

This study demonstrates that postsynaptic heteromeric nicotinic acetylcholine receptors facilitate the excitation of hippocampus CA1 pyramidal neurons. This receptor function is developmentally regulated with its strongest influence on neuron excitability occurring for mice during early postnatal life. Given the role of nicotinic signaling to mediate neuron physiological and morphological development, these findings show that postsynaptic heteromeric nicotinic acetylcholine receptors are positioned to influence the maturation of CA1 pyramidal neurons within hippocampal learning and memory networks.
Keywords: hippocampus, CA1 pyramidal neuron, nicotinic receptor, electrophysiology, brain development
Abstract
The hippocampus plays a key role in learning and memory. The normal development and mature function of hippocampal networks supporting these cognitive functions depends on afferent cholinergic neurotransmission mediated by nicotinic acetylcholine receptors. Whereas it is well-established that nicotinic receptors are present on GABAergic interneurons and on glutamatergic presynaptic terminals within the hippocampus, the ability of these receptors to mediate postsynaptic signaling in pyramidal neurons is not well understood. We use whole cell electrophysiology to show that heteromeric nicotinic receptors mediate direct inward currents, depolarization from rest and enhanced excitability in hippocampus CA1 pyramidal neurons of male mice. Measurements made throughout postnatal development provide a thorough developmental profile for these heteromeric nicotinic responses, which are greatest during the first 2 wk of postnatal life and decrease to low adult levels shortly thereafter. Pharmacological experiments show that responses are blocked by a competitive antagonist of α4β2* nicotinic receptors and augmented by a positive allosteric modulator of α5 subunit-containing receptors, which is consistent with expression studies suggesting the presence of α4β2 and α4β2α5 nicotinic receptors within the developing CA1 pyramidal cell layer. These findings demonstrate that functional heteromeric nicotinic receptors are present on CA1 pyramidal neurons during a period of major hippocampal development, placing these receptors in a prime position to play an important role in the establishment of hippocampal cognitive networks.
NEW & NOTEWORTHY
This study demonstrates that postsynaptic heteromeric nicotinic acetylcholine receptors facilitate the excitation of hippocampus CA1 pyramidal neurons. This receptor function is developmentally regulated with its strongest influence on neuron excitability occurring for mice during early postnatal life. Given the role of nicotinic signaling to mediate neuron physiological and morphological development, these findings show that postsynaptic heteromeric nicotinic acetylcholine receptors are positioned to influence the maturation of CA1 pyramidal neurons within hippocampal learning and memory networks.
the hippocampus is a specialized component of the corticolimbic system that plays an important role in higher order cognitive processes such as learning, memory, and attention (Eichenbaum et al. 1992; Kim 2015; Scoville and Milner 1957; Squire and Zola 1996). Alterations to the morphology and/or function of the hippocampus are implicated in the etiology for a number of neurodevelopmental disorders that involve deficits in these cognitive processes, including autism spectrum disorder (Loveland et al. 2008; Schumann et al. 2004), attention deficit hyperactivity disorder (Li et al. 2014; Plessen et al. 2006), and the teratogenic effects of developmental exposure to ethanol (McGoey et al. 2003; Sutherland et al. 1997) and nicotine (Damborsky et al. 2012). Proper hippocampal function depends on afferent cholinergic neurotransmission originating in the medial septum/diagonal band of Broca (MSDB) within the basal forebrain, since hippocampal acetylcholine (ACh) levels increase in animals exploring novel objects and environments (Aloisi et al. 1997; Anzalone et al. 2009; Ceccarelli et al. 1999; Stanley et al. 2012) and selective lesions of the septohippocampal cholinergic neurons decrease markers of cholinergic activity within the hippocampus and also impair spatial learning and object recognition memory (Berger-Sweeney et al. 2001; Cai et al. 2012; Easton et al. 2011).
Pharmacological studies demonstrate that ACh acts at the nicotinic class of acetylcholine receptor (nAChR) within the hippocampus to support learning and memory (Davis et al. 1997; Felix and Levin 1997; Kenney et al. 2012; Nott and Levin 2006). These nAChRs are predominantly of the homomeric α7 and heteromeric α4β2* subclasses, and it is now understood that their modulation of both GABAergic and glutamatergic signaling influences the net impact of ACh on hippocampal networks (McQuiston 2014; Placzek et al. 2009; Yakel 2012). Functional nAChRs of both subclasses have been identified in hippocampal GABAergic interneurons (Alkondon and Albuquerque 2001; Bell et al. 2011; Frazier et al. 1998; Ji and Dani 2000; Jones and Yakel 1997; Khiroug et al. 2003; McQuiston and Madison 1999; Sudweeks and Yakel 2000), and their activation can mediate local inhibition or disinhibition responses (Alkondon et al. 1999; Bell et al. 2015; Ji and Dani 2000). The role of nAChRs in hippocampal pyramidal neurons is not as well understood. Electrophysiological experiments in rodents age postnatal day 11 (P11) and older show no direct nicotinic responses (Frazier et al. 1998; Jones and Yakel 1997; Khiroug et al. 2003; Sudweeks and Yakel 2000) or nicotinic responses that occur in a low proportion of pyramidal neurons (Hefft et al. 1999; McQuiston and Madison 1999), and there is conflicting evidence for the contribution of heteromeric nAChRs toward responses that are observed (He et al. 2013; Hefft et al. 1999; McQuiston and Madison 1999; Tu et al. 2009). Expression analyses of whole hippocampal tissue (Shacka and Robinson 1998; Zhang et al. 1998) and specifically of the cornu ammonis area 1 (CA1) pyramidal neuron layer (Didier et al. 1995; Machaalani et al. 2010; Winzer-Serhan and Leslie 2005) suggest that the content for heteromeric nAChR subunits is developmentally regulated with a peak during the first 2 wk of postnatal life, followed by a significant decline shortly thereafter. However, to the best of our knowledge, the function of heteromeric nAChRs has not been measured in developing hippocampal pyramidal neurons earlier than P11.
The objective of this current study was to determine whether functional heteromeric nAChRs are present in developing mouse hippocampus CA1 pyramidal neurons. Whole cell electrophysiological recording of visually identified CA1 pyramidal neurons located within acute hippocampal slices of mice aged P5–10 consistently identified postsynaptic current responses to bath-applied ACh (in the presence of blockers to both muscarinic ACh receptors and α7 subunit-containing nAChRs) that also mediated depolarization from rest and increased the rate of action potential firing in these same neurons. Nicotinic current responses were resistant to the inhibition of synaptic transmission and were significantly reduced by a selective antagonist of α4β2* nAChRs, suggesting that they were mediated by this class of receptor located directly on recorded CA1 pyramidal neurons. The measurement of nicotinic responses throughout postnatal development revealed that the developmental regulation of heteromeric nAChR function is consistent with previous subunit expression analyses, with nicotinic responses being significantly greater during the early P5–10 period than at later maturational stages of P15–20 (juvenile), P25–30 (adolescent), and P60–100 (young adult). Because the rodent early postnatal period is characterized by significant positive neurotransmission that drives hippocampal neuron development, these findings suggest that postsynaptic heteromeric nAChRs may contribute to these processes in CA1 pyramidal neurons to influence the development and maturation of corticolimbic learning and memory networks.
MATERIALS AND METHODS
Experimental animals.
Timed-pregnant female CD1 strain mice were purchased from Charles River Canada (Saint-Constant, QC, Canada) and housed individually in plastic cages measuring 47 cm × 25 cm × 15 cm with ad libitum access to water and food (Tekland Global 14% Protein Rodent Maintenance Diet; Harlan Laboratories, Mississauga, ON, Canada). Mice were kept in a secure vivarium with an ambient temperature of 21–24°C and a 12:12-h reverse light cycle with lights on at 8:00 PM. The day of birth was considered to be postnatal day 0 (P0) for that litter. Litters were left in the same cage until weaning on P30, at which time offspring were separated on the basis of sex, housed in groups of up to 5 mice per cage in plastic cages measuring 29 cm × 19 cm × 13 cm, and provided with ad libitum access to water and food. Male offspring were analyzed at four developmental ages: P5–10 (young postnatal), P15–20 (juvenile), P25–30 (adolescent), and P60–100 (young adult). Experimental animals were cared for according to the principles and guidelines of the Canadian Council on Animal Care, and the experimental protocol was approved by the University of Guelph Animal Care Committee.
Electrophysiology.
Mice were killed by decapitation under isoflurane anesthesia. Brains were rapidly excised from the skull in 4°C oxygenated sucrose artificial cerebrospinal fluid (ACSF: 254 mM sucrose, 10 mM d-glucose, 26 mM NaHCO3, 2 mM CaCl2, 2 mM MgSO4, 3 mM KCl, and 1.25 mM NaH2PO4, pH 7.4). Coronal slices containing the rostral/dorsal hippocampus were cut at 400-μm thickness using a Leica VT1200 vibrating microtome (Leica Microsystems, Richmond Hill, ON, Canada) from approximately Bregma −1.46 mm to Bregma −2.18 mm (Paxinos and Franklin 2001). Slices were transferred to oxygenated ACSF (128 mM NaCl, 10 mM d-glucose, 26 mM NaHCO3, 2 mM CaCl2, 2 mM MgSO4, 3 mM KCl, and 1.25 mM NaH2PO4, pH 7.4), maintained at 30°C in a prechamber, and allowed to recover for at least 2 h before being used for whole cell recordings. Slices were transferred to a modified chamber (Warner Instruments, Hamden, CT) mounted on the stage of an Axioskop FS2 microscope (Carl Zeiss Canada, Toronto, ON, Canada). ACSF was oxygenated with carbogen (95% O2-5% CO2) and superfused over the slice at room temperature at a rate of 3–4 ml/min. Whole cell recording of hippocampal CA1 pyramidal neurons was performed using glass pipette electrodes (2–5 MΩ) containing 120 mM K-gluconate, 5 mM KCl, 2 mM MgCl2, 4 mM K2-ATP, 400 μM Na2-GTP, 10 mM Na2-phosphocreatine, 33 μM Alexa Fluor 488 hydrazide (Life Technologies, Burlington, ON, Canada), and 10 mM HEPES buffer (adjusted to pH 7.3 with KOH). Individual CA1 pyramidal neurons were visualized using infrared differential interference contrast microscopy and identified on the basis of location within the pyramidal cell layer and the presence of a prominent proximal apical dendrite. Pyramidal neuron cell type was confirmed visually once the Alexa Fluor 488 hydrazide from the intracellular recording electrode solution had diffused throughout the neuron, allowing for the epifluorescent visualization of the full apical dendrite morphology. All recordings were made using a Multiclamp 700B amplifier, acquired at 20 kHz, low-pass filtered at 2 kHz using a Digidata 1440A data acquisition system (Molecular Devices, Sunnyvale, CA), and corrected for the liquid junction potential.
Neuron electrophysiological properties were first recorded in current-clamp mode by measuring responses to positive and negative current steps (see Table 1). All experiments were performed in the continuous presence of 200 nM atropine and 10 nM methyllycaconitine (MLA) to block muscarinic receptors and α7 subunit-containing nicotinic receptors, respectively. Nicotinic receptor-mediated responses were probed by the addition of 1 mM ACh for 15 s in the ACSF bath following a constant baseline of recording. ACh was allowed to wash out of the slices for at least 5 min between applications. All responses were measured using Clampfit 10.3 software (Molecular Devices). For the measurement of depolarizing and current responses, the mean membrane potential or holding current value for each 1-s period of an experiment was calculated as the average of the 20,000 data values recorded during that period. Depolarizing responses were measured in current-clamp mode by subtracting the mean membrane potential at the peak of the response from the mean resting membrane potential at baseline. Acceleration of action potential firing was measured in current-clamp mode by first injecting a positive current to elicit an ∼1-Hz baseline firing frequency. The rate of firing was measured over a 15-s period during the peak ACh response and reported as a percent increase in frequency from the baseline frequency for each cell. Current responses were assessed in voltage-clamp mode with neurons held at −75 mV. This approach was employed to mitigate the potential for nicotinic-driven presynaptic GABA release and subsequent activation of postsynaptic GABAA receptors to induce chloride currents in recorded neurons. Because −75 mV is near the equilibrium potential for chloride in this preparation, there is minimal flux of chloride ions and there are no observable chloride currents under these recording conditions. Inward current responses were measured by subtracting the mean holding current at the peak of the ACh response from the mean holding current at baseline.
Table 1.
Electrophysiological properties for developing CA1 pyramidal neurons
| P5–10 | P15–20 | P25–30 | P60–100 | P Value | |
|---|---|---|---|---|---|
| Resting membrane potential, mV | −73.4 ± 0.5 | −72.7 ± 0.5 | −73.3 ± 0.4 | −73.8 ± 0.6 | 0.6 |
| Input resistance, MΩ | 318.3 ± 11.1* | 222.9 ± 8.9† | 160.7 ± 7.6‡ | 156.2 ± 4.1‡ | <0.0001 |
| Spike amplitude, mV | 75.5 ± 1.3* | 83.0 ± 2.5† | 91.8 ± 1.9‡ | 82.5 ± 1.6† | <0.0001 |
Values are means ± SE at postnatal days 5–10 (P5–10; n = 62), P15–20 (n = 35), P25–30 (n = 33), and P60–100 (n = 35). P values were determined by 1-way ANOVA. *†‡Different symbols indicate statistically significant differences among data sets (Tukey's post hoc test).
Additional pharmacological experiments (see Fig. 2) were performed using a cocktail of synaptic blockers applied to the bath comprising 2 μM tetrodotoxin (TTX) to block voltage-gated sodium channels, 20 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) to block AMPA and kainate glutamate receptors, 50 μM d-(−)-2-amino-5-phosphonopentanoic acid (APV) to block NMDA glutamate receptors, and 10 μM bicuculline to block GABAA receptors. The α4β2* nAChR competitive antagonist dihydro-β-erythroidine (DHβE) was applied to the bath at 3 μM for a subset of experiments (see Fig. 2). Nicotine desensitization of nAChR function was assessed (see Fig. 3) by measuring the inward current response to 1 mM ACh, allowing for 5 min of washout, and then applying either 100, 300, or 500 nM nicotine in the bath for 10 min. After a 10-min washout, the 1 mM ACh response was measured again, and the percent desensitization was calculated for that cell as [(ACh current before nicotine − ACh current after nicotine)/ACh current before nicotine] × 100. Developmental effects of acetylcholinesterase and extracellular matrix density were assessed (see Fig. 5) using 1 mM carbamoylcholine chloride (carbachol). The incorporation of α5 subunits into nAChRs was assessed by first inactivating acetylcholinesterase in the slice via 10-min preexposure to 20 μM diisopropyl fluorophosphate (DFP) and then measuring the inward current response to 10 μM ACh before and after 10-min application of 0.1 μM galanthamine. The percent potentiation was calculated as [(ACh current after galanthamine − ACh current before galanthamine)/ACh current before galanthamine] × 100. TTX citrate was obtained from Alomone Labs (Jerusalem, Israel). ACh chloride, carbachol chloride, atropine, and nicotine hydrogen bitartrate were purchased from Sigma Aldrich (Oakville, ON, Canada). CNQX disodium, APV, bicuculline methiodide, DHβE, and galanthamine hydrobromide were purchased from Tocris Bioscience/Bio-Techne (Minneapolis, MN). DFP was purchased from BioShop Canada (Burlington, ON, Canada). All drugs were stored in stock solutions at −20°C.
Fig. 2.

In young postnatal mice, excitation of CA1 pyramidal neurons by ACh is mediated by postsynaptic α4β2* nicotinic receptors. All recordings were made using mice aged postnatal day 5–10 in the continuous presence of 200 nM atropine to block muscarinic acetylcholine receptors and 10 nM MLA to block α7 subunit-containing nicotinic acetylcholine receptors. A: current responses were not desensitized by repeated application of 1 mM ACh for 15 s (paired t-test, P = 0.8). B: similarly, current responses to 1 mM ACh (15 s) were not significantly affected by 10-min pretreatment with the following synaptic blockers: 2 μM TTX, 20 μM CNQX, 50 μM APV, and 10 μM bicuculline (P = 0.8). C: current responses to 1 mM ACh (15 s) were significantly reduced by 10-min pretreatment with the α4β2* nicotinic receptor competitive antagonist dihydro-β-erythroidine (DHβE; 3 μM) (*P = 0.002). D: for neurons induced to fire action potentials at 1 Hz by positive current injection, the increase in firing frequency following application of 1 mM ACh (15 s) was significantly reduced by 10-min pretreatment with 3 μM DHβE (*P < 0.0001). All values are means ± SE. Representative voltage-clamp or current-clamp traces are shown at right for 1 neuron from each pharmacological experiment with ACh application marked by gray horizontal bars.
Fig. 3.
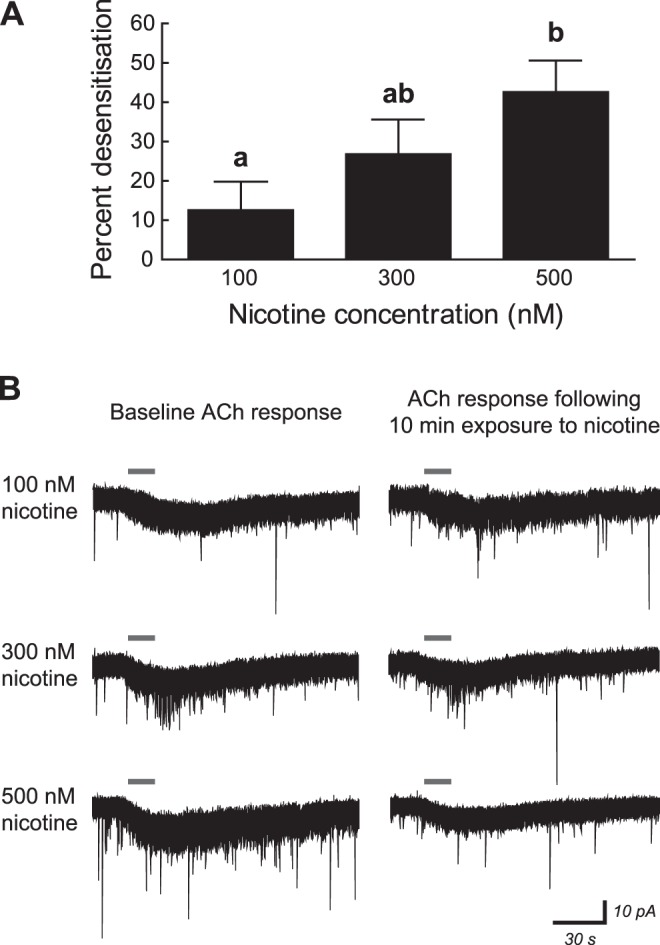
Acute nicotine exposure desensitizes nicotinic receptors in young postnatal CA1 pyramidal neurons in a concentration-dependent manner. A: receptor desensitization by nicotine was calculated in neurons of mice aged postnatal day 5–10 by measuring the peak inward current response to 1 mM ACh (15 s; in the presence of 200 nM atropine and 10 nM MLA) before and after a 10-min exposure to 100, 300, or 500 nM nicotine. Nicotine was allowed to wash out of the brain slice for 10 min before ACh was reapplied, and nicotine was applied only once to each brain slice. The percent decrease/desensitization for the ACh response was significantly affected by nicotine concentration (1-way ANOVA, P = 0.04), with percent desensitization following 500 nM nicotine application being greater than that following 100 nM nicotine application (Tukey's post hoc test, P = 0.03). All values are means ± SE. a,bDifferent letters indicate statistically significant differences among data sets. B: representative voltage-clamp traces showing inward current responses to ACh (gray horizontal bars) before and after 10-min exposure to 100, 300, or 500 nM nicotine.
Fig. 5.
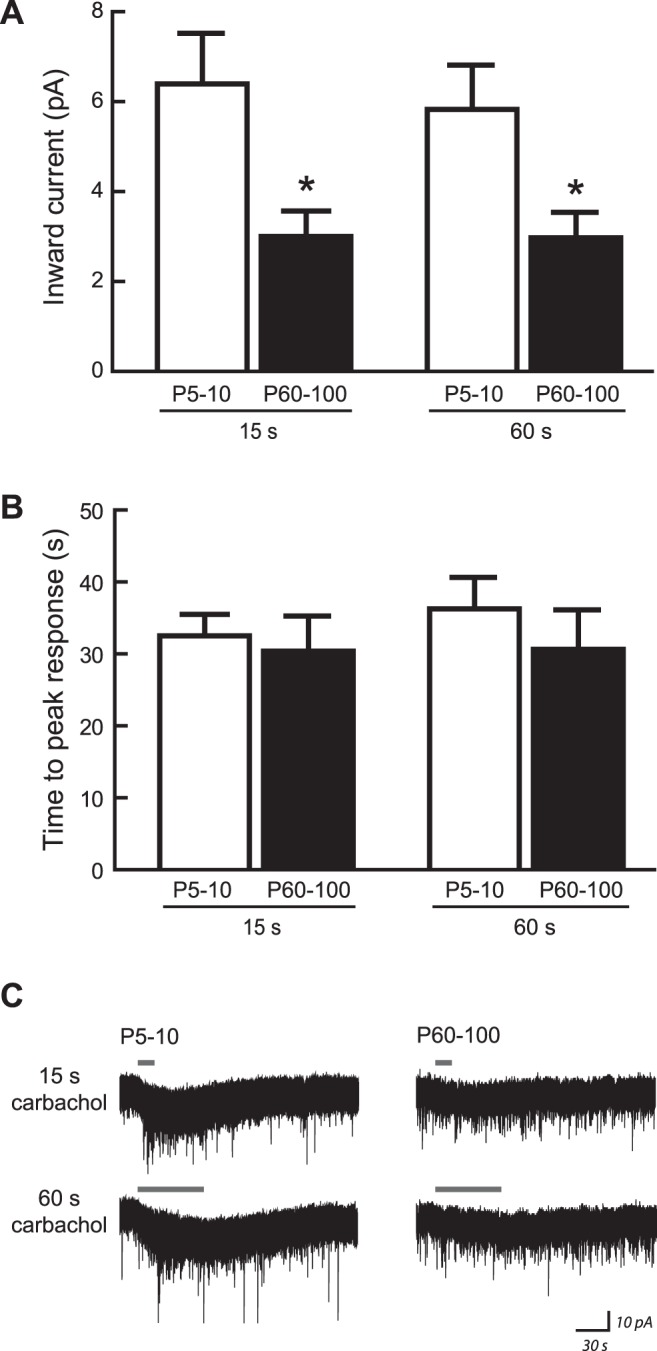
Developmental changes to CA1 nicotinic responses do not result from altered acetylcholinesterase activity or extracellular matrix density. Inward current responses were measured at postnatal day 5–10 (P5–10) and P60–100 following application of carbachol, which is a nicotinic receptor agonist that is not metabolized by endogenous acetylcholinesterase present within brain slices. A: magnitude of current responses to 1 mM carbachol (applied for 15 or 60 s in the presence of 200 nM atropine and 10 nM MLA) was significantly affected by age [2-way ANOVA: F(1,14) = 7.11, P = 0.02], where currents were greater at P5–10 than at P60–100 (Bonferroni's post hoc test, all *P < 0.05). Response magnitude was not affected by length of carbachol application [F(1,14) = 1.28, P = 0.3], and there was no interaction between effects of age and application length [F(1,14) = 1.04, P = 0.3]. B: in this experiment, the time required for carbachol to wash into the slice and elicit a maximal current response was not affected by age [F(1,14) = 0.42, P = 0.5] or length of carbachol application [F(1,14) = 0.70, P = 0.4], and there was no interaction between effects of these two factors [F(1,14) = 0.54, P = 0.5]. C: representative voltage-clamp traces showing inward current responses to 1 mM carbachol at each age. All values are means ± SE.
Neuron morphology.
A subset of all recorded hippocampus CA1 pyramidal neurons were patched and recorded in acute brain slices as described above, with the exception that the intracellular recording electrode also contained 0.3% (wt/vol) biocytin (Tocris Bioscience). Neurons were held for at least 40 min to allow for biocytin to diffuse into the cell, and recording electrodes were then withdrawn slowly to allow resealing of neuronal cell membranes. Slices were fixed overnight at 4°C in a solution containing 4% (wt/vol) paraformaldehyde in 100 mM phosphate buffer (pH 7.5). Free-floating slices were washed 3 times for 10 min each with Tris-buffered saline (TBS: 100 mM Tris and 150 mM NaCl, pH 7.5) and then treated with 0.5% (vol/vol) H2O2 and 0.25% (vol/vol) Triton X-100 in TBS for 15 min to suppress endogenous peroxidase activity. Slices were washed 3 times for 10 min each with TBS and then incubated in a solution made from Vectastain Elite ABC avidin/biotinylated horseradish peroxidase complexes (Vector Laboratories, Burlington, ON, Canada; prepared according to the manufacturer's recommendations) and 0.25% (vol/vol) Triton X-100 in TBS overnight at room temperature. Slices were washed 3 times for 10 min each with TBS and incubated in a solution containing 0.05% (wt/vol) 3,3-diaminobenzidine (DAB), 0.04% (wt/vol) nickel chloride, and 0.25% (vol/vol) Triton X-100 in TBS for 5 min, and then in this same solution that also contained 0.001% (vol/vol) H2O2 for 15 min. Slices were washed with TBS 3 times for 10 min each, mounted onto microscope slides, and allowed to dry overnight. They were then dehydrated using a series of ethanol gradients, cleared, and coverslipped using Permount (Fisher Scientific, Ottawa, ON, Canada).
Neurons were imaged in brightfield using an Olympus BX53 upright microscope equipped with an Olympus UPlanSApo ×4 0.16-NA objective and an Olympus UPlanSApo ×30, 1.05-NA silicone-immersion objective (Olympus, Richmond Hill, ON, Canada). For high-resolution photomicrographs, image stacks that overlapped in the x- and y-axes and separated in the z-axis by 1 μm per image were captured using the ×30 objective and a 3-axis motorized stage under the control of Neurolucida software (MicroBrightField, Williston, VT). Image stacks were collapsed into two-dimensional minimum-intensity images that were then stitched together in the x- and y-axes.
Statistical analysis.
All data are means ± SE for individual neurons within each experimental group. Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA), and a level of P < 0.05 was required to indicate statistical significance. Effects of synaptic blockers and receptor antagonists on ACh responses were analyzed using two-tailed paired t-tests. Effects of age on nicotinic current, depolarization, and excitability responses and effects of nicotine concentration on receptor desensitization were analyzed by one-way ANOVA followed by Tukey's post hoc test. Comparisons between regular firing and burst firing pyramidal neurons and effects of age and application time on carbachol responses were performed using two-way ANOVA followed by Bonferroni's post hoc test. The effect of age on galanthamine potentiation of ACh currents was measured by two-tailed unpaired t-test.
RESULTS
Excitation of young postnatal CA1 pyramidal neurons by postsynaptic heteromeric nicotinic receptors.
Several in situ studies have shown that mRNA for the nicotinic α4, β2, and α5 subunits is expressed within the CA1 pyramidal neuron layer of mice (Heath et al. 2010; Hsu et al. 2013; Marks et al. 1992; Salas et al. 2003), rats (Sudweeks and Yakel 2000), and humans (Machaalani et al. 2010) and that subunit expression is greater during early postnatal periods than in adulthood (Didier et al. 1995; Machaalani et al. 2010; Winzer-Serhan and Leslie 2005). We first sought to determine whether functional heteromeric nAChRs are present on young postnatal CA1 pyramidal neurons by using whole cell recording within acute brain slices from mice aged P5–10. The pyramidal morphology of each recorded neuron was confirmed by the addition of Alexa Fluor 488 hydrazide in the pipette solution and live epifluorescent visualization of its prominent apical dendrite, and a subset of neurons were also analyzed in greater detail using biocytin in the pipette solution and post hoc chromogenic visualization. Photomicrographs of an exemplary recorded neuron are shown in Fig. 1A. Its response to the injection of positive and negative current steps is shown in Fig. 1B, which is typical of responses in regular-firing CA1 pyramidal neurons (Graves et al. 2012). Heteromeric nAChRs were isolated pharmacologically in this preparation by the continuous bath application of atropine (200 nM, to block muscarinic acetylcholine receptors) and MLA (10 nM, to block α7 subunit-containing nAChRs). Bath application of 1 mM ACh for 15 s resulted in positive inward currents in neurons held at −75 mV for all neurons examined (108/108 neurons) and positive depolarization responses from rest for all neurons examined (25/25 neurons). Exemplary responses are shown in Fig. 1, C (current response) and D (depolarization response). This nAChR activation has a profound impact on the function of active neurons, because the same application of 1 mM ACh increased the action potential firing frequency by ∼250% in all neurons tested (39/39 neurons). An exemplary recording of ACh acceleration of action potential firing is shown in Fig. 1E. We applied ACh at 1 mM because it is actively metabolized by endogenous acetylcholinesterase while washing into the slice preparation (Bailey et al. 2010). To facilitate comparison of our data with that from cell culture and synaptosome studies, we verified in a separate set of experiments that current responses to 1 mM ACh in our slice preparation (9.0 ± 1.6 pA, n = 4) were not different from current responses to 100 μM ACh measured after acetylcholinesterase had been inactivated by 10-min exposure to 20 μM DFP (7.6 ± 1.0 pA, n = 7; 2-tailed unpaired t-test, P = 0.4).
Fig. 1.
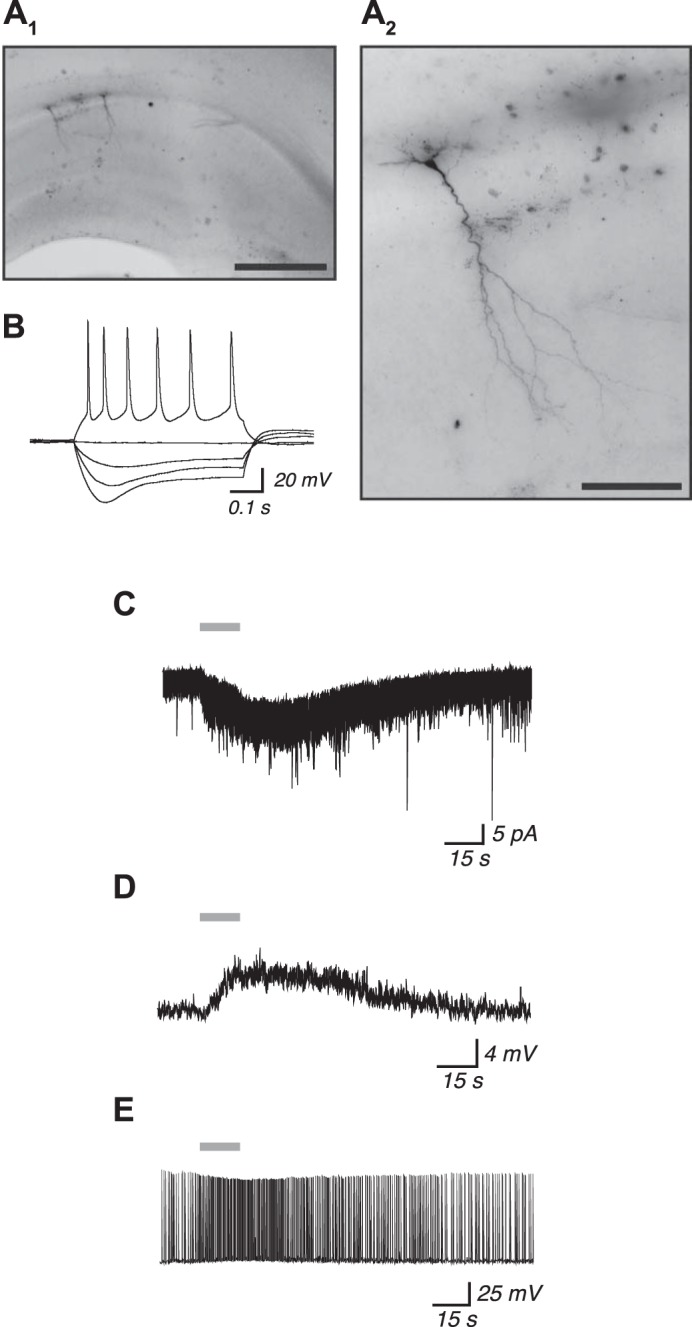
Activation of heteromeric nicotinic acetylcholine receptors facilitates excitation of young postnatal mouse CA1 pyramidal neurons. A1: representative photomicrograph of the dorsal hippocampus from a mouse aged postnatal day 10 showing 2 CA1 pyramidal neurons that were analyzed for electrophysiology and visualized post hoc using biocytin. Scale bar, 500 μm. A2: magnified view of the neuron at left in A1 shows its characteristic CA1 pyramidal neuron morphology. Scale bar, 100 μm. B: current-clamp recording from the neuron in A2 showing changes to membrane potential following the injection of 500-ms depolarizing and hyperpolarizing current steps, which are consistent with typical responses from CA1 pyramidal neurons. Application of 1 mM ACh for 15 s (gray horizontal bars) results in an inward current response (C), depolarization from rest (D), and an acceleration of action potential firing (E). All recordings were made in the continuous presence of 200 nM atropine to block muscarinic acetylcholine receptors and 10 nM MLA to block α7 subunit-containing nicotinic acetylcholine receptors.
We next sought to characterize these nicotinic responses in CA1 neurons from young postnatal mice. Nicotinic receptors did not desensitize to repeated application of 1 mM ACh, which is consistent with heteromeric β2 subunit-containing receptors (Fig. 2A; first application: 7.6 ± 1.4 pA; second application: 7.7 ± 1.5 pA; n = 8; 2-tailed paired t-test, P = 0.8). Nicotinic current responses to ACh were not affected by the inhibition of synaptic transmission with the use of a cocktail of blockers for voltage-gated sodium channels (2 μM TTX), ionotropic glutamate receptors (20 μM CNQX and 50 μM APV), and GABAA receptors (10 μM bicuculline) (Fig. 2B; baseline: 10.8 ± 1.4 pA; after 10-min exposure to synaptic blockers: 10.6 ± 1.0 pA; n = 7; P = 0.8). However, the α4β2* nAChR competitive antagonist DHβE significantly suppressed ACh-induced nicotinic currents (Fig. 2C; baseline: 10.0 ± 1.6 pA; after 10-min exposure to 3 μM DHβE: 1.7 ± 1.2 pA; n = 5; P = 0.002). Moreover, although the ACh-induced nicotinic currents before exposure to DHβE were significantly different from the root mean square (RMS) current noise in these neurons of 2.5 ± 0.3 pA (2-tailed unpaired t-test, P = 0.002), currents measured after exposure to DHβE were not significantly different from the baseline RMS current noise (P = 0.5). Exposure to DHβE also significantly suppressed the ACh-induced increase in action potential firing frequency for neurons already firing action potentials at ∼1 Hz (Fig. 2D; baseline: 311.4 ± 33.5% increase; after 10-min exposure to 3 μM DHβE: 78.8 ± 15.3% increase; n = 9; P < 0.0001). The results from this set of experiments are consistent with the activation of α4β2* nAChRs located directly on CA1 pyramidal neurons of young postnatal mice.
Nicotine desensitization of nicotinic receptors in young postnatal CA1 pyramidal neurons.
Nicotine binds with high affinity to α4β2* nAChRs in the human brain (Brody et al. 2006) and also within the mouse hippocampus (Horti et al. 1997; Lomazzo et al. 2010). It possesses both agonist and desensitizing abilities at nAChRs, and this complex pharmacological profile is influenced by the specific subunit composition and posttranslational modification of the heteromeric nAChR pentamer (Bailey et al. 2010, 2014; Grady et al. 2012; Quick and Lester 2002; Wageman et al. 2014). We tested the ability of multiple nicotine concentrations to desensitize nAChRs in CA1 pyramidal neurons from mice aged P5–10 by measuring inward current responses to 1 mM ACh (15-s application) before and after a 10-min exposure to 100, 300, or 500 nM nicotine. Nicotine was allowed to wash out of the brain slice for 10 min before ACh was reapplied, and nicotine was applied only once to each brain slice. Nicotine at 100 nM did not significantly reduce the ACh response (initial response: 5.5 ± 0.6 pA; after 100 nM nicotine: 4.6 ± 0.5 pA; n = 9; paired t-test, P = 0.1). However, nicotine at 300 and 500 nM, which are concentrations similar to those expected in the blood and brain after a cigarette is smoked (Henningfield et al. 1993; Rose et al. 2010), both significantly reduced ACh current responses at the nAChR. Values were 9.1 ± 2.2 (initial response) vs. 6.6 ± 1.7 pA (after 300 nM nicotine; n = 7; P = 0.02) and 8.5 ± 1.0 (initial response) vs. 4.6 ± 0.6 pA (after 500 nM nicotine; n = 10; P = 0.001). These values are expressed in Fig. 3 as the percentage by which nicotine desensitized the initial ACh in each neuron and were significantly affected by nicotine concentration (1-way ANOVA, P = 0.04). Tukey's post hoc test showed that percent desensitization following 500 nM nicotine was greater than that following 100 nM nicotine (P = 0.03).
Developmental changes to nicotinic receptor function.
Because developmental analyses show the expression of heteromeric nAChR subunits to be greater in the CA1 pyramidal neuron layer during young postnatal life than at later ages (Didier et al. 1995; Machaalani et al. 2010; Winzer-Serhan and Leslie 2005), we next sought to determine whether the electrophysiological function of these receptors is developmentally regulated within CA1 pyramidal neurons. This was done by measuring whole cell current and depolarization responses to ACh in mice aged P5–10 (young postnatal), P15–20 (juvenile), P25–30 (adolescent), and P60–100 (young adult). Heteromeric nAChRs were isolated pharmacologically in all experiments by the bath application of 200 nM atropine and 10 nM MLA. Inward current responses to 1 mM ACh (15 s) showed a significant effect of age (Fig. 4A; 1-way ANOVA, P < 0.0001), where currents in neurons from mice aged P5–10 (9.5 ± 1.0 pA, n = 59) were more than three times greater than the RMS baseline current noise of 2.6 ± 0.2 pA and significantly greater than currents in neurons from mice aged P15–20 (4.1 ± 0.4 pA, n = 26), P25–30 (3.3 ± 0.6 pA, n = 28), and P60–100 (2.2 ± 0.5 pA, n = 30; Tukey's post hoc test, P ≤ 0.0003 for each comparison). A similar effect of age was observed on the ability of 1 mM ACh (15 s) to depolarize neurons from rest (P < 0.0001), as shown in Fig. 4B. Nicotinic depolarization responses were significantly greater in neurons from mice aged P5–10 (3.5 ± 0.8 mV, n = 25) than in neurons from mice aged P25–30 (1.0 ± 0.2 mV, n = 23) and P60–100 (0.6 ± 0.1 mV, n = 32; P ≤ 0.0009 for each comparison). The influence of these nAChR responses on neuron excitability was measured by applying 1 mM ACh (15 s) to neurons that had been previously induced to fire action potentials at a frequency of 1 Hz by positive current injection. The amount of positive current injection required to induce the 1-Hz firing frequency was recorded for the majority of the neurons in this experiment and was found to vary across development with values of 48.7 ± 7.7 pA (n = 20) at P5–10, 22.2 ± 2.3 pA (n = 22) at P15–20, 26.9 ± 4.6 pA (n = 24) at P25–30, and 47.2 ± 11.1 pA (n = 23) at P60–100 (Kruskal-Wallis test, P = 0.002). This amount of current was significantly greater at P5–10 than at P15–20 and P25–30 (Dunn's post hoc test, P < 0.02 for each comparison). There was an effect of age on the percent increase in firing frequency following ACh application that was similar to that seen for current and depolarization responses (Fig. 4C; P < 0.0001), where the ACh response was significantly greater in neurons from mice aged P5–10 (256.9 ± 38.5% increase, n = 30) than in neurons from mice aged P15–20 (105.2 ± 12.9% increase, n = 32), P25–30 (53.9 ± 9.3% increase, n = 30), and P60–100 (41.2 ± 8.1% increase, n = 29; P < 0.0001 for each comparison). Basic electrophysiological properties for neurons in this experiment are shown in Table 1. During postnatal development, resting membrane potential did not change (1-way ANOVA, P = 0.6), input resistance decreased (P < 0.0001), and spike amplitude increased (P < 0.0001).
Fig. 4.
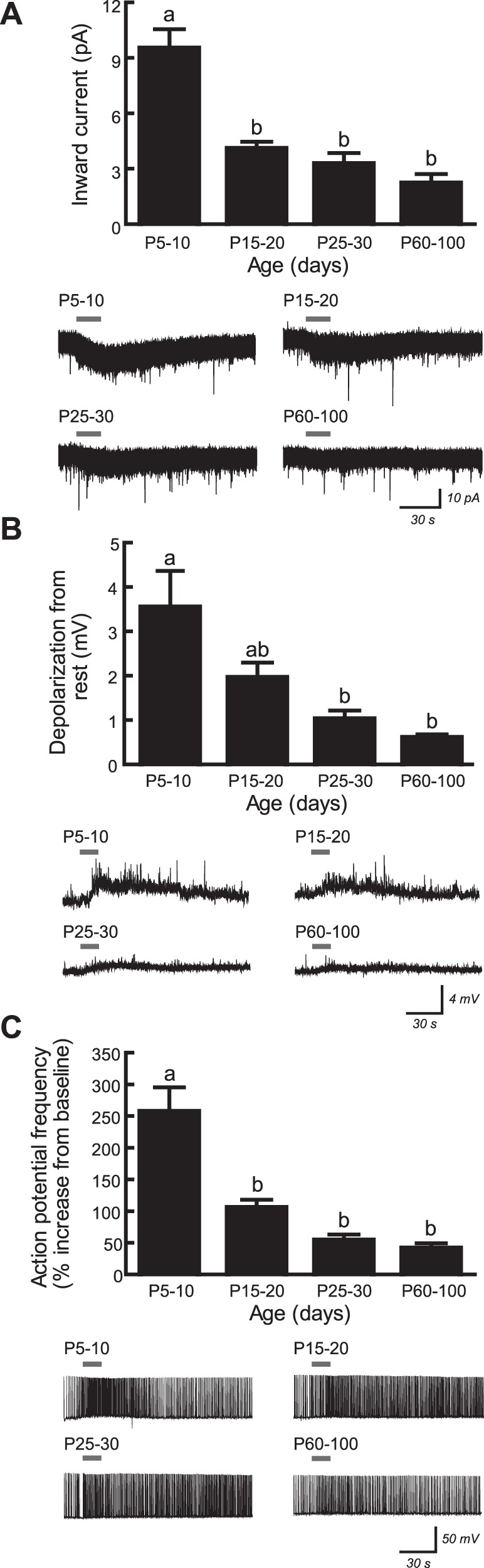
Nicotinic stimulation of CA1 pyramidal neurons is developmentally regulated and greatest in young postnatal mice. A: inward current responses to 1 mM ACh (15 s) were significantly affected by age (1-way ANOVA, P < 0.0001), where currents were greater at postnatal day 5–10 (P5–10) than at each of P15–20, P25–30, and P60–100 (Tukey's post hoc test, all P ≤ 0.0003). Exemplary voltage-clamp traces at each age are shown below the histogram plot. B: depolarization from rest following 1 mM ACh application (15 s) was significantly affected by age (P < 0.0001), where the response at P5–10 was significantly greater than that at P25–30 and P60–100 (both P ≤ 0.0009). Exemplary current-clamp traces are located below the histogram plot and show typical depolarization responses at each age. C: in neurons that had been induced to fire action potentials at 1 Hz, acceleration of firing frequency following application of 1 mM ACh (15 s) was significantly affected by age (P < 0.0001), where the response was greater at P5–10 than at P15–20, P25–30, and P60–100 (all P < 0.0001). Exemplary current-clamp traces showing the typical changes in firing frequency at each age are shown below the histogram plot. All recordings were made in the continuous presence of 200 nM atropine and 10 nM MLA. All values are means ± SE. a,bDifferent letters indicate statistically significant differences among data sets.
Two distinct classes of CA1 pyramidal neurons have been identified that display either regular-firing or burst-firing patterns of action potential activity (Graves et al. 2012). Neurons from this experiment were classified on the basis of threshold-level current injection, where regular-firing neurons were identified based on their initial action potentials having an instantaneous frequency less than 100 Hz and burst-firing neurons were identified by the presence of two or more initial action potentials having an instantaneous frequency greater than 100 Hz riding on a distinct slow depolarizing envelope (Graves et al. 2012; Su et al. 2001). There were more regular-firing neurons than burst-firing neurons at each age, and the proportion of each neuron type was not affected by age (P5–10: regular 55, burst 7; P15–20: regular 24, burst 11; P25–30: regular 26, burst 7; and P60–100: regular 26, burst 9; χ2 test, P = 0.1). Two-factor analysis of the developmental data for effects of age and neuron type showed that nicotinic receptor function was not different and also showed similar developmental changes among the two neuron types. Inward currents were affected by age [2-way ANOVA: F(3,135) = 7.62, P < 0.0001] but not by neuron type [F(1,135) = 0.008, P = 0.9], and there was no interaction between these two factors [F(3,135) = 0.05, P = 1.0]. Depolarization responses from rest showed a strong trend toward an effect of age [F(3,97) = 2.64, P = 0.05] but no effect of neuron type [F(1,97) = 1.52, P = 0.2] or interaction between the two factors [F(3,97) = 1.62, P = 0.2]. Nicotinic acceleration of action potential firing in active neurons showed an effect of age [F(3,111) = 11.82, P < 0.0001] but no effect of neuron type [F(1,111) = 0.00003, P = 1.0] or interaction between these two factors [F(3,111) = 0.40, P = 0.8]. Resting membrane potential [F(1,157) = 1.02, P = 0.3] and input resistance [F(1,152) = 0.32, P = 0.6] were not affected by neuron type. Spike amplitude was significantly greater in regular-firing neurons than in burst-firing neurons [F(1,156) = 5.16, P = 0.02].
Influence of endogenous acetylcholinesterase and extracellular matrix density on observed developmental changes to CA1 nicotinic receptor function.
Acetylcholinesterase is active in the early postnatal mouse hippocampus (Virgili et al. 1991), and endogenous acetylcholinesterase can metabolize exogenous ACh as it washes into brain slices (Bailey et al. 2010). Because hippocampus acetylcholinesterase activity increases during rodent postnatal development (Cermak et al. 1999; Virgili et al. 1991), we sought to determine whether this underlies the observed developmental changes to CA1 nAChR function using the ACh analog carbachol that is not rapidly metabolized by acetylcholinesterase or, to the best of our knowledge, by another enzyme present within brain slices (Goodman et al. 1985; Molitor 1936). Current responses to 1 mM carbachol (in the presence of 200 nM atropine and 10 nM MLA) were measured at P5–10 and P60–100. Carbachol was applied in the bath for either 15 or 60 s to also determine whether the kinetics of drug wash-in to the receptor are affected by potential developmental changes to extracellular matrix density. Similar to the experiment shown in Fig. 4A in which ACh was used, the magnitude of nicotinic currents elicited by carbachol was significantly affected by age [Fig. 5A; 2-way ANOVA: F(1, 14) = 7.11, P = 0.02] with currents being greater at P5–10 than at P60–100 (Bonferonni's post hoc test, all P < 0.05). However, the magnitude of nicotinic currents was not affected by the length of carbachol application [F(1,14) = 1.28, P = 0.3], and there was no interaction between effects of age and length of carbachol application [F(1,14) = 1.04, P = 0.3], suggesting that postnatal age does not affect the ability of drug to wash into the slice in our preparation. The kinetics of drug wash-in for this experiment were further investigated by measuring the time required for carbachol to elicit a peak current response in each neuron. The time to peak response was not affected by age [Fig. 5B; 2-way ANOVA: F(1,14) = 0.42, P = 0.5] or length of carbachol application [F(1,14) = 0.70, P = 0.4], and there was no interaction between effects of these two factors [F(1,14) = 0.54, P = 0.5]. These results suggest that observed developmental changes to nAChR function do not depend on altered acetylcholinesterase activity or extracellular matrix density.
Influence of α5 subunit incorporation toward developmental changes to CA1 nicotinic receptor function.
The nicotinic α5 subunit is present in ∼35% of heteromeric nAChRs within hippocampus tissue samples (Mao et al. 2008), and its incorporation is known to augment nAChR function (Kuryatov et al. 2008; Tapia et al. 2007). Because the expression of α5 subunit mRNA within the rat CA1 layer peaks during the first postnatal week and decreases thereafter, similar to our observed developmental pattern for nAChR function in CA1 pyramidal neurons, we sought to determine pharmacologically whether the proportion of nAChRs containing the α5 subunit changes during development. Incorporation of α5 subunits was probed at P5–10 and P60–100 using the α5 subunit-selective positive allosteric modulator galanthamine (Kuryatov et al. 2008). Peak current responses to 10 μM ACh (15 s, in the presence of 200 nM atropine and 10 nM MLA) were measured before and after a 10-min exposure to 0.1 μM galanthamine. Experiments were performed after endogenous acetylcholinesterase within the slice was inactivated by a 10-min exposure to 20 μM DFP, because galanthamine inhibits this enzyme at high concentrations. Although current responses were greater at P5–10 than at P60–100, the percentage by which galanthamine potentiated ACh responses was not different between these two ages (Fig. 6; 2-tailed unpaired t-test, P = 0.8). These data suggest that the α5 subunit is incorporated into a similar proportion of heteromeric nAChRs in both young postnatal and adult CA1 pyramidal neurons.
Fig. 6.

Potentiation of CA1 nicotinic responses by galanthamine, a positive modulator of α5 subunit-containing α4β2* nicotinic receptors, is not affected by postnatal age. Nicotinic current responses to 10 μM ACh (15 s) were measured before and after a 10-min exposure to 0.1 μM galanthamine. All recordings were made following the inactivation of acetylcholinesterase by diisopropyl fluorophosphate and in the continuous presence of 200 nM atropine and 10 nM MLA. The percent increase for ACh responses was not affected by age (unpaired t-test, P = 0.8). All values are means ± SE. Exemplary voltage-clamp traces for current responses to ACh before and after galanthamine exposure are shown at right.
DISCUSSION
Our findings provide evidence that CA1 pyramidal neurons of the mouse hippocampus are transiently excited by postsynaptic heteromeric nAChRs during early postnatal development. This conclusion is based on a series of physiological and pharmacological experiments performed using whole cell electrophysiological recording of visually identified CA1 pyramidal neurons within acute hippocampal brain slices collected at distinct developmental ages. Nicotinic stimulation in young postnatal animals produced direct inward currents and increased neuron excitability in a manner that is consistent with the presence of functional α4β2* nAChRs located directly on recorded neurons. By characterizing these responses across postnatal development, we now present a cohesive developmental profile for heteromeric nAChR function in mouse CA1 pyramidal neurons from P5 to P100, with the greatest function occurring during the first 2 wk of postnatal life. This strong developmental regulation suggests that heteromeric, putative α4β2* nAChRs are present and active selectively during a period when they may contribute to the cholinergic regulation of CA1 pyramidal neuron development within hippocampal learning and memory networks.
Heteromeric nicotinic receptors within the hippocampus.
The proper development and mature function of the hippocampus depends on cholinergic innervation from the MSDB (Aloisi et al. 1997; Anzalone et al. 2009; Berger-Sweeney et al. 2001; Cai et al. 2012; Ceccarelli et al. 1999; Easton et al. 2011; Liu et al. 2006; Lozada et al. 2012; Stanley et al. 2012), with ACh activating nAChRs located on local GABAergic interneurons and glutamatergic excitatory neurons (McQuiston 2014; Placzek et al. 2009; Yakel 2012). It is well-established that both α7 and α4β2* nAChRs mediate nicotinic currents in GABAergic interneurons to regulate the excitability of hippocampal circuits (Adams et al. 2002; Alkondon and Albuquerque 2001; Alkondon et al. 1999; Bell et al. 2011; Bell et al. 2015; Frazier et al. 1998; Ji and Dani 2000; Jones and Yakel 1997; Khiroug et al. 2003; McQuiston and Madison 1999; Sudweeks and Yakel 2000). Conversely, it is only more recently that studies have shown evidence for the presence of functional α7 (Grybko et al. 2011; Ji et al. 2001; Kalappa et al. 2010) and α4β2* nAChRs (He et al. 2013; Tu et al. 2009) located directly on hippocampus pyramidal neurons. We provide in this study comprehensive in situ evidence that heteromeric, putative α4β2* nAChRs are present on CA1 pyramidal neurons, with their greatest function occurring during hippocampal development. In our slice preparation, nicotinic current responses to bath-applied ACh were maintained following the blockade of synaptic transmission using inhibitors of voltage-gated sodium channels (TTX), glutamatergic signaling (CNQX and APV), and GABAergic signaling (bicuculline), suggesting that they were mediated by postsynaptic nAChRs. Currents were likely not mediated by α7 homomeric nAChRs because of their rapid desensitization kinetics (Quick and Lester 2002) and also because we conducted all experiments with α7 nAChRs blocked using the selective antagonist MLA. MLA also has been demonstrated to block the more recently discovered α7β2 heteromeric nAChR that could potentially form in pyramidal neurons (Liu et al. 2012; Murray et al. 2012). We found that ACh-induced postsynaptic currents and enhanced neuron excitability were both inhibit ed significantly by the α4β2* nAChR-selective antagonist DHβE, suggesting that responses were mediated by this class of nAChR located directly on recorded pyramidal neurons. Further evidence to support our findings may be generated in future studies using the complementary electrophysiological approach of recording responses to the stimulation of heteromeric nicotinic receptors in acutely dissociated CA1 pyramidal neurons and by labeling receptor subunit protein on CA1 pyramidal neurons within high-resolution immunohistochemical experiments.
Analysis of whole hippocampus tissue reveals expression of the α2, α3, α4, α5, β2 and β4 heteromeric nAChR subunits and shows that the predominant assembled isoforms are those containing α4 and β2 subunits only (comprising 40% of heteromeric nAChRs) and those containing α4 and β2 subunits along with the α5 accessory subunit (comprising 35% of heteromeric nAChRs) (Lomazzo et al. 2010; Mao et al. 2008). In situ analysis confirms that the α4, β2, and α5 subunits are expressed in the CA1 pyramidal neuron layer (Heath et al. 2010; Marks et al. 1992; Salas et al. 2003; Winzer-Serhan and Leslie 2005) and within the pyramidal neurons themselves (Sudweeks and Yakel 2000). Because the developmental profile for heteromeric nAChR function in this current study is consistent with that for expression of the α4 (Didier et al. 1995) and α5 subunits (Winzer-Serhan and Leslie 2005) within the CA1 pyramidal neuron layer, it is likely that our measured responses were mediated by α4β2 and α4β2α5 receptors. Further evidence for the incorporation of α5 subunits into α4β2* nAChRs comes from the nicotine desensitization and galanthamine potentiation experiments. The degree by which nicotine desensitized nAChR function in this current study was similar to that reported previously for prefrontal cortex pyramidal neurons that express α4, β2, and α5 subunits, but not for prefrontal cortex pyramidal neurons that lack the α5 subunit (Bailey et al. 2010). CA1 neuron nAChR currents were also potentiated by the α5 subunit-selective positive allosteric modulator galanthamine. The degree by which galanthamine potentiated nAChR currents was similar at P5–10 and P60–100, suggesting that a similar proportion of nAChRs contain the α5 subunit at each age. Given that the presence of an α5 subunit augments nAChR activity (Bailey et al. 2010; Kuryatov et al. 2008; McClure-Begley et al. 2009; Tapia et al. 2007), these data further suggest that our observed developmental decrease in nAChR function does not result from a selective decrease in the number of α4β2α5 nAChRs only.
We also investigated whether factors in our preparation other than receptor number could explain the observed developmental decrease in nAChR function. Increased acetylcholinesterase activity in the slices of older mice (Virgili et al. 1991) could have metabolized exogenous ACh as it washed into the slice toward nAChRs on the recorded neuron. However, we found that the developmental decrease to nAChR function was also present for CA1 neurons when we used the receptor agonist carbachol, which is not rapidly metabolized by acetylcholinesterase. The interpretation that this result confirms a developmental decrease in nAChR function for CA1 pyramidal neurons depends on carbachol not being rapidly metabolized by enzymes other than acetylcholinesterase within brain slices, which is consistent with available literature (Goodman et al. 1985; Molitor 1936). It was also possible that increased extracellular matrix density in older mice impeded the ability of exogenous ACh to wash in and maximally activate nAChRs when applied for 15 s. We found in the carbachol experiment that 15– and 60-s application of carbachol elicited similar nAChR currents and that the same developmental decrease in nAChR function was observed using both lengths of application. Moreover, the time required for carbachol to wash in and elicit a maximal current response was not affected by application length or age of the animal. The input resistance for CA1 pyramidal neurons decreased significantly as mice aged, which may have contributed to the observed developmental profiles for excitability responses measured in current-clamp mode. This is especially true for the depolarizing responses from rest (Fig. 4B), which show a relative developmental profile similar to that observed for input resistance. Our findings also suggest, however, that although the augmentation of excitability in firing neurons (Fig. 4C) may be influenced by input resistance and active electrophysiological properties (e.g., afterhyperpolarization), nicotinic currents appear to play a greater role here. For example, neurons at P5–10 required ∼2.5 times greater injected current than neurons at P15–20 to achieve the 1-Hz baseline firing (see results for data) and also received ∼2.5 times greater inward current than neurons at P15–20 following ACh application (Fig. 4A). However, instead of providing similar effects on neuron firing, nicotinic stimulation increased firing frequency at P5–10 by an amount that was ∼2.5 times greater than that measured at P15–20.
Nicotinic signaling in normal and aberrant hippocampal development.
Markers for cholinergic neurotransmission appear within the rat hippocampus during late gestation and are widespread by P3-5 (Happe and Murrin 1992; Milner et al. 1983). Expression of heteromeric nAChR subunits follows a similar early profile to peak during the first 2 wk before declining shortly thereafter (Didier et al. 1995; Shacka and Robinson 1998; Winzer-Serhan and Leslie 2005; Zoli et al. 1995). The ontogenic profile for these neurochemical markers and for the observed peak in CA1 neuron heteromeric nAChR function in this current study coincides with a period of local GABA-mediated excitatory neurotransmission resulting from high intracellular concentrations of chloride ions flowing through GABAA receptor channels to form primitive network oscillations known as giant depolarizing potentials (Ben-Ari et al. 1989; Garaschuk et al. 1998). Nicotinic signaling at α7 and β2* nAChRs contributes to the switch in GABAergic signaling from excitation to inhibition by mediating increased expression of the KCC2 Cl− transporter that maintains a low intracellular concentration of Cl− (Liu et al. 2006). Because this switch occurs in rat hippocampus during the first 2 wk of postnatal life (Garaschuk et al. 1998; Tyzio et al. 2006, 2007) and depends on postsynaptic Ca2+ transients (Ganguly et al. 2001), nicotinic signaling at the more Ca2+-permeable α4β2α5 nAChR is positioned spatially and temporally to contribute toward this developmental phenomenon in CA1 pyramidal neurons.
Nicotinic signaling plays an important role in neuron morphological development. Experiments in genetically-modified mice demonstrate that nAChRs containing the β2 subunit contribute to the production of dendritic spines in vivo for pyramidal neurons of the cerebral cortex and CA1 area of the hippocampus (Ballesteros-Yanez et al. 2010; Lozada et al. 2012). Nicotinic signaling regulates dendrite growth during neuron development, although its net effect may depend on cell type and timing. In cultured neurons, signaling at nAChRs limits neurite outgrowth (Lipton et al. 1988; Nordman and Kabbani 2012; Pugh and Berg 1994) and mediates neurite retraction in a Ca2+-dependent manner (Pugh and Berg 1994). Consistent with these effects, heteromeric nAChRs containing the α5 subunit appear to mediate the retraction of apical dendrites for medial prefrontal layer VI pyramidal neurons during postnatal maturation (Bailey et al. 2012). Genetic deletion of the β2 nAChR subunit leads to either increased or decreased dendritic fields for layer II/III pyramidal neurons depending on the cortical region examined (Ballesteros-Yanez et al. 2010). Moreover, for cultured rat olfactory bulb neurons and for hippocampal adult-born neurons, nAChR activation has been found to promote neurite/dendrite elongation (Campbell et al. 2010; Coronas et al. 2000). The postnatal development of CA1 pyramidal neurons is characterized by increased dendrite complexity and length over the first 2–4 wk for basal dendrites (Nishimura et al. 2011) and over at least the first 5 wk for apical dendrites (Jacobson et al. 1988). However, to the best of our knowledge, the influence of nAChRs on hippocampal dendrite complexity has not been examined.
The presence of functional heteromeric nAChRs on CA1 pyramidal neurons may have important implications for our understanding of aberrant hippocampus development. Several studies have demonstrated that perinatal exposure to nicotine in rodents and humans impairs cognitive functions that are supported by the hippocampus, such as learning, memory, and attention (Dwyer et al. 2009; Ernst et al. 2001; Fried and Watkinson 2001; Johns et al. 1982; Levin et al. 1999; Li et al. 2015; Pauly and Slotkin 2008; Poorthuis et al. 2013; Portugal et al. 2012; Schneider et al. 2011), and alters neurotransmission in the CA1 region (Damborsky et al. 2012; Parameshwaran et al. 2012, 2013). We show in this current study that nicotine at concentrations relevant to those seen in the brain of smokers desensitizes heteromeric nAChRs in young postnatal CA1 pyramidal neurons. This functional inhibition may alter the role of nicotinic signaling in the maturation of CA1 pyramidal neurons, as it does for medial prefrontal pyramidal neurons (Bailey et al. 2014), to alter their function within mature hippocampal networks. Autism spectrum disorder (ASD) and attention deficit hyperactivity disorder (ADHD) are associated with deficits in learning, memory and attention (Allen and Courchesne 2001; Loveland et al. 2008; Mayes and Calhoun 2007; Sowerby et al. 2011), and also with increased hippocampus size (Plessen et al. 2006; Schumann et al. 2004). Both nicotine and α4β2* nAChR agonists can improve attention and working memory in ADHD (Plessen et al. 2006; Wilens and Decker 2007; Wilens et al. 2006) and the augmentation of nicotinic neurotransmission is a proposed therapeutic strategy for ASD (Deutsch et al. 2010; Ghaleiha et al. 2013). Moreover, the α4β2* nAChR currently is a target of interest for a number of neurological disorders that involve deficits to hippocampus-dependent cognitive functions, including the neurodevelopmental disorders ASD, ADHD, and depression and the neurodegenerative disorder Alzheimer's disease (Arneric et al. 2007; Dineley et al. 2015; Hurst et al. 2013; Taly et al. 2009).
Conclusion.
Our results demonstrate that heteromeric nAChRs mediate postsynaptic nicotinic signaling in developing CA1 pyramidal neurons. This places heteromeric nAChRs in a position to directly influence the physiological and morphological maturation of CA1 pyramidal neurons and determine their function within mature hippocampal cognitive circuits. This work provides insight into the normal development of the hippocampus and may also inform efforts for pharmaceutical development aimed to mitigate or treat multiple neurodevelopmental disorders involving the hippocampal nicotinic system.
GRANTS
This work was supported by Natural Sciences and Engineering Research Council of Canada Discovery Grant 436190 (to C.D.C. Bailey) and Canada Foundation for Innovation Project no. 30381 (to C.D.C. Bailey). B.Y.T. Chung was the recipient of an Ontario Graduate Scholarship and an OVC Scholarship from the Ontario Veterinary College at the University of Guelph. D.L. Jacklin was the recipient of an Ontario Graduate Scholarship.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.Y.T.C. and C.D.C.B. conception and design of research; B.Y.T.C., W.B., and D.L.J. performed experiments; B.Y.T.C. and C.D.C.B. analyzed data; B.Y.T.C., B.D.W., and C.D.C.B. interpreted results of experiments; B.Y.T.C. and C.D.C.B. prepared figures; B.Y.T.C. drafted manuscript; B.Y.T.C., B.D.W., and C.D.C.B. edited and revised manuscript; B.Y.T.C., W.B., D.L.J., B.D.W., and C.D.C.B. approved final version of manuscript.
REFERENCES
- Adams CE, Broide RS, Chen Y, Winzer-Serhan UH, Henderson TA, Leslie FM, Freedman R. Development of the alpha7 nicotinic cholinergic receptor in rat hippocampal formation. Brain Res Dev Brain Res 139: 175–187, 2002. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Nicotinic acetylcholine receptor α7 and α4β2 subtypes differentially control GABAergic input to CA1 neurons in rat hippocampus. J Neurophysiol 86: 3043–3055, 2001. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci 19: 2693–2705, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen G, Courchesne E. Attention function and dysfunction in autism. Front Biosci 6: D105–D119, 2001. [DOI] [PubMed] [Google Scholar]
- Aloisi AM, Casamenti F, Scali C, Pepeu G, Carli G. Effects of novelty, pain and stress on hippocampal extracellular acetylcholine levels in male rats. Brain Res 748: 219–226, 1997. [DOI] [PubMed] [Google Scholar]
- Anzalone S, Roland J, Vogt B, Savage L. Acetylcholine efflux from retrosplenial areas and hippocampal sectors during maze exploration. Behav Brain Res 201: 272–278, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arneric SP, Holladay M, Williams M. Neuronal nicotinic receptors: a perspective on two decades of drug discovery research. Biochem Pharmacol 74: 1092–1101, 2007. [DOI] [PubMed] [Google Scholar]
- Bailey CD, Alves NC, Nashmi R, De Biasi M, Lambe EK. Nicotinic alpha5 subunits drive developmental changes in the activation and morphology of prefrontal cortex layer VI neurons. Biol Psychiatry 71: 120–128, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CD, De Biasi M, Fletcher PJ, Lambe EK. The nicotinic acetylcholine receptor alpha5 subunit plays a key role in attention circuitry and accuracy. J Neurosci 30: 9241–9252, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CD, Tian MK, Kang L, O'Reilly R, Lambe EK. Chrna5 genotype determines the long-lasting effects of developmental in vivo nicotine exposure on prefrontal attention circuitry. Neuropharmacology 77: 145–155, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros-Yanez I, Benavides-Piccione R, Bourgeois JP, Changeux JP, DeFelipe J. Alterations of cortical pyramidal neurons in mice lacking high-affinity nicotinic receptors. Proc Natl Acad Sci USA 107: 11567–11572, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell KA, Shim H, Chen CK, McQuiston AR. Nicotinic excitatory postsynaptic potentials in hippocampal CA1 interneurons are predominantly mediated by nicotinic receptors that contain alpha4 and beta2 subunits. Neuropharmacology 61: 1379–1388, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell LA, Bell KA, McQuiston AR. Acetylcholine release in mouse hippocampal CA1 preferentially activates inhibitory-selective interneurons via alpha4beta2* nicotinic receptor activation. Front Cell Neurosci 9: 115, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol 416: 303–325, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger-Sweeney J, Stearns NA, Murg SL, Floerke-Nashner LR, Lappi DA, Baxter MG. Selective immunolesions of cholinergic neurons in mice: effects on neuroanatomy, neurochemistry, and behavior. J Neurosci 21: 8164–8173, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D, Jou J, Allen V, Tiongson E, Chefer SI, Koren AO, Mukhin AG. Cigarette smoking saturates brain alpha 4 beta 2 nicotinic acetylcholine receptors. Arch Gen Psychiatry 63: 907–915, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Gibbs RB, Johnson DA. Recognition of novel objects and their location in rats with selective cholinergic lesion of the medial septum. Neurosci Lett 506: 261–265, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell NR, Fernandes CC, Halff AW, Berg DK. Endogenous signaling through alpha7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J Neurosci 30: 8734–8744, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli I, Casamenti F, Massafra C, Pepeu G, Scali C, Aloisi AM. Effects of novelty and pain on behavior and hippocampal extracellular ACh levels in male and female rats. Brain Res 815: 169–176, 1999. [DOI] [PubMed] [Google Scholar]
- Cermak JM, Blusztajn JK, Meck WH, Williams CL, Fitzgerald CM, Rosene DL, Loy R. Prenatal availability of choline alters the development of acetylcholinesterase in the rat hippocampus. Dev Neurosci 21: 94–104, 1999. [DOI] [PubMed] [Google Scholar]
- Coronas V, Durand M, Chabot JG, Jourdan F, Quirion R. Acetylcholine induces neuritic outgrowth in rat primary olfactory bulb cultures. Neuroscience 98: 213–219, 2000. [DOI] [PubMed] [Google Scholar]
- Damborsky JC, Griffith WH, Winzer-Serhan UH. Chronic neonatal nicotine exposure increases excitation in the young adult rat hippocampus in a sex-dependent manner. Brain Res 1430: 8–17, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Bliss TV, Dutrieux G, Laroche S, Errington ML. Induction and duration of long-term potentiation in the hippocampus of the freely moving mouse. J Neurosci Methods 75: 75–80, 1997. [DOI] [PubMed] [Google Scholar]
- Deutsch SI, Urbano MR, Neumann SA, Burket JA, Katz E. Cholinergic abnormalities in autism: is there a rationale for selective nicotinic agonist interventions? Clin Neuropharmacol 33: 114–120, 2010. [DOI] [PubMed] [Google Scholar]
- Didier M, Bix G, Berman SA, Bursztajn S. Expression of the alpha 4 neuronal nicotinic acetylcholine receptor subunit in the developing mouse hippocampus. Int J Dev Neurosci 13: 703–713, 1995. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Pandya AA, Yakel JL. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci 36: 96–108, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer JB, McQuown SC, Leslie FM. The dynamic effects of nicotine on the developing brain. Pharmacol Ther 122: 125–139, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton A, Fitchett AE, Eacott MJ, Baxter MG. Medial septal cholinergic neurons are necessary for context-place memory but not episodic-like memory. Hippocampus 21: 1021–1027, 2011. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H, Otto T, Cohen NJ. The hippocampus–what does it do? Behav Neural Biol 57: 2–36, 1992. [DOI] [PubMed] [Google Scholar]
- Ernst M, Moolchan ET, Robinson ML. Behavioral and neural consequences of prenatal exposure to nicotine. J Am Acad Child Adolesc Psychiatry 40: 630–641, 2001. [DOI] [PubMed] [Google Scholar]
- Felix R, Levin ED. Nicotinic antagonist administration into the ventral hippocampus and spatial working memory in rats. Neuroscience 81: 1009–1017, 1997. [DOI] [PubMed] [Google Scholar]
- Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an alpha-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci 18: 1187–1195, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried PA, Watkinson B. Differential effects on facets of attention in adolescents prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol 23: 421–430, 2001. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105: 521–532, 2001. [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Hanse E, Konnerth A. Developmental profile and synaptic origin of early network oscillations in the CA1 region of rat neonatal hippocampus. J Physiol 507: 219–236, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaleiha A, Ghyasvand M, Mohammadi MR, Farokhnia M, Yadegari N, Tabrizi M, Hajiaghaee R, Yekehtaz H, Akhondzadeh S. Galantamine efficacy and tolerability as an augmentative therapy in autistic children: A randomized, double-blind, placebo-controlled trial. J Psychopharmacol 28: 677–685, 2013. [DOI] [PubMed] [Google Scholar]
- Goodman LS, Gilman A, Gilman AG. Goodman and Gilman's The Pharmacological Basis of Therapeutics. New York: Macmillan, 1985, p. xvi. [Google Scholar]
- Grady SR, Wageman CR, Patzlaff NE, Marks MJ. Low concentrations of nicotine differentially desensitize nicotinic acetylcholine receptors that include alpha5 or alpha6 subunits and that mediate synaptosomal neurotransmitter release. Neuropharmacology 62: 1935–1943, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves AR, Moore SJ, Bloss EB, Mensh BD, Kath WL, Spruston N. Hippocampal pyramidal neurons comprise two distinct cell types that are countermodulated by metabotropic receptors. Neuron 76: 776–789, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grybko MJ, Hahm ET, Perrine W, Parnes JA, Chick WS, Sharma G, Finger TE, Vijayaraghavan S. A transgenic mouse model reveals fast nicotinic transmission in hippocampal pyramidal neurons. Eur J Neurosci 33: 1786–1798, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happe HK, Murrin LC. Development of high-affinity choline transport sites in rat forebrain: a quantitative autoradiography study with [3H]hemicholinium-3. J Comp Neurol 321: 591–611, 1992. [DOI] [PubMed] [Google Scholar]
- He YX, Wu MN, Zhang H, Qi JS. Amyloid beta-protein suppressed nicotinic acetylcholine receptor-mediated currents in acutely isolated rat hippocampal CA1 pyramidal neurons. Synapse 67: 11–20, 2013. [DOI] [PubMed] [Google Scholar]
- Heath CJ, King SL, Gotti C, Marks MJ, Picciotto MR. Cortico-thalamic connectivity is vulnerable to nicotine exposure during early postnatal development through alpha4/beta2/alpha5 nicotinic acetylcholine receptors. Neuropsychopharmacology 35: 2324–2338, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Hulo S, Bertrand D, Muller D. Synaptic transmission at nicotinic acetylcholine receptors in rat hippocampal organotypic cultures and slices. J Physiol 515: 769–776, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend 33: 23–29, 1993. [DOI] [PubMed] [Google Scholar]
- Horti A, Scheffel U, Stathis M, Finley P, Ravert HT, London ED, Dannals RF. Fluorine-18-FPH for PET imaging of nicotinic acetylcholine receptors. J Nucl Med 38: 1260–1265, 1997. [PubMed] [Google Scholar]
- Hsu YW, Tempest L, Quina LA, Wei AD, Zeng H, Turner EE. Medial habenula output circuit mediated by alpha5 nicotinic receptor-expressing GABAergic neurons in the interpeduncular nucleus. J Neurosci 33: 18022–18035, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst R, Rollema H, Bertrand D. Nicotinic acetylcholine receptors: from basic science to therapeutics. Pharmacol Ther 137: 22–54, 2013. [DOI] [PubMed] [Google Scholar]
- Jacobson CD, Antolick LL, Scholey R, Uemura E. The influence of prenatal phenobarbital exposure on the growth of dendrites in the rat hippocampus. Brain Res Dev Brain Res 44: 233–239, 1988. [DOI] [PubMed] [Google Scholar]
- Ji D, Dani JA. Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J Neurophysiol 83: 2682–2690, 2000. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron 31: 131–141, 2001. [DOI] [PubMed] [Google Scholar]
- Johns JM, Louis TM, Becker RF, Means LW. Behavioral effects of prenatal exposure to nicotine in guinea pigs. Neurobehav Toxicol Teratol 4: 365–369, 1982. [PubMed] [Google Scholar]
- Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J Physiol 504: 603–610, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalappa BI, Gusev AG, Uteshev VV. Activation of functional alpha7-containing nAChRs in hippocampal CA1 pyramidal neurons by physiological levels of choline in the presence of PNU-120596. PLoS One 5: e13964, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Raybuck JD, Gould TJ. Nicotinic receptors in the dorsal and ventral hippocampus differentially modulate contextual fear conditioning. Hippocampus 22: 1681–1690, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khiroug L, Giniatullin R, Klein RC, Fayuk D, Yakel JL. Functional mapping and Ca2+ regulation of nicotinic acetylcholine receptor channels in rat hippocampal CA1 neurons. J Neurosci 23: 9024–9031, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. Encoding and retrieval along the long axis of the hippocampus and their relationships with dorsal attention and default mode networks: the HERNET model. Hippocampus 25: 500–510, 2015. [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Onksen J, Lindstrom J. Roles of accessory subunits in alpha4beta2(*) nicotinic receptors. Mol Pharmacol 74: 132–143, 2008. [DOI] [PubMed] [Google Scholar]
- Levin ED, Christopher NC, Weaver T, Moore J, Brucato F. Ventral hippocampal ibotenic acid lesions block chronic nicotine-induced spatial working memory improvement in rats. Brain Res Cogn Brain Res 7: 405–410, 1999. [DOI] [PubMed] [Google Scholar]
- Li J, Bo L, Zhang P, Gao Q, Li L, Tang J, Wu C, Li D, Xiao J, Chen J, Tao J, Mao C, Xu Z. Exposure to nicotine during pregnancy and altered learning and memory in the rat offspring. Nicotine Tob Res 17: 661–666, 2015. [DOI] [PubMed] [Google Scholar]
- Li Y, Li F, He N, Guo L, Huang X, Lui S, Gong Q. Neural hyperactivity related to working memory in drug-naive boys with attention deficit hyperactivity disorder. Prog Neuropsychopharmacol Biol Psychiatry 53: 116–122, 2014. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Frosch MP, Phillips MD, Tauck DL, Aizenman E. Nicotinic antagonists enhance process outgrowth by rat retinal ganglion cells in culture. Science 239: 1293–1296, 1988. [DOI] [PubMed] [Google Scholar]
- Liu Q, Huang Y, Shen J, Steffensen S, Wu J. Functional alpha7beta2 nicotinic acetylcholine receptors expressed in hippocampal interneurons exhibit high sensitivity to pathological level of amyloid beta peptides. BMC Neurosci 13: 155, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Neff RA, Berg DK. Sequential interplay of nicotinic and GABAergic signaling guides neuronal development. Science 314: 1610–1613, 2006. [DOI] [PubMed] [Google Scholar]
- Lomazzo E, MacArthur L, Yasuda RP, Wolfe BB, Kellar KJ. Quantitative analysis of the heteromeric neuronal nicotinic receptors in the rat hippocampus. J Neurochem 115: 625–634, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveland KA, Bachevalier J, Pearson DA, Lane DM. Fronto-limbic functioning in children and adolescents with and without autism. Neuropsychologia 46: 49–62, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu Z, Berg DK. Induction of dendritic spines by beta2-containing nicotinic receptors. J Neurosci 32: 8391–8400, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaalani R, Kashi PK, Waters KA. Distribution of nicotinic acetylcholine receptor subunits alpha7 and beta2 in the human brainstem and hippocampal formation. J Chem Neuroanat 40: 223–231, 2010. [DOI] [PubMed] [Google Scholar]
- Mao D, Perry DC, Yasuda RP, Wolfe BB, Kellar KJ. The alpha4beta2alpha5 nicotinic cholinergic receptor in rat brain is resistant to up-regulation by nicotine in vivo. J Neurochem 104: 446–456, 2008. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci 12: 2765–2784, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayes SD, Calhoun SL. Learning, attention, writing, and processing speed in typical children and children with ADHD, autism, anxiety, depression, and oppositional-defiant disorder. Child Neuropsychol 13: 469–493, 2007. [DOI] [PubMed] [Google Scholar]
- McClure-Begley TD, King NM, Collins AC, Stitzel JA, Wehner JM, Butt CM. Acetylcholine-stimulated [3H]GABA release from mouse brain synaptosomes is modulated by alpha4beta2 and alpha4alpha5beta2 nicotinic receptor subtypes. Mol Pharmacol 75: 918–926, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGoey TN, Reynolds JN, Brien JF. Chronic prenatal ethanol exposure-induced decrease of guinea pig hippocampal CA1 pyramidal cell and cerebellar Purkinje cell density. Can J Physiol Pharmacol 81: 476–484, 2003. [DOI] [PubMed] [Google Scholar]
- McQuiston AR. Acetylcholine release and inhibitory interneuron activity in hippocampal CA1. Front Synaptic Neurosci 6: 20, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV. Nicotinic receptor activation excites distinct subtypes of interneurons in the rat hippocampus. J Neurosci 19: 2887–2896, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner TA, Loy R, Amaral DG. An anatomical study of the development of the septo-hippocampal projection in the rat. Brain Res 284: 343–371, 1983. [DOI] [PubMed] [Google Scholar]
- Molitor H. A comparative study of the effects of five choline compounds used in therapeutics–acetylcholine chloride, acetyl beta-methyl-choline chloride, carbaminoyl choline ethyl ether beta-methylcholine chloride carbaminoyl beta-methylcholine chloride. J Pharmacol Exp Ther 58: 337–360, 1936. [Google Scholar]
- Murray TA, Bertrand D, Papke RL, George AA, Pantoja R, Srinivasan R, Liu Q, Wu J, Whiteaker P, Lester HA, Lukas RJ. alpha7beta2 nicotinic acetylcholine receptors assemble, function, and are activated primarily via their alpha7-alpha7 interfaces. Mol Pharmacol 81: 175–188, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Gu X, Swann JW. Seizures in early life suppress hippocampal dendrite growth while impairing spatial learning. Neurobiol Dis 44: 205–214, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordman JC, Kabbani N. An interaction between alpha7 nicotinic receptors and a G-protein pathway complex regulates neurite growth in neural cells. J Cell Sci 125: 5502–5513, 2012. [DOI] [PubMed] [Google Scholar]
- Nott A, Levin ED. Dorsal hippocampal alpha7 and alpha4beta2 nicotinic receptors and memory. Brain Res 1081: 72–78, 2006. [DOI] [PubMed] [Google Scholar]
- Parameshwaran K, Buabeid MA, Bhattacharya S, Uthayathas S, Kariharan T, Dhanasekaran M, Suppiramaniam V. Long term alterations in synaptic physiology, expression of beta2 nicotinic receptors and ERK1/2 signaling in the hippocampus of rats with prenatal nicotine exposure. Neurobiol Learn Mem 106: 102–111, 2013. [DOI] [PubMed] [Google Scholar]
- Parameshwaran K, Buabeid MA, Karuppagounder SS, Uthayathas S, Thiruchelvam K, Shonesy B, Dityatev A, Escobar MC, Dhanasekaran M, Suppiramaniam V. Developmental nicotine exposure induced alterations in behavior and glutamate receptor function in hippocampus. Cell Mol Life Sci 69: 829–841, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly JR, Slotkin TA. Maternal tobacco smoking, nicotine replacement and neurobehavioural development. Acta Paediatr 97: 1331–1337, 2008. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Academic, 2001, p. xxv. [Google Scholar]
- Placzek AN, Zhang TA, Dani JA. Nicotinic mechanisms influencing synaptic plasticity in the hippocampus. Acta Pharmacol Sin 30: 752–760, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessen KJ, Bansal R, Zhu H, Whiteman R, Amat J, Quackenbush GA, Martin L, Durkin K, Blair C, Royal J, Hugdahl K, Peterson BS. Hippocampus and amygdala morphology in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry 63: 795–807, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorthuis RB, Bloem B, Verhoog MB, Mansvelder HD. Layer-specific interference with cholinergic signaling in the prefrontal cortex by smoking concentrations of nicotine. J Neurosci 33: 4843–4853, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portugal GS, Wilkinson DS, Turner JR, Blendy JA, Gould TJ. Developmental effects of acute, chronic, and withdrawal from chronic nicotine on fear conditioning. Neurobiol Learn Mem 97: 482–494, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh PC, Berg DK. Neuronal acetylcholine receptors that bind alpha-bungarotoxin mediate neurite retraction in a calcium-dependent manner. J Neurosci 14: 889–896, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol 53: 457–478, 2002. [DOI] [PubMed] [Google Scholar]
- Rose JE, Mukhin AG, Lokitz SJ, Turkington TG, Herskovic J, Behm FM, Garg S, Garg PK. Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc Natl Acad Sci USA 107: 5190–5195, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Orr-Urtreger A, Broide RS, Beaudet A, Paylor R, De Biasi M. The nicotinic acetylcholine receptor subunit alpha 5 mediates short-term effects of nicotine in vivo. Mol Pharmacol 63: 1059–1066, 2003. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Anderson DW, Sonnenahalli H, Vadigepalli R. Sex-based differences in gene expression in hippocampus following postnatal lead exposure. Toxicol Appl Pharmacol 256: 179–190, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann CM, Hamstra J, Goodlin-Jones BL, Lotspeich LJ, Kwon H, Buonocore MH, Lammers CR, Reiss AL, Amaral DG. The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J Neurosci 24: 6392–6401, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry 20: 11–21, 1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Robinson SE. Postnatal developmental regulation of neuronal nicotinic receptor subunit alpha 7 and multiple alpha 4 and beta 2 mRNA species in the rat. Brain Res Dev Brain Res 109: 67–75, 1998. [DOI] [PubMed] [Google Scholar]
- Sowerby P, Seal S, Tripp G. Working memory deficits in ADHD: the contribution of age, learning/language difficulties, and task parameters. J Atten Disord 15: 461–472, 2011. [DOI] [PubMed] [Google Scholar]
- Squire LR, Zola SM. Structure and function of declarative and nondeclarative memory systems. Proc Natl Acad Sci USA 93: 13515–13522, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EM, Wilson MA, Fadel JR. Hippocampal neurotransmitter efflux during one-trial novel object recognition in rats. Neurosci Lett 511: 38–42, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Alroy G, Kirson ED, Yaari Y. Extracellular calcium modulates persistent sodium current-dependent burst-firing in hippocampal pyramidal neurons. J Neurosci 21: 4173–4182, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudweeks SN, Yakel JL. Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J Physiol 527: 515–528, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland RJ, McDonald RJ, Savage DD. Prenatal exposure to moderate levels of ethanol can have long-lasting effects on hippocampal synaptic plasticity in adult offspring. Hippocampus 7: 232–238, 1997. [DOI] [PubMed] [Google Scholar]
- Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov 8: 733–750, 2009. [DOI] [PubMed] [Google Scholar]
- Tapia L, Kuryatov A, Lindstrom J. Ca2+ permeability of the (alpha4)3(beta2)2 stoichiometry greatly exceeds that of (alpha4)2(beta2)3 human acetylcholine receptors. Mol Pharmacol 71: 769–776, 2007. [DOI] [PubMed] [Google Scholar]
- Tu B, Gu Z, Shen JX, Lamb PW, Yakel JL. Characterization of a nicotine-sensitive neuronal population in rat entorhinal cortex. J Neurosci 29: 10436–10448, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzio R, Cossart R, Khalilov I, Minlebaev M, Hubner CA, Represa A, Ben-Ari Y, Khazipov R. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314: 1788–1792, 2006. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Holmes GL, Ben-Ari Y, Khazipov R. Timing of the developmental switch in GABAA mediated signaling from excitation to inhibition in CA3 rat hippocampus using gramicidin perforated patch and extracellular recordings. Epilepsia 48, Suppl 5: 96–105, 2007. [DOI] [PubMed] [Google Scholar]
- Virgili M, Contestabile A, Barnabei O. Postnatal maturation of cholinergic markers in forebrain regions of C57BL/6 mice. Brain Res Dev Brain Res 63: 281–285, 1991. [DOI] [PubMed] [Google Scholar]
- Wageman CR, Marks MJ, Grady SR. Effectiveness of nicotinic agonists as desensitizers at presynaptic alpha4beta2- and alpha4alpha5beta2-nicotinic acetylcholine receptors. Nicotine Tob Res 16: 297–305, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilens TE, Decker MW. Neuronal nicotinic receptor agonists for the treatment of attention-deficit/hyperactivity disorder: focus on cognition. Biochem Pharmacol 74: 1212–1223, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilens TE, Verlinden MH, Adler LA, Wozniak PJ, West SA. ABT-089, a neuronal nicotinic receptor partial agonist, for the treatment of attention-deficit/hyperactivity disorder in adults: results of a pilot study. Biol Psychiatry 59: 1065–1070, 2006. [DOI] [PubMed] [Google Scholar]
- Winzer-Serhan UH, Leslie FM. Expression of alpha5 nicotinic acetylcholine receptor subunit mRNA during hippocampal and cortical development. J Comp Neurol 481: 19–30, 2005. [DOI] [PubMed] [Google Scholar]
- Yakel JL. Nicotinic ACh receptors in the hippocampus: role in excitability and plasticity. Nicotine Tob Res 14: 1249–1257, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Liu C, Miao H, Gong ZH, Nordberg A. Postnatal changes of nicotinic acetylcholine receptor alpha 2, alpha 3, alpha 4, alpha 7 and beta 2 subunits genes expression in rat brain. Int J Dev Neurosci 16: 507–518, 1998. [DOI] [PubMed] [Google Scholar]
- Zoli M, Le Novere N, Hill JA Jr, Changeux JP. Developmental regulation of nicotinic ACh receptor subunit mRNAs in the rat central and peripheral nervous systems. J Neurosci 15: 1912–1939, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]