO mice (Fig. 3 and Supplemental Fig. 5D). To determine the impact of long-term Orai1 ablation on muscle strength and endurance in vivo, we quantified basal activity (Supplemental Fig. 6), motor coordination (Fig. 4A), sustained grip strength (Fig. 4B), and long-term endurance (Fig. 4C, D) in WT and cOrai-KO mice. Open field test measurements revealed no difference in basal activity among WT and cOrai-KO mice (Supplemental Fig. 6). Using an established rotarod assay to assess balance and motor coordination, the latency to fall at all speeds was not different between WT and cOrai-KO mice (Fig. 4A). Consistent with ex vivo data, sustained grip strength, measured as the latency to fall from an inverted wire grid, was reduced >70% in cOrai-KO mice compared to WT littermates (Fig. 4B). Additionally, both prolonged rotarod (Fig. 4C) and treadmill running (Fig. 4D) assays revealed significantly reduced endurance in cOrai-KO mice compared to age-matched WT controls.
Figure 4.
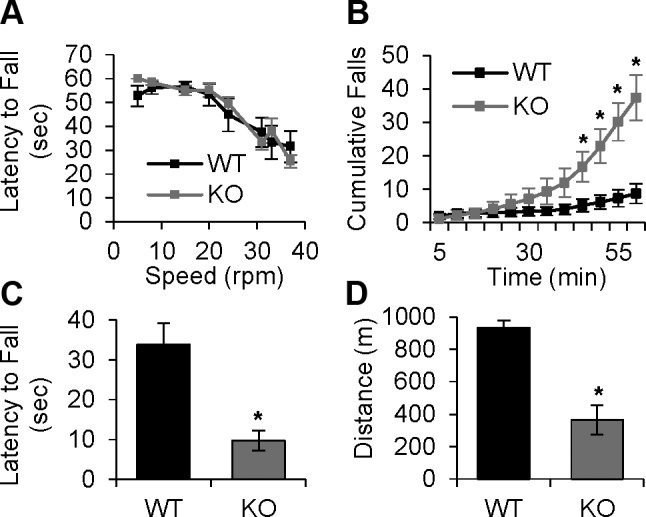
Loss of Orai1 in skeletal muscle does not alter motor coordination but results in reduction in sustained grip strength and treadmill endurance. A) Motor coordination quantified from average latency to fall from rotarod at indicated speeds in WT and cOrai-KO mice (n = 11–12 animals). B) Sustained grip strength, measured from average latency to fall from inverted grid. Bars represent average latency to fall per trial averaged over 3 d of experiments (n = 8–10 animals). C) Cumulative number of falls during rotarod endurance task in WT and cOrai-KO mice (n = 10–12 animals). D) Summary of average total distance run by WT and Orai-KO mice during 1 h treadmill endurance protocol (n = 7–8 animals).
Tam administration in adulthood induces a significant reduction in Orai1 transcript
A role for active SOCE in limiting muscle fatigue is controversial (8, 12, 25, 26). In resting muscle, the free Ca2+ concentration in the SR is maintained at ∼0.5 to 1 mM by the presence of moderate affinity, high-capacity Ca2+ buffering proteins (27). Given the Ca2+ binding affinity of the STIM1 luminal EF hand (∼200 µM) (28, 29), physiologic activation of Orai1 channels in skeletal muscle would be expected to occur under conditions that promote massive SR Ca2+ release sufficient to lead to transient store depletion (e.g., prolonged high-frequency tetanic stimulation). Although ex vivo muscle contractility (Fig. 3) and in vivo behavioral data (Fig. 4) from adult WT and cOrai-KO mice indicate that Orai1 is required for maximal specific force development and sustained muscle function, the relative degree to which the observed defects in muscle performance and endurance in cOrai-KO mice are due to the impact of Orai1 on muscle fiber type and/or Ca2+ homeostasis (e.g., maintaining SR Ca2+ store content) is unclear. To address the role of Orai1 channel activation during muscle use, we generated an inducible, muscle-specific Orai-KO mouse model wherein we were able to measure muscle function in the absence of fiber type changes.
Inducible, muscle-specific Orai-KO mice were generated by crossing floxed Orai1 mice (Orai1fl/fl) animals with mice expressing Cre recombinase driven by the HSA promoter (19, 20). In this model, Cre recombinase was flanked on either side by sequences encoding Mer ligand-binding domains (i.e., HSA-MCM), to allow for translocation of the translated Cre enzyme into the nucleus and loxP recombination only on administration of the estrogen receptor ligand Tam (20). One month after 5 Tam injections at 4 mo of age (Fig. 5A), Orai1 transcript was reduced over 75% in TA, EDL, and Sol muscles compared to CO controls (Fig. 5B), and Orai1 function in SOCE was reduced by 80% (Fig. 5C).
Figure 5.

Despite significant reductions in Orai1 transcript, force generation and whole-animal fatigue do not differ at 1 mo after Tam administration. A) Four-month-old animals were given 5 injections on 5 consecutive days of Tam (40 mg/kg/d) or equivalent volume of CO (vehicle), then collected and measured 1 mo later. B) Orai1 transcript normalized to Cacna1s transcript in Sol, EDL, and TA muscles isolated from mice 1 mo after 5 injections of CO or Tam (n = 3 animals). C) Representative Mn2+ quench traces (top; scale bars: x = 100 s, y = 4 × 105 counts) and maximal rate of Mn2+ quench in isolated FDB fibers (n = 6–9 cells from 2 animals). D, F) Representative traces of ex vivo force generation by Sol (D) and EDL (F) stimulated at 150 Hz for 500 ms. E, G) Maximal specific force-frequency summary generated by excised Sol (E) and EDL (G) muscles (n = 3–7 muscles). H, I) Total treadmill distance run (H) and cumulative number of rests taken (I) by CO- and Tam-injected mice during 1 h endurance treadmill protocol (n = 3–4 animals).
Maximal force generation and whole-body fatigue are not altered in muscles from Tam-treated animals
Both Sol and EDL muscles isolated from cOrai-KO mice exhibited significant reductions in maximal specific force during high-frequency stimulation (Fig. 3). To establish the role of acute Orai1 activation in this phenotype, we measured the force–frequency relationship in EDL and Sol muscles isolated from inducible mice given 5 CO or Tam injections at 4 mo of age, 1 mo before collection. At this time point, there were no measurable changes in myosin expression or fiber CSA (Supplemental Fig. 7A, B). Despite significantly reduced Orai1 transcript and function, the force–frequency profile in these muscles was not different compared to CO-injected controls (Fig. 5D–G). Additionally, there was no measurable decay in force during fused tetani in EDL muscles from Tam-treated animals (Fig. 5F). To measure the in vivo effects of acute Orai1 activation in endurance exercise, mice underwent a 1 h, 1 km treadmill task. Unlike cOrai-KO mice, both CO- and Tam-treated animals could complete the entire endurance protocol (mean ± sem, CO 970 ± 30 m, Tam 1000 ± 0 m) (Fig. 5H). In fact, the number of rests taken by Tam animals, defined by the number of times the mouse rested on the platform at the rear of the treadmill for more than 3 s, was not greater than CO-treated animals (Fig. 5I).
Orai1-mediated Ca2+ entry does not facilitate long-term slow, type I myosin expression in adulthood
Whole-muscle myosin measurement in Sol muscles revealed a significant reduction in the contribution of type I myosin (Supplemental Fig. 2). In many cell types, activation of Orai1 channels in response to store depletion has been shown to activate the calcineurin-NFAT signaling axis (19, 30, 31), and in skeletal muscle, activation of this axis is a known regulator of slow, type I myosin expression (4). To address the role of Orai1 activation in maintenance of type I myosin expression, inducible Orai-KO mice were given a series of 5 injections of CO or Tam at 4 mo of age, and muscles were collected 4 to 6 mo later (Supplemental Fig. 8A). In the absence of any additional treatments, mice given Tam maintained a >70% reduction in Orai1 transcript and a >90% reduction in maximal Mn2+ quench rate compared to CO controls (Supplemental Fig. 8B, C). However, no differences were observed in Sol fiber CSA, relative fiber type distribution (Supplemental Fig. 8D, E), or Casq1 expression (Supplemental Fig. 8F, G) between CO- and Tam-treated animals.
DISCUSSION
Role of Orai1 in muscle development and postnatal myosin expression
In this study, we generated constitutive and inducible muscle-specific KO mouse models to differentiate the acute and long-term developmental roles of Orai1 in skeletal muscle. Similar to muscle-specific dnOrai1 transgenic mice (8), cOrai-KO mice are also viable and fertile, and they live a normal life span. This is in stark contrast to the perinatal lethality observed for muscle-specific STIM1-KO mice (18). Differences observed between the 2 models could reflect the multiple roles and targets of STIM1 that extend beyond Orai1 activation (32). Interestingly, while dnOrai1 animals are significantly smaller than WT littermates from birth, cOrai-KO mice follow the same growth curve as their WT littermates for the first 16 wk of life, and only thereafter exhibit moderately reduced whole body and muscle mass (Figs. 1B and 2A). This apparent discrepancy may arise from 2 key differences between the models. First, as dnOrai1 mice are a transgenic model, the overexpressed dnOrai1 protein could aberrantly incorporate into and block other channels (e.g., TRPC1 channels) involved in muscle development and hypertrophy (33, 34). In contrast, muscle-specific ablation of Orai1 should result in a selective loss of Orai1 function. A second key difference is that the dnOrai1 and Cre recombinase transgenes in the 2 models are driven by different promoters (HSA and MCK, respectively). Thus, temporal and/or fiber type differences in transgene expression could also contribute to the different phenotypes observed (35, 36).
Despite marked reductions in Orai1 mRNA expression and SOCE function in muscle from young cOrai-KO mice (Fig. 1C), relative fiber type distribution and CSA were normal in Sol muscles from these animals (Fig. 2C–E). However, by 3 mo of age, a significant reduction in the number and reduced CSA of unique type I expressing fibers was observed in Sol muscles from cOrai-KO animals (Fig. 2G). This reduction in unique type I fibers can be interpreted in 2 ways. One explanation is that Orai1 is necessary for muscle fibers to undergo postnatal transition from fast to slow myosin expression, and that transitioning fibers are being suspended in a hybrid state (37). The other is that in the absence of Orai1, type I myosin cannot be maintained and type IIA myosin is aberrantly up-regulated in otherwise unique type I expressing fibers (38). Both explanations converge on a role for Orai1 in promoting proper myosin expression in adult muscle.
Orai1 in maintenance of type I expression and oxidative fiber hypertrophy
In slow-twitch muscle fibers, chronic patterns of low-frequency motor neuron activity lead to Ca2+-induced activation of calcineurin, dephosphorylation, and nuclear translocation of NFAT transcription factors as well as transcription of genes specific for the slow fiber program (2). Using the inducible Orai-KO model, we tested the hypothesis that Orai1 in muscle, similar to other cell types, is upstream in the calcineurin-NFAT signaling axis to modulate homeostatic type I myosin expression in adult muscle (19, 30, 31). However, contrary to this hypothesis, no measurable changes in myosin distribution were found in Sol muscles 4 to 6 mo after Tam-induced Orai1 ablation in adult mice (Supplemental Fig. 8).
These findings suggest that Orai1 is required for proper perinatal muscle remodeling (37), which is further supported by careful analysis of myosin isoform distribution patterns in young versus old Sol (Fig. 2D, G). In muscles from young WT animals, a small population of fibers initially express type I myosin while a larger population of fibers are positive for type IIA myosin. By 3 mo, an increase in type I fibers concomitant with a decrease in type IIA fibers is observed. In muscles from cOrai-KO muscles, a similar decrease in unique type IIA fibers occurs without the appropriate increase in unique type I fibers, suggesting that Orai1 is required to turn off type IIA expression in these transitory fibers.
One consistent phenotype of muscles from STIM1-KO, dnOrai1, and cOrai-KO mice is a significant reduction in average fiber CSA (Fig. 2B) (8, 18). In fact, Sol muscles from adult dnOrai1 and cOrai-KO mice exhibit identical reductions in oxidative fiber CSA (Fig. 2; unpublished results). Interestingly, this phenotype is different from that of a human patient with an Orai1 loss-of-function mutation whose muscle exhibited relative, fast fiber-specific atrophy and predominance of type I fibers (23). This discrepancy may result in part from Orai1 loss-of-function effects in nonmuscle cell types in the human patient that are absent in muscle-specific Orai-KO mice and/or side effects of drug treatment in the patient (39). Despite this difference, our findings in cOrai-KO mice are consistent with previous reports of muscle hypotonia, reduced distance walking, and postural muscle weakness in patients with loss-of-function mutations in ORAI1 or STIM1 (23, 40).
Loss of Orai1 function does not result in an increase in centrally nucleated fibers in either human or mouse muscle (Supplemental Fig. 1) (23), indicating the absence of significant muscle regeneration. However, loss of Orai1 function results in decreased muscle fiber CSA in both humans and cOrai-KO mice, consistent with an increase in muscle atrophy or reduction in muscle hypertrophy. Our finding that changes in unique type I myosin content and muscle fiber CSA do not manifest over time in muscles of Tam-inducible Orai-KO mice (Supplemental Fig. 8) suggests that the underlying signaling processes are likely initiated at an early developmental time point. Unlike muscle-specific STIM1-KO mice (18), preliminary results in Sol muscles from adult cOrai-KO mice did not reveal a significant change in basal Akt activation, a component of the primary hypertrophy signaling axis in muscle (data not shown). However, it remains possible that Orai1 is required for activation of this signaling axis at an early developmental time point. Future studies are needed to systematically evaluate the Cn-NFAT and Akt signaling pathways during the precise developmental time window when Orai1 is required to drive subsequent changes in type I myosin levels and fiber hypertrophy.
Orai1 promotes development of SR Ca2+ store content
Similar to that previously reported for muscle fibers from dnOrai1 transgenic mice and muscle-specific STIM1-KO mice (8, 18), FDB fibers from adult but not young cOrai1-KO mice exhibit reduced releasable Ca2+ store content (Fig. 1H). In addition, we found that EDL muscle contractile decay was increased during 500 ms stimuli for all frequencies above 75 Hz (Fig. 1H), consistent with Orai1-dependent Ca2+ entry contributing to sustained force production during high-frequency stimulation. However, the degree to which this increase in contractile fade in EDL muscles from cOrai-KO mice results from dynamic changes in SR Ca2+ during repetitive stimulation is unclear. Although we did not measure store content after Tam-induced Orai1 ablation in adult mice, maximal force generation during tetanic stimulation was normal in muscles from these animals, consistent with the absence of significant store depletion (Fig. 5) (24). These results suggest that Orai1 is not required for store refilling during brief (500 ms), high-frequency tetanic stimulation. Further, we hypothesize that the reduction in total Ca2+ store content observed in fibers from adult cOrai-KO mice (Fig. 1H) reflects a decrease in total SR Ca2+ store capacity due in part to reduced Casq1 expression (Supplemental Fig. 4).
Orai1-dependent SOCE is not required for maximal force generation or exercise endurance
Ex vivo muscle contractility (Fig. 3) and in vivo behavioral data (Fig. 4) from adult cOrai-KO mice, consistent with published findings in dnOrai1 mice (8), indicate that Orai1 is necessary for maximal specific force development and sustained muscle function. Previous publications have proposed that Orai1 limits fatigue during repetitive stimulation by promoting acute activation for myoplasmic Ca2+ delivery and store refilling (8, 25, 41). In contrast, we found that force decay rates during repetitive high-frequency stimulation were similar in muscles isolated from WT and cOrai-KO mice (Fig. 3I, J), arguing against a critical role for acute Orai1-mediated Ca2+ influx in limiting muscle fatigue. Additionally, using an inducible Orai-KO mouse model, we found that despite significant reductions in Orai1 transcript and function, these mice do not exhibit the reduced force generation and whole body fatigue observed in adult cOrai-KO mice (Fig. 5). These data, along with ex vivo measurements from young cOrai-KO animals (Supplemental Fig. 5), indicate that the deficits in maximal force generation and endurance exercise observed in cOrai-KO mice are due to reduced oxidative fatigue-resistant fiber content and Ca2+ store capacity and not to an acute loss of store-operated Ca2+ entry.
ACKNOWLEDGMENTS
This work was supported by a U.S. National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases grant (AR059646 to R.T.D.), an M. D. Anderson Cancer Center grant (MDA275574 to R.T.D.), and a National Science Foundation Graduate Research Fellowship Award (DGE-1419118 to E.M.C.).
Glossary
- 6-FAM
fluorescein
- AM
fura-2 acetoxymethyl ester
- Cacna1s
calcium channel, voltage-dependent, L type, α 1S subunit
- Casq
calsequestrin
- CO
corn oil
- cOrai1-KO
constitutive Orai1-knockout
- CRAC
calcium release-activated calcium
- CSA
cross-sectional area
- dn
dominant negative
- EDL
extensor digitorum longus
- ER
endoplasmic reticulum
- FDB
flexor digitorum brevis
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HSA
human skeletal actin
- KO
knockout
- MCK
muscle creatine kinase
- MCM
Mer-Cre-Mer
- Mer
mutated estrogen receptor
- NFAT
nuclear factor of activated T cell
- SOCE
store-operated calcium entry
- Sol
soleus
- SR
sarcoplasmic reticulum
- STIM1
stromal interaction molecule 1
- TA
tibialis anterior
- Tam
tamoxifen
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
E. M. Carrell, R. T. Dirksen, and H. J. McBride designed research; E. M. Carrell and A. R. Coppola analyzed data; E. M. Carrell and A. R. Coppola performed research; E. M. Carrell, R. T. Dirksen and H. J. McBride wrote the paper; and H. J. McBride contributed new reagents.
REFERENCES
- 1.Schiaffino S., Reggiani C. (2011) Fiber types in mammalian skeletal muscles. Physiol. Rev. 91, 1447–1531 [DOI] [PubMed] [Google Scholar]
- 2.Chin E. R., Allen D. G. (1996) The role of elevations in intracellular [Ca2+] in the development of low frequency fatigue in mouse single muscle fibres. J. Physiol. 491, 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dolmetsch R. E., Lewis R. S., Goodnow C. C., Healy J. I. (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386, 855–858 [DOI] [PubMed] [Google Scholar]
- 4.Chin E. R., Olson E. N., Richardson J. A., Yang Q., Humphries C., Shelton J. M., Wu H., Zhu W., Bassel-Duby R., Williams R. S. (1998) A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 12, 2499–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prakriya M., Lewis R. S. (2015) Store-operated calcium channels. Physiol. Rev. 95, 1383–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoth M., Penner R. (1992) Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355, 353–356 [DOI] [PubMed] [Google Scholar]
- 7.Lyfenko A. D., Dirksen R. T. (2008) Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J. Physiol. 586, 4815–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei-Lapierre L., Carrell E. M., Boncompagni S., Protasi F., Dirksen R. T. (2013) Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 4, 2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vig M., DeHaven W. I., Bird G. S., Billingsley J. M., Wang H., Rao P. E., Hutchings A. B., Jouvin M. H., Putney J. W., Kinet J. P. (2008) Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 9, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gwack Y., Srikanth S., Oh-Hora M., Hogan P. G., Lamperti E. D., Yamashita M., Gelinas C., Neems D. S., Sasaki Y., Feske S., Prakriya M., Rajewsky K., Rao A. (2008) Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol. Cell. Biol. 28, 5209–5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guzman R., Valente E. G., Pretorius J., Pacheco E., Qi M., Bennett B. D., Fong D. H., Lin F. F., Bi V., McBride H. J. (2014) Expression of ORAII, a plasma membrane resident subunit of the CRAC channel, in rodent and non-rodent species. J. Histochem. Cytochem. 62, 864–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stiber J., Hawkins A., Zhang Z. S., Wang S., Burch J., Graham V., Ward C. C., Seth M., Finch E., Malouf N., Williams R. S., Eu J. P., Rosenberg P. (2008) STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 10, 688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yarotskyy V., Dirksen R. T. (2012) Temperature and RyR1 regulate the activation rate of store-operated Ca2+ entry current in myotubes. Biophys. J. 103, 202–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacruz R. S., Feske S. (2015) Diseases caused by mutations in ORAI1 and STIM1. Ann. N. Y. Acad. Sci. 1356, 45–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H., Ding X., Lopez J. R., Takeshima H., Ma J., Allen P. D., Eltit J. M. (2010) Impaired Orai1-mediated resting Ca2+ entry reduces the cytosolic [Ca2+] and sarcoplasmic reticulum Ca2+ loading in quiescent junctophilin 1 knock-out myotubes. J. Biol. Chem. 285, 39171–39179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darbellay B., Arnaudeau S., König S., Jousset H., Bader C., Demaurex N., Bernheim L. (2009) STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J. Biol. Chem. 284, 5370–5380 [DOI] [PubMed] [Google Scholar]
- 17.Cully T. R., Launikonis B. S. (2013) Store-operated Ca²⁺ entry is not required for store refilling in skeletal muscle. Clin. Exp. Pharmacol. Physiol. 40, 338–344 [DOI] [PubMed] [Google Scholar]
- 18.Li T., Finch E. A., Graham V., Zhang Z. S., Ding J. D., Burch J., Oh-hora M., Rosenberg P. (2012) STIM1-Ca(2+) signaling is required for the hypertrophic growth of skeletal muscle in mice. Mol. Cell. Biol. 32, 3009–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somasundaram A., Shum A. K., McBride H. J., Kessler J. A., Feske S., Miller R. J., Prakriya M. (2014) Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J. Neurosci. 34, 9107–9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarthy J. J., Srikuea R., Kirby T. J., Peterson C. A., Esser K. A. (2012) Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skelet. Muscle 2, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Talmadge R. J., Roy R. R. (1993) Electrophoretic separation of rat skeletal muscle myosin heavy-chain isoforms. J. Appl. Physiol. 75, 2337–2340 [DOI] [PubMed] [Google Scholar]
- 22.Hakim C. H., Li D., Duan D. (2011) Monitoring murine skeletal muscle function for muscle gene therapy. Methods Mol. Biol. 709, 75–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCarl C. A., Picard C., Khalil S., Kawasaki T., Rother J., Papolos A., Kutok J., Hivroz C., Ledeist F., Plogmann K., Ehl S., Notheis G., Albert M. H., Belohradsky B. H., Kirschner J., Rao A., Fischer A., Feske S. (2009) ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. 124, 1311–1318.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dutka T. L., Cole L., Lamb G. D. (2005) Calcium phosphate precipitation in the sarcoplasmic reticulum reduces action potential-mediated Ca2+ release in mammalian skeletal muscle. Am. J. Physiol. Cell Physiol. 289, C1502–C1512 [DOI] [PubMed] [Google Scholar]
- 25.Launikonis B. S., Murphy R. M., Edwards J. N. (2010) Toward the roles of store-operated Ca2+ entry in skeletal muscle. Pflugers Arch. 460, 813–823 [DOI] [PubMed] [Google Scholar]
- 26.Germinario E., Esposito A., Midrio M., Peron S., Palade P. T., Betto R., Danieli-Betto D. (2008) High-frequency fatigue of skeletal muscle: role of extracellular Ca(2+). Eur. J. Appl. Physiol. 104, 445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beard N. A., Laver D. R., Dulhunty A. F. (2004) Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 85, 33–69 [DOI] [PubMed] [Google Scholar]
- 28.Stathopulos P. B., Li G. Y., Plevin M. J., Ames J. B., Ikura M. (2006) Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 281, 35855–35862 [DOI] [PubMed] [Google Scholar]
- 29.Brandman O., Liou J., Park W. S., Meyer T. (2007) STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131, 1327–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srikanth S., Gwack Y. (2013) Orai1-NFAT signalling pathway triggered by T cell receptor stimulation. Mol. Cells 35, 182–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W., Halligan K. E., Zhang X., Bisaillon J. M., Gonzalez-Cobos J. C., Motiani R. K., Hu G., Vincent P. A., Zhou J., Barroso M., Singer H. A., Matrougui K., Trebak M. (2011) Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury. Circ. Res. 109, 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hooper R., Samakai E., Kedra J., Soboloff J. (2013) Multifaceted roles of STIM proteins. Pflugers Arch. 465, 1383–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao Y., Erxleben C., Abramowitz J., Flockerzi V., Zhu M. X., Armstrong D. L., Birnbaumer L. (2008) Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc. Natl. Acad. Sci. USA 105, 2895–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zanou N., Shapovalov G., Louis M., Tajeddine N., Gallo C., Van Schoor M., Anguish I., Cao M. L., Schakman O., Dietrich A., Lebacq J., Ruegg U., Roulet E., Birnbaumer L., Gailly P. (2010) Role of TRPC1 channel in skeletal muscle function. Am. J. Physiol. Cell Physiol. 298, C149–C162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miniou P., Tiziano D., Frugier T., Roblot N., Le Meur M., Melki J. (1999) Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 27, e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyons G. E., Mühlebach S., Moser A., Masood R., Paterson B. M., Buckingham M. E., Perriard J. C. (1991) Developmental regulation of creatine kinase gene expression by myogenic factors in embryonic mouse and chick skeletal muscle. Development 113, 1017–1029 [DOI] [PubMed] [Google Scholar]
- 37.Butler-Browne G. S., Whalen R. G. (1984) Myosin isozyme transitions occurring during the postnatal development of the rat soleus muscle. Dev. Biol. 102, 324–334 [DOI] [PubMed] [Google Scholar]
- 38.McCullagh K. J., Calabria E., Pallafacchina G., Ciciliot S., Serrano A. L., Argentini C., Kalhovde J. M., Lømo T., Schiaffino S. (2004) NFAT is a nerve activity sensor in skeletal muscle and controls activity-dependent myosin switching. Proc. Natl. Acad. Sci. USA 101, 10590–10595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta A., Gupta Y. (2013) Glucocorticoid-induced myopathy: pathophysiology, diagnosis, and treatment. Indian J. Endocrinol. Metab. 17, 913–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S. H., Tanasa B., Hogan P. G., Lewis R. S., Daly M., Rao A. (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185 [DOI] [PubMed] [Google Scholar]
- 41.Dirksen R. T. (2009) Checking your SOCCs and feet: the molecular mechanisms of Ca2+ entry in skeletal muscle. J. Physiol. 587, 3139–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]