

Abstract
Gene therapy for HIV-1 infection is a promising alternative to lifelong combination antiviral drug treatment. Chemokine receptor 5 (CCR5) is the coreceptor required for R5-tropic HIV-1 infection of human cells. Deletion of CCR5 renders cells resistant to R5-tropic HIV-1 infection, and the potential for cure has been shown through allogeneic stem cell transplantation with naturally occurring homozygous deletion of CCR5 in donor hematopoietic stem/progenitor cells (HSPC). The requirement for HLA-matched HSPC bearing homozygous CCR5 deletions prohibits widespread application of this approach. Thus, a strategy to disrupt CCR5 genomic sequences in HSPC using zinc finger nucleases was developed. Following discussions with regulatory agencies, we conducted IND-enabling preclinical in vitro and in vivo testing to demonstrate the feasibility and (preclinical) safety of zinc finger nucleases-based CCR5 disruption in HSPC. We report here the clinical-scale manufacturing process necessary to deliver CCR5-specific zinc finger nucleases mRNA to HSPC using electroporation and the preclinical safety data. Our results demonstrate effective biallelic CCR5 disruption in up to 72.9% of modified colony forming units from adult mobilized HSPC with maintenance of hematopoietic potential in vitro and in vivo. Tumorigenicity studies demonstrated initial product safety; further safety and feasibility studies are ongoing in subjects infected with HIV-1 (NCT02500849@clinicaltrials.gov).
Introduction
HIV is a chronic virus infection that is currently managed by life-long antiretroviral therapy (ART). At a practical level, lifetime antiretroviral therapy is a less-than-optimum therapeutic modality, and the development of alternative approaches to treatment is warranted. Cell-based therapeutic strategies, such as creating an HIV-1-resistant immune system by transplantation of genetically modified T-cells, have shown promise in preclinical models and limited early clinical trials.1,2
The CCR5 chemokine receptor is expressed on macrophages, T cells, dendritic cells, microglial cells, and interacts with the chemokine ligands Rantes, MIP-1α, and MIP-1β.3 CCR5 also functions as an obligate coreceptor for R5-tropic HIV-1 enabling viral entry into cells.4–8 Individuals homozygous for a 32-bp deletion in the CCR5 gene (CCR5Δ32/Δ32) that results in a dysfunctional receptor are resistant to R5-tropic HIV-1 infection,9 while heterozygotes have a slower progression to acquired immunodeficiency syndrome (AIDS) compared with individuals carrying the wild-type CCR5 gene.10 Reducing the HIV-1 viral load through CCR5 inhibition has been demonstrated with small molecule inhibitors, such as Maraviroc. Furthermore, “sterilizing cure” has been achieved in an individual who underwent allogeneic stem cell transplantation with CCR5Δ32/Δ32 HSPC11,12 and has been off ART for more than 8 years, with undetectable HIV-1 RNA and proviral DNA in the peripheral blood, bone marrow, and rectal mucosa.12 Despite the promising outcome, the widespread application of allogeneic stem cell transplantations is limited by the availability of HLA-matched CCR5Δ32/Δ32 donors and the unacceptably high risk of morbidity and mortality.13
As an alternative, HIV-1 immunity can be engineered using zinc finger nucleases (ZFN) to create a CCR5Δ32/Δ32-like immune system. Specialized ZFNs (SB-728) designed to target the CCR5 gene in human cells and thereby disrupt the CCR5 receptor have been developed and tested in humans.2 In preclinical studies, genetic modification of either transformed or primary CD4+ T cells or CD34+ HSPC via transient exposure to ZFNs targeting the CCR5 locus has been shown to result in cells and/or progeny (CD4+ T cells derived from edited CD34+ HSPCs) that are resistant to HIV infection.14–16
SB-728 was cloned into an Ad5/35 pseudotyped adenoviral vector (Ad5/35-SB-728) and used to generate CCR5-modified autologous CD4+ T cells (SB-728-T) for phase 1/2 testing in HIV-1 infected subjects (NCT01044654@clinicaltrials.gov and NCT00842634@clinicaltrials.gov).2 Early clinical results demonstrated that modified SB-728-T cells are safe, engraft, persist over time, and home to the gut-associated lymphoid tissues. Furthermore, these studies demonstrated that loss of CCR5 per se did not result in an overt pathophysiological phenotype in humans. A clinical study (NCT01543152@clinicaltrials.gov) with escalating doses of cyclophosphamide to enhance SB-728-T engraftment in subjects infected with HIV-1 is ongoing.
We previously reported on the evaluation of Ad5/35-SB-728 to modify adult mobilized peripheral blood (CD34+) HSPC for clinical use.15 However, the cytotoxicity of adenoviral vectors on HSPC prevented their use in our intended clinical study. Alternatively, the delivery of mRNA to cells by electroporation is common and has been adapted to the production of dendritic cells17,18 and CAR T-cells19 for clinical use. Large scale methods for electroporation of nucleic acids into hematopoietic stem cells have also been developed and are compatible with good manufacturing practices (GMP).20 In support of our current application, ZFNs have been shown to be effective in disrupting genomic targets when expressed from mRNA after intracellular delivery by electroporation.21 Based on these results, methods for the use of SB-728 mRNA (SB-728mR) were developed to support the clinical-scale manufacture of CCR5-disrupted autologous CD34+ HSPC (SB-728mR-HSPC). We describe here the preclinical development of CCR5-modified CD34+ HSPC from healthy adult growth factor-mobilized peripheral blood using electroporation mediated delivery of SB-728 mRNA.
Results
Optimization of ZFN-based CCR5 gene disruption in HSPC
The dose of SB-728mR was titrated on HSPC isolated from a healthy donor to characterize the relationship between dose, on and off target genome disruption, cell recovery, viability, and biological function. A G-CSF-mobilized hematopoietic progenitor cell apheresis product (HPC-A) was purchased from a commercial vendor and shipped to City of Hope (COH) by overnight courier. CD34+ HSPC were enriched from the HPC-A by positive selection as previously reported.15 CD34-enriched cells were incubated overnight in SCF, Flt-3L, TPO, and IL-6 (SFT6), as described in “Materials and Methods”, then washed and resuspended in electroporation buffer with 0, 50, 75, 100 or 150 µg/ml SB-728mR. Both research grade (rSB-728mR) and GMP compliant (SB-728mR) mRNA was tested. Cells were electroporated with the MaxCyte GT Transfection System using a preprogrammed pulse condition previously identified by the manufacturer for mRNA transfection of CD34+ HSPC.20 After electroporation, samples were incubated for 20 minutes at 37°C and then placed at 30°C overnight (16–20 hours).22 These day-1 postelectroporation cells were then transferred to 37°C for another 24-hour incubation prior to analysis and cryopreservation.
Cells were cultured in bulk for up to 7 days and tested for CCR5 disruption three times during the first 5 days of bulk culture (days 1, 2, and 5) using two independent analyses of the samples, i.e., targeted genome sequencing (MiSeq, Illumina, San Diego, CA) and Surveyor nuclease assay (Cel-1). We observed high levels of CCR5 disruption (40–60%) at all rSB-728mR concentrations tested at all time points with no significant differences in the estimated frequency of disruption between the two assay methods (MiSeq vs Cel-1 in Figure 1a).
Figure 1.

Evaluation of the effects of electroporation of HSPC with rSB-728mR. (a) Extent of CCR5 disruption as estimated by (open box) MiSeq and (closed box) Surveyor nuclease assay (Cel-1) after electroporation with varying concentrations of research grade CCR5-specific ZFN mRNA (rSB-728mR). Regarding to MiSeq data, ~ 3,000–30,000 total sequence reads per sample were obtained and used for calculation of % CCR5 disruption (% indels). (b) Extent of modification of CCR5 and next four top off-target sequences after electroporation with varying concentrations of rSB-728mR. (c) Viability of HSPC on day 1 (D1) and day 2 (D2) after electroporation (EP). (d) The effects of electroporation on hematopoietic potential measured as colony forming units (CFU) of HSPC plated 2 days after electroporation with 50 or 150 μg/ml rSB-728mR or150 μg/ml GMP Grade SB-728 mRNA. Controls were treated with or without 150 μg/ml rSB-728mR but without electroporation (No EP). A total of 500 HSPC were plated from each sample, and CFUs were read at 14 days. *P < 0.05, **P < 0.01. CCR5, chemokine receptor 5; HSPCs, hematopoietic stem/progenitor cells; ZFNs, zinc finger nucleases.
Bulk culture analysis for off-target modification at four previously identified sites (CCR2, KRR1, FBXL11, and ZCCHC14) demonstrated that off-target modification increased with the SB-728 mRNA concentrations and was higher in KRR1 and CCR2 than FBXL11 and ZCCHC14 (Figure 1b). The frequency of cells with CCR5 disruption did not substantially increase using rSB-728mR doses between 50 and 150 µg/ml. Some HSPC were lost during the process, but overall recovery was between 50 and 60% of input cell number at all rSB-728mR concentrations (data not shown), with high viability (>70%) in all samples 1 and 2 days after electroporation (Figure 1c). A small but significant (P < 0.05) reduction in viability of cells was observed 2 days after electroporation using 150 µg/ml rSB-728mR.
We also compared the relative effect of electroporation on hematopoietic potential using standard clonogenic hematopoietic colony forming unit (CFU) assays. Following electroporation and 2 days of recovery as described above, 500 viable CD34+ HSPC were plated in triplicate in complete methylcellulose media (MethoCult Stem Cell Technologies, Vancouver, British Columbia, Canada) and total colonies were counted after 14 days of culture. No significant reduction in colony formation was observed when cells were electroporated in the presence of 50 μg/ml of rSB-728mR compared with untreated controls. A modest but significant reduction in the number of colonies was observed following electroporation in the presence of 150 μg/ml of either rSB-728mR or SB-728mR (P < 0.01 and P < 0.05, respectively) (Figure 1d). No significant reduction in colonies was seen when cells were incubated in the presence of 150 μg/ml SB-728mR without electroporation, demonstrating a lack of toxicity of the SB-728mRNA alone.
Individual colonies derived from 2-week CFU cultures of HSPC (treated as described above) were also evaluated for the extent of monoallelic and biallelic disruption of CCR5 alleles. Samples were first screened for the presence of any CCR5 disruption using the Surveyor Nuclease assay. Clones with detectable CCR5 disruption were further characterized by targeted genome sequencing (MiSeq). Of the 192 colonies analyzed for each condition, 139 and 138 clones (from 50 and 150 µg/ml rSB-728mR treated cells, respectively) were further analyzed for mono- and biallelic disruption rates. The results are shown in Table 1. Overall, 54.7 and 69.6% of colonies (from 50 or 150 µg/ml rSB-728mR treated cells, respectively) had at least one CCR5 allele disrupted, slightly higher than but consistent with the bulk culture estimate of the same samples. However, 72.9% of modified colonies derived from HSPC electroporated in the presence of 150 µg/ml rSB-728mR contained biallelic disruption as compared with 39.5% of modified colonies derived from HSPC electroporated in the presence of 50 µg/ml rSB-728mR. These results are not evident from bulk culture screening and demonstrate a significant ZFN potency increase at 150 µg/ml mRNA concentrations albeit with decreased viability and CFU count.
Table 1. Disruption of CCR5 alleles in CFU from rSB-728mR treated HSPC.
| 50 µg/ml rSB-728mR | 150 µg/ml rSB-728mR | |
|---|---|---|
| Bulk Cel-1 (%) | 41 | 57 |
| Total CFUs (number) | 192 | 192 |
| CFUs with valid sequence (number) | 139 | 138 |
| Wild-type (number; %) | 63 (45.3) | 42 (30.4) |
| Modified CFUs (number; %) | 76 (54.7) | 96 (69.6) |
| Heterozygous (number; %) | 46 (33.1) | 26 (18.8) |
| Biallelic disruption (number; %) | 30 (21.6) | 70 (50.7) |
| Modified CFUs with biallelic disruption (%) | 39.5 | 72.9 |
CCR5, Chemokine receptor 5; CFU, colony forming units; HSPC, hematopoietic stem/progenitor cells.
Samples of HSPC electroporated with 50 or 150 µg/ml rSB-728mR were evaluated for overall CCR5 disruption in bulk 2-week cultures (Bulk Cel-1%) or for the extent of monoallelic and biallelic disruption in colonies in colonies derived from 2-week CFU cultures. Colonies were first screened for CCR5 disruption by Cel-1 then clones were assayed by rapid sequence analysis. The number and frequency, N (% of total), of colonies with wild type sequences (Wild type), total modified CFU (Modified CFU), those CFU with one allele modified (Heterozygous) and with both alleles modified (Biallelic disruption) are shown. The percentage of all modified colonies with biallelic disruption is shown at the bottom for each RNA concentration. CFUs with no sequence information or a phenotype corresponding to mixed CFUs were excluded from final analysis.
In vivo assessment of engraftment and lineage differentiation
In order to determine the effect on in vivo engraftment and lineage potential of HSPC treated with rSB-728mR, we transplanted immunodeficient NSG mice with 1 million cells that were either nonelectroporated (control, n = 5) HSPC or HSPC from each of two individual healthy donors treated with 150 µg/ml of rSB-782mR (treated, n = 5), as described in “Materials and Methods”. Animals were maintained for 20 weeks and then necropsied to determine levels of engraftment and lineage differentiation in bone marrow and spleen. The average engraftment (% CD45+ cells) in the bone marrow of animals receiving treated HSPC from donor 1 was higher than the control engraftment of bone marrow from the same donor (P < 0.05) (Figure 2a). No differences were observed in the average bone marrow engraftment from donor 2 or in the engraftment of the spleen of either donor. We did not assess CCR5 disruption in these samples.
Figure 2.
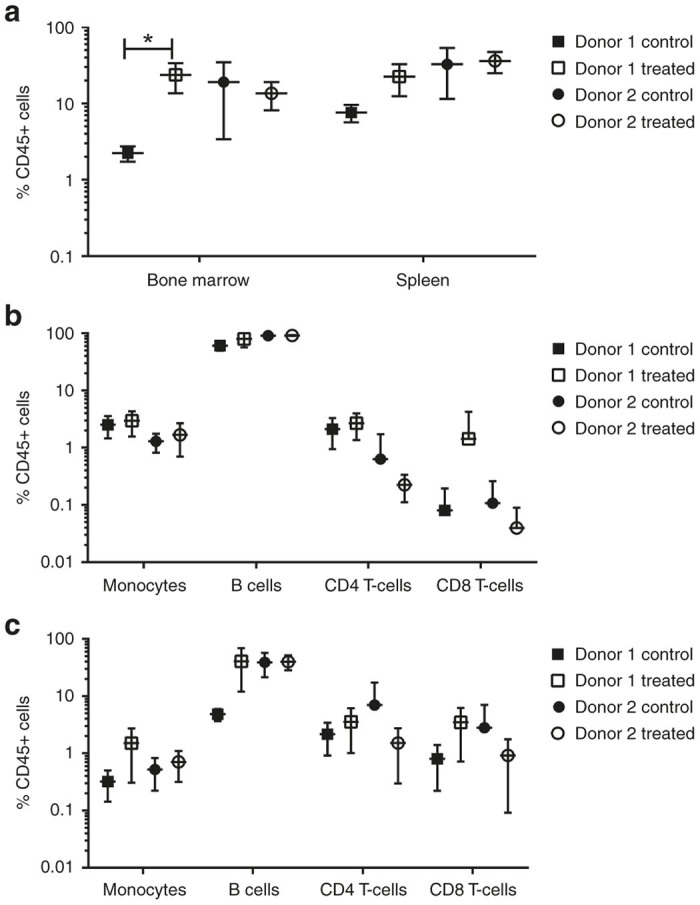
Engraftment studies. CD34+ cells from two healthy donors were stimulated overnight and subjected to electroporation using 150 µg/ml of the GMP grade mRNA (SB-728mR). (a) The %CD45+ cells in bone marrow and spleen, analyzed at 20 weeks, are shown with error bars. The frequency (%) of the engrafted human CD45+ cells that were CD14+ monocytes, CD19+ B cells, CD4+ T cells and CD8+ T cells in bone marrow (b) and in spleen (c) are shown with error bars. *P < 0.05.
The bone marrow (Figure 2b) and spleen (Figure 2c) of each animal was analyzed for the relative frequency of CD14+ monocytes, CD19+ B-cells, CD4+ T-cells and CD8+ T-cells among the CD45+ human cells (% of CD45+ cells) in control and treated animals. No significant differences in the engraftment of any lineage were observed between control and treated groups for either donor. These results indicated that treatment with rSB-728mR HSPC under the described conditions did not lead to a reduction in the level of engraftment or changes in the lineage potential of the cells.
Qualification of manufacturing process
Based on the results from our development studies and previous clinical experience, we designed a plan for production of CCR5-disrupted HSPC for an anticipated clinical trial (NCT02500849@clinicaltrials.gov) (Supplementary Figure S1). We performed full-scale process qualification runs using G-CSF-mobilized HPC-A (12–15 L apheresis) from healthy donors as starting material. CD34+ HSPC were isolated from the HPC-A from four independent donors according to Standard Operating Procedures created for the proposed clinical trial. Cells from these engineering runs were also used to perform tumorigenicity/toxicity testing (see below). We electroporated the HSPC with 150 µg/ml SB-728 mRNA, in each process qualification run in order to enhance our ability to detect the effects of off-target modification and possible tumor-inducing activity in the safety studies.
Process yields were calculated to address the feasibility of preparing sufficient product for an anticipated dosing of 2 × 106 CD34+ cells/kg patient weight when considering all potential process related losses and a fixed amount of starting material (HPC-A). All proposed in-process and release tests were run on each product. The results from the in-process testing of yield from the four products prepared are shown in Table 2. The overall process recovery from cell washing, labeling, CD34-selection and electroporation, was ~70%. These data drove the specification to collect ≥7.5 × 106 CD34+ cells/kg to allow for production of ≥ 2 × 106 SB728mRNA treated CD34+ cells/kg patient weigh with sufficient material for release testing, sample archiving and a backup product of 2.5 × 106 unmanipulated CD34+ cells/kg patient weight to be infused if the patient fails to engraft with the investigational product.
Table 2. Process yields during CD34 cell enrichment, culture and electroporation from the four qualification runs performed at clinical scale.
| Run# | CD34+ collected | % Recovery during selection | # of cells stimulated | # of cell electroporated | # of cells post-electroporated | % of cells post-electroporated | % Overall recovery |
|---|---|---|---|---|---|---|---|
| GLP Run 1 | 2.32E+08 | 100 | 2.21E+08 | 2.01E+08 | 1.41E+08 | 70 | 70 |
| GLP Run 2 | 1.70E+08 | 100 | 1.71E+08 | 1.46E+08 | 1.08E+08 | 74 | 74 |
| GLP Run 3 | 1.48E+08 | 84 | 1.40E+08 | 1.18E+08 | 1.04E+08 | 88 | 74 |
| GLP Run 4 | 4.87E+08 | 100 | 1.91E+08 | 1.66E+08 | 1.30E+08 | 78 | 68 |
GLP, ; HSPCs, hematopoietic stem/progenitor cells.
CD34+ HSPCs were isolated from healthy adult donors, prestimulated overnight and subjected to electroporation with 150 µg/ml of rSB-728mR. GLP run indicates individual process run number. Total CD34+ cell collected and percentage of cells recovered from the CD34-selection column are shown. The yield of cells before and after electroporation as well as overall process recovery is shown.
The results of release testing for viability, Mycoplasma, sterility, endotoxin, CFU assay, CCR5 modification, and phenotypic analysis from the three products used for the tumorigenicity study are shown in Table 3. All products met or exceeded release specifications set for the SB-728mR-HSPC product with the exception of Good Laboratory Practices (GLP) run #4 in which the CD34 purity of the final product was 76.71% largely due to a 15% contamination with Glycophorin A+ red blood cells. Karyotyping was performed on 20 mitotic cells from each of the three products (60 cells total). There were no significant chromosomal abnormalities and no structural aberrations of any chromosome detectable within the limits of resolution of this assay (data not shown).
Table 3. Release testing results for the qualification runs.
| Assay | Specifications | GLP#1 | GLP#2 | GLP#3 | GLP#4 |
|---|---|---|---|---|---|
| Viability | ≥ 70% Viable cells | 82% | 89% | 92% | 92% |
| Viable cell count | Report result | 1.28E+08 | 1.08E+08 | 1.03E+08 | 9.35E+07 |
| Mycoplasma PCR | Negative | Negative | Negative | Negative | Negative |
| Sterility, modified USP | Negative for bacteria and fungus after 28 days | Negative | Negative | Negative | Negative |
| Endotoxin-LAL | ≤ 1 EU/ml | < 0.500 Eu/ml | < 0.500 Eu/ml | < 0.500 Eu/ml | < 0.500 Eu/ml |
| CFU | >100 CFU/500 cells plated | 277 | 253 | 256 | 169 |
| Surveyor nuclease assay (Cel-1) | Detectable CCR5 disruption | Detected | Detected | Detected | Detected |
| 54.18 (Cel-1) | 63.8 (Cel-1) | ||||
| 52.19 (MiSeq) | 24.90 (MiSeq) | 70.83 (MiSeq) | 56.16 (MiSeq) | ||
| Phenotypic analysis | >85% CD34+ | CD34+: 93.9% | CD34+: 95.68% | CD34+: 92.4% | CD34+: 76.71% |
| CD3+: 0.74% | CD3+: 1.03% | CD3+: 1.73% | CD3+: 1.99% | ||
| CD14+: 1.26% | CD14+: 0.07% | CD14+: 1.08% | CD14+: 0.75% | ||
| CD15+: 0.62% | CD15+: 3.45% | CD15+: 1.73% | CD15+: 0.98% | ||
| CD19+: 0.37% | CD19+: 0.38% | CD19+: 1.08% | CD19+: 1.48% | ||
| GlyA+: 1.75% | GlyA+: 3.45% | GlyA+: 3.63% | GlyA+: 15.09% |
CCR5, chemokine receptor 5; CFU, colony forming units; GLP, ; PCR, polymerase chain reaction.
The four process qualification runs were subjected to full release testing as specified for clinical materials. All products passed identity, safety, potency, and sterility testing. The three GLP runs (GLP#1, 3, and 4) with the greatest level of CCR5 modification were used for the tumorigenicity study. Two of three products passed released tests for CCR5 disruption using either enzymatic (Cel-1) or sequence (MiSeq) analysis. Phenotypic markers for lineage were reported for information (product characterization) only.
Stability studies were also performed on the final products for the qualification runs for up to 18 months, as we anticipate this is the maximal time between product manufacturing and infusion in the proposed clinical study. For this purpose, aliquots from each run were cryopreserved in CryoStor CS5 (Biolife Solutions, Bothell, WA), frozen in a controlled rate freezer and evaluated at months 1, 2, and 18 for viability and potency, i.e., hematopoietic potential by CFU (see Supplementary Table S1). All products passed endotoxin, fungal, and Mycoplasma testing. Since products were all sterile at the time of the release, we did not test sterility further.
CCR5 and off-target modification
We have previously reported four off-target sites in CD34+ HSPC that include a site in the coding region of CCR2, an intergenic region on Chromosome 12 (KRR1 being the nearest gene), and intronic sites in the genes FBXL11 and ZCCHC14.15,16,23 To evaluate the level of off-target genome modification in human CD34+ HSPC, these cells were collected from mobilized donors and electroporated with 75 μg/ml or 150 μg/ml SB-728mR. Samples were collected 7 days postelectroporation and subjected to genomic DNA (gDNA) purification and sequencing analysis (MiSeq) to evaluate genome modification, i.e., insertion/deletion (indel) score (the deviation from wild-type sequence reads), at CCR5 target site. As shown in Supplementary Table S2, each of the 23 potential target sites predicted by SELEX-based in silico bioinformatics modeling (Bioinf)16 or by integration deficient lentivirus end-capture-based integration site analysis23 were analyzed by MiSeq and reported as total valid sequence reads and % indel.
Following modification (process qualification runs GLP#1–#4), cells were placed in CFU (methylcellulose) cultures as described in “Materials and Methods” or cultured for 2 weeks in bulk culture growth media containing SFT6 to assess biological activity and levels of CCR5 and off-target gene modification. Individual clones were genotyped to calculate the percentage of total colonies with CCR5 modifications, the percentage of total CFU with biallelic modifications, and percent CCR5-modified CFU having biallelic modification. High levels of modification, including biallelic CCR5 modification, were detected in all samples (Figure 3a). Analysis of off-target modifications was performed on bulk cultures and again revealed high levels of CCR5 modification with <20% modification of CCR2 and decreasing levels at the KRR1, FBLX11 and ZCCHC14 loci (Figure 3b).
Figure 3.

On-target and off-target sequence disruption. CD34+ cells were electroporated with 150 µg/ml SB-728mR and placed in bulk culture or methyl cellulose for CFU assay, as described in “Materials and Methods”. (a) Bulk cultures and single colonies were analyzed by miSeq and the percent of bulk or total CFU modified, percent of CFU with biallelic modifications among all CFU and percent of CFU with biallelic among all modified CFU is shown (b) The frequency of disruption among on/off target sequences are shown. CFU, colony forming units.
Tumorigenicity study
As part of the IND-enabling studies, we carried out a 20 week tumorigenicity/toxicology study in NSG (immunodeficient NOD/scid/γc−/−) mice to evaluate the safety of SB-728mR-HSPC. CD34+ HSPC derived from three of the healthy donors (GLP #1, #3, and #4) were processed according to our clinical Standard Operating Procedures with the exception that HSPC were electroporated with 150 µg/ml RNA, twice as much as the concentration proposed for use in the clinical studies to maximize ZFN activity and any potential ZFN-induced genotoxicity in the HSPC. The cells were cryopreserved while release tests were performed to simulate conditions expected for clinical use. Prior to injection of mice, cells were thawed and cultured overnight in media containing cytokines (SFT6) to enhance engraftment (DD personal observation). GLP#2 was not used in the tumorigenicity study because cells had CCR5 disruption of <30% at the time of release and we deemed this level not representative of our general findings nor likely to be indicative of potential for generating cells with tumorigenic potential. At the time of treatment, the on-target gene modification profile (extent of CCR5 allele modification) in SB-728mR-HSPC from GLP#1, #3, and #4 ranged from 54% to 67% indels for CCR5. As mentioned above, the CD34+ cell purity of GLP run #4 was below release criteria of 85% CD34+. The contaminants were predominantly Glycophorin A+ red blood cells and were not expected to interfere with engraftment. NSG mice were subject to nonlethal radiation (250cGy) up to 6 hours prior to injection to promote efficient engraftment of human CD34+ HSPC. A single dose administration of 106 CD34+ SB-728mR-HSPC per mouse was given via retro-orbital injection on day 0, whereas control animals received the same dose of nonelectroporated HSPC from the same donor (control group).
A total of 158 NSG mice (75 females and 84 males) received SB-728mR-HSPC and 60 control animals (30 females and 30 males) received nonelectroporated CD34+ HSPC. The animals were injected with the combined equivalent of a full human cell dose totaling 150 × 106 SB-728mR-HSPC. As with the mRNA concentration used for electroporation, this high-dose cell product was meant to uncover any potential toxicity associated with the use of gene-modified HSPC and was agreed upon by the FDA pharmacology/toxicology reviewers of the IND. Mice underwent clinical observations and evaluations of body weight, HSPC engraftment, lineage development, gene modification, gross and microscopic pathology.
Tumorigenicity study: general findings
Survival to the time-point of tumorigenicity study termination was 95% (57/60) in the control group and 86% (136/158) in the mice that received SB-728mR- HSPC; this difference did not reach statistical significance (P = 0.2; Mann–Whitney U-test). Of the 25 mice that died early, 16 developed septicemia, likely related to radiation conditioning, 14 of these in the first 3 weeks of the study. Five deaths were attributed to neoplasia (one control group animal and four test article group animals) and one death attributed to renal failure. The cause of death was undetermined from three mice (two control group animals and one test article group animal). All animals received a complete macroscopic and microscopic necropsy ~5 months after engraftment. Control and CCR5-modified HSPC cells were generally well tolerated, with no evidence of adverse effects on clinical observations, body weight or findings at necropsy. No statistically significant absolute or relative organ weight differences were recorded between controls and SB-728mR-HSPC-treated mice when data from all GLP lots were combined. Small statistically significant decreases in organ/body weight ratios were observed for several tissues (brain, heart, liver, and kidneys) in SB-728mR-HSPC-treated male mice, but lacked histologic correlates that did not occur in females or other donor groups, and were therefore attributed to gender-specific variation.
Tumorigenicity study: engraftment and gene editing
Both SB-728mR-HSPC and nonelectroporated control HSPC successfully engrafted in mice (100% of surviving mice at 4 weeks postinjection), with human hematopoietic progeny measured in blood and bone marrow in all animals during the course of the study. We evaluated the extent of engraftment by human cells in blood from animals transplanted with each donor throughout the study (Figure 4a) and in bone marrow at necropsy (Figure 4b). Bone marrow samples were lost from one control and one SB-728mR-HSPC animal due to operator error, and all 20 week-22 GLP#4 control blood samples and 8 of 25 SB-728mR-HSPC blood samples were lost due to technical issues. Animals from the GLP#4 group showed a statistically significant increase in engraftment of peripheral blood with SB-728mR-HSPC at 12 weeks in both male and female mice (P < 0.05), compared with controls. A similar increase in engraftment was seen in the bone marrow of female mice treated with SB-728mR-HSPC (GLP#1 and GLP#4), compared to controls. Collectively, mean engraftment levels in blood peaked at 12 weeks for males (2.55 ± 4.15%) and females (3.90 ± 4.59%) administered SB-728mR-HSPC, and at 4 weeks for males (0.86 ± 0.84%) and at 22 weeks for females (5.07 ± 18.83%) in the control groups. At necropsy, the percentage of animals engrafted (above background) with human cells was 78% (29 of 37 animals) in the combined control groups and 88% (112 of 128 animals) in the combined SB-728mR-HSPC groups. Similarly, the percentage of animals with human cells in the bone marrow was 98% (51 of 52 animals) in the combined control groups and 96% (120 of 125 animals) in the combined SB-728mR-HSPC groups. Together, these data demonstrated that electroporation of HSPC with SB-728mR had no negative effect on the frequency of engrafted animals at the doses used in this study.
Figure 4.

SB-728mR has no impact on HSPC engraftment. Cohorts of NSG mice were transplanted with 106 HSPC per mouse from three independent donors (GLP#1, GLP#3, and GLP#4) following electroporation in the presence of 150 µg/ml SB-728mR or no electroporation and followed for percent CD45+ cells in peripheral blood at (a) 4-, 12, and 22-weeks and (b) in bone marrow (BM) at necropsy. *P < 0.05; **P < 0.01. Dotted line shows minimal threshold for defining engraftment with human cells. Note: BM samples were lost from one control and one SB-728mR-HSPC animal due to operator error. All week-22 GLP#4 control blood samples and 8 of 25 SB-728mR-HSPC blood samples were lost due to technical issues. HSPCs, hematopoietic stem/progenitor cells.
At the time of administration into the NSG mice, the CCR5 modification profiles for the three qualifications runs (GLP#1, #3, and #4) were between 54 and 67%. MiSeq analysis was conducted on gDNA isolated from the blood (weeks 4 and 12; at necropsy) and bone marrow (at necropsy; weeks 20–22) for the same genome loci (CCR2, Chromosome 12, FBXL11, and ZCCHC14) to evaluate the stability of modification following transplantation. Control animals showed no levels of CCR5 disruption, whereas for all SB-728mR-HPSC products administered, the peak level of CCR5 disruption in blood cells occurred at week 4, with a mean value of 20.43% ± 8.67, 23.86% ± 16.52, and 27.51% ± 10.46 indels for GLP#1, #3, and #4, respectively (Figure 5). CCR5 disruption levels decreased to ~10–20% by the end of the study. At necropsy, bone marrow showed 22.11 ± 15.26%, 30.79 ± 19.45%, and 18.07 ± 10.07% indels, for the GLP#1, #3, and #4, respectively. The reduction in the frequency of CCR5 modified cells in the blood of mice at 4 and 20 weeks is likely reflective of differences in the frequency of modification of long term repopulating cells versus short term engrafting cells in the starting HSPC population. Of note, the off-target gene modifications previously observed from in vitro studies (Supplementary Table S2 and Figure 1b) were not observed above background in blood collected at necropsy from control or SB-728mR-HSPC groups. A fraction of indel levels was seen in bone marrow for CCR2 and CHR.12/KRR1 (~7% indels for each) and in bone marrow for FBXL11 (2.6% indels). These low levels of off-target gene modification in bone marrow cells from animals administered SB-728mR-HSPC did not result in findings of bone marrow toxicity as measured by histopathology evaluation.
Figure 5.
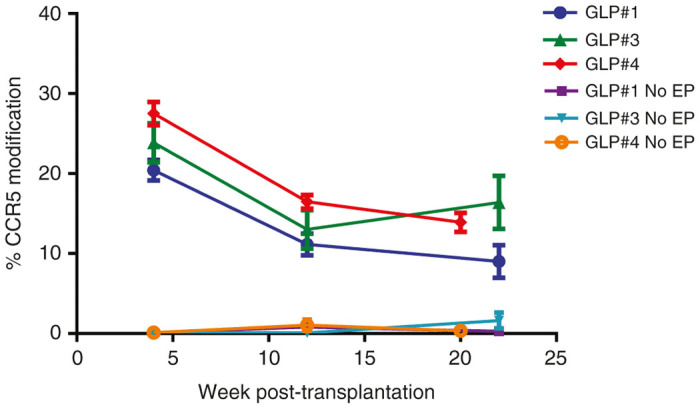
Percent CCR5 gene modification in mice transplanted with SB-728mR HSPC. NSG mice were transplanted with 106 HSPC from three independent donors (GLP#1, GLP#3, and GLP#4) following electroporation (EP) in the presence of 150 µg/ml SB-728mR or no electroporation (No EP) and followed for percent CD45+ cells in peripheral blood mononuclear cells at 4-, 12-, and 22-weeks. CCR5, chemokine receptor 5; HSPCs, hematopoietic stem/progenitor cells.
Tumorigenicity study: histopathologic findings
Neoplasms accounted for death or early termination in five animals, including one with lymphoma (SB-728mR-HSPC-treated female), one with a salivary gland adenocarcinoma (male, control), and three SB-728mR-HSPC-treated mice with osteosarcoma (two males and one female). All findings present in control animals were consistent with incidental findings commonly seen in irradiated NSG mice. There were no notable or consistent differences in body weights or body weight gains during the in-life phase of the study or in terminal body weights among the treatment groups or donor groups.
Histopathologic analysis did not detect any test-article related toxicological findings. Microscopic findings deemed to be potentially attributable to SB-728mR-treatment were limited to spleen (increased proliferation of large pale cells tabulated as “hyperplasia, lymphoid”) and sternal bone marrow (increased presence of large pleomorphic pale cells, tabulated as “infiltrate, lymphohistiocytic”), both findings were seen primarily in one group of animals, i.e., females who received cells from the same GLP #1 cell lot. Further immunohistochemistry (IHC) evaluation, described in more detail below, showed that the large pale cells in bone marrow and spleen were of human origin (human CD45+), and typical of engrafted human stem cells in NSG mice. These cells were described as engrafted, orderly, and maturing human stem cells. In addition, evaluation of blood smears revealed no unusual findings and no evidence of hematogenous neoplasia.
In the spleen, the large pale cells were identified to be of human origin using IHC as they were consistently positive for human CD45, with smaller numbers of cells also positive for human CD3 or human CD19. The cells were negative for human CD68, a macrophage/monocyte-specific marker. These cells were identified based on location, morphology and staining characteristics as lymphocytes of human stem cell origin which localized to areas consistent with splenic white pulp following engraftment in NSG mice. These large pale cells in the white pulp were human CD68-negative, and IHC evaluation of the spleen also revealed human CD45/CD68-stained cells lining the red pulp sinusoids. Based on location, morphology and staining characteristics, these cells were identified as macrophages of human origin that localized to the red pulp sinusoids following engraftment in NSG mice. There was no IHC evidence of neoplastic transformation of any of the engrafted human stem cells in the spleen.
In bone marrow, the lymphohistiocytic cells were organized into small foci or multifocal clusters and were identified as human in origin as they were consistently positive for human CD45 with intermediate staining for human CD68, limited staining for human CD3, and very little staining for CD19. IHC staining also revealed human CD68-positive myelomonocytic series cells. Based on location, morphology, and staining characteristics, these cells were identified as human CD45/human CD68-positive myeloid and/or monocytoid cells of human stem cell origin that localized to the bone marrow and were evident in small focal to multifocal clusters. There was no IHC evidence of neoplastic transformation of any of the engrafted human cells in bone marrow.
Eleven neoplasms were found during necropsy or on microscopic evaluation, including three in the control group (duodenum adenoma, adenocarcinoma, and lung adenoma) and eight in the SB-728mR-HSPC group (three lung adenomas, spleen hemangioma, three osteosarcomas, and thymic lymphoma). These equate to incidences of 5.0% in the controls (3/60 animals) and 5.1% (8/158 animals) in the SB-728mR-HSPC treated groups, respectively. IHC staining (human CD45+) demonstrated that none of the tumors were of human origin or originated from human HSPC.
To further confirm the species-of-origin for malignant cell type of the thymic lymphoma and osteosarcomas, we performed dual color fluorescence in situ hybridization (FISH) (Cytogenetic Core, COH, CA) targeting human chromosome 2 and mouse chromosome 2 on formalin-fixed paraffin embedded (FFPE) sections. The tests were performed using normal mouse spleen and thymus tissue from C57BL/6 and NSG mice and normal human CD3+ cells as controls. All tumor tissue sections evaluated had > 98% of cells with two copies of mouse chromosome 2, indicating that all tissues were of mouse origin, with no evidence of human tumor or tissue detectable (Supplementary Figure S2a,b).
Based on the results of this study, SB-728mR-HSPC modified at the CCR5 locus were well tolerated, showed successful engraftment and no evidence of neoplastic transformation of the engrafted human stem cells in the 5-month NSG mouse model. Therefore, SB-728mR-HSPC did not demonstrate potential for tumorigenesis or leukemagenesis using the described NSG mouse model.
Discussion
The development of new treatment modalities for patients with HIV-1 infections is warranted by the chronic toxicity of a life-long regimen of antiretroviral drugs. The ability to create a single, potentially curative treatment for HIV-1 infection using autologous HSPC fulfills several tenets for new drug development. First, it would address unmet medical need and one of the biggest challenges with HIV-1 treatment, i.e., patient compliance with treatment regimen. A single infusion of gene modified cells that provides lifelong resistance to HIV infection and prevents progression to AIDS obviates the need for ongoing intervention or lifelong compliance with drug dosing. Second, a single treatment could potentially be administered at a cost that is substantially lower than lifelong provision of drugs and clinical visits for drug-related sequelae and progressive disease.1 Thus, a single transplant of HIV-1 resistant blood cells could result in significant improvement in the clinical management and cost of treating this disease.
Several clinical trials have been reported using genetically modified T-cells or hematopoietic stem cells to transplant HIV-infected patients who either are currently failing HIV-1 drug therapy or undergoing autologous stem cell transplantation as part of their treatment for HIV-related lymphoma.2,24 On-going studies in patients treated with Ad5/F35SB-728 modified T-cells have demonstrated the safety of the procedure, persistence of infused cells and apparent control of virus in a patient who was naturally heterozygous for deleted CCR5. In such a patient, every cell has only one wild-type copy of the CCR5 gene, and an additional (ZFN induced) allelic disruption would rendered these cells CCR5−/−. However, a growing body of evidence points to monocyte-derived macrophages as a potential long term reservoir of latent virus,25–27 and a few reports suggest that some hematopoietic stem cells may also be infected with HIV-1.28,29 The transplantation of autologous CCR5-deleted HSPC has the potential to result in a (lifetime) supply of both HIV-1-resistant CD4+ T-cells as well as HIV-1 resistant CD4+ monocytes. The case of a single patient apparently cured of HIV-1 following stem cell transplantation using HIV-1-resistant (CCR5∆32/∆32) donor cells has already provided proof of concept for the HSPC transplantation approach.11 The obvious concern is whether autologous HSPC can be genetically edited with sufficient efficiency to reproduce the allogeneic experience.
In order for a stem cell approach to engineering an HIV resistant immune system to be successful, sufficient number of HSPC must be harvested, genetically modified and returned to the patient without significant loss of hematopoietic potential or induction of tumorigenic properties. Establishing the safety of any investigational product, especially first in class products such as the genetically modified HSPC described here, is of paramount importance prior to initiating clinical studies. The studies described herein address the practicality and safety of a proposed process in which HSPC undergo minimal manipulations to achieve the desired genomic disruption. We have previously reported on attempts to modify HSPC to confer HIV-1 resistance using viral vectors to deliver HIV resistance genes or CCR5-specific ZFN sequences.15,24 The vectors were expensive to produce (lentivirus vectors), resulted in limited and variable modification of HSPC or exhibited high cytotoxicity on the target cells (adenoviral vectors).
To circumvent these potential limitations, we have developed a nonviral platform for the delivery of messenger RNA encoding CCR5-specific ZFN sequences (SB-728mR) leading to significant levels of mono- and bi-allelic disruption of CCR5 in clones of HSPC. The RNA can be manufactured according to cGMP requirements, is relatively nontoxic and can be readily delivered by electroporation at doses required to achieve high, but not toxic dose levels. By transiently expressing ZFNs from mRNA, we have not only demonstrated the efficient biallelic disruption of CCR5 genomic sequences but also reduced the risk of off target modification and potential risk of toxicity using adenoviral vectors to modify HSPC that we previously observed.15 We have demonstrated in these studies that our manipulations allow for sufficient recovery of viable cells with in vitro colony forming potential and provide long term multilineage engraftment in immunodeficient animals without detectable tumor formation. Although we observed a small but significant reduction in CFU formation from HSPC treated with 150 µg/ml SB-728-mR and reduction in the frequency of CCR5 modified cells in the blood of mice between 4 and 20 weeks, there was no statistically significant reduction in the overall human cell engraftment between treated and untreated animals. The progressive reduction in frequency of CCR5 modification among CD45+ cells is likely reflective of differences in the frequency of modification between short term repopulating (progenitor) cells and long term repopulating (stem) cells in the starting HSPC population. While it is also possible that cells with CCR5 modification may have a selective disadvantage, there is no evidence that CCR5 gene expression is required for hematopoiesis and we are unable to distinguish between these two possibilities without prospective isolation of a more refined stem cell population as a substrate for gene modification. Previous studies using this model system demonstrated that cord blood HSPC modified by electroporation using SB-728 DNA, provided lasting protection of CD4+ T-cells and were able to reduce serum levels of virus by at least one log.14 Taken together these data suggests that the use of CCR5 modified HSPC as described here will result in hematopoietic engraftment with an HIV-resistant immune repertoire capable of controlling viremia and restoring immune function.
We have now optimized our procedures and established the feasibility of manufacturing clinical doses of equivalent material using adult HSPC-derived cell products. The general safety and sufficient efficacy (biallelic disruption rates) warrant the movement of this approach into clinical trials. In the clinical trial, HSPC will be isolated from patients mobilized with G-CSF (filgrastim) and plerixafor (Mozobil/AMD3100) to ensure sufficient numbers of HSPC are harvested for both a research and a backup product. Plerixafor enhances mobilization of HSPC, and this treatment has been successfully used to optimize HSPC collection from multiple myeloma and lymphoma patients who failed to collect sufficient HSPC for transplant using standard protocols.30,31 A minimal collection of ≥ 7.5 × 106 CD34+ cells/kg will be required, of which the first 2.5 × 106 CD34+ cells/kg patient weight would be collected as an unmanipulated, cryopreserved back-up product to be infused if a patient fails to engraft with the investigational product. The remaining harvest would be used to manufacture SB-728mR-HSPC adequate to ensure that the target dose of ≥ 5 × 106 CD34+ cells/kg will be available for infusion after accounting for process losses and release testing. This target dose of CD34 cells was selected based on evidence that the cut-off for more rapid hematopoietic recovery post-transplant occurs with a dose ≥ 2 × 106 CD34+ cells/kg (reviewed in Vose et al.32). For the validation runs that established our proposed manufacturing method, we used only G-CSF-mobilized HSPC, and this was sufficient to meet FDA expectations for preclinical development of the candidate therapeutic. There is no evidence to suggest that the engraftment of a CD34 cell is different when mobilized by G-CSF alone versus G-CSF plus plerixafor.33 Despite the differences in molecular signature and phenotype of CD34+ cells mobilized by G-CSF versus G-CSF plus plerixafor,34 there is convincing evidence that the two CD34+ cell products perform similarly in the setting of HSPC transplantation.32,35 Thus, we rationalized that the cost and risk of complications with G-CSF and plerixafor in healthy donors was not justifiable for these studies.
In addition, to test whether the proposed treatment of CD34+ HSPC would induce significant tumorigenic effects, a dose of 150 µg/ml of SB-728mR was chosen (twice that to be used in the clinical trial) to enhance the likelihood of detecting genotoxicity/tumorigenicity in SB728mRNA treated HSPC. As shown in Figure 1b, the 75 µg/ml SB-728mR dose, the planned clinical dose, was nearly as effective at inducing overall CCR5 disruption but may result in a lower level of biallelic disruption. If we fail to see control of viremia in the first proposed clinical trial (NCT02500849) using the 75 µg/ml SB-728mR dose, then the higher dose of 150 µg/ml will have been shown to be safe and could be substituted in later patient cohorts.
There was no evidence of gross chromosomal abnormalities due to SB-728mR treatment based on karyotypic analysis of a limited number of metaphase spreads (n = 60). While more in depth methods for chromosomal analysis such as chromosomal microarray assay are useful as a clinical tool in the evaluation of patients with developmental problems,36 there is lack of experience with this assay as a validated toxicologic tool. In vivo assays were used to determine if any genomic alterations (not detected by analysis of metaphase spreads) led to loss of lineage potential or tumor formation. This approach was sufficient to meet FDA expectations for preclinical safety in the context of our proposed trial.
The major off-target of SB-238mR ZFN is CCR2, a gene that is important in monocyte trafficking to tissue.37 In murine models, disruption of CCR2 function has been associated with worsening of West Nile virus (WNV) infection38 but with less severity of pulmonary influenza infection.39 It is presumed that the disproportionately small fraction of monocytes that might have CCR2 disruption will not be sufficient to produce any biologic effects in our research patients. Similarly, the other off-targeting by SB-728mR ZFN, because it involves the intergenic sequences of FBXL11, ZCCHC14, and Chr 12/KRR1 and not the exons within modified cells, have little likelihood for producing biologic consequences. The on-target effect of CCR5 disruption, on the other hand, could have an effect on WNV infection since this has been shown in persons homozygous for the CCR5 delta 32 mutations who have a greater risk for symptomatic WNV infection.40 During the clinical trial, subjects will be counseled to maintain mosquito precautions when seasonal WNV activity is high.
These results represent significant progress toward clinical translation of this approach for several reasons. First, attaining high levels of biallelic disruption is significant since, although cells possessing single CCR5 disruptions are less susceptible to HIV infection, only cells containing biallelic CCR5 disruptions are completely resistant to R5-tropic viral infections.9 Second, the elimination of viral vectors significantly reduces the cost of production of clinical materials. Third, the process is scalable, highly reproducible and is not dependent on batch specific characteristics (e.g., viral titer) that often confound reproducibility. All of these parameters are best addressed early in the product development lifecycle as we have done here.
Finally, we have developed a genomic editing methodology to modify a target locus in human HSPC that appears to be safe, efficient, and reproducible. As with any preclinical studies, the methods used were designed to assess the safety, efficiency, and reproducibility only of the proposed manufacturing process. Certain changes to cell mobilization protocol and the use of in vitro and animal models of engraftment reflect practical and safety limitations encountered during the development of cell therapy products and are thus of limited use in predicting outcomes when patient samples are prepared in a similar fashion. Nonetheless, these studies were sufficient to meet the preclinical requirements for the proposed trial and were in part developed based on our collective prior experience with HSPC and T-cell gene therapy using T-cells and during pre-IND discussion with the FDA and other regulatory and process development experts. These studies obviously do not replace the need for phase 1 safety testing in the clinic and were meant to inform the risk:benefit analysis required to initiate phase 1 clinical trials.
The methodologies used here are not limited to CCR5 disruption, but may be applied to the genetic disruption of other genes of stem cells implicated in disease. Clinical studies in this area will undoubtedly pave the way for future applications of genome editing technologies and represent a major advance in the development of cell based genetic therapy for disease.
Materials and Methods
pSB-728 expression plasmid and mRNA product preparation
The plasmid pSB-728mR was constructed at Sangamo BioSciences (Richmond, CA, www.sangamo.com). The CCR5 ZFN expression cassette encodes for two ZFNs (8267-FokEL and 8196z-FokKK) separated by a 2A peptide sequence and a 64 nt polyadenylation sequence (poly A) on the 3′-end. This bicistronic mRNA permits expression of both ZFNs from the same transgene in nearly equivalent proportions. The final plasmid encoding the SB-728mR expression cassette was generated by cloning the whole transcription unit (from the T7 promoter to the polyadenylation sequence) into the pVax vector (Life Technologies, Carlsbad, CA, www.lifetechnologies.com). SB-728mR is a capped messenger RNA (mRNA) synthesized from the plasmid pSB-728mR by in vitro transcription using T7 RNA polymerase. For this study, a 10 mg process qualification lot (Batch No. 13-25-02) was produced at EUFETS GmbH using the same process and plasmid DNA template used for GMP manufacturing. The same lot of the SB-728mR was used to generate all three lots of SB-728mR HSPC.
The manufacturing process for SB-728mR (mRNA) used in the tumorigenicity study and for the clinical studies is similar, with differences mainly attributed to larger scale manufacturing of the GMP material, as described below.
Clinical grade SB-728mR manufacturing
Based on guidance received from the FDA during a pre-IND teleconference, the cGMP manufacturing plan for CCR5 ZFN mRNA (SB-728mR) involved deriving a bacterial Master Cell Bank (MCB) and producing a cGMP plasmid lot of pSB-728mR at Althea Technologies (San Diego, CA). The plasmid manufacturing project at Althea was initiated with process development (PD) and host strain selection for the MCB. Four bacterial host strains were transformed with the plasmid construct to make Research Cell Banks (RCB). Evaluated for bacterial growth, plasmid yields, and product purity, two RCB were further evaluated in 5l fermentation scouting runs. The best performing strain (DH10β) was chosen to generate the MCB. MCB production yielded a total of 200 vials: four were used for QC testing, and 196 went to inventory. Two 30l GMP manufacturing runs were initiated. The downstream purification, final formulation and vial filling of pSB-728mR plasmid were completed, and the final lot was released. This pSB-728mR GMP lot was then transferred to EUFETS GmbH (Idar-Oberstein, Germany) for cGMP production of the SB-728mR. Concurrently, EUFETS carried out PD runs for production of SB-728mR mRNA.
A 10 mg PD lot of SB-728mR mRNA has been made at EUFETS, and the material was evaluated in the CD34 HSPC manufacturing process to determine if similar levels of CCR5 disruption are obtained in comparison to previously tested research lots of SB-728mR mRNA. Both the mRNA production yield and potency by ZFN activity in CD34+ HSPC were found to be satisfactory. This 10 mg lot of SB-728mR mRNA was used to develop the final CD34+ HSPC manufacturing process and to manufacture the cells for the non-GLP toxicity study (qualification, GLP runs #1–#4).
The cGMP production of SB-728mR at EUFETS production yielded ~360 mg of SB-728mR mRNA. After removing vials for QC testing, retains and stability, 238 vials (1 mg/vial) went to long term storage and will be used for clinical studies. This amount of material would be sufficient for manufacturing 238 lots of SB-728mR-HSPC.
CD34+ cell processing
G-CSF mobilized HPC-A products were ordered using a commercial provider, Progenitor Cell Therapy (Mountain View, CA), and shipped via overnight courier to COH. Informed consent was obtained for each donor by the vendors according to vendor-specific protocols and Institutional Review Board (IRB) review. HPC-A products were washed and CD34+ cells were enriched using the Miltenyi CliniMACS device according to manufacturer’s directions. All products were processed within 36 hours of collection.
Pre-stimulation of CD34+ cells
CD34+ cells are enriched from the HSPC products by closed bag processing and magnetic bead selection using a Miltenyi CliniMACS cell selection device (Miltenyi Biotec., Auburn, CA, www.miltenyibiotec.com) in the process development laboratories of COH. Enriched CD34+ cells undergo overnight prestimulation in Serum-free Stem Cell Growth Media (SCGM, CellGenix, Freiburg, Germany, www.cellgenix.com) formulated with 2 mmol/l of L-glutamine (Mediatech, Manassas, VA, www.cellgro.com) and hematopoietic cytokines that include the following; 100 ng/ml stem cell factor (SCF), Fms-like tyrosine kinase receptor ligand (Flt-3L), interleukin-6 (IL-6), (Invitrogen, Carlsbad, CA, www.invitrogen.com) and 10 ng/ml of thrombopoietin (Tpo), (CellGenix, Freiburg, Germany, www.cellgenix.com).
Electroporation of CD34+ cells
Electroporation was performed using a preconfigured setting on a MaxCyte GT Transfection System in a CL1.1 process assembly (MaxCyte, Gaithersburg, MD). Cells are electroporated using a proprietary electroporation buffer in sterile, single use processing assemblies. Postelectroporation, cells were transferred to a VueLife bag (Saint Gobain, Gaithersburg, MD) and incubated for 20 minutes in a 37°C, 5% CO2 incubator. The cells were then diluted in 10 volumes of SCGM with hematopoietic cytokines as previously described and transiently incubated at 30°C for 16–18 hours to enhance ZFN expression. Cells were given additional media with cytokines and were transferred to a 37°C, 5% CO2 incubator for an additional 24 hours. At 48 hours post-electroporation, the SB-728mRNA modified CD34+ cells were washed three times in PBS:HSA and resuspended in CryoStor CS5 cryoprotectant (BioLife Solutions, Bothell, WA) and frozen using a controlled rate freezer. Modified cells were stored in the vapor phase of a LN2. Prior to freezing, cell viability (Guava ViaCount) was checked to ensure >70% viability. The nontransfected control CD34+ HSPC were cultured and stored identically, but were neither exposed to SB-728mR nor electroporated.
Methylcellulose culture
Unmodified controls and electroporated cells were harvested 16–24 hours after electroporation. A total of 500 cells were plated per plate in triplicate in MethoCult H4435-enriched methylcellulose medium according to the manufacturer’s instructions (StemCell Technologies, http://www.stemcell.com). Total colonies and differential lineage colonies were enumerated under inverted microscope 12–14 days after incubation.
Surveyor nuclease assay
The percentage of CCR5 alleles disrupted by ZFN treatment was measured by Surveyor nuclease assay which was previously qualified by Sangamo to meet FDA requirements. In this assay, genomic DNA was extracted using the MasterPure Complete Purification kit (Epicentre, Madison, WI) according to manufacturer’s instructions. The purified genomic DNA was used as a template to amplify a fragment of the CCR5 gene using the primers C5_Cel_160_F1: AAGATGGATTATCAAGTGTCAAGTCC; and C5_Cel_160_R1: CAAAGTCCCACTGGGCG in the presence of a 32P-dATP and 32P-dCTP. The polymerase chain reaction products were then heated, allowed to reanneal followed by treatment with the mismatch-sensitive Surveyor nuclease (Transgenomic, Omaha, NE) as described in order to detect insertions and deletions caused by nonhomologous end joining. The resulting radiolabeled products digested with the Surveyor nuclease were resolved by polyacrylamide gel electrophoresis (PAGE), and the ratio of cleaved to uncleaved products was calculated to give a measure of the frequency of gene disruption. The assay is sensitive enough to detect single-nucleotide changes.
Targeted genome sequencing
Gene modification efficiency was assessed at Sangamo BioSciences by deep DNA sequencing. The locus of interest (the ZFN binding site within the CCR5 gene and the four known off-target sites (CCR2, FBXL11, ZCCHC14, and Chr. 12/KRR1)) was polymerase chain reaction-amplified and the level of modification was determined by paired-end deep sequencing on an Illumina MiSeq. Paired sequences were merged via SeqPrep (John St. John, https://github.com/jstjohn/SeqPrep, unpublished). A Needleman-Wunsch (Needleman, 1970) alignment was performed between the wild-type sequence and the obtained query sequence to map insertions and deletions (indels). Gene modification was calculated by dividing the total number of indel sequence reads by the total number of sequence reads. A range of 3,000–30,000 total sequence reads per sample was obtained for most samples. However, depth of sequencing varied among different applications. Specifically, ~100,000 sequences per sample were obtained for the off-target analysis (Supplementary Table S2). About 1,000 or more sequences per sample were obtained for CFU genotyping analysis (Figure 3a and Table 1).
SB-728mR-HSPC preparation for use in the tumorigenicity study
The same manufacturing process used to generate SB-728mR-HSPC for clinical studies was used to generate SB-728mR-HSPC for the toxicology study, with the exception that twice the amount of SB-728mR (twofold the clinical dose) was utilized for electroporation of the HSPC in the toxicology study and cells were prestimulated for 1 day after thawing and prior to infusion, to ensure engraftment in the mice. The test article, SB-728mR-HSPC, was prepared in the Laboratory for Cellular Medicine at COH, Duarte in four independent qualification runs, GLP#1, #2, #3, and #4, using CD34+ HSPC isolated by apheresis from four G-CSF mobilized healthy volunteer donors as described above. An aliquot of cells from each lot was frozen separately and evaluated by MiSeq deep sequencing to determine the level of CCR5 disruption and ensure adequate ZFN activity (≥30% CCR5 gene modification) was achieved.
Prior to injection, the cryopreserved cells from GLP#1, #3, and #4 were thawed, washed, and cultured for 24 hours in media containing a cytokine cocktail as described previously. Viability was assessed to ensure cell viability remained >70%. The cells were then harvested, formulated in phosphate-buffered saline (PBS) with 1% heparin, and injected intravenously via retro-orbital injection (50 μl) into NSG mice NSG mice were subject to nonlethal radiation (250 cGy) up to 6 hours prior to injection to promote efficient engraftment of human CD34+ HSPC.
Tumorigenicity study methods
Adult male and female NOD.Cg-Prkcdscid Il2rgtm1Wjl/SzJ (NOD/SCID/IL2Rγnull, or NSG) mice were originally purchased from Jackson Labs (Bar Harbor, ME), and a breeding colony was established at the USC Health Sciences Campus. About 218 NSG mice were used in this study, separated into control HSPC (30 males, 30 female; total 60 animals) and SB-728mR-HSPC (83 males, 75 females; total 158 animals) groups. Animals were approximately 7–10 weeks of age at the time of treatment, and weighed ~16–24 g (females) and 20–30 g (males).
All animal experiments were carried out according to the National Institute of Health (NIH) guidelines for the care and use of laboratory animals and the USC Standard Operating Procedures, and were approved by the IACUC of the USC Keck School of Medicine. At all times, AALAC standards for the care and treatment of animals were followed.
Adult NSG mice received nonlethal full body irradiation (250 cGy) prior to engraftment. Within 6 hours of being irradiated mice were anesthetized using 2.0–2.5% isofluorane. Once anesthetized, the animals were injected intravenously via retro-orbital injection with 106 CD34+ HSPC, formulated in 50 μl PBS/1% heparin, using a 28 and 1/2 gauge needle and U-110 insulin syringe. Dose volume was 50 μl/mouse. Postinjection, mice were monitored daily for general health, appearance, behavioral abnormalities, and weighed weekly.
Animals were observed for up to 6 hours following HSPC dosing for clinical signs of toxicity.
Thereafter, cages were checked daily for mortality or moribundity. Mice were observed for any visible indicators of changing health using standard clinical observations. If any animal showed an overt change, a more detailed inspection was performed in a biological safety cabinet and the specific ailment noted. If the condition was not an indication for euthanasia, the animal was monitored closely throughout the day to evaluate if the condition was improving or deteriorating.
If the condition was worsening, a determination was made to kill the animal.
Once weekly, individual mice were weighed and evaluated for any signs of hunching and/or lethargy, inability to ambulate, or other signs of obvious distress. A gentle palpation of the animal was performed to inspect for tumor growth. Additionally, the eyes, skin, and general appearance were inspected for signs of neurological or autonomic issues. Any specific symptoms were noted.
All unscheduled deaths were recorded in the study report. In accordance with USC’s IACUC guidelines, animals that had evidence of sustained, severe graft versus host disease, >15% weight loss and/or severe hair loss, or were hunching and/or lethargic, unable to ambulate, or other signs of obvious distress or lethargy were euthanized prior to scheduled sacrifice (20–22 weeks). All USC IACUC endpoint guidelines were followed.
Retro-orbital blood sampling was performed under anesthetic (2.0–2.5% isofluorane), and the blood was collected using sterile heparinized micro-hematocrit capillary tubes.
Blood (50–100 μl) was collected 4 and 12 weeks post-treatment and at necropsy (week 20–22) via retro-orbital bleed to measure the percentage of human CD45+ cells by fluorescence-activated cell sorting (FACS).
Blood (50–100 μl) was collected at necropsy via retro-orbital bleed to isolate genomic DNA for PCR analysis to detect the presence of human genomic DNA using human-specific primers, to analyze modification at CCR5 and the major off-target sites by MiSeq deep sequencing, and to produce blood smears for staining with Wrights-Giemsa and differential count assessment.
Mouse engraftment and human cell analysis
Prior to injection, the cryopreserved cells from GLP#1, #3, and #4 were thawed, washed, and cultured for 24 hours in media containing a cytokine cocktail as described previously. Viability was assessed to ensure cell viability remained >70%.
A total of 105 female and 138 male 7–10 weeks old NOD.Cg-Prkcdscid Il2rgtm1Wjl/SzJ (NSG) mice were weighed and separated into cages based on sex at 1 week prior to engraftment. On the day of engraftment, mice were subjected to nonlethal radiation (250 cGy) up to 6 hours prior to injection to promote efficient engraftment of human CD34+ HSPC. HSPC were suspended at a concentration of 2 × 107 cells/ml in PBS containing 1% heparin and each mouse received 106 cells by retro-orbital injection as previously described.14
Mice were monitored daily for signs of distress defined as ruffled fur, excessive grooming, hunched postures, or lethargy. Animals in distress were further observed and if improvement was not observed within 24 hours, mice were killed and preserved in formaldehyde for necropsy at study completion. Weights of mice were obtained every week postengraftment and any mouse exhibiting a >15% drop in body weight was killed and preserved in formaldehyde for necropsy at study completion.
Peripheral blood (70 μl) was sampled every 4 weeks after engraftment for 20 weeks, and a fraction of the spleen and bone marrow were isolated at necropsy. Necropsies were performed by Vet Path Services (Mason, OH). Mouse blood and tissue samples were also subjected to genomic DNA purification using NucleoSpin Tissue XS kits (Macherey-Nagel, Bethlehem, PA) and subsequent molecular analysis as described above.
For flow cytometric analysis, whole blood and tissue samples were blocked in FCS (Denville) and stained with the following antibody-fluorophore conjugates: CD4-FITC (RPA-T4), CD3-PE (UCHT1), CD19-APC (HIB19), and CD45-PerCP (TUI16) (BD Biosciences) for 15 minutes at room temperature. Red blood cells were lysed after staining by incubation in BD Pharm Lyse buffer (BD Biosciences), lysis was halted by the addition of PBS, and cells were analyzed by flow cytometry using a Cyan ADP Flow Analyzer (Beckman Coulter). Compensation samples were created with BD CompBeads (BD Biosciences) and compensation for signal overlap was performed at the time of acquisition. Analysis of flow cytometry data was performed using FlowJo software version 9.5.3 or version X (Treestar, Ashland, OR). Initial gating was performed as forward scatter height versus forward scatter area to obtain the single cell population; the resulting population was plotted on a side scatter area versus forward scatter area grid to gate for live lymphocyte populations.
All animal experiments were carried out according to the National Institute of Health (NIH) guidelines for the care and use of laboratory animals and were performed with the approval of the University of Southern California Institutional Animal Care and Use Committee.
Acknowledgments
The studies published here were funded by the California Institute for Regenerative Medicine grants DR1-01490 for the USC, Sangamo and COH teams and by CIRM grant SP3A-07536 for the Sangamo BioSciences and COH teams. Research reported in this publication included work performed in the Cytogenetics Core, supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The following are employees of Sangamo Biosciences: M.C.H., J.W., G.L., P.D.G., K.A.K., S.B.H., and K.M. No COH or University of Southern California Keck School of Medicine employees had a conflict of interest in this research.
References
- DiGiusto, DL, Stan, R, Krishnan, A, Li, H, Rossi, JJ and Zaia, JA (2013). Development of hematopoietic stem cell based gene therapy for HIV-1 infection: considerations for proof of concept studies and translation to standard medical practice. Viruses 5: 2898–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas, P, Stein, D, Tang, WW, Frank, I, Wang, SQ, Lee, G et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370: 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raport, CJ, Gosling, J, Schweickart, VL, Gray, PW and Charo, IF (1996). Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP-1beta, and MIP-1alpha. J Biol Chem 271: 17161–17166. [DOI] [PubMed] [Google Scholar]
- Deng, H, Liu, R, Ellmeier, W, Choe, S, Unutmaz, D, Burkhart, M et al. (1996). Identification of a major co-receptor for primary isolates of HIV-1. Nature 381: 661–666. [DOI] [PubMed] [Google Scholar]
- Dragic, T, Litwin, V, Allaway, GP, Martin, SR, Huang, Y, Nagashima, KA et al. (1996). HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381: 667–673. [DOI] [PubMed] [Google Scholar]
- Doranz, BJ, Rucker, J, Yi, Y, Smyth, RJ, Samson, M, Peiper, SC et al. (1996). A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85: 1149–1158. [DOI] [PubMed] [Google Scholar]
- Choe, H, Farzan, M, Sun, Y, Sullivan, N, Rollins, B, Ponath, PD et al. (1996). The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85: 1135–1148. [DOI] [PubMed] [Google Scholar]
- Feng, Y, Broder, CC, Kennedy, PE and Berger, EA (1996). HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272: 872–877. [DOI] [PubMed] [Google Scholar]
- Liu, R, Paxton, WA, Choe, S, Ceradini, D, Martin, SR, Horuk, R et al. (1996). Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86: 367–377. [DOI] [PubMed] [Google Scholar]
- Zimmerman, PA, Buckler-White, A, Alkhatib, G, Spalding, T, Kubofcik, J, Combadiere, C et al. (1997). Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk. Mol Med 3: 23–36. [PMC free article] [PubMed] [Google Scholar]
- Allers, K, Hütter, G, Hofmann, J, Loddenkemper, C, Rieger, K, Thiel, E et al. (2011). Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood 117: 2791–2799. [DOI] [PubMed] [Google Scholar]
- Yukl, SA, Boritz, E, Busch, M, Bentsen, C, Chun, TW, Douek, D et al. (2013). Challenges in detecting HIV persistence during potentially curative interventions: a study of the Berlin patient. PLoS Pathog 9: e1003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan, A and Zaia, JA (2014). HIV-associated non-Hodgkin lymphoma: viral origins and therapeutic options. Hematology Am Soc Hematol Educ Program 2014: 584–589. [DOI] [PubMed] [Google Scholar]
- Holt, N, Wang, J, Kim, K, Friedman, G, Wang, X, Taupin, V et al. (2010). Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol 28: 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L, Krymskaya, L, Wang, J, Henley, J, Rao, A, Cao, LF et al. (2013). Genomic editing of the HIV-1 coreceptor CCR5 in adult hematopoietic stem and progenitor cells using zinc finger nucleases. Mol Ther 21: 1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, EE, Wang, J, Miller, JC, Jouvenot, Y, Kim, KA, Liu, O et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26: 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabot, S, Rosazza, C, Golzio, M, Zumbusch, A, Teissié, J and Rols, MP (2013). Nucleic acids electro-transfer: from bench to bedside. Curr Drug Metab 14: 300–308. [DOI] [PubMed] [Google Scholar]
- Van Tendeloo, VF, Ponsaerts, P and Berneman, ZN (2007). mRNA-based gene transfer as a tool for gene and cell therapy. Curr Opin Mol Ther 9: 423–431. [PubMed] [Google Scholar]
- Morgan, RA and Kakarla, S (2014). Genetic modification of T cells. Cancer J 20: 145–150. [DOI] [PubMed] [Google Scholar]
- Li, L, Allen, C, Shivakumar, R and Peshwa, MV (2013). Large volume flow electroporation of mRNA: clinical scale process. Methods Mol Biol 969: 127–138. [DOI] [PubMed] [Google Scholar]
- Watanabe, M, Umeyama, K, Matsunari, H, Takayanagi, S, Haruyama, E, Nakano, K et al. (2010). Knockout of exogenous EGFP gene in porcine somatic cells using zinc-finger nucleases. Biochem Biophys Res Commun 402: 14–18. [DOI] [PubMed] [Google Scholar]
- Doyon, Y, Choi, VM, Xia, DF, Vo, TD, Gregory, PD and Holmes, MC (2010). Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat Methods 7: 459–460. [DOI] [PubMed] [Google Scholar]
- Gabriel, R, Lombardo, A, Arens, A, Miller, JC, Genovese, P, Kaeppel, C et al. (2011). An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29: 816–823. [DOI] [PubMed] [Google Scholar]
- DiGiusto, DL, Krishnan, A, Li, L, Li, H, Li, S, Rao, A et al. (2010). RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med 2: 36ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, JH, Hearps, AC, Martin, GE, Williams, KC and Crowe, SM (2014). The importance of monocytes and macrophages in HIV pathogenesis, treatment, and cure. AIDS 28: 2175–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A, Abbas, W and Herbein, G (2014). HIV-1 latency in monocytes/macrophages. Viruses 6: 1837–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeycutt, JB, Wahl, A, Baker, C, Spagnuolo, RA, Foster, J, Zakharova, O et al. (2016). Macrophages sustain HIV replication in vivo independently of T cells. J Clin Invest 126: 1353–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara, LA and Collins, KL (2011). Hematopoietic stem/precursor cells as HIV reservoirs. Curr Opin HIV AIDS 6: 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara, LA, Ganesh, JA and Collins, KL (2012). Latent HIV-1 infection occurs in multiple subsets of hematopoietic progenitor cells and is reversed by NF-κB activation. J Virol 86: 9337–9350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating, GM (2011). Plerixafor: a review of its use in stem-cell mobilization in patients with lymphoma or multiple myeloma. Drugs 71: 1623–1647. [DOI] [PubMed] [Google Scholar]
- Yannaki, E, Anagnostopoulos, A, Karponi, G, Zervou, F, Constantinou, V, Psatha, N et al. (2013). Superior Stem Cell Mobilization by a Combination of Plerixafor + G-CSF in Patients with Thalassemia. In: American Society of Gene & Cell Therapy 16th Annual Meeting. American Society for Cell & Gene Therapy Salt Lake City, Utah. [Google Scholar]
- Vose, JM, Ho, AD, Coiffier, B, Corradini, P, Khouri, I, Sureda, A et al. (2009). Advances in mobilization for the optimization of autologous stem cell transplantation. Leuk Lymphoma 50: 1412–1421. [DOI] [PubMed] [Google Scholar]
- Alexander, ET, Towery, JA, Miller, AN, Kramer, C, Hogan, KR, Squires, JE et al. (2011). Beyond CD34+ cell dose: impact of method of peripheral blood hematopoietic stem cell mobilization (granulocyte-colony-stimulating factor [G-CSF], G-CSF plus plerixafor, or cyclophosphamide G-CSF/granulocyte-macrophage [GM]-CSF) on number of colony-forming unit-GM, engraftment, and Day +100 hematopoietic graft function. Transfusion 51: 1995–2000. [DOI] [PubMed] [Google Scholar]
- Donahue, RE, Jin, P, Bonifacino, AC, Metzger, ME, Ren, J, Wang, E et al. (2009). Plerixafor (AMD3100) and granulocyte colony-stimulating factor (G-CSF) mobilize different CD34+ cell populations based on global gene and microRNA expression signatures. Blood 114: 2530–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brave, M, Farrell, A, Ching Lin, S, Ocheltree, T, Pope Miksinski, S, Lee, SL et al. (2010). FDA review summary: Mozobil in combination with granulocyte colony-stimulating factor to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation. Oncology 78: 282–288. [DOI] [PubMed] [Google Scholar]
- Miller, DT, Adam, MP, Aradhya, S, Biesecker, LG, Brothman, AR, Carter, NP et al. (2010). Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86: 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boring, L, Gosling, J, Chensue, SW, Kunkel, SL, Farese, RV Jr, Broxmeyer, HE et al. (1997). Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest 100: 2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, JK, Obara, CJ, Rivollier, A, Pletnev, AG, Kelsall, BL and Murphy, PM (2011). Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J Immunol 186: 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, KL, Sweeney, S, Kang, BD, Ramsburg, E and Gunn, MD (2011). CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J Immunol 186: 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, JK, Louie, CY, Glaser, C, Jean, C, Johnson, B, Johnson, H et al. (2008). Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta-analysis of 4 cohorts in the US epidemic. J Infect Dis 197: 262–265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.