

Abstract
Whereas the cerebral cortex has long been regarded by neuroscientists as the major locus of cognitive function, the white matter of the brain is increasingly recognized as equally critical for cognition. White matter comprises half of the brain, has expanded more than gray matter in evolution, and forms an indispensable component of distributed neural networks that subserve neurobehavioral operations. White matter tracts mediate the essential connectivity by which human behavior is organized, working in concert with gray matter to enable the extraordinary repertoire of human cognitive capacities. In this review, we present evidence from behavioral neurology that white matter lesions regularly disturb cognition, consider the role of white matter in the physiology of distributed neural networks, develop the hypothesis that white matter dysfunction is relevant to neurodegenerative disorders, including Alzheimer's disease and the newly described entity chronic traumatic encephalopathy, and discuss emerging concepts regarding the prevention and treatment of cognitive dysfunction associated with white matter disorders. Investigation of the role of white matter in cognition has yielded many valuable insights and promises to expand understanding of normal brain structure and function, improve the treatment of many neurobehavioral disorders, and disclose new opportunities for research on many challenging problems facing medicine and society.
Keywords: cognition, dementia, glial cells, myelin, white matter
the central importance of the cerebral cortex in the elaboration of human behavior enjoys such wide popularity that this perspective has become axiomatic. The billions of cortical neurons in the human brain, and the trillions of synapses linking them, are regularly regarded as comprising the singular repository of cognitive function, so much so that neurologists routinely invoke the term “higher cortical function” to describe the study of brain-behavior relationships. Similarly, neuroscientists retain such firm allegiance to the hegemony of the cerebral cortex that the term “corticocentric myopia” has been used to describe the relative paucity of neuroscientific investigation devoted to other brain areas that may contribute to cognitive function (Parvizi 2009).
Yet roughly half the brain is occupied by white matter, and it is reasonable to ask in what way this collection of myelinated tracts might inform the study of cognition (Fields 2008). The vast extent of white matter is an obvious datum meriting consideration: in one adult, the millions of myelinated fibers coursing throughout the brain have a combined length that would encircle the Earth more than three times (Walhovd et al. 2014). Evidence from clinical neurology is now indicating that white matter serves a critical role in the organization of the distributed neural networks that are now conceptualized as the structural basis of evolved human behaviors (Catani et al. 2012; Mesulam 1990). The advent of magnetic resonance imaging (MRI) in the last three decades has enabled the study of white matter in vivo (Catani et al. 2012), illuminating how lesions of white matter tracts disturb cognition. Many cellular changes in white matter can influence MRI, including differences in axon diameter, packing, or tortuosity, myelin, astrocytes, oligodendrocytes, and vasculature. MRI has also shown how differences in normal white matter structure correlate with level of performance in a wide variety of cognitive functions, such as facility with arithmetic (Matejko and Ansari 2015) and musical performance (Ullén 2009). In parallel, experimental advances have occurred that elucidate basic aspects of white matter structure and function (Fields 2008; Walhovd et al. 2014). Together, these developments have introduced a new multidisciplinary approach to the study of normal and abnormal cognition by focusing on a long-overlooked region of the brain. In this review, we consider recent clinical and laboratory evidence informing the understanding of white matter-cognition relationships in health and disease.
The Contribution of White Matter to Cognition
The neural basis of human cognition has been largely established by the clinical-pathological study of diseases that disrupt cognitive function. Behavioral neurology relies heavily on the “lesion method” (Damasio 1984), which considers cognitive dysfunction in relation to brain lesions, and for most of the history of medicine postmortem analysis of the brain was the only means available to examine the lesions produced by neurological disorders such as stroke, traumatic brain injury (TBI), and degenerative dementia. Together with gray matter, white matter was included in these studies, and classic observations in the nineteenth century identified neurobehavioral syndromes such as conduction aphasia, pure alexia, and ideomotor apraxia in which focal vascular lesions of white matter were found to play a pivotal role. These and other syndromes were famously summarized by the behavioral neurologist Norman Geschwind in his seminal 1965 paper on disconnection syndromes (Geschwind 1965). According to Geschwind, disconnection of cerebral regions by white matter damage merited consideration equal to that given to focal lesions of the cerebral cortex.
Modern neuroimaging dramatically advanced the study of brain-behavior relationships, first in the 1970s with computed tomography (CT), when for the first time neurologists could see structural brain pathology in vivo (Bradley 1986). MRI, however, proved far more useful than CT in the visualization of white matter and its lesions (Bradley 1986), allowing for correlation of these abnormalities with cognitive dysfunction during life (Tanridag and Kirshner 1987). In the early years of the MRI era, white matter lesions were often called “unidentified bright objects” because of uncertainty about what specific neuropathology could be responsible (Kertesz et al. 1988), but with time it became clear that these white matter hyperintensities reflected the appearance of water accumulating in the presence of myelin disruption from many etiologies (Anderson et al. 2014). One of the most common MRI abnormalities was termed leukoaraiosis (LA), referring to scattered hyperintense lesions in the cerebral white matter of older people on T2 and fluid-attenuated inversion recovery (FLAIR) images (Hachinski et al. 1987). The origin of LA was determined to be most likely related to ischemia (Pantoni and Garcia 1997), leading investigators to consider the possibility that LA is an early phase of the well-known vascular white matter dementia known as Binswanger's disease (BD; Babikian and Ropper 1987). Indeed, evolving consensus opinion increasingly supports the view that LA is a clinically mild precursor state to the progressive dementia of BD (Rosenberg et al. 2016; Schmidt et al. 2016). Figure 1 shows axial MR brain images of older individuals to demonstrate the scattered ischemic white matter lesions of LA (Fig. 1A) and confluent ischemic white matter hyperintensity consistent with BD (Fig. 1B). The study of LA (a new observation) in the context of BD (an old disease) is but one example of how white matter-cognition relationships rapidly gained momentum with the advent of MRI (Catani et al. 2012).
Fig. 1.

Axial fluid-attenuated inversion recovery (FLAIR) MRI scans showing the scattered white matter lesions of leukoaraiosis (A) and confluent white matter hyperintensity (B) consistent with Binswanger's disease.
The focal neurobehavioral syndromes related to white matter lesions (Geschwind 1965) offered an important clue that disruption of distributed neural networks could be a general phenomenon underlying cognitive dysfunction, but the emergence of Alzheimer's disease (AD) as a major medical challenge (Katzman 1976) focused much investigation on the cerebral cortex, where the signature microscopic lesions of this disease are found (Katzman 1976; Querfurth and LaFerla 2010). As work on AD soon dominated the study of dementia, cognitive decline came to be widely conceptualized as a result of cortical disease (Geldmacher and Whitehouse 1996), and pathology in subcortical regions received relatively little attention.
The cerebral cortex, however, occupies only the outermost 1–3 mm of the brain, and many subcortical structures play a crucial role in cognition. The idea of subcortical dementia, which gained prominence in the 1970s (Albert et al. 1974; McHugh and Folstein 1975), proposed that diseases of the subcortical gray matter such as Parkinson's disease and Huntington's disease—with major pathology in the substantia nigra and caudate, respectively—feature cognitive loss because of disturbance in the timing and activation of cognitive functions. Whereas criticism was initially raised regarding the characterization of clinical and neuropathological features of subcortical dementia (Brown and Marsden 1988; Whitehouse 1986), the concept persists as clinically helpful in characterizing the phenomenology of dementing illness in relation to specific regions of pathological involvement (Bonelli and Cummings 2008; Ropper et al. 2014).
White matter is the other constituent of the subcortex. To illustrate the widespread distribution of myelinated systems, Fig. 2 shows a diffusion tensor imaging (DTI) depiction of normal white matter tracts. Comprising roughly half the total brain volume, white matter tracts course throughout the brain to unite cortical and subcortical gray matter regions into functional neuronal ensembles subserving cognition and emotion (Catani et al. 2012; Mesulam 1990; Schmahmann and Pandya 2006). White matter thus forms an essential component of the human connectome, the structural description of the human brain (Sporns et al. 2005). An important observation is that white matter has actually expanded more in evolution than gray matter, reflecting the requirement for more myelination as cortical expansion leads to greater distance between neuronal cell bodies (Zhang and Sejnowski 2000). The selective expansion of white matter, in turn, appears to contribute to the singular intelligence of Homo sapiens: while other intelligent animals such as whales and elephants actually have larger brains, with nearly as many cortical neurons, humans have the most extensive cerebral myelination (Roth and Dicke 2005). Thus human cognition depends as much on brain connectivity as on the activity of cortical neurons.
Fig. 2.
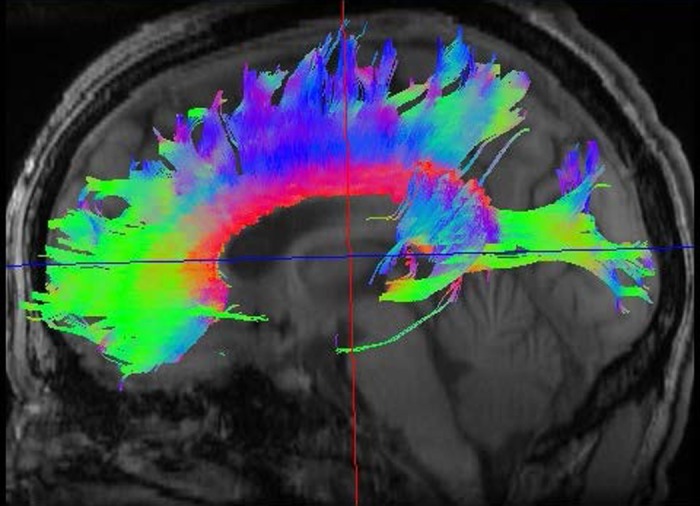
DTI scan of a normal adult brain showing 3 white matter tracts. Color coding permits the demonstration of tracts oriented within the right-left, anterior-posterior, and superior-inferior planes: red indicates the corpus callosum, green represents the arcuate fasciculus, and blue depicts the corticospinal tract. DTI image provided by M. Brown, Univ. of Colorado.
These considerations are consistent with prior observations on focal disconnection syndromes (Geschwind 1965) and further predict that diffuse lesions of the white matter can reliably produce cognitive deficits that may even reach the severity of dementia. Evidence for this notion is steadily mounting, based primarily on the correlation of cognitive loss with MRI white matter lesions (Filley 2012). A wide variety of white matter disorders can compromise cognition (Filley 2012), and because these disorders typically produce widespread or diffuse neuropathology, the parallel involvement of multiple distributed neural networks is presumed responsible. MRI has been transformative in this context, and a newer and particularly appealing neuroimaging technology being investigated is DTI, which enables the detailed depiction of white matter microstructure by assessing the diffusion of water along myelinated tracts (Zhang et al. 2012). The most common DTI parameters, fractional anisotropy (FA) and mean diffusivity (MD), are used to quantitate the degree of abnormal (isotopic, or random) water diffusion within white matter, but DTI is not yet capable of generating specific information as to whether axonal, myelin, or other pathology is involved (Zhang et al. 2012). This technique, however, holds much promise for the noninvasive identification and characterization of white matter pathology.
A convincing example of the potential for white matter neuropathology to produce dementia is toluene leukoencephalopathy (TL), a dementia syndrome resulting from intense and prolonged exposure to inhaled toluene as a result of solvent vapor abuse (Filley 2013; Filley et al. 1990, 2004; Hormes et al. 1986; Rosenberg et al. 1988). This common but underappreciated form of substance abuse can produce severe neurotoxicity because of daily exposure to inhaled toluene that may be pursued for many years (Hormes et al. 1986). The high lipophilicity of toluene accounts for its predilection to damage myelin, and toxic exposure produces widespread cerebral myelin loss with concomitant dementia (Rosenberg et al. 1988). The degree of cerebral white matter injury correlates with the severity of cognitive impairment (Filley et al. 1990), supporting the notion that cerebral white matter injury in TL can be sufficient to cause dementia. The white matter regions specifically involved in the pathogenesis of dementia appear to be the large tracts of the cerebral hemispheres, as intracortical myelin has been noted to be unaffected (Filley et al. 2004; Rosenberg et al. 1988) or relatively spared (Kornfeld et al. 1994). Indeed, a systematic review of 30 empirical studies found that toluene can produce dementia by preferentially affecting white matter relative to gray matter and that periventricular and subcortical white matter is most vulnerable (Yücel et al. 2008).
Based on the observations of TL, other neurological disorders with prominent white matter involvement have been observed to manifest a similar clinical picture (Schmahmann et al. 2008). In addition to leukotoxic injury as exemplified by TL, vascular, traumatic, demyelinative, inflammatory, infectious, metabolic, hydrocephalic, neoplastic, and genetic disorders can all damage brain white matter and produce similar cognitive effects (Filley 2012). Table 1 displays these 10 categories and an example of a specific disorder within each. Many of these disorders feature coexistent gray matter involvement, and in many cases an admixture of gray and white matter pathology likely accounts for cognitive dysfunction and dementia. Yet all the entities in Table 1 share a substantial burden of white matter pathology, and evidence is accumulating that supports a selective role of white matter injury in cognitive dysfunction and dementia (Schmahmann et al. 2008). To highlight the potential for white matter disorders to produce this often devastating outcome, the term “white matter dementia” (WMD) was introduced in 1988 to help organize thinking about white matter dysfunction in relation to cognitive decline (Filley et al. 1988).
Table 1.
Disorders with cognitive impairment and prominent white matter pathology
| Category | Disorder |
|---|---|
| Toxic | Toluene leukoencephalopathy |
| Vascular | Binswanger's disease |
| Traumatic | Traumatic brain injury |
| Demyelinative | Multiple sclerosis |
| Inflammatory | Systemic lupus erythematosus |
| Infectious | Human immunodeficiency virus infection |
| Metabolic | Vitamin B12 deficiency |
| Hydrocephalic | Normal-pressure hydrocephalus |
| Neoplastic | Gliomatosis cerebri |
| Genetic | Metachromatic leukodystrophy |
A unique profile of cognitive deficits has been seen to characterize WMD and reflects the physiological role of white matter in normal cognition (Filley 2012; Filley et al. 1988). The most important feature of WMD is cognitive slowing, an expected result of slowed impulse transmission in the brain resulting from damage to myelin, and in some cases axons as well. Other deficits include executive dysfunction, memory retrieval dysfunction, sustained attention deficit, visuospatial impairment, and various psychiatric disorders; conversely, language, extrapyramidal function, and procedural memory are relatively preserved (Filley 2012). This profile differs from that of both the cortical dementia of AD—in which amnesia, aphasia, apraxia, and agnosia are typical (Geldmacher and Whitehouse 1996)—and subcortical dementia, in which procedural memory is impaired (Saint-Cyr et al. 1998). Table 2 displays core cognitive differences between cortical, white matter, and subcortical dementia. These differences are most evident in the early stages of dementia, as with the progression of neuropathology all cognitive functions are eventually lost and the dementias become indistinguishable. Yet the clinical distinctions in Table 2 are not merely academic; the profile of deficits and strengths revealed at an early stage can be very helpful in diagnosis and treatment. From the available evidence, therefore, it appears that diffuse damage to the white matter can produce a distinctive dementia syndrome, recognition of which can be clinically valuable (Filley 2012; Filley et al. 1989; Lafosse et al. 2007; Schmahmann et al. 2008).
Table 2.
Cognitive features of cortical, white matter, and subcortical dementia
| Cortical | White Matter | Subcortical |
|---|---|---|
| Amnesia | Cognitive slowing | Executive dysfunction |
| Aphasia | Executive dysfunction | Cognitive slowing |
| Apraxia | Memory retrieval deficit | Memory retrieval deficit |
| Agnosia | Normal procedural learning | Impaired procedural learning |
It should be noted, however, that dementia is generally encountered only with a heavy burden of white matter lesions. In contrast, many individuals harbor lesser degrees of white matter neuropathology, and have a less severe clinical syndrome. For example, the relatively mild white matter lesion burden of LA shown in Fig. 1A produces cognitive slowing and executive dysfunction but not dementia, whereas the more advanced white matter disease shown in Fig. 1B often culminates in the progressive dementia of BD (Filley 2012). The early cognitive syndrome seen with mild white matter disease on conventional MRI may even result from involvement of what has been called the normal-appearing white matter (NAWM) (Filley 2012), which can be found with the use of advanced neuroimaging technologies such as MR spectroscopy (MRS) and DTI. This more subtle syndrome, which has been termed mild cognitive dysfunction (MCD; Kozora and Filley 2011), features a profile of cognitive slowing, inattention, and executive dysfunction, qualitatively similar to the pattern seen in WMD. MCD has clinical implications as a plausible precursor of WMD, which may appear later as the burden of white matter pathology worsens (Filley 2012). The constructs of WMD and MCD need more study, but thus far these ideas are consistent with what is known of white matter structure and function in health and disease. That is, the structural alteration of multiple white matter tracts by various neuropathologies can disrupt the normal function of these tracts in a manner that begins with the subtle cognitive disturbance of MCD and then, if unchecked, advances to the disabling neurobehavioral syndrome of WMD. In clinical neurology, MCD could provide a much-needed framework for the early identification of cognitive loss from white matter involvement that can potentially be treated before irreversible disability supervenes.
The Role of White Matter in Distributed Neural Networks
In general, white matter can be seen as providing for the transfer of information within distributed neural networks, while gray matter subserves information processing (Filley 2012). Accordingly, primary white matter damage results most prominently in cognitive slowing, whereas primary gray matter disease leads to more specific cognitive deficits, most apparent in the cortical dementia AD, which features amnesia, aphasia, apraxia, and amnesia related to regional neuronal and synaptic loss (Geldmacher and Whitehouse 1996; Table 2). However, as discussed below, suboptimal conduction velocity can also impair information processing; a good example of this phenomenon is optic nerve demyelination in acute optic neuritis that impairs visual acuity in multiple sclerosis (MS) (Thurtell et al. 2009). Neurons are the fundamental units of all networks, and distinctions between the operations of white and gray matter are not absolute. Disease may begin in the white matter or the gray, or progress from one tissue to the other, and complex interactions between the two are typical. As myelocentrism can be as limiting as corticocentrism, a balanced view of the representation of cognitive function is crucial. In this section, we consider physiological aspects of white matter underlying both information transfer and processing as a basis for appreciating the effects of myelin and axonal dysfunction.
An understanding of white matter function begins with the details of its microscopic anatomy. Figure 3 displays key aspects of white matter structure. Precision of spike time arrival is critical for information processing and synaptic plasticity. Temporal summation of postsynaptic membrane potentials from multiple synaptic inputs onto a dendrite requires millisecond precision to depolarize the postsynaptic neuron to threshold for initiating a spike. In addition, the strength of synapses can be increased or decreased by the degree of coincidence of synaptic input relative to postsynaptic action potential firing (spike timing-dependent synaptic plasticity; Dan and Poo 2006). That is, synapses that fire coincidently with or a few milliseconds before action potential firing in the postsynaptic neuron become strengthened, but synaptic strength is weakened in synapses firing a few milliseconds after the action potential. Despite the high temporal precision required for synaptic function in gray matter, conduction delays over long-range axons in the mammalian brain (and especially large-brained primates) are on the order of tens of milliseconds or longer. Thus cognitive function can be impaired by suboptimal conduction times through white matter tracts connecting synaptic relay points, and complete impulse conduction failure following demyelination is not required for functional impairment.
Fig. 3.

White matter structure. A: ∼40 μm × 60 μm × 60 μm volume of white matter (mouse optic nerve) reconstructed from serial block face electron microscopy, showing the composition of axons, myelin, astrocytes, oligodendrocytes, and vasculature. Changes in any of these components or in the tortuosity of the fibers will influence diffusional MRI, where the voxel volume is 100 μm × 100 μm × 100 μm in high-resolution MRI of rodents and typically 2 mm × 2 mm × 2 mm in human brain imaging. B: optic nerve axons in cross section showing multilaminar wrapping of compact myelin. C: 3-dimensional reconstruction of a node of Ranvier in mouse optic nerve from serial block face electron microscopy: myelin (purple), paranodal loops (salmon), perinodal astrocyte (green and blue). Inset: longitudinal slice through the node of Ranvier revealing the nodal gap (gray) between the paranodal loops of myelin containing high-density voltage-sensitive sodium channels. White, axon. D: illustration of myelinated axons, showing the multilaminar myelin sheath and electrogenic node of Ranvier. Scale bars: 10 μm in A, 1 μm in B–D.
Coordination of neural activity in large assemblies of neurons through phase and amplitude coupling is critical for cognition and consciousness (Buzsáki 2006; Singer 2009), and the conduction time between synaptic relay points is an important parameter affecting the coherence and frequency of brain wave activity. Brain waves, which represent the oscillations of neural activity at appropriate frequencies, and the coupling of oscillatory activity across long-distance cortical networks, are critical for cognitive function, gating of sensory information, and the binding of cognitive operations. White matter disease can disrupt normal electroencephalography (EEG) coherence patterns in association with impaired cognitive function (Nunez et al. 2015). Recent mathematical modeling predicts that conduction delays will have a profound effect on coupling oscillatory activity in the brain and that even small changes in myelination can produce substantial effects on coupling oscillatory activity in the brain (Pajevic et al. 2014). Indeed, this modeling predicts that biological mechanisms for adaptively modulating conduction delays must exist to prevent hypo- and hypersynchrony of coupled oscillators in the brain and that changes in myelin would be one of the most effective means for such plasticity (Pajevic et al. 2014). A number of neuropsychiatric disorders are associated with disruption in brain rhythms that may derive from changes in myelin (Mathalon and Sohal 2015; Uhlhaas and Singer 2015), including autism (Welsh and Ahn 2005), schizophrenia, obsessive-compulsive disorder, depression (Ferrarelli et al. 2012; Schulman et al. 2011; Xu et al. 2013), and dyslexia (Ucles et al. 2009). These examples highlight the possibility that idiopathic disorders with cognitive dimensions may be understood by considering myelin disturbances that result in network dysfunction. The connectopathy so produced may include both psychiatric and neurological dysfunction, as illustrated by the dysmyelinative disease metachromatic leukodystrophy (MLD), in which a typical progression from early psychosis to later dementia has been identified, presumably as a result of advancing white matter disease (Black et al. 2003).
In addition to myelin defects degrading impulse conduction, the myelin sheath provides metabolic support for axons (Fünfschilling et al. 2013; Lee et al. 2012). Demyelination and the resulting impaired trophic support for axons by oligodendrocytes can cause axonal and neuronal degeneration (Lee et al. 2012), further contributing to cognitive deficits in disorders such as MS (Koenig et al. 2014) and TBI (Armstrong et al. 2016). Axonal dysfunction in white matter disorders is well recognized to confer a worse prognosis than that implied by myelin damage alone (Medana and Esiri 2003; Trapp et al. 1998). Thus whereas myelin damage slows network activity, the superimposed loss of axons may preclude any neural conduction and render the network inoperative.
White Matter and Neurodegenerative Disease
An intriguing implication of the relationship between white matter and cognition is the etiopathogenesis of neurodegenerative dementia. The diseases within this group, the most common of which is AD, remain idiopathic, incurable, and a major threat to medicine and society. While the neurological disorders capable of producing WMD do not include those in the category of neurodegeneration, white matter dysfunction has in fact been correlated with cognitive dysfunction in one such disease, fragile X tremor ataxia syndrome (FXTAS) (Filley et al. 2015), and no a priori reason exists as to why idiopathic neurodegenerative diseases should necessarily implicate selective gray matter involvement. The idea that white matter dysfunction may prove important in understanding the origin of neurodegenerative disease warrants consideration.
The problem of AD remains particularly disturbing because, despite much effort over the past three decades, no disease-modifying treatment is available. Reflecting the corticocentric bias that exerts a powerful influence on the study of dementia (Parvizi 2009), AD is widely regarded as a cortical disease in which neuritic plaques and neurofibrillary tangles are entirely responsible for the dementia syndrome (Geldmacher and Whitehouse 1996; Querfurth and LaFerla 2010). Amyloid is thought to be the primary cause of cortical injury, followed in pathogenesis by tau neurotoxicity (Querfurth and LaFerla 2010). The amyloid hypothesis currently dominates the AD field, postulating the centrality of amyloid β42 (Aβ) and its oligomers in producing cortical damage, and this notion has stimulated a great deal of work on identifying agents that can treat the disease by ridding the brain of this presumably toxic protein (Hardy and Selkoe 2002). Although the pathogenetic role of Aβ in autosomal dominant early-onset AD offers support for the amyloid hypothesis (Querfurth and LaFerla 2010), it is sobering to consider that many normal elders harbor sufficient amyloid to qualify for the diagnosis of AD, that the normal function of amyloid and its relative amyloid precursor protein (APP) are unknown, and that all efforts to treat AD by targeting brain amyloid have thus far failed (Castellani and Perry 2014). Nevertheless, amyloid and tau may plausibly disrupt cortical structure, and ongoing studies of anti-Aβ therapies may yet find that amyloid reduction can be effective at some point in the course of AD. However, the idea has recently been offered that cortical pathology occurs as a downstream event in AD pathogenesis, and that early white matter injury triggers an adaptive response that produces the deposition of cortical amyloid and tau (Bartzokis 2011; Castellani and Perry 2014). In this context, recent MRI evidence has supported the notion that white matter hyperintensities are prominent early features of both late-onset (Brickman 2013) and dominantly inherited (Lee et al. 2016) AD. Moreover, DTI has shown that AD is associated with disrupted connectivity between various cortical and subcortical regions (Bozzali et al. 2010). Thus the “myelin model of AD” proposes that upstream events within the cerebral white matter, such as vascular disease and head trauma, initiate the deposition of amyloid and then tau as later end products (Bartzokis 2011). Indeed, evidence exists for both vascular (Marnane et al. 2016) and traumatic white matter (Scott et al. 2016) damage promoting amyloid deposition in the cerebral cortex. If the myelin model proves to be correct, preventive intervention long before the onset of cognitive dysfunction may be profoundly important.
Cholinergic deficit is a prominent feature of AD, and acetylcholinesterase inhibitors (AChEIs) such as donepezil and rivastigmine are used therapeutically in patients with AD. While AChEIs are known to act at the cholinergic synapse, the benefits of AChEI treatment in AD patients have also been linked to direct effects on oligodendrocytes and myelination (Bartzokis 2007). A paradoxical increase in white matter connectivity in the internal capsule of AD patients is associated with AChEI treatment (Bozzali et al. 2012), and donepezil-induced oligodendrocyte differentiation is inhibited by the nicotinic acetylcholine receptor antagonist mecamylamine but not by the muscarinic acetylcholine receptor antagonist scopolamine (Imamura et al. 2015). Interestingly, butyrylcholinesterase (BChE), an enzyme closely related to AChE, is found mainly in white matter and glia, and a BChE genotype influences white matter loss in AD (Lane and He 2013). BChE becomes expressed in association with cerebral cortical Aβ plaques (Darvesh 2016), linking acetylcholine to both gray and white matter pathology in AD.
Another neurodegenerative disease attracting much attention is chronic traumatic encephalopathy (CTE), in which repetitive mild TBI in athletes and soldiers is proposed to lead to a dementia syndrome consequent to cortical tauopathy that appears many years later (McKee et al. 2013). The highly visible publicity surrounding CTE should not obscure significant controversy over its very existence, as critics have pointed out that tauopathy has not been definitively shown to cause dementia and that the clinical features of CTE can be explained by alternative diagnoses such as depression and frontotemporal dementia (Iverson et al. 2015). However, because mild TBI regularly involves diffuse axonal injury (DAI) spread broadly throughout the white matter (Alexander 1995), it is plausible that the insult of repetitive concussion—or even repeated subconcussive blows (Bailes et al. 2013)—may activate a neuropathological cascade that manifests later as hyperphosphorylated tau accumulation in the cortex that produces dementia. In health, tau is a normal protein primarily associated with microtubules within brain axons, and observations of boxers with dementia pugilistica, a neurodegenerative disease very similar to CTE, have documented tau hyperphosphorylation as a result of repetitive mild TBI (Bartzokis 2011). Although tauopathy is emphasized as the centerpiece of CTE neuropathology, white matter injury is present in all stages of the disease (McKee et al. 2013), implying that DAI may be the trigger for progressive tau deposition. The progressive accumulation of abnormal tau that is thought to occur, possibly via a mechanism similar to prion propagation (Prusiner 2013), might therefore actually be a downstream phenomenon in CTE, similar to the proposed role of amyloid in the myelin model of AD (Bartzokis 2011). While CTE remains highly controversial, the possible long-term effects of traumatic white matter injury have far-reaching implications for both civilians and the military.
Prevention and Treatment of White Matter Lesions
Many opportunities can be considered for preventing and treating white matter lesions that can produce cognitive dysfunction and dementia. Medical intervention and public policy strategies merit attention because many white matter disorders are related to vascular risk factors, head injury, and intoxication with a variety of leukotoxins (Filley 2012). Standard medical care clearly plays a role, offering much benefit in terms of the control of hypertension, diabetes, hyperlipidemia, metabolic syndrome, cigarette smoking, and obesity and by preventing TBI and substance abuse. Attention to vascular risk factors is crucial, as these problems are strongly associated with white matter ischemia because of the selective atherosclerotic vulnerability of small penetrating arterioles irrigating cerebral white matter. Hypertension, for example, is a powerful risk factor for white matter ischemic lesions and stroke (Debette and Markus 2010), and a large prospective study found that antihypertensive therapy was not only effective for stroke prevention but also reduced the incidence of dementia (Forette et al. 2002), raising the possibility of an effect on ischemic white matter disease. Prospective studies evaluating the efficacy of primary cerebrovascular care and physical exercise are underway to address the hypothesis that targeting white matter ischemia can prevent dementia (Prins and Scheltens 2015). Disease-specific medical treatment can also contribute, as in the demyelinative disease MS, in which a variety of medications have been shown to reduce white matter disease burden (Wingerchuk and Carter 2014). A novel line of inquiry now attracting considerable attention is the microbiome, and recent findings in mice that normal gut microbiota promote normal prefrontal cortical myelination (Hoban et al. 2016) may have clinical relevance. Public policy intervention is also important, improving the health of white matter by policies intended to enhance access to medical care, enact seat belt and helmet laws, support education to encourage lifelong cognitive engagement, and promote physical fitness.
Another approach to prevention and treatment involves the exploitation of intrinsic white matter repair mechanisms. Plasticity in the brain, meaning the capacity of neurons to be modified as a result of experience, has traditionally been considered a function of the gray matter, but recent findings have found that white matter also exhibits this phenomenon (Fields 2008; Zatorre et al. 2012). Clinical and basic science investigations have been foundational in elaborating this novel idea (Fields 2015).
White matter plasticity has been shown in both normal individuals, such as piano players whose pyramidal tract integrity correlated with number of hours practiced (Bengtsson et al. 2005), and neurological patients, such as those with Broca's aphasia in whom right arcuate fasciculus volume increased as melodic intonation therapy improved language performance (Schlaug et al. 2009). Human and experimental animal studies using DTI have found that structural changes indicative of plasticity may occur in the fornix, as well as the hippocampus, within 2 h after engagement in a learning task (Hofstetter et al. 2013). Physical activity also appears to be salutary, as aerobic exercise has been found to improve white matter integrity in community-dwelling older adults (Voss et al. 2013). Better performance in inhibitory control is associated with white matter structure (increased FA) in prefrontal and frontostriatal tracts in healthy subjects (Forstmann et al. 2008) and in children with attention-deficit hyperactivity disorder (Liu et al. 2012). In animal studies, social isolation in mice can impair myelination of the forebrain (Liu et al. 2012; Makinodan et al. 2012), demonstrating experience-dependent plasticity of myelination. Motor skill learning on a treadmill requires oligodendrogenesis in adult mice (McKenzie et al. 2014). The exploitation of white matter plasticity may thus find clinical utility as a method of both maintaining normal tract function and repairing damaged tracts to restore normal cognition. New findings in white matter neurobiology may open new avenues for preventing or improving cognitive dysfunction, and for clinicians and policy makers, the prospect of making evidence-based recommendations such as playing a musical instrument, learning a new language, and staying fit could indeed be appealing.
In the last 10 years, considerable advances have been made in identifying molecular mechanisms regulating development of oligodendrocytes and myelination, clarifying how cognitive impairments associated with white matter disorders may be prevented and treated. Brain myelination is an extremely complex process, regulated by multiple intrinsic and extrinsic signals during development and throughout life. Myelination encompasses a broad scope of biological processes, including appropriate cell specification from progenitors, precise control of cell proliferation, selective survival and apoptosis of cells in the oligodendrocyte lineage, cell migration, differentiation, identification of the proper cellular targets to be myelinated (only appropriate axons are myelinated, not other cellular structures, and not all axons are myelinated), the formation of specialized axon-glial membrane complexes providing intercellular communication to induce myelin formation, intricate cytokinesis and synthesis of enormous amounts of lipid and membrane protein, and compaction of multilaminar axonal ensheathments into a compact spiral sheath of membrane that is unlike any other membrane specialization or cell-cell contact known.
In addition, because myelination requires the formation of nodes of Ranvier to enable saltatory conduction, an intricate paranodal apparatus is needed to segregate appropriate ion channels in the axonal membrane that are essential for impulse conduction. This segregation is achieved by precise subcellular localization of unique cell adhesion molecules—a trimolecular complex of Caspr, Contactin, and NF155, and others (Zonta et al. 2008)—that are organized into septate junctions that can be visualized only by high-resolution transmission electron microscopy. Thus many cellular signals are involved in the process of myelin formation, myelin maintenance, and remyelination, and this complexity increases vulnerability to various genetic and environmental factors that can cause abnormal myelination and dysfunction. As these signaling molecules become identified, however, new potential approaches to therapeutic intervention become available.
A number of membrane receptors are implicated in oligodendrocyte differentiation and myelination (Table 3). Many of these molecules are of potential relevance to neurological disorders affecting cognition. In hypothyroidism, for example, one of the most sought-after and commonly treated causes of dementia, cognitive deficits may in fact be related to cerebral white matter involvement because thyroid hormones are important for myelination (Barres et al. 1994). Many implications for the treatment of myelin damage are also apparent. Neuregulin is centrally involved in myelination and has been proposed as a possible myelin repair strategy in older people with ischemic or traumatic white matter injury (Bartzokis 2011). In younger people with MS, agents targeting LINGO-1 activity are being tested in clinical trials as a means of enhancing remyelination based on the role of LINGO-1 in inhibiting myelination by oligodendrocytes (Mi et al. 2005).
Table 3.
Molecules important for oligodendrocyte differentiation and myelination
| Cellular Function | Signaling Molecule | Reference |
|---|---|---|
| Cell adhesion | Contactin | Hu et al. 2003 |
| Cell adhesion | Integrins | Decker and ffrench-Constant 2004 |
| Cell adhesion | L1-CAM | Stevens et al. 1998 |
| Cell adhesion (extracellular matrix) | Tenascin-C | Frost et al. 1996; Garcion et al. 2001 |
| Growth factor | Insulin-like growth factor (IGF-I) | Carson et al. 1993; Zeger et al. 2007 |
| Growth factor | PDGF-α | Barres and Raff 1993 |
| Growth factor | Epidermal growth factor receptor (EGFR) | Aguirre et al. 2007 |
| Growth factor | FGF | Fumagalli et al. 2011 |
| Growth factor | BDNF | Fulmer et al. 2014; Miyamoto et al. 2015 |
| Receptor tyrosine kinase | Neuregulin 1-ErbB | Brinkmann et al. 2008 |
| Signal transduction | Neurogenic locus notch homolog protein (Notch 1), membrane receptor for Jagged and Delta | Genoud et al. 2002; Wang et al. 1998 |
| Signal transduction | Wnt (Frizzled family receptor, β-catenin translocation to nucleus) | Fancy et al. 2009 |
| G protein-coupled receptor | Gpr 17 | Chen et al. 2009 |
| Intercellular signaling | Leucine-rich repeat neuronal protein 1 (LINGO-1) | Aguirre et al. 2007 |
| Thyroid hormone | Thyroxine/triiodothyronine | Barres et al. 1994 |
| Neurotransmitter | ATP/adenosine | Ishibashi et al. 2006; Stevens et al. 2002 |
| Neurotransmitter | GABA | Zonouzi et al. 2015 |
| Neurotransmitter | Glutamate | Lundgaard et al. 2013; Wake et al. 2011, 2015 |
| Neurotransmitter | Acetylcholine | Abiraman et al. 2015; Deshmukh et al. 2013; Mei et al. 2014 |
Cell adhesion molecules and extracellular matrix molecules also influence oligodendrocyte differentiation and myelination, notably integrins (Decker and ffrench-Constant 2004), Tenascin-C (Frost et al. 1996; Garcion et al. 2001), and others. Interestingly, experiments in which dorsal root ganglion neurons are stimulated to fire action potentials in different frequencies show that mRNA abundance for different cell adhesion molecules on axons (NCAM, N-cadherin, and L1-CAM) is differentially regulated by specific patterns of action potential firing (Itoh et al. 1995, 1997). Moreover, specific frequencies of firing that lower expression of L1-CAM, an adhesion molecule that is necessary for myelin wrapping, reduce the amount of myelin that forms (Stevens et al. 1998). Mechanisms regulating myelination according to the pattern of action potential activity in neural circuits are especially important with respect to involvement of white matter plasticity in cognitive function.
Indeed, neurotransmitter signaling has been found to be particularly important in modifying development of myelinating glia in an activity-dependent manner. Among these neurotransmitters are ATP (Stevens and Fields 2000), adenosine (Ishibashi et al. 2006; Stevens et al. 2002), GABA (Zonouzi et al. 2015), glutamate (Lundgaard et al. 2013; Wake et al. 2011, 2015), and acetylcholine (Abiraman et al. 2015; Deshmukh et al. 2013; Mei et al. 2014). Neurotransmitters can be released along axons firing action potentials by both vesicular and nonvesicular release mechanisms (Fields 2011; Fields and Ni 2010). GABA (Zonouzi et al. 2015) and glutamate (Mangin et al. 2012) have been shown to influence oligodendrocyte proliferation and differentiation. Inhibitors of muscarinic acetylcholine receptors are effective in improving myelination in animal models of MS (Abiraman et al. 2015; Deshmukh et al. 2013; Mei et al. 2014) and in phase II MS clinical trials (Green 2016). In addition to these developmental effects, activation of NMDA and glutamate (mGluR) receptors on oligodendrocyte cell processes stimulates the formation of an axo-glial signaling complex that generates intracellular calcium transients in the oligodendrocyte cell process adjacent to the axon in response to action potential firing. This in turn initiates myelination selectively on electrically active axons by stimulating the local synthesis of myelin basic protein from mRNA transported from the cell body into individual processes of oligodendrocytes (Wake et al. 2011). A consequence of this activity-dependent signaling would be that axons that are electrically active would become preferentially myelinated. This has been confirmed by recent research showing that, when given a choice, oligodendrocytes will form functional contacts preferentially with electrically active axons. Oligodendrocytes then form myelin selectively on these electrically active axons while avoiding interaction with axons in the same vicinity in which glutamate release has been blocked by treatment with botulinum toxin (Wake et al. 2015). Studies in zebrafish show that although initiation of myelination is not affected, stabilization of the nascent myelin sheath is promoted by vesicle release from axons in zebrafish (Hines et al. 2015; Mensch et al. 2015). Activity-dependent specification of myelination would have significant effects on network function by the ∼50 times increase in conduction velocity of myelinated axons compared with unmyelinated axons.
To summarize, activity-dependent plasticity of both synapses in gray matter and myelin in white matter is increasingly appreciated as being of fundamental importance for central nervous system function and cognition. Several medical implications of activity-dependent myelination are apparent. First, the block of axonal conduction in demyelinated axons may impair remyelination, particularly in view of the many aspects of myelination that are stimulated by impulse activity, and axonal loss is one reason spontaneous remyelination is limited in patients with MS. Second, functional recovery after axotomy requires not only axon regeneration but also remyelination, suggesting that treatments to promote remyelination could promote recovery after axon injury. Finally, functional activity or electrical stimulation may be helpful therapeutically in promoting remyelination after demyelination and other white matter injury, as shown by the increased numbers of oligodendrocytes in rodents after spinal cord injury (Becker et al. 2010) and in cell culture (Gary et al. 2012) when electrical stimulation is delivered.
Summary and Future Directions
White matter provides the structural connectivity between gray matter regions throughout the brain, and the importance of myelinated systems in cognitive function derives support from both behavioral neurology and basic neuroscience. Complementing the information processing of the cerebral cortex, white matter tracts provide for the information transfer within the brain, enabling the rapid and efficient integrative capacity of neural systems necessary for the highly evolved cognitive operations of H. sapiens. Distributed neural networks are organized to mediate critical aspects of cognition such as attention, memory, language, visuospatial skills, and executive function, all of which depend critically on the structural connectivity provided by myelinated systems. Disrupted white matter has potentially profound effects, including dementia, and from this perspective a host of clinical and basic science implications become readily apparent.
Advances in this field can be facilitated by a focus on many unresolved questions. First, because white and gray matter lesions are often commingled in disease states, more specific study of the effects of white matter dysfunction in isolation would be useful, particularly as it evolves over time from subtle NAWM changes to grossly visible macrostructural lesions on conventional MRI. Second, research on early vascular and traumatic injuries of white matter, initially mild or even inapparent but much more significant in the years to follow, may lead to crucial insights into the etiopathogenesis and treatment of presently irreversible degenerative dementias. Finally, the prevention and treatment of white matter disease merits much more study, as there appear to be many opportunities to focus on this area with medical and public policy intervention and by exploiting the emerging field of white matter plasticity.
GRANTS
This work was supported by funds for intramural research from the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
C.M.F. and R.D.F. conception and design of research; C.M.F. analyzed data; C.M.F. interpreted results of experiments; C.M.F. prepared figu res; C.M.F. and R.D.F. drafted manuscript; C.M.F. edited and revised manuscript; C.M.F. approved final version of manuscript.
REFERENCES
- Abiraman K, Pol SU, O'Bara MA, Chen GD, Khaku ZM, Wang J, Thorn D, Vedia BH, Ekwegbalu EC, Li JX, Salvi RJ, Sim FJ. Anti-muscarinic adjunct therapy accelerates functional human oligodendrocyte repair. J Neurosci 35: 3676–3688, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci 10: 990–1002, 2007. [DOI] [PubMed] [Google Scholar]
- Albert ML, Feldman RG, Willis AL. The “subcortical dementia” of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 37: 121–130, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander MP. Mild traumatic brain injury: pathophysiology, natural history, and clinical management. Neurology 45: 1253–1260, 1995. [DOI] [PubMed] [Google Scholar]
- Anderson VC, Obayashi JT, Kaye JA, Quinn JF, Berryhill P, Riccelli LP, Peterson D, Rooney WD. Longitudinal relaxographic imaging of white matter hyperintensities in the elderly. Fluids Barriers CNS 11: 24, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RD, Mierzwa AJ, Marion CM, Sullivan GM. White matter involvement after TBI: clues to axon and myelin repair capacity. Exp Neurol 275: 328–333, 2016. [DOI] [PubMed] [Google Scholar]
- Babikian V, Ropper AH. Binswanger's disease: a review. Stroke 18: 2–12, 1987. [DOI] [PubMed] [Google Scholar]
- Bailes JE, Petraglia AL, Omalu BI, Nauman E, Talavage T. Role of subconcussion in repetitive mild traumatic brain injury. J Neurosurg 119: 1235–1245, 2013. [DOI] [PubMed] [Google Scholar]
- Barres B, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development 120: 1097–1108, 1994. [DOI] [PubMed] [Google Scholar]
- Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 361: 258–260, 1993. [DOI] [PubMed] [Google Scholar]
- Bartzokis G. Acetylcholinesterase inhibitors may improve myelin integrity. Biol Psychiatry 62: 294–301, 2007. [DOI] [PubMed] [Google Scholar]
- Bartzokis G. Alzheimer's disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging 32: 1341–1371, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker D, Gary DS, Rosenzweig ES, Grill WM, McDonald JW. Functional electrical stimulation helps replenish progenitor cells in the injured spinal cord of adult rats. Exp Neurol 222: 211–218, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson SL, Nagy Z, Skare S, Forsman L, Forssberg H, Ullén F. Extensive piano practicing has regionally specific effects on white matter development. Nat Neurosci 8: 1148–1150, 2005. [DOI] [PubMed] [Google Scholar]
- Black DN, Taber KH, Hurley RA. Metachromatic leukodystrophy: a model for the study of psychosis. J Neuropsychiatry Clin Neurosci 15: 289–293, 2003. [DOI] [PubMed] [Google Scholar]
- Bonelli RM, Cummings JL. Frontal-subcortical dementias. Neurologist 14: 100–107, 2008. [DOI] [PubMed] [Google Scholar]
- Bozzali M, Parker GJ, Serra L, Embleton K, Gili T, Perri R, Caltagirone C, Cercignani M. Anatomical connectivity mapping: a new tool to assess brain disconnection in Alzheimer's disease. Neuroimage 54: 2045–2051, 2011. [DOI] [PubMed] [Google Scholar]
- Bozzali M, Parker GJ, Spano B, Serra L, Giulietti G, Perri R, Magnani G, Marra C, Vita MG, Caltagirone C, Cercignani M. Brain tissue modifications induced by cholinergic therapy in Alzheimer's disease. Hum Brain Mapp 34: 3158–3167, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley WG., Jr Magnetic resonance imaging in the central nervous system: comparison with computed tomography. Magn Reson Annu 1986: 81–122, 1986. [PubMed] [Google Scholar]
- Brickman AM. Contemplating Alzheimer's disease and the contribution of white matter hyperintensities. Curr Neurol Neurosci Rep 13: 415, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann BG, Agarwal S, Sereda MW, Garratt AN, Müller T, Wende H, Stassart RM, Nawaz S, Humml C, Velanac V, Radyushkin K, Goebbels S, Fischer TM, Franklin RJ, Lai C, Ehrenreich H, Birchmeier C, Schwab MH, Nave KA. Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron 59: 581–595, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RG, Marsden CD. “Subcortical dementia”: the neuropsychological evidence. Neuroscience 25: 363–387, 1988. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Rhythms of the Brain. Oxford, UK: Oxford Univ. Press, 2006. [Google Scholar]
- Carson MJ, Behringer RR, Brinster RL, McMorris FA. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron 10: 729–740, 1993. [DOI] [PubMed] [Google Scholar]
- Casey BJ, Epstein JN, Buhle J, Liston C, Davidson MC, Tonev ST, Spicer J, Niogi S, Millner AJ, Reiss A, Garrett A, Hinshaw SP, Greenhill LL, Shafritz KM, Vitolo A, Kotler LA, Jarrett MA, Glover G. Frontostriatal connectivity and its role in cognitive control in parent-child dyads with ADHD. Am J Psychiatry 164: 1729–1736, 2007. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Perry G. The complexities of the pathology-pathogenesis relationship in Alzheimer disease. Biochem Pharmacol 88: 671–676, 2014. [DOI] [PubMed] [Google Scholar]
- Catani M, Dell'acqua F, Bizzi A, Forkel SJ, Williams SC, Simmons A, Murphy DG, Thiebaut de Schotten M. Beyond cortical localization in clinico-anatomical correlation. Cortex 48: 1262–1287, 2012. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wu H, Wang S, Koito H, Li J, Ye F, Hoang J, Escobar SS, Gow A, Arnett HA, Trapp BD, Karandikar NJ, Hsieh J, Lu QR. The oligodendrocyte-specific G protein-coupled receptor GR17 is a cell-intrinsic timer of myelination. Nat Neurosci 12: 1398–1406, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damasio AR. Behavioral neurology; research and practice. Semin Neurol 4: 117–119, 1984. [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev 86: 1033–1048, 2006. [DOI] [PubMed] [Google Scholar]
- Darvesh S. Butyrylcholinesterase as a diagnostic and therapeutic target for Alzheimer's disease. Curr Alzheimer Res 13: 1173–1177, 2016. [DOI] [PubMed] [Google Scholar]
- Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ 341: c3666, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker L, ffrench-Constant C. Lipid rafts and integrin activation regulate oligodendrocyte survival. J Neurosci 24: 3816–3825, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ, Padmanabhan K, Swoboda JG, Ahmad I, Kondo T, Gage FH, Theofilopoulos AN, Lawson BR, Schultz PG, Lairson LL. A regenerative approach to the treatment of multiple sclerosis. Nature 502: 327–332, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH. Dysregulation of the Wnt pathway inhibits timely myelination and remylenation iin the mammalian CNS. Genes Dev 23: 1571–1585, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarelli F, Sarasso S, Guller Y, Riedner BA, Peterson MJ, Bellesi M, Massimini M, Postle BR, Tononi G. Reduced natural oscillatory frequency of frontal thalamocortical circuits in schizophrenia. Arch Gen Psychiatry 69: 766–774, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD. White matter in learning, cognition and psychiatric disorders. Trends Neurosci 31: 361–370, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD. Nonsynaptic and nonvesicular ATP release from neurons and relevance to neuron-glia signaling. Semin Cell Dev Biol 22: 214–219, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD. A new mechanism of nervous system plasticity: activity-dependent myelination. Nat Rev Neurosci 16: 756–767, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Ni Y. Nonsynaptic communication through ATP release from volume-activated anion channels in axons. Sci Signal 3: ra73, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filley CM. The Behavioral Neurology of White Matter (2nd ed). New York: Oxford Univ. Press, 2012. [Google Scholar]
- Filley CM. Toluene abuse and white matter: a model of toxic leukoencephalopathy. Psychiatr Clin North Am 36: 293–302, 2013. [DOI] [PubMed] [Google Scholar]
- Filley CM, Brown MS, Onderko K, Ray M, Bennett RE, Berry-Kravis E, Grigsby J. White matter disease and cognitive impairment in FMR1 premutation carriers. Neurology 84: 2146–2152, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filley CM, Franklin GM, Heaton RK, Rosenberg NL. White matter dementia: clinical disorders and implications. Neuropsychiatry Neuropsychol Behav Neurol 1: 239–254, 1988. [Google Scholar]
- Filley CM, Halliday W, Kleinschmidt-DeMasters BK. The effects of toluene on the central nervous system. J Neuropathol Exp Neurol 63: 1–12, 2004. [DOI] [PubMed] [Google Scholar]
- Filley CM, Heaton RK, Nelson LM, Burks JS, Franklin GM. A comparison of dementia in Alzheimer's disease and multiple sclerosis. Arch Neurol 46: 157–161, 1989. [DOI] [PubMed] [Google Scholar]
- Filley CM, Rosenberg NL, Heaton RK. White matter dementia in chronic toluene abuse. Neurology 40: 532–534, 1990. [DOI] [PubMed] [Google Scholar]
- Forette R, Seux ML, Staessen J, Thijs L, Babarskiene MR, Babeanu S, Bossini A, Fagard R, Gil-Extremera B, Laks T, Kobalava Z, Sarti C, Tuomilehto J, Vanhanen H, Webster J, Yodfat Y, Birkenhäger WH, Systolic Hypertension in Europe Investigators. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch Intern Med 162: 2046–2052, 2002. [DOI] [PubMed] [Google Scholar]
- Forstmann BU, Jahfari S, Scholte HS, Wolfensteller U, van den Wildenberg WP, Ridderinkhof KR. Function and structure of the right interior frontal cortex predict individual differences in response inhibition: a model-based approach. J Neurosci 28: 9790–9796, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost E, Kiernan BW, Faissner A, ffrench-Constant C. Regulation of oligodendrocyte precursor migration by extracellular matrix: evidence for substrate-specific inhibition of migration by tenascin-C. Dev Neurosci 18: 266–273, 1996. [DOI] [PubMed] [Google Scholar]
- Fulmer CG, VonDran WM, Stillman AA, Huang Y, Hampstead BL, Dreyfus CF. Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J Neurosci 34: 8186–8196, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Daniele S, Lecca D, Lee PR, Parravicini C, Fields RD, Rosa P, Antonucci F, Verderio C, Trincavelli ML, Bramanti P, Martini C, Abbracchio MP. Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. J Biol Chem 286: 10593–10604, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, Nave KA. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485: 517–521, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Dupree JL, Nave KA, Bansal R. Fibroblast growth factor receptor signaling in oligodendrocytes regulates myelin sheath thickness. J Neurosci 32: 6631–6641, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcion E, Faissner A, ffrench-Constant C. Knockout mice reveal a contribution of the extracellular matrix molecule tenascin-C to neural precursor proliferation and migration. Development 128: 2485–2496, 2001. [DOI] [PubMed] [Google Scholar]
- Gary DS, Malone M, Capestany P, Houdayer T, McDonald JW. Electrical stimulation promotes the survival of oligodendrocytes in mixed cortical cultures. J Neurosci Res 90: 72–83, 2012. [DOI] [PubMed] [Google Scholar]
- Geldmacher DS, Whitehouse PJ. Evaluation of dementia. N Engl J Med 335: 330–336, 1996. [DOI] [PubMed] [Google Scholar]
- Genoud S, Lappe-Siefke C, Goebbels S, Radtke F, Aguet M, Scherer SS, Suter U, Nave KA, Mantei N. Notch1 control of oligodendrocyte differentiation in the spinal cord. J Cell Biol 158: 709–718, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind N. Disconnexion syndromes in animals and man. Brain 88: 237–294, 585–644, 1965. [DOI] [PubMed] [Google Scholar]
- Green AJ. Assessment of clemastine fumarate as a remyelinating agent in multiple sclerosis. ClinicalTrials.gov Identifier: NCT02040298. Last verified, April 2016. https://clinicaltrials.gov/ct2/show/NCT02040298?term=NCT02040298&rank=1. [Google Scholar]
- Hachinski VC, Potter P, Merskey H. Leuko-araiosis. Arch Neurol 44: 21–23, 1987. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356, 2002. [DOI] [PubMed] [Google Scholar]
- Hines JH, Ravanelli AM, Schwindt R, Scott EK, Appel B. Neuronal activity biases axon selection for myelination in vivo. Nat Neurosci 108: 332–336, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban AE, Stilling RM, Ryan FJ, Shanahan F, Dinan TG, Claesson MJ, Clarke G, Cryan JF. Regulation of prefrontal cortex myelination by the microbiota. Transl Psychiatry 6: e774, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstetter S, Tavor I, Tzur Moryosef S, Assaf Y. Short-term learning induces white matter plasticity in the fornix. J Neurosci 33: 12844–12850, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormes JT, Filley CM, Rosenberg NL. Neurologic sequelae of chronic solvent vapor abuse. Neurology 36: 698–702, 1986. [DOI] [PubMed] [Google Scholar]
- Hu QD, Ang BT, Karsak M, Hu WP, Cui XY, Duka T, Takeda Y, Chia W, Sankar N, Ng YK, Ling EA, Maciag T, Small D, Trifonova R, Kopan R, Okano H, Nakafuku M, Chiba S, Hirai H, Aster JC, Schachner M, Pallen CJ, Watanabe K, Xiao ZC. F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell 115: 163–175, 2003. [DOI] [PubMed] [Google Scholar]
- Imamura O, Arai M, Dateki M, Ogata T, Uchida R, Tomoda H, Takishima K. Nicotinic acetylcholine receptors mediate donepezil-induced oligodendrocyte differentiation. J Neurochem 135: 1086–1098, 2015. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. Astrocytes promote myelination in response to electrical impulses. Neuron 49: 823–832, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Ozaki M, Stevens B, Fields RD. Activity-dependent regulation of N-cadherin in DRG neurons: differential regulation of N-cadherin, NCAM, and L1 by distinct patterns of action potentials. J Neurobiol 33: 735–748, 1997. [DOI] [PubMed] [Google Scholar]
- Itoh K, Stevens B, Schachner M, Fields RD. Regulated expression of the neural cell adhesion molecule L1 by specific patterns of neural impulses. Science 270: 1369–1372, 1995. [DOI] [PubMed] [Google Scholar]
- Iverson GL, Gardner AJ, McCrory P, Zafonte R, Castellani RJ. A critical review of chronic traumatic encephalopathy. Neurosci Biobehav Rev 56: 276–293, 2015. [DOI] [PubMed] [Google Scholar]
- Katzman R. The prevalence and malignancy of Alzheimer disease. A major killer. Arch Neurol 33: 217–218, 1976. [DOI] [PubMed] [Google Scholar]
- Kertesz A, Black SE, Tokar G, Benke T, Carr T, Nicholson L. Periventricular and subcortical hyperintensities on magnetic resonance imaging. “Rims, caps, and unidentified bright objects.” Arch Neurol 45: 404–408, 1988. [DOI] [PubMed] [Google Scholar]
- Koenig KA, Sakaie KE, Lowe JJ, Lin J, Stone L, Bermel RA, Beall EB, Rao SM, Trapp BD, Phillips MD. Hippocampal volume is related to cognitive decline and fornicial diffusion measures in multiple sclerosis. Magn Reson Imaging 32: 354–358, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld M, Moser AB, Moser HW, Kleinschmidt-DeMasters B, Nolte K, Phelps A. Solvent vapor abuse leukoencephalopathy. Comparison to adrenoleukodystrophy. J Neuropathol Exp Neurol 53: 389–398, 1994. [DOI] [PubMed] [Google Scholar]
- Kozora E, Filley CM. Cognitive dysfunction and white matter abnormalities in systemic lupus erythematosus. J Int Neuropsychol Soc 17: 385–392, 2011. [DOI] [PubMed] [Google Scholar]
- Lafosse JM, Corboy JR, Leehey MA, Seeberger LC, Filley CM. MS vs. HD: can white matter and subcortical gray matter pathology be distinguished neuropsychologically? J Clin Exp Neuropsychol 29: 142–154, 2007. [DOI] [PubMed] [Google Scholar]
- Lane RM, He Y. Butyrylcholinesterase genotype and gender influence Alzheimer's disease phenotype. Alzheimers Dement 9: e1–e73, 2013. [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487: 443–448, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TL, Marcus DS, Fagan AM, Goate A, Fox NC, Cairns NJ, Holtzman DM, Buckles V, Ghetti B, McDade E, Martins RN, Saykin AJ, Masters CL, Ringman JM, Ryan NS, Förster S, Laske C, Schofield PR, Sperling RA, Salloway S, Correia S, Jack C Jr, Weiner M, Bateman RJ, Morris JC, Mayeux R, Brickman AM, Dominantly Inherited Alzheimer Network. White matter hyperintensities are a core feature of Alzheimer's disease: evidence from the dominantly inherited Alzheimer network. Ann Neurol 79: 929–939, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Dietz K, DeLoyht JM, Kelkar D, Kaur J, Vialou V, Lobo MK, Dietz DM, Nestler EJ, Dupree J, Casaccia P. Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nat Neurosci 15: 1621–1623, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgaard I, Luzhynskaya A, Stockley JH, Wang Z, Evans KA, Swire M, Volbracht K, Gautier HO, Franklin RJ, Ffrench-Constant C, Attwell D, Káradóttir RT. Neuregulin and BDNF induce a switch to NMDA receptor-dependent myelination by oligodendrocytes. PLoS Biol 11: e1001743, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinodan M, Rosen KM, Ito S, Corfas G. A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 337: 1357–1360, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangin JM, Li P, Scafidi J, Gallo V. Experience-dependent regulation of NG2 progenitors in the developing barrel cortex. Nat Neurosci 15: 1192–1194, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marnane M, Al-Jawadi OO, Mortazavi S, Pogorzelec KJ, Wang BW, Feldman HH, Hsiung GY, Alzheimer's Disease Neuroimaging Initiative. Periventricular hyperintensities are associated with elevated cerebral amyloid. Neurology 86: 535–543, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matejko AA, Ansari D. Drawing connections between white matter and numerical and mathematical cognition: a literature review. Neurosci Biobehav Rev 48: 35–52, 2015. [DOI] [PubMed] [Google Scholar]
- Mathalon DH, Sohal VS. Neural oscillations and synchrony in brain dysfunction and neuropsychiatric disorders: it's about time. JAMA Psychiatry 72: 840–844, 2015. [DOI] [PubMed] [Google Scholar]
- McHugh PR, Folstein MF. Psychiatric syndromes of Huntington's chorea. In: Psychiatric Aspects of Neurologic Disease, edited by Benson DF, Blumer D. New York: Grune and Stratton, 1975. [Google Scholar]
- McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC. The spectrum of disease in chronic traumatic encephalopathy. Brain 136: 43–64, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, Richardson WD. Motor skill learning requires active central myelination. Science 346: 318–322, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain 126: 515–530, 2003. [DOI] [PubMed] [Google Scholar]
- Mei F, Fancy SP, Shen YA, Niu J, Zhao C, Presley B, Miao E, Lee S, Mayoral SR, Redmond SA, Etxeberria A, Xiao L, Franklin RJ, Green A, Hauser SL, Chan JR. Micropillar arrays as high-throughput screening platform for therapeutics in multiple sclerosis. Nat Med 20: 954–960, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensch S, Baraban M, Almeida R, Czopka T, Ausborn J, El Manira A, Lyons DA. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat Neurosci 18: 628–630, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM. Large-scale neurocognitive networks and distributed processing for attention, memory, and language. Ann Neurol 28: 597–613, 1990. [DOI] [PubMed] [Google Scholar]
- Mi S, Miller RL, Lee X, Scott Shulag-Morskaya ML, Shao ZS, Chang J, Thill G, Levesque M, Zhang M, Hession C, Sah D, Trapp B, He Z, Jung V, McCoy JM, Pepinsky RB. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci 8: 745–751, 2005. [DOI] [PubMed] [Google Scholar]
- Miyamoto N, Maki T, Shindo A, Liang AC, Maeda M, Egaw N, Itoh K, Lo EK, Lok J, Ihara M, Arai K. Astrocytes promote oligodendrogenesis after white matter damage via brain-derived neurotrophic factor. J Neurosci 35: 14002–14008, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez PL, Srinivasan R, Fields RD. EEG functional connectivity, axon delays and white matter disease. Clin Neurophysiol 126: 110–120, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajevic S, Basser PJ, Fields RD. Role of myelin plasticity in oscillations and synchrony of neuronal activity. Neuroscience 276: 135–147, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke 28: 652–659, 1997. [DOI] [PubMed] [Google Scholar]
- Parvizi J. Corticocentric myopia: old bias in new cognitive sciences. Trends Cogn Sci 13: 354–359, 2009. [DOI] [PubMed] [Google Scholar]
- Prins ND, Scheltens P. White matter hyperintensities, cognitive impairment and dementia: an update. Nat Rev Neurol 11: 157–165, 2015. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 47: 601–623, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med 362: 329–344, 2010. [DOI] [PubMed] [Google Scholar]
- Ropper AH, Samuels MA, Klein JP. Adams and Victor's Principles of Neurology (10th ed). New York: McGraw-Hill, 2014. [Google Scholar]
- Rosenberg GA, Wallin A, Wardlaw JM, Markus HS, Montaner J, Wolfson L, Iadecola C, Zlokovic BV, Joutel A, Dichgans M, Duering M, Schmidt R, Korczyn AD, Grinberg LT, Chui HC, Hachinski V. Consensus statement for diagnosis of subcortical small vessel disease. J Cereb Blood Flow Metab 36: 6–25, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NL, Kleinschmidt-DeMasters BK, Davis KA, Dreisbach JN, Hormes JT, Filley CM. Toluene abuse causes diffuse central nervous system white matter changes. Ann Neurol 23: 611–614, 1988. [DOI] [PubMed] [Google Scholar]
- Roth G, Dicke U. Evolution of the brain and intelligence. Trends Cogn Sci 9: 250–257, 2005. [DOI] [PubMed] [Google Scholar]
- Saint-Cyr JA, Taylor AE, Lang AE. Procedural learning and neostriatal dysfunction in man. Brain 111: 941–959, 1988. [DOI] [PubMed] [Google Scholar]
- Schlaug G, Marchina S, Norton A. Evidence for plasticity in white-matter tracts of patients with chronic Broca's aphasia undergoing intense intonation-based speech therapy. Ann NY Acad Sci 169: 385–394, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmahmann JD, Pandya DN. Fiber Pathways of the Brain. New York: Oxford Univ. Press, 2006. [Google Scholar]
- Schmahmann JD, Smith EE, Eichler RS, Filley CM. Cerebral white matter: neuroanatomy, clinical neurology, and neurobehavioral correlates. Ann NY Acad Sci 1142: 266–309, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's disease. Acta Neuropathol 101: 518–524, 2001. [DOI] [PubMed] [Google Scholar]
- Schmidt R, Seiler S, Loitfelder M. Longitudinal change of small-vessel disease-related brain abnormalities. J Cereb Blood Flow Metab 36: 26–39, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman JJ, Cancro R, Lowe SF, Lu KD, Walton KD, Llinás RR. Imaging of thalamocortical dysrhythmia in neuropsychiatry. Front Hum Neurosci 5: 69, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott G, Ramlackhansingh AF, Edison P, Hellyer P, Cole J, Veronese M, Leech R, Greenwood RJ, Turkheimer FE, Gentleman SM, Heckemann RA, Matthews PM, Brooks DJ, Sharp DJ. Amyloid pathology and axonal injury after brain trauma. Neurology 86: 821–828, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer W. Distributed processing and temporal codes in neuronal networks. Cogn Neurodyn 3: 189–196, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporns O, Tononi G, Kötter R. The human connectome: a structural description of the human brain. PLoS Comput Biol 1: e42, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Fields RD. Response of Schwann cells to action potentials in development. Science 287: 2267–2271, 2000. [DOI] [PubMed] [Google Scholar]
- Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 36: 855–868, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Tanner S, Fields RD. Control of myelination by specific patterns of neural impulses. J Neurosci 18: 9303–9311, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanridag O, Kirshner HS. Magnetic resonance imaging and CT scanning in neurobehavioral syndromes. Comparative neuroradiologic findings. Psychosomatics 28: 517–528, 1987. [DOI] [PubMed] [Google Scholar]
- Taveggia C, Thaker P, Petrylak A, Caporaso GL, Toews A, Falls DL, Einheber S, Salzer JL. Type III neuregulin-1 promotes oligodendrocyte myelination. Glia 56: 284–294, 2008. [DOI] [PubMed] [Google Scholar]
- Thurtell MJ, Bala E, Yaniglos SS, Rucker JC, Peachey NS, Leigh RJ. Evaluation of optic neuropathy in multiple sclerosis using low-contrast visual evoked potentials. Neurology 73: 1849–1857, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338: 278–285, 1998. [DOI] [PubMed] [Google Scholar]
- Ucles P, Mendez M, Garay J. Low-level defective processing of non-verbal sound in dyslexic children. Dyslexia 15: 72–85, 2009. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Oscillations and neuronal dynamics in schizophrenia: the search for basic symptoms and translational opportunities. Biol Psychiatry 77: 1001–1009, 2015. [DOI] [PubMed] [Google Scholar]
- Ullén F. Is activity regulation of late myelination a plastic mechanism in the human nervous system? Neuron Glia Biol 5: 29–34, 2009. [DOI] [PubMed] [Google Scholar]
- Voss MW, Heo S, Prakash RS, Erickson KI, Alves H, Chaddock L, Szabo AN, Mailey EL, Wójcicki TR, White SM, Gothe N, McAuley E, Sutton BP, Kramer AF. The influence of aerobic fitness on cerebral white matter integrity and cognitive function in older adults: results of a one-year exercise intervention. Hum Brain Mapp 34: 2972–2985, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Lee PR, Fields RD. Control of local protein synthesis and initial events in myelination by action potentials. Science 333: 1647–1651, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Ortiz FC, Woo DH, Lee PR, Angulo MC, Fields RD. Nonsynaptic junctions on myelinating glia promote preferential myelination of electrically active axons. Nat Commun 6: 7844, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhovd KB, Johansen-Berg H, Káradóttir RT. Unraveling the secrets of white matter—bridging the gap between cellular, animal and human imaging studies. Neuroscience 276: 2–13, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, Barres BA. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 21: 63–75, 1998. [DOI] [PubMed] [Google Scholar]
- Welsh JP, Ahn ES, Placantonakis DG. Is autism due to brain desynchronization? Int J Dev Neurosci 23: 253–263, 2005. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ. The concept of subcortical and cortical dementia: another look. Ann Neurol 19: 1–6, 1986. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc 89: 225–240, 2014. [DOI] [PubMed] [Google Scholar]
- Yücel M, Takagi M, Walterfang M, Lubman DI. Toluene misuse and long-term harms: a systematic review of the neuropsychological and neuroimaging literature. Neurosci Biobehav Rev 3 2: 910–926, 2008. [DOI] [PubMed] [Google Scholar]
- Xu T, Stephane MM, Parhi KK. Multidimensional analysis of the abnormal neural oscillations associated with lexical processing in schizophrenia. Clin EEG Neurosci 44: 135–143, 2013. [DOI] [PubMed] [Google Scholar]
- Zatorre RJ, Fields RD, Johansen-Berg H. Plasticity in gray and white: neuroimaging changes in brain structure during learning. Nat Neurosci 15: 528–536, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeger M, Popken G, Zhang J, Xuan S, Lu QR, Schwab MH, Nave KA, Rowitch D, D'Ercole AJ, Ye P. Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normal in vivo oligodendrocyte development and myelination. Glia 55: 400–411, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Aggarwal M, Mori S. Structural insights into the rodent CNS via diffusion tensor imaging. Trends Neurosci 35: 412–421, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Sejnowski TJ. A universal scaling law between gray matter and white matter of cerebral cortex. Proc Natl Acad Sci USA 97: 5621–5626, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL, Harvey L, Sun D, Hübner CA, Cull-Candy SG, Farrant M, Gallo V. GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat Neurosci 18: 674–682, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta B, Tait S, Melrose S, Anderson H, Harroch S, Higginson J, Sherman DL, Brophy PJ. Glial and neuronal isoforms of Neurofascin have distinct roles in the assembly of nodes of Ranvier in the central nervous system. J Cell Biol 181: 1169–1177, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]