

Abstract
We describe a new catalytic method for accessing carbocation intermediates via the mesolytic cleavage of alkoxyamine radical cations. In this process, electron transfer between an excited state oxidant and a TEMPO-derived alkoxyamine substrate gives rise to a radical cation with a remarkably weak C-O bond. Spontaneous scission results in the formation of the stable nitroxyl radical TEMPO• as well as a reactive carbocation intermediate that can be intercepted by a wide range of nucleophiles. Notably, this process occurs under neutral conditions and at comparatively mild potentials, enabling catalytic cation generation in the presence of both acid sensitive and easily oxidized nucleophilic partners.
Keywords: carbocation, mesolytic cleavage, radical cation, alkoxyamine, photoredox catalysis, TEMPO
Graphical abstract
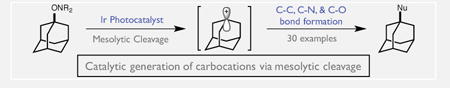
Though carbocations are classical intermediates in synthetic chemistry,[1] their applications in complex target synthesis and asymmetric catalysis remain limited by the methods required for their generation. Conventional approaches rely on the use of either strong Lewis or Brønsted acids[2] or stoichiometric silver reagents.[3] In turn, these protocols place restrictions on the scope of nucleophiles that can be successfully employed. More recently, facilitated ionization mediated by thioureas and other related hydrogen-bond donor catalysts have led to tremendous advances in asymmetric carbocation reactivity.[4] However, unstabilized carbocations are often difficult to access using these methods. In light of these constraints, we reasoned that new catalytic methods for the generation of carbocations under neutral conditions might provide a significant synthetic benefit, and enable more extensive use of these versatile electrophiles in complex contexts. We report here a novel method for the catalytic generation of simple benzylic and tertiary alkyl carbocations based on the mesolytic cleavage of TEMPO-derived alkoxyamine radical cations and their efficient capture by a wide range of nucleophiles. The design, development and mechanistic features of this protocol are described herein.
Our interest in the mesolytic cleavage of radical cations stems from the fact that bonds proximal to the unpaired electron in these intermediates are dramatically destabilized.[5,[6] In certain cases these bonds are sufficiently weakened such that they undergo spontaneous scission, resulting in the formation of two new intermediates – a neutral free radical and a carbocation. While pioneering studies from Arnold,[7] Floreancig,[8] Mariano,[9] and Albini[10] have demonstrated the feasibility and value of these methods, mesolytic cleavage-based strategies for simple carbocation generation remain underutilized.
To design a practical system for cation generation, we first sought to understand the molecular features that govern the efficiency of mesolytic cleavage. Foremost, to enable bond breaking, the strength of the scissile bond in the radical cation must be reduced to near 0 kcal/mol. The extent of bond weakening associated with one electron oxidation of any substrate can be readily calculated using the thermochemical cycle shown in Scheme 1. In this scheme, the difference in bond strengths between the neutral substrate and the radical cation is equal to the potential difference between the (R-X/R-X+•) and (R+/R•) couples (ΔBDFE = ΔE0). As the potential required for cation reduction is decoupled from the identity of the dissociated radical fragment, the strength of the scissile bond in the radical cation is principally a function of two variables – the BDFE of the scissile bond in the starting material and the potential required for generation of the radical cation. From a synthetic perspective, we sought to design a system in which substrate oxidation would occur at a mild potential to ensure compatibility with more complex substrates and a wide range of nucleophiles. However, confining this couple to a less positive potential requires that the strength of the scissile bond in the starting material be weak – a feature which could potentially result in substrate instability.
Scheme 1.

Reaction design and thermochemistry of bond weakening in radical cations. BDFE = bond dissociation free energy in kcal/mol.
In seeking to balance these competing requirements, we were drawn to the idea that alkoxyamines derived from TEMPO might be ideal substrates for mesolytic cleavage. As a result of the high stability of TEMPO radical, the C-O bond strengths in these compounds are unusually weak. For example, the benzylic C-O BDFE in the TEMPO adduct of isopropyl benzene is only 26 kcal/mol,[11] in comparison to a C-O BDFE of 81 kcal/mol for the corresponding 3° benzylic alcohol.[12] However, these adducts are also relatively robust and can typically be chromatographed and stored at room temperature for extended periods without decomposition. Moreover, the lone pair on nitrogen in these compounds can be oxidized at potentials significantly less positive (Ep/2 ∼ 0.7 V vs. Fc/Fc+ in MeCN) than those of many common nucleophiles, providing a possible mechanism for selective carbocation generation in their presence. These alkoxyamines are well known and have been extensively studied in the context of atom transfer radical polymerization, but their use as cation precursors is to the best of our knowledge, unprecedented.[13]
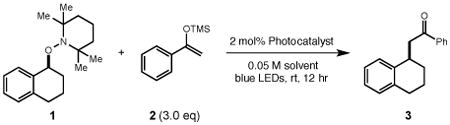
To evaluate these ideas, we elected to explore the mesolytic cleavage of alkoxylamine 1 in the presence of silyl enol ether nucleophile 2 and a variety of visible light photoredox catalysts. In this process, we imagined that the excited state of the catalyst would remove an electron from the nitrogen lone pair of 1, resulting in the formation of a radical cation 1+•. As outlined above, the C-O bond strength in 1+• is expected to be weak,[14] resulting in facile mesolytic cleavage to furnish a benzylic carbocation and TEMPO radical. Importantly, the cation and neutral radical are not expected to remain electrostatically associated in solution, which may increase both the lifetime and reactivity of the nascent electrophile. Trapping of the cation by 2 would follow, resulting in C-C bond formation. Reduction of TEMPO• by the reduced form of the photocatalyst and silyl transfer would furnish product 3 and a silyl-TEMPO derivative, and return the active form of the redox catalyst. Notably, direct outer sphere electron transfer to reduce TEMPO• to TEMPO anion is very challenging (E1/2 = −1.95 V vs. Fc/Fc+ in MeCN).[15] However, it is well known that in the presence of protons this reduction step can occur via a PCET process at potentials that are significantly less negative.[16] We reasoned that the silyl group may play a similar role, with silyl-coupled ET to TEMPO• resulting in direct formation of neutral closed shell TEMPO-SiR3.
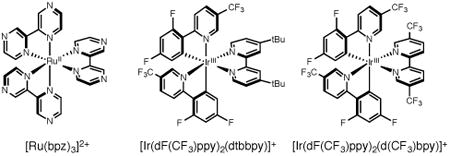
Preliminary experiments demonstrated that use of [Ru(bpy)3](PF6)2 did not provide any product, consistent with the fact that its excited state reduction potential is 320 mV less positive than that of substrate 1 (Table 1, entry 1). Gratifyingly, the use of more oxidizing photocatalysts, such as [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 or [Ru(bpz)3](BArF)2 proved more effective, with the latter furnishing the desired product 3 in 73% yield (entries 2,3). Further improvements were realized through use of [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 which provided 3 in 78% yield (entry 5). A further evaluation of reaction solvents with [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 demonstrated that nitromethane was optimal, generating 3 in 95% yield after 12 hours at room temperature (entry 11). In control reactions lacking either the Ir photocatalyst or visible light irradiation no product formation was observed (entries 12, 13). We also questioned whether silyl cations or Brønsted acids generated in situ during the course of the reaction might catalyze cation formation. However, addition of 50 mol% of either TMSOTf or HBF4•Et2O to the reaction in the absence of photocatalyst resulted in 7% and 0% yields of product 3, respectively (entries 14, 15).
Table 1.
Optimization studies.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Photocatalyst | E0 (V vs Fc)[a] | Solvent | Yield (%)[b] |
| 1 | [Ru(bpy)3](PF6)2 | +0.39 | CH2Cl2 | 0 |
| 2 | [Ir(dF-CF3-ppy)2(dtbbpy)]PF6 | +0.28 | CH2Cl2 | 19 |
| 3 | [Ru(bpz)3](BArF)2 | +1.07 | CH2Cl2 | 73 |
| 4 | 2,4,6-triphenylpyrilium | +1.92 | CH2Cl2 | 7 |
| 5 | [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 | +0.98 | CH2Cl2 | 78 |
| 6 | [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 | +1.26 | C6H6 | 0 |
| 7 | [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 | +1.26 | dioxane | 4 |
| 8 | [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 | +1.26 | DME | 21 |
| 9 | [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 | +1.26 | TFE | 40 |
| 10 | [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 | +1.26 | MeCN | 43 |
| 11 | [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 | +1.26 | MeNO2 | 95 |
|
| ||||
| Entry | Change from best conditions (Entry 11) | Solvent | Yield (%)[b] | |
|
| ||||
| 12 | No Light | MeNO2 | 0 | |
| 13 | No Photocatalyst | MeNO2 | 0 | |
| 14[c] | HBF4•OEt2 (50 mol%) | MeNO2 | 0 | |
| 15[c] | TMSOTf (50 mol%) | MeNO2 | 7 | |
Reduction potentials of Mn*/Mn-1 redox couple in MeCN
Yields determined by GC analysis using internal standard Ph2O.
No photocatalyst, no light.
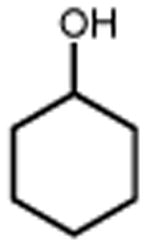
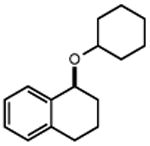
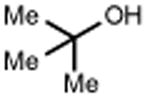
With these optimized conditions in hand, we next explored the scope of this process. Using alkoxyamine 1 as a model electrophile, together with 2 mol% [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 in MeNO2 solution, we evaluated a diverse range of nucleophiles (Table 2). In addition to 2, numerous other silyl enol ethers and allyl silanes could be accommodated, furnishing alkylation products in good yields (5-13, 68–89% yield). Similarly, a vinyl trifluoroborate salt was coupled successfully to furnish C-C coupled product 15 (61% yield). Direct Friedel-Crafts arylation with an indole nucleophile was also successful (17, 82% yield). Notably, within this set of carbon nucleophiles, three different electrophiles – silicon, boron, and proton – were all competent to facilitate TEMPO reduction and enable catalytic turnover. Turning our attention to heteroatom nucleophiles, we found that a variety of nitrogen groups could be introduced efficiently, including azide, methyl carbamate, and sulfonamide derivatives (19, 21, 23, 58-85% yield). Also, aniline and diphenylamine nucleophiles reacted to furnish N-alkylated products 25 and 27 (67% and 65% yield), respectively. Alcohol nucleophiles were also examined with both cyclohexanol and tert-butanol providing C-O coupled compounds in good yields (29 and 31, 77% and 82% yield). Hindered ethers such as these are often challenging to form via classical methods, and suggest that this method may prove useful in the production of congested C-O linkages. Lastly, we found that the reaction can also be adapted for use in intramolecular settings, as demonstrated by the etherification and arylation reactions leading to products 33 and 35.
Table 2. Nucleophile scope.
| |||
|---|---|---|---|
|
| |||
| Nucleophile | Product | Nucleophile | Product |
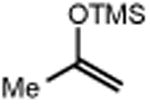
|
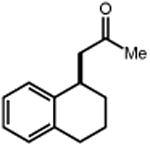
|
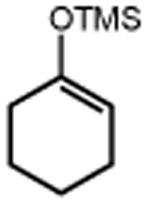
|
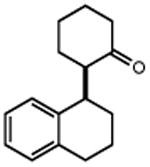
|
| 4 | 5 74% | 6 | 7 85% (d.r.1:1) 70%b |
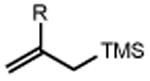
|
|
|
|
|
8 R = H 10 R = Me 12 R = Ph |
9 68% 11 89% 13 86% |
14 | 15 61% |
|
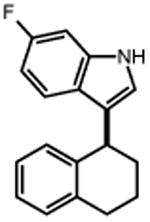
|
TMS–N3 |
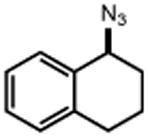
|
| 16 | 17 82% | 18 | 19 78% |
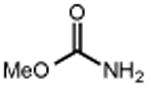
|
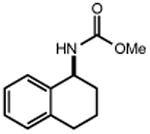
|
Ts–NH2 |
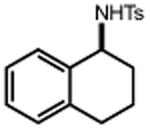
|
| 20 | 21 85% | 22 | 23 58% |
|
|
|
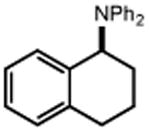
|
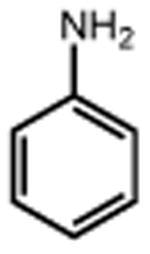
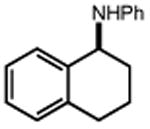
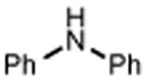
| 24 | 25 67% | 26 | 27 65% |
|
|
|
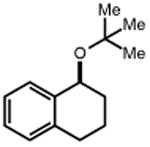
|
| 28 | 29 77% | 30 | 31 82%c |
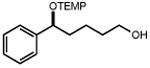
|
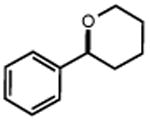
|
||
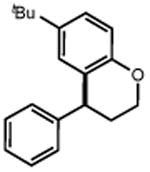
| 32 | 33 82% | ||
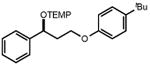
|
|
||
| 34 | 35 86% | ||
Reaction run on 0.5 mmol scale. Yields and diastereoselectivity are determined for isolated material following chromatography and are the avrage of two runs.
With electrophile 42.
6.0 equivalents of tert-butanol.
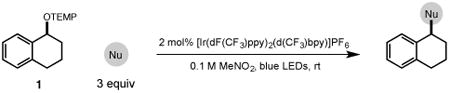
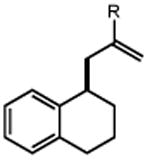
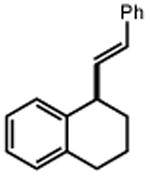
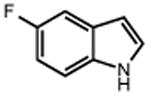
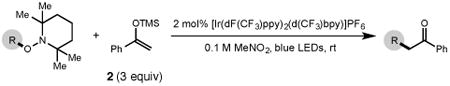
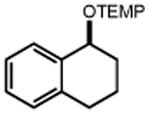
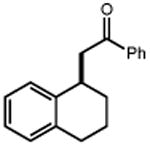
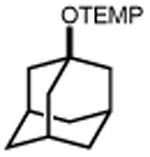
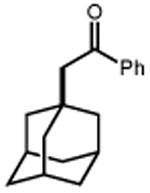
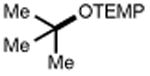
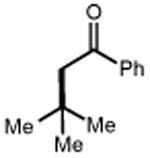
Next, we evaluated the scope of carbocations that can be generated using this protocol. Under the optimal conditions, a variety of cyclic and acyclic benzylic cations could be formed and subsequently alkylated with 2 (Table 3), furnishing products in good yields. In the acyclic series, both secondary and tertiary cations could be alkylated, as could a variety of substituted phenyl derivatives (37-57, 67-93% yield). Perhaps unsurprisingly, arenes bearing strong electron withdrawing groups were not amenable to carbocation formation using this protocol, though halogen substituents were readily accommodated. Allylic cations and oxocarbenium ions could also be generated and alkylated in good yields (59 and 61, 83% and 55% yield). Gratifyingly, even tertiary alkyl cations could be accessed using this method, as evidenced by the alkylation of both adamantyl and tert-butyl derived substrates, though the yield of the latter is likely diminished by competitive deprotonation of the cation intermediate (63 and 65, 85% and 21% yield). With respect to limitations, this method is currently unable to generate unstabilized 2° carbocations, such as the cyclohexyl cation, as their alkoxyamine precursors exhibit significantly stronger C-O bonds.[11b] Efforts to address this limitation through variation of the key properties discussed above are currently ongoing.
Table 3. Electrophile scope.
| |||
|---|---|---|---|
|
| |||
| Electrophile | Product | Electrophile | Product |
|
|
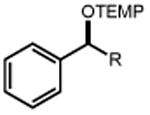
|
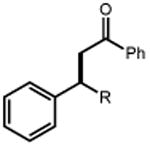
|
| 1 | 3 92% |
36 R = Me 38 R = i-Pr 40 R = Ph |
37 89% 39 71% 41 84% |
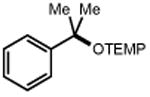
|
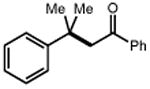
|
||
| 42 | 43 92% |
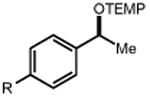
|
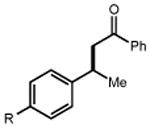
|
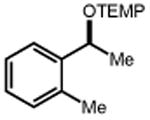
|
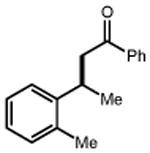
|
||
| 56 | 57 88% |
44 R = Me 46 R = t-Bu 48 R = OMe 50 R = F 52 R = Cl 54 R = Br |
45 90% 47 91% 49 90% 51 93% 53 75% 55 67% |
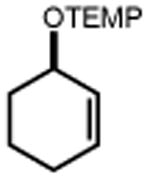
|
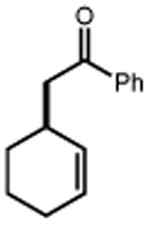
|
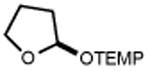
|
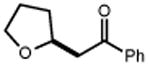
|
| 58 | 59 83% | 60 | 61 55% |
|
|
|
|
| 62 | 63 85% | 64 | 65 21%b |
Reaction run on 0.5 mmol scale. Yields are determined for isolated material following chromatography and are the average of two runs.
GC yield
To better understand the details of the oxidatively induced mesolytic cleavage step, we evaluated both of its constituent elementary steps. First, luminescence-quenching assays revealed concentration-dependent quenching of the excited state of [Ir(dF(CF3)ppy)2(d(CF3)bpy)]PF6 by alkoxyamine 1, consistent with single-electron oxidation (KSV = 86 M-1). Thermodynamically, this electron transfer process is exergonic by 550 mV. Next, we studied the feasibility of C-O cleavage following one-electron oxidation using voltammetric techniques. Specifically, initial CV scans of substrate 1 in MeCN revealed an irreversible oxidation event at 0.71 V vs. Fc/Fc+, which was assigned to the oxidation of the nitrogen lone pair of 1. However, the return scan in the reducing direction also exhibited a new peak at 0.21 V vs Fc/Fc+. Subsequent experiments demonstrated that this current feature occurs at an identical potential as the TEMPO•/oxoammonium redox couple,[15] suggesting that TEMPO• is generated in solution during the oxidative sweep. Together these results are consistent with a process wherein oxidatively induced mesolytic cleavage of the C-O bond occurs following single-electron oxidation of 1. In support of this reasoning, the C-O BDE of 1+• is calculated to be only 10 kcal/mol using the thermochemical cycle presented in Scheme 1. We also determined the quantum yield for the reaction of 1 with enolsilane 2 to be 0.43.
In conclusion, we have described a new photocatalytic method for accessing carbocation intermediates via the mesolytic cleavage of alkoxyamine radical cations. This process occurs under mild, Brønsted-neutral catalytic conditions and enables efficient alkylation reactions of a wide variety of nucleophiles. We are optimistic that this work will find use in complex settings that are not amenable to traditional cation generation schemes, as well as lead to the rational design of new mesolytic cleavage partners with an expanded substrate scope.
Scheme 2. Proposed catalytic cycle.

Acknowledgments
We gratefully acknowledge the NIH (R01 GM120530) for support of this work. We also acknowledge Ryan Evans (Princeton) for the original synthesis of the Ir catalyst used in this work.
References
- 1.a) Olah GA, Prakash GKS. Carbocation Chemistry. Wiley; Hoboken, NJ: 2004. [Google Scholar]; b) Naredle RR, Klumpp DA. Chem Rev. 2013;113:6905–6948. doi: 10.1021/cr4001385. [DOI] [PubMed] [Google Scholar]
- 2.a) Mori K, Sueoka S, Akiyama T. J Am Chem Soc. 2011;133:2424–2426. doi: 10.1021/ja110520p. [DOI] [PubMed] [Google Scholar]; b) Li H, Li W, Liu W, He Z, Li Z. Angew Chem Int Ed. 2011;50:2975–2978. doi: 10.1002/anie.201006779. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2011;123:3031–3034. [Google Scholar]; c) Lv J, Zhang Q, Zhong X, Luo S. J Am Chem Soc. 2015;137:15576–15583. doi: 10.1021/jacs.5b11085. [DOI] [PubMed] [Google Scholar]; d) Zhang F, Das S, Walkinshaw AJ, Casitas A, Taylor M, Suero MG, Gaunt MJ. J Am Chem Soc. 2014;136:8851–8854. doi: 10.1021/ja504361y. [DOI] [PubMed] [Google Scholar]; e) Nitsch D, Huber SM, PÖthig A, Narayanan A, Olah GA, Prakash GKS, Bach T. J Am Chem Soc. 2014;136:2851–2857. doi: 10.1021/ja411772n. [DOI] [PubMed] [Google Scholar]; f) Rueping M, Uria U, Lin MY, Atodiresei I. J Am Chem Soc. 2011;133:3732–3735. doi: 10.1021/ja110213t. [DOI] [PubMed] [Google Scholar]
- 3.For a remarkable recent example, see: Loach RP, Fenton OS, Movassaghi M. J Am Chem Soc. 2016;138:1057–1064. doi: 10.1021/jacs.5b12392.
- 4.a) Brak K, Jacobsen EN. Angew Chem Int Ed. 2013;52:534–561. doi: 10.1002/anie.201205449. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:558–588. [Google Scholar]; b) Mahlau M, List B. Angew Chem Int Ed. 2013;52:518–533. doi: 10.1002/anie.201205343. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:540–556. [Google Scholar]; c) Phipps RJ, Hamilton GL, Toste FD. Nat Chem. 2012;4:603–614. doi: 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]; d) Mittal N, Lippert KM, Kanta De C, Klauber EG, Emge TJ, Schreiner PR, Seidel D. J Am Chem Soc. 2015;137:5748–5758. doi: 10.1021/jacs.5b00190. [DOI] [PubMed] [Google Scholar]
- 5.McNaught AD, Wilkinson A. IUPAC Compendium of Chemical Terminology, 2nd ed (the “Gold Book”) Blackwell Scientific Publications, Oxford; 1997. [Google Scholar]
- 6.a) Schmittel M, Burghart A. Angew Chem Int Ed. 1997;36:2550–2589. [Google Scholar]; Angew Chem. 1997;109:2658–2699. [Google Scholar]; b) Gaillard ER, Whitten DG. Acc Chem Res. 1996;29:292–297. [Google Scholar]; c) Maslak P, Narvaez JN. Angew Chem Int Ed. 1990;29:283–285. [Google Scholar]; Angew Chem. 1990;102:302–304. [Google Scholar]; d) Baciocchi E, Bietti M, Lanzalunga O. Acc Chem Res. 2000;33:243–251. doi: 10.1021/ar980014y. [DOI] [PubMed] [Google Scholar]; e) Fagnoni M, Dondi D, Ravelli D, Albini A. Chem Rev. 2007;107:2725–2756. doi: 10.1021/cr068352x. [DOI] [PubMed] [Google Scholar]
- 7.a) Borg RM, Arnold DR, Cameron TS. Can J Chem. 1984;62:1785–1802. [Google Scholar]; b) Popielarz R, Arnold DR. J Am Chem Soc. 1990;112:3068–3082. [Google Scholar]
- 8.a) Kumar VS, Floreancig PE. J Am Chem Soc. 2001;123:3842–3843. doi: 10.1021/ja015526d. [DOI] [PubMed] [Google Scholar]; b) Floreancig PE. Synlett. 2007;2:191–192. [Google Scholar]; c) Wang L, Seiders JR, Floreancig PE. J Am Chem Soc. 2004;126:12596–12603. doi: 10.1021/ja046125b. [DOI] [PubMed] [Google Scholar]
- 9.a) Lim SH, Nahm K, Ra CS, Cho DW, Yoon UC, Latham JA, Dunaway-Mariano D, Mariano PS. J Org Chem. 2013;78:9431–9443. doi: 10.1021/jo401680z. [DOI] [PubMed] [Google Scholar]; b) Cho DW, Yoon UC, Mariano PS. Acc Chem Res. 2011;44:204–215. doi: 10.1021/ar100125j. [DOI] [PubMed] [Google Scholar]
- 10.a) Fagnoni M, Albini A. Acc Chem Res. 2005;38:713–721. doi: 10.1021/ar0402356. [DOI] [PubMed] [Google Scholar]; b) Protti S, Ravelli D, Mannucci B, Albini A, Fagnoni M. Angew Chem Int Ed. 2012;51:8577–8580. doi: 10.1002/anie.201202794. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2012;124:8705–8708. [Google Scholar]; c) Fasani E, Mella M, Alibini A. J Chem Soc Perkin Trans. 1995;2:449–452. [Google Scholar]
- 11.a) Kothe T, Marque S, Martschke R, Popov M, Fischer H. J Chem Soc, Perkin Trans. 1998;2:1553–1560. [Google Scholar]; b) Marsal P, Roche M, Tordo P, de Sante Claire P. J Phys Chem A. 1999;103:2899–2905. [Google Scholar]
- 12.Luo YR. Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton, FL: 2007. p. 323. [Google Scholar]
- 13.Hawker CJ, Bosman AW, Harth E. Chem Rev. 2001;101:3661–3688. doi: 10.1021/cr990119u. [DOI] [PubMed] [Google Scholar]
- 14.C-O Bond strength calculated from the equation in Scheme 1, oxidative potential of radical to cation from: Wayner DDM, McPhee DJ, Griller D. J Am Chem Soc. 1988;110:132–137.
- 15.Warren JJ, Tronic TA, Mayer JM. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warren JJ, Mayer JM. J Am Chem Soc. 2011;133:8544–8551. doi: 10.1021/ja201663p. [DOI] [PMC free article] [PubMed] [Google Scholar]