

Abstract
Purpose.
Traumatic brain injury (TBI) frequently leads to chronic visual dysfunction. The purpose of this study was to investigate the effect of TBI on retinal ganglion cells (RGCs), and to test whether treatment with the novel neuroprotective compound P7C3-S243 could prevent in vivo functional deficits in the visual system.
Methods.
Blast-mediated TBI was modeled using an enclosed over-pressure blast chamber. The RGC physiology was evaluated using a multielectrode array and pattern electroretinogram (PERG). Histological analysis of RGC dendritic field and cell number were evaluated at the end of the study. Visual outcome measures also were evaluated based on treatment of mice with P7C3-S243 or vehicle control.
Results.
We show that deficits in neutral position PERG after blast-mediated TBI occur in a temporally bimodal fashion, with temporary recovery 4 weeks after injury followed by chronically persistent dysfunction 12 weeks later. This later time point is associated with development of dendritic abnormalities and irreversible death of RGCs. We also demonstrate that ongoing pathologic processes during the temporary recovery latent period (including abnormalities of RGC physiology) lead to future dysfunction of the visual system. We report that modification of PERG to provocative postural tilt testing elicits changes in PERG measurements that correlate with a key in vitro measures of damage: the spontaneous and light-evoked activity of RGCs. Treatment with P7C3-S243 immediately after injury and throughout the temporary recovery latent period protects mice from developing chronic visual system dysfunction.
Conclusions.
Provocative PERG testing serves as a noninvasive test in the living organism to identify early damage to the visual system, which may reflect corresponding damage in the brain that is not otherwise detectable by noninvasive means. This provides the basis for developing an earlier diagnostic test to identify patients at risk for developing chronic CNS and visual system damage after TBI at an earlier stage when treatments may be more effective in preventing these sequelae. In addition, treatment with the neuroprotective agent P7C3-S243 after TBI protects from visual system dysfunction after TBI.
Keywords: multielectrode array, retinal ganglion cell, pattern ERG, blast injury, neuroprotection
This paper describes abnormal RGC physiology after blast-mediated TBI and tests a neuroprotective compound to prevent in vivo RGC decrements.
Introduction
Blast-mediated traumatic brain injury (TBI) is a highly-prevalent cause of progressive lifelong impairment in visual and central nervous system (CNS) functioning.1 Visual and ocular symptoms include increased light sensitivity, retinopathy, optic neuropathy, dysfunctional optic motility, and visual field loss, while other CNS symptoms consist of cognitive and motor disruption.2–5 Patients with mild TBI also frequently report nonspecific visual difficulties that, while not detected on routine clinical eye exam or testing, ultimately lead to significantly decreased visual quality of life.6 These visual consequences of TBI result from early damage to the eye and brain, and likely herald future progression to more serious chronic damage. Thus, there is a significant need to develop novel testing strategies to objectively identify patients experiencing early damage to their visual system after TBI, to guide therapeutic intervention and monitor therapeutic response, as well as to develop new pharmacologic treatments that will prevent damage to the CNS and visual system after TBI.
Recent reports, including ours, have demonstrated that the key consequences of human TBI also are present in rodent models.7,8 For example, we have reported recently that blast-mediated TBI in mice results in visual-, cognitive-, motor-, and mental illness–related phenotypes that recapitulate those sustained by patients after blast-exposure.8,9 We also have shown that retinal ganglion cells (RGCs) are highly susceptible to damage and death after blast-mediated TBI.8 The RGCs are CNS neurons located near the inner surface of the retina that extend axons to form the optic nerve, which transmits visual information from the retina to other regions of the brain for higher-level processing. Because RGC function is measured easily in living animals and people, we hypothesized that these neurons might provide a novel and noninvasive target for monitoring early CNS damage after blast-mediated TBI.
Here, we showed that acquisition of early RGC deficits after blast-mediated injury correlates with a readily measurable RGC response to light in living animals, the pattern electroretinogram (PERG). The PERG is a measure of RGC function determined by the summation of RGC depolarization in response to patterned light exposure. Specifically, the amplitude of voltage change is measured noninvasively at the cornea or eyelid in response to an alternating, reversing black and white grid.10–12 Conducted with the subject in prone position with eyes at the level of the heart, known as neutral position PERG, reflects the function of RGCs under homeostatic conditions. However, tilting the mouse 60° into the head down position with its eyes below the level of the heart, termed provocative position PERG, measures the function of RGCs under increased IOP.
We hypothesized that provocative PERG might enable detection of subclinical damage to the visual system that is not normally observed on routine clinical eye exam. If so, then this would provide an opportunity to identify at-risk patients at an early time point for rehabilitative intervention, before the onset of chronic irreversible damage to the visual system. In addition to identifying early visual system dysfunction after blast-mediated TBI, we also sought to evaluate whether initiation of treatment at this time with the neuroprotective agent P7C3-S243 would serve to preserve visual system function after TBI. The P7C3 class of molecules originally was identified through a target-agnostic in vivo discovery screen for small, drug-like molecules capable of safely increasing the net magnitude of hippocampal neurogenesis in adult mice.13 The vast majority of newborn hippocampal neurons die before structurally and functionally integrating into hippocampal circuitry, and the original P7C3 molecule was identified as an aminopropyl carbazole that blocked death of newborn hippocampal neurons without altering their rate of proliferation.13–16 The P7C3 compounds also potently block cell death of mature neurons located in other regions of the central and peripheral nervous system, as shown through testing in animal models of stress-associated depression,17 Parkinson's disease,15,18 amyotrophic lateral sclerosis,19 peripheral nerve crush injury,20 and TBI.9,21 In all of these cases, protection of neurons from cell death by P7C3 compounds correlated with preserved neurological function. Recently, the molecular mechanism of action of the P7C3 class of compounds has been identified as direct activation of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in nicotinamide adenine dinucleotide salvage.22 Characterization of the new highly active derivative known as P7C3-S243 also has been reported recently.15 This agent was shown recently to preserve cognitive and motor function after blast-mediated TBI, and here we tested whether it also could protect the visual system as well after this injury.
Materials and Methods
Animals
All animal studies were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and were approved by the Iowa City Department of Veterans Affairs and The University of Iowa Institutional Animal Care and Use Committees. Male 12-week-old C57BL/6J mice, or mice with YFP expressing RGCS, B6.Cg-Tg(Thy1-YFP)HJrs/J, were used in this study (95 total mice).
Blast Injury Induction
An enclosed blast chamber was used, one half of which was pressurized, with a 13-cm opening between the sides of the chamber, as described previously.8 A Mylar membrane (Mylar A, 0.00142 gauge; Country Plastics, Ames, IA, USA) was placed over the opening on the pressurized side of the chamber. The unpressurized side of the tank contained a padded polyvinyl chloride (PVC) protective restraint for positioning of an anesthetized mouse 30 cm from the Mylar membrane. Compressed air was pumped into the pressurized side of the tank to 20 psi, at which point the membrane ruptured and created a blast wave. Mice were anesthetized with a combination of ketamine (0.03 mg/g, intraperitoneal [IP]) and xylazine (0.005 mg/g, IP) and positioned within the blast chamber with the left side of the head and eye oriented toward the source of the blast wave. To ensure that the primary effect of the blast wave was at the level of the head, only the head of the mouse was exposed to the blast wave, with the rest of the body shielded. The head of the mouse was allowed to move freely and was not fixed in position. Immediately following exposure to the blast wave, mice were placed on a heating pad to facilitate recovery from general anesthesia and to prevent hypothermia. Xylazine anesthesia was reversed with Yohimbine chloride (0.001 mg/g, IP) to speed the recovery from anesthesia. Control mice used in this study were anesthetized, treated with analgesic, and placed in the blast chamber, but did not receive a blast exposure. Blasted and sham-blasted mice received analgesic via subcutaneous injection (0.1 mL/20 g body weight) of buprenorphine (0.003 mg/mL) immediately after the blast or sham-blast, respectively.
Pattern Evoked Electroretinography
Pattern-evoked electroretinography (PERG) was used to objectively measure the function of RGCs by recording the amplitude of the PERG waveform following TBI. Mice were anesthetized with a combination of ketamine (0.03 mg/g, IP) and xylazine (0.005 mg/g, IP), and then placed on a heated recording table to maintain body temperature. Neutral position PERG responses were evoked using alternating, reversing, and black and white vertical stimuli delivered on a monitor with a Roland Consult ERG system (Roland Consult, Brandenburg, Germany). To record the PERG response, commercially available mouse corneal gold ring electrodes were used (S&V Technologies AG, Hennigsdorf, Germany). A reference needle electrode was placed at the base of the head, and a ground electrode was placed at the base of the tail to complete the circuit. Each animal was placed at the same fixed position in front of the monitor to prevent recording variability due to animal placement. Stimuli (18° radius visual angle subtended on full field pattern, 2 reversals per second, 300 averaged signals with cut off filter frequencies of 1 to 30 Hz, 98% contrast, 80 cd/m2 average monitor illumination intensity) were delivered under mesopic conditions without dark adaptation to exclude the possible effect of direct photoreceptor-derived evoked responses. A diffuser placed over the pattern on the monitor also did not elicit a measurable evoked potential, further ensuring that the electrical responses were elicited from RGCs. The PERG response was evaluated by measuring the amplitude (peak to trough) of the waveform, as we have described previously.8,23 The PERG response was recorded at baseline, and 1, 4, and 16 weeks following blast injury.
Provocative PERG testing was performed 4 weeks after blast exposure by placing the mouse in a 60° head-down position as described previously24 using a custom-made PERG system using the same stimulation parameters as described above, with 372 averaged signals (Jorvec, Miami, FL, USA). Mice were left in this position for 15 minutes before initiation of the recording and remained in this position throughout the duration of the recording.
Spectral-Domain Optical Coherence Tomography (SD-OCT)
We performed SD-OCT analysis using a Spectralis SD-OCT (Heidelberg Engineering, Vista, CA, USA) imaging system coupled with a 25 diopter (D) lens for mouse ocular imaging (Heidelberg Engineering). Mice were anesthetized with a combination of ketamine (0.03 mg/g, IP) and xylazine (0.005 mg/g, IP) and placed on a heating pad to maintain body temperature. Pupils were dilated using a 1% tropicamide solution. The cornea was moisturized with a saline solution, which was applied every 20 to 30 seconds. Volume scans (49 line dense array) positioned directly over the optic nerve head were performed to quantify the RGC complex thickness (RGC bodies + axons + dendrites). Scans were analyzed by an individual masked to the treatment of the mouse in the superior retina, along a line raster scan placed approximately 150 μm from the peripapillary region. All scans were analyzed by excluding blood vessels from the RGC complex thickness calculation, since blood vessels in rodents are almost completely embedded in the RNFL.25
Treatment With P7C3-S243
Mice were injected intraperitoneally twice daily in divided doses for a total of 10 mg/kg/d P7C3-S243, which was synthesized according to previously published methods.15 The P7C3-S243 was solubilized in 2.5% dimethyl sulfoxide (DMSO) diluted in 10% Koliphor P 188, a thickening agent, (Sigma-Aldrich, St. Louis, MO, USA) and brought to a final working volume by diluting with 5% dextrose. Treatment was initiated immediately after blast-injury, within 5 minutes of removing the mouse from the chamber and continued daily for the duration of the experiment.
Ocular P7C3-S243 Concentration Pharmacokinetic Analysis
The C57BL/6J mice dosed IP with P7C3-S243 at 10 mg/kg were used for pharmacokinetic (PK) analysis of total vitreous humor drug levels. The P7C3-S243 was formulated for administration as described above. At various times post dose, animals were sacrificed with an inhalation overdose of CO2 and vitreous humor was collected. Plasma was prepared from blood by centrifugation at 9300g for 10 minutes at 4°C and was stored along with the brain tissue and vitreous humor at −80°C until analysis. Brain homogenates were prepared by homogenizing the tissues in a 3-fold volume of PBS. Total brain homogenate volume was estimated as volume of PBS added plus volume of brain in milliliters. Vitreous humor homogenates were prepared by homogenizing the humor in a 5-fold volume of PBS. Total vitreous homogenate volume was estimated as volume of PBS added plus volume of humor in milliliters. Compound was extracted from vitreous humor by a simple protein precipitation method involving the addition of 100 μL of acetonitrile containing formic acid and the internal standard (IS) n-benzylbenzamide (IS final concentration = 25 ng/mL; final formic acid concentration 0.1%) to 50 μL of vitreous humor. The samples were vortexed for 15 seconds, incubated at room temp for 10 minutes and spun twice for 5 minutes at 4°C 16,100g in a standard microcentrifuge. The supernatant then was analyzed by liquid chromatography tandem mass spectrometry (LC/MS/MS). Compound was extracted from plasma and brain homogenates by passage over a Phenomenex (Torrence, CA, USA) Phree Phospholipid Removal tabbed 1 mL tube to remove phospholipids as well as proteins. Briefly, 100 μL of brain homogenate or 50 μL of plasma was loaded in the center of the Phree tube on a vacuum manifold. Either 300 μL (brain) or 400 μL (plasma) of acetonitrile, containing formic acid and the n-benzylbenzamide IS to yield the final concentrations listed above, then was added and the mixture pipeted gently twice to mix. A vacuum was applied and the flow through collected. An additional 300 μL (brain) or 400 μL (plasma) of acetonitrile solution was passed over the tube and the flow through added to the original material. The flow through was vortex mixed and 700 μL taken for analysis by LC-MS/MS.
Nontreated mice were used to collect blank brain homogenate for standards and quality controls (QCs). Standard curves were prepared by addition of the appropriate compound to plasma, brain homogenate, or vitreous humor homogenate and processing as described above. A value of 3× above the signal obtained in the blank plasma or brain or vitreous humor homogenate was designated the limit of detection (LOD). The limit of quantitation (LOQ) was defined as the lowest concentration at which back calculation yielded a concentration within 20% of the theoretical value and above the LOD signal. The LOQ for plasma was 5 ng/mL, and for brain and vitreous humor it was 1 ng/mL. Compound levels were monitored by LC/MS/MS using an AB/Sciex (Framingham, MA, USA) 3200 Qtrap mass spectrometer coupled to a Shimadzu (Columbia, MD, USA) Prominence LC. The P7C3A20 was detected with the mass spectrometer in multiple reaction monitoring (MRM) mode by following the precursor to fragment ion transition 507.0 → 204.1 (positive mode; M+H+). The IS, n-benzylbenzamide, was monitored using a 212.1 → 91.1 transition (positive mode; M+H+). An Agilent (Santa Clara, CA, USA) XDB C18 column (50 × 4.6 mm, 5 μ packing) was used for chromatography with the following conditions: Buffer A, 0.1% formic acid in water; Buffer B, 0.1% formic acid in methanol, 0 to 1 at 0 minutes 50% B, 1.0 to 1.5 minutes gradient to 100% B, 1.5 to 3.0 minutes 100% B, 3.0 to 3.1 minutes gradient to 0% B, 3.1 to 4.5 minutes 0% B.
Multi-Electrode Array Analysis of Single RGC Physiology
Spontaneous activity and light-evoked responses of RGCs were recorded from control mice and from mice exposed to blast 1, 5, and 16 weeks after blast injury using multielectrode array (MEA) techniques as reported previously.26 Dark-adapted retinas from the left eye, directly exposed to blast, (4 retinas for each time point) were positioned with the ganglion cell layer (GCL) down onto a multielectrode array with 10-μm contacts spaced 200-μm apart (Multichannel Systems, Reutlingen, Germany). The array was mounted on a microscope (Axioplan; Zeiss, Gottingen, Germany) and perfused with 37°C oxygenated Ringer medium at 2.5 to 4 mL/min (in mM: 124 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 22 glucose). Preparations were stabilized for 1 hour before testing (Bionic Technologies, Salt Lake City, UT, USA). Full-field 1-second flash stimuli were displayed at 5-second intervals, and responses were averaged over 20 trials. Action potential (spike) waveforms accepted for analysis were ≥60 μV in amplitude and ≥1.85 times the root mean square of the background signal. Responses from different cells on the same electrode were distinguished by supervised principle components analysis (Offline Sorter; Plexon, Dallas, TX, USA). Accepted data demonstrated a refractory period of >1 ms (typically 2–5 ms) and did not display recognizable noise patterns (60 Hz, >10 kHz transients, or sinusoidal oscillations). One to three cells were identified per microelectrode by this procedure. For each recorded cell, the spontaneous mean firing rate was determined as the number of spikes divided by the length of the recording period. The activity maps show the average spontaneous firing rates for the RGCs on each electrode plotted as contour maps. Light-evoked responses were quantified as the total number of action potentials occurring within 1 second following a light transition (ON or OFF), after subtracting the background spontaneous firing rate over the 1 second preceding the stimulus. These responses were averaged over 10 or 20 trials, and amplitude was expressed as a net mean firing rate (spikes/s). For such calculations, ganglion cells were included if the net response to the maximum stimulus (34 μW/cm2 retinal irradiance) was at least 1 spike/s. The transiency index is the fraction of each response occurring in the first 200 ms after stimulus ON-set or OFF-set. Means ± SEM are presented for the parameters above. Given their nonnormal distribution, however, central values for many of these parameters also were expressed as medians and were compared using the Mann-Whitney U test in Graphpad Prism (GraphPad Software, Inc., La Jolla, CA, USA).
Quantification of Retinal Whole Mounts
Perfusion fixed eyes (4% paraformaldehyde in PBS) were flat-mounted and stained with hematoxylin and eosin (H&E) using standard methodology. Using a light microscope (BX52; Olympus, Tokyo, Japan) equipped with a digital camera (DP72; Olympus), 2 nonoverlapping ×20 images were collected at the peripheral, mid peripheral, and central positions of each of 4 retinal petals, yielding a total of 24 images per retina. Retinal regions were defined as peripheral (outer edge of the petal to one-third of the distance to the optic nerve head), central (outside edge of optic nerve head to one-third the distance to the outer edge of the petal), or mid peripheral (area between the peripheral and central retina). Quantitative analyses of cell counts on image files were performed using a semiautomated macro for Image J Imaging Software (National Institutes of Health [NIH], Bethesda, MD, USA). Nuclei in the GCL were identified in color images using a threshold segmentation approach followed by quantitative analysis using the particle analyzer tools available in ImageJ. Inclusion criteria for morphology were set to include GCL-nuclei within a size range of 5 to 125 μm2 and circularity of 0.69 to 1.00. Nuclei falling outside these parametric ranges, including the vast majority of vascular endothelial cell nuclei, were excluded from counts. For cells included in the counts, cells with nuclei within an area of 25 to 49.9 um2 were classified as displaced amacrine cells (DACs) and all other cells were classified as RGCs.
Dendritic Analysis of RGCs
Whole-mounted Thy1-YFP mouse retinas were visualized using the Carl Zeiss LSM 710 with Argon 514 nm laser (Carl Zeiss Meditec, Dublin, CA, USA) configured for EYFP at 20% laser line attenuator transmission. Z-stack images were captured with Zen 2011 imaging software at ×40 magnification in 2 × 2 tile scans. Dendritic fields were traced and their area determined as a polygon around the distal tips of the dendrites using Aperio ImageScope and Aperio Digital Slide Studio software (Aperio Group LLC, Sausalito, CA, USA).
IOP Measurement
The IOP was measured in the neutral (0°) position after mice were anesthetized using a Tonolab rebound tonometer (Icare, Helsinki, Finland). Mice subsequently were placed in the 60° head-down position for 15 minutes and the IOP was again recorded. Five measurements were taken for each eye in each position and averaged to give one measurement per position.
Statistics
The specific statistical tests used for each method are detailed in each section. Statistical analysis was conducted using GraphPad Prism Software v6.0.
Results
Blast Exposure Induces Temporally Bimodal Deficits in Neutral Position PERG
To begin, we measured PERG amplitude in living mice after blast-mediated TBI to determine the response of RGC activity to blast over time. In sham-blast (sham) mice, baseline neutral position PERG amplitude was 20.58 ± 1.10 μV (n = 11). The PERG measurements in these same mice at 1 (22.46 ± 1.58 μV) and 4 (21.83 ± 1.37 μV) weeks later showed no significant differences in amplitude from baseline (Fig. 1a). Sham mice at 16 weeks after sham-blast exhibited a small, but significant increase in PERG response (24.78 ± 1.45 μV, P < 0.05) (Fig. 1a), reflecting a slightly suppressed baseline response, since artificial increases in PERG function have not been reported. By contrast, blast-injury mice exhibited a decrease in PERG response from 23.85 ± 0.96 μV (n = 12) at baseline to 19.92 ± 1.42 μV 1 week after blast (P < 0.05). The PERG response of blast mice was restored temporarily, however, 4 weeks after blast injury (23.60 ± 1.21 μV), and then observed to be substantially decreased 16 weeks after blast injury (18.20 ± 0.54 μV, P < 0.01, Fig. 1b). Together, these results demonstrated that neutral position PERG deficits observed acutely after injury temporarily resolve by 4 weeks after blast injury, followed by a latent period of temporary recovery. A subsequent decrement in the PERG response then is observed 16 weeks after induction of TBI.
Figure 1.

Blast-induced functional RGC damage in C57BL/6J mice. Sham mice not exposed to a blast wave did not display any evidence of functional RGC deficits at 1 week (1w), 4 weeks (4w), or 16 weeks (16w) after blast exposure. (A). The PERG analysis after injury revealed deficits at 1w and 16w (B). All PERG recordings are measured in the neutral position. *Significant difference compared to baseline recordings using ANOVA with Dunnett's posttest; P < 0.05.
Blast Exposure Induces Permanent Thinning of the RGC Layer
To determine whether decreased PERG in blast mice might be related to the structural integrity of the retina, we used SD-OCT to analyze in vivo thickness of the RGC complex. Sham mice had a baseline RGC-complex thickness of 68.39 ± 0.92 μm, which remained unchanged 1 (67.07 ± 0.73), 4 (67.68 ± 0.44 μm), and 16 (68.95 ± 0.88 μm; Figs. 2a, 2b) weeks later. By contrast, blast injury elicited an acute decrease in RGC complex thickness, from 66.92 ± 0.66 μm at baseline to 63.18 ± 0.66 μm 1 week after exposure (P < 0.05). Comparable levels 4 weeks (63.77 ± 0.43 μm, P < 0.05) and 16 weeks later also were observed (64.44 ± 0.83 μm, P < 0.05; Figs. 2c, 2d, repeated measures ANOVA with Dunnett's multiple comparison test). These results suggested a considerable amount of functional plasticity in the visual system after blast, as the structural measures of RGC morphology decrease permanently and immediately following blast exposure, yet neutral position PERG response varies in a bimodal fashion with a latent period of functional recovery.
Figure 2.

Mice exposed to sham-blast did not display any decrease in the RGC complex thickness over time (A, B). Mice exposed to blast-mediated TBI had a decreased RGC complex thickness beginning 1 week after blast-induction, which persisted for the duration of the experiment (C, D). *Significant difference compared to baseline recordings using ANOVA with Dunnett's posttest; P < 0.05. The SD-OCT images in sham (B) and blast-exposed (D) mice are from 16 weeks after induction.
Functional Plasticity of RGC Spontaneous Activity and Response to Light After Blast Injury
To investigate the apparent mismatch between normal retinal functioning reflected by neutral-position PERG and reduced retinal thickness detected by SD-OCT at the 4-week time point after blast-injury, we used in vitro MEA analysis in acutely dissected retina. This technique measures spontaneous and light-evoked responses of individual RGCs. The RGCs elicit action potentials spontaneously and randomly in the dark. We observed that the average spontaneous firing rate of RGCs in the dark was significantly increased at 1 (P < 0.0001, n = 220 RGCs), 5 (P < 0.0001, n = 317 cells), and 16 (P = 0.0002, n = 273 cells) weeks after blast-injury, compared to sham mice (n = 312 cells; Figs. 3, 4). This spontaneous hyperactivity was evident not only in the average RGC firing rate (Fig. 4a), but also in the cumulative firing rate of RGCs (the proportion of all RGCs firing above a given rate; Figs. 4b, 4c).
Figure 3.

The activity maps show increase in the spontaneous activity at all times probed post blast. The average spontaneous firing rates of RGC's per electrode were plotted as contour maps for control retinas (a) and retinas collected at 1 (b), 5 (c), and 16 (d) weeks post blast. The average rates in spikes/s are plotted on a logarithmic (log10) scale and the color scale is included in the figure. Circles in the figure indicate the position of the optic nerve head.
Figure 4.
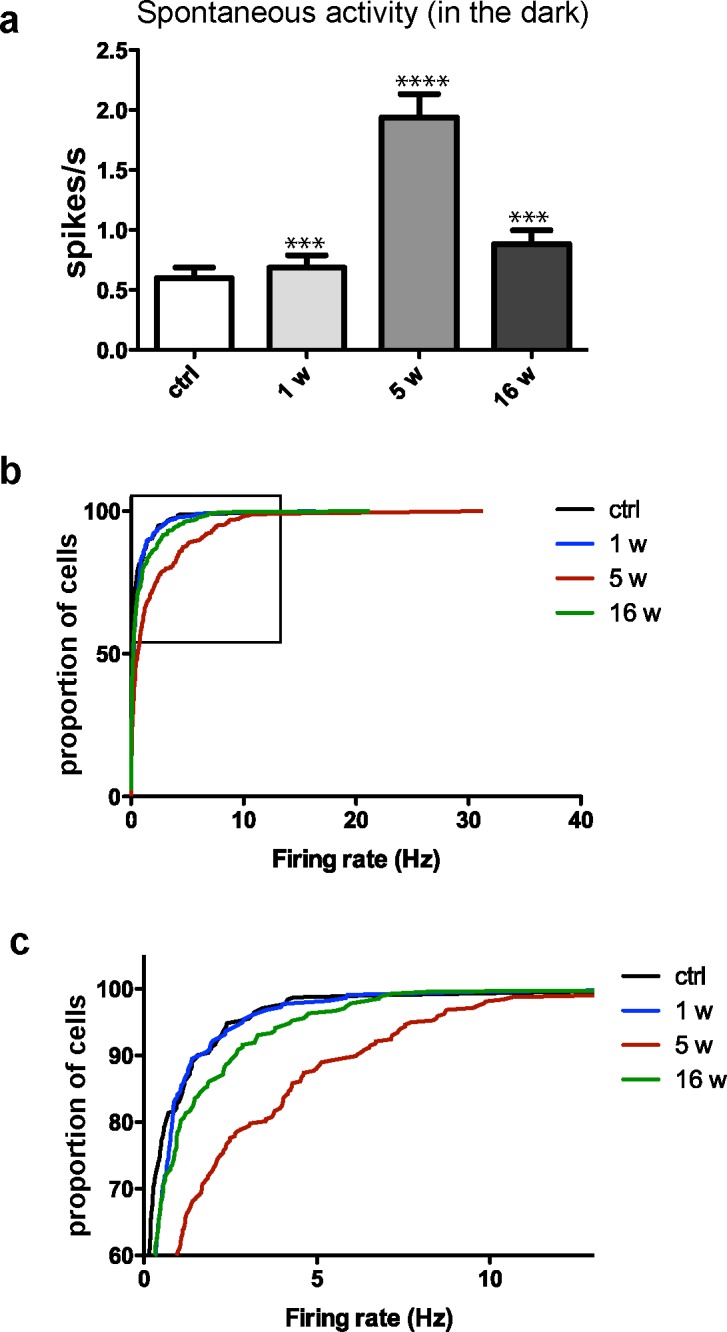
Spontaneous activity of RGCs increases after exposure to blast. (a) Average spontaneous firing rates of RGCs in control (ctrl) retinas and in retinas exposed to blast at 1 (1 w), 5 (5w), and 16 (16w) weeks post blast. (b) Cumulative frequency histograms showing the distribution of mean average rates for ctrl and blast-exposed RGC's. (c) Enlarged insert from (b). Statistical analysis performed by Mann-Whitney U test. Significant difference compared to controls; ***P < 0.001, ****P < 0.0001.
The RGCs are the first neurons in the visual transduction cascade that fire an action potential in response to light, and are divided into “ON RGCs” that respond to onset of light and “OFF RGCs” that respond to offset of light. The responses of these classes of RGCs to full-field light flashes in acute retinal whole-mounts were significantly greater 5 weeks after blast-exposure (n = 368 cells) when compared to acute retinal preparations from sham mice (n = 247 cells; Figs. 5a, 5b; total response, P < 0.0001; ON, P = 0.0185; OFF, P < 0.0001; Mann-Whitney U test). However, no statistically significant differences from sham mice were present in blast-injured retina 1 or 16 weeks after injury (Figs. 5a, 5b), indicating that RGC light responsiveness increases temporarily following an initial latent period after blast trauma, and then returns to normal. Closer analysis reveals that the change in light responsiveness 5 weeks after injury includes, on average, a decrease in the relative proportion of responses elicited by light onset (ON/total response ratio; P = 0.0093 versus RGCs of control retinas), with a corresponding increase in OFF/total ratio; P = 0.0093, Figure 5c. That is, for analysis of this population of RGCs, the relative responsiveness to light onset versus offset is more evenly balanced at this interval after blast injury, whereas at other time points there is a disparity in the ON versus OFF response of RGCs.
Figure 5.

Light-evoked responses change in a time-dependent manner after exposure to blast. (a) The total median response amplitude for RGCs in control (ctrl) retinas and at 1 (1w), 5 (5w), and 16 (16w) weeks after blast-exposure. (b) Average amplitude of response to maximal full-field stimulus of RGCs from control retinas and in retinas at 1 w, 5w, and 16w post blast. (c) The ON/Total ratio decreased significantly at 5w post blast, while the OFF/Total ratio increased significantly, and no significant difference is present at 1w and 16w post-blast exposure. (d) The transiency index of the response to the ON-set of light increased significantly at 1w and 5w post-blast, while the transiency index for the OFF-set of light increased 1w post blast. Statistical analysis performed by Mann-Whitney U test. Significant difference compared to controls; *P < 0.05, **P < 0.01, ****P < 0.0001.
We also have observed changes in the duration of RGC responses to the onset and offset of light after blast injury by means of transiency index (TI), which is the fraction of each response occurring in the first 200 ms after stimulus onset or offset.27–29 The mean TI of ON and OFF responses increased significantly at 7 days post blast (ON, P < 0.0001; OFF, P = 0.0011), while ON responses were more sustained (lower TI, P < 0.0001) 5 weeks after blast (Figs. 5d, 6). Examination of the size and structure of RGC receptive fields after injury, estimated using random white noise checkerboard stimulus and reverse correlation techniques,27–29 showed that some RGC receptive fields were substantially larger 5 weeks after injury (Fig. 7), in contrast to what was observed at 1 or 16 weeks post blast.
Figure 6.

Responses to maximal full-field light flash (34 μV/cm2, 1 second) shown as peristimulus time histograms (PSTHs) change in a time-dependent manner after exposure to blast. Stimulus timing is indicated by the white background. The response length in ON cells decreases 1 week after blast, increases 5 weeks after blast, and is normal at 16 weeks after blast compared to controls (a). The OFF response length is decreased 1 week post blast (b).
Figure 7.

Exposure to blast affects the receptive fields of RGCs as determined by multielectrode array analysis. Examples of normal (a) and altered (b) receptive fields, 5 weeks after blast-mediated injury.
Blast Exposure Results in RGC Loss but Does Not Affect DACs
To determine whether blast-mediated TBI elicits pure RGC dysfunction without cell loss, as opposed to retinal dysfunction concomitant with RGC death, retinas from blast and sham mice were collected 16 weeks after induction, whole mounted, stained, and analyzed using an automated, objective counting method that we have developed and manually verified to recognize the nuclear features of RGCs and DACs. Because the spatial pattern of RGC death is an important hallmark in other optic neuropathies, such as glaucoma, the density of RGCs was quantified in three distinct spatial regions (central, mid peripheral, and peripheral) of each retina. The density of all RGCs in sham mice was calculated for central (7556 ± 284.3 cells/mm2, mean ± SEM), mid peripheral (6679 ± 267.9 cells/mm2), and peripheral (6269 ± 235.9 cells/mm2) regions. A significant decrease in RGC density was observed in all regions (central, 6471 ± 287.3 cells/mm2; mid peripheral, 5737 ± 220.0 cells/mm2, and peripheral, 5236 ± 207.0 cells/mm2; P < 0.05, 2-tailed heteroscedastic t-test, Figs. 8a–c) 16 weeks following blast exposure. The spatial pattern of RGC loss occurred throughout the entire retina, which suggests that RGC loss after blast-mediated TBI is distinct from other forms of optic neuropathies that are typically associated with focal patterns of RGC loss.
Figure 8.

Quantification of RGC loss after blast-mediated TBI. A significant decrease in the density of RGCs was observed 16 weeks after blast-mediated TBI, compared to mice receiving sham-injury (a). We have shown that blast-injury does not affect the density of DACs in the RGC layer (b). There was a significant shift in the density of RGCs:DACs in blast-exposed eyes (c). *Significant difference as determined by 2-tailed Students t-test (a, d, e).
We next sought to determine whether other cell types within the retina also were affected by blast injury, such as DACs. The DACs are amacrine cells that normally reside in the RGC layer of the retina and function to provide input to RGCs, mediate lateral interactions between RGCs, and modify RGC receptive fields. Specifically, we quantified the density of DACs and examined the ratio of RGCs:DACs to determine whether damage to the RGC layer affected these other types of cells. The density of DACs in sham mice (9904 ± 366.9 cells/mm2) was not significantly different than in blast mice (9517 ± 251.5 cells/mm2, P = 0.3958, Student's t-test, Fig. 8d). However, the ratio of the total number of RGCs/DACs in blast mice (0.81 ± 0.06) was significantly lower than in sham mice (0.96 ± 0.02, P = 0.0347, Student's t-test, Fig. 8e), indicating a selective loss of RGCs.
Blast Injury Results in Dendritic Rearrangement of RGCs
The area covered by the dendritic arborization of RGCs was calculated in mice exposed to sham- or blast-injury, 1, 4, or 16 weeks after exposure (Fig. 9). No change was observed in the dendritic field area of blast mice at 1-week (29,648 ± 2268 μm2, n = 128 cells), 4-week (36,770 ± 2849 μm2, n = 82 cells), or 16-week (25,885 ± 2279 μm2, n = 98 cells) time points, compared to sham mice (28,951 ± 3391 μm2, n = 66 cells). Average dendritic field area, however, was significantly larger at 4 weeks compared to 16 weeks after blast-injury (P < 0.05, ANOVA with Tukey's multiple comparison test) within the blast-injury group. Taken together, we observed an increased dendritic field at 4 weeks after blast injury that corresponds with elevation of the MEA response at 5 weeks, despite apparently reduced thickness of the RGC layer around this time. At 16 weeks after injury, however, functional measures of PERG and dendritic field analysis were reduced, compatible with progression of significant neuronal damage and RGC loss.
Figure 9.

Dendritic arborization of individual RGCs. There is no significant change in the dendritic arborization of RGCs 1 (1w), 4 (4w), or 16 (16w) weeks after induction of blast-mediated TBI. There was, however, a significant change in the dendritic arborization between 4 and 16 weeks post injury. *P < 0.05, ANOVA with Tukey's multiple comparison test.
Provocative PERG Testing Reveals RGC Dysfunction During the Temporary Recovery Latent Period That Correlates With Abnormal RGC Physiology
Provocative PERG testing, recorded after tilting the mouse's head down to increase IOP, can be used to reveal latent RGC dysfunction30,31 that is not otherwise detectable by routine clinical eye exam or neutral position PERG testing. Importantly, this technique can be applied to rodents and people. We measured provocative PERG 4 weeks after blast-injury (Fig. 10A) based on the hypothesis that abnormal RGC firing activity 5 weeks after blast may reflect latent RGC dysfunction. Furthermore, we sought to test whether the neuroprotective agent P7C3-S243, previously reported to preserve cognitive and motor dysfunction after blast-mediated TBI9 and other neurodegenerative diseases,13,15,18,19 also could prevent RGC dysfunction after TBI. Because of the blood-retina barrier, we conducted ocular pharmacokinetic analysis after intraperitoneal injection of P7C3-S243 (10 mg/kg). The maximum vitreous concentration of P7C3-S243 occurred 90 minutes after injection at a concentration of 234.2 ng/g. These results indicated that P7C3-S243 is able to cross the blood–retina barrier.
Figure 10.

Provocative PERG analysis and neuroprotection of RGCs with P7C3-S243 4 weeks after blast (A). Artificially increasing the IOP reveals RGC abnormalities in blast-exposed mice that are not apparent in sham mice not exposed to a blast-wave. The IOP was evaluated in mice exposed to blast injury treated with vehicle or P7C3-S243. There was a significant increase in IOP in the head-down provocative test position in the vehicle- and P7C3-S243–treated groups (P < 0.001) 4 weeks after blast exposure (B). There was no significant difference in the IOP when vehicle- and P7C3-S243–treated mice were compared. Treatment with P7C3-S243 (10 mg/kg daily) prevented RGC deficits, which were present in mice not treated with P7C3-S243 (C). *Significant difference compared to baseline values using ANOVA, P < 0.05.
Rebound tonometry was performed to determine whether treatment with P7C3-S243 affected IOP regulation, since provocative PERG analysis is dependent on the response of RGCs to transiently increased IOP. Treatment with P7C3-S243 had no effect on IOP dynamics in blast-exposed mice (Fig. 10B). The IOP increased with provocative testing to a similar degree in TBI+Veh and TBI-S243 mice, and baseline and provoked IOP were not significantly different between TBI+Veh and TBI- P7C3-S243 mice. The TBI+ P7C3-S243–treated mice had a baseline IOP of 13.33 ± 0.43 mm Hg, which increased significantly to 19.53 ± 0.91 mm Hg during provocative testing (P < 0.001, 1-way ANOVA with Tukey's multiple comparison test). TBI+Veh treated mice had a baseline IOP of 12.17 ± 0.54 mm Hg, which significantly increased to 20.67 ± 1.29 mm Hg during provocative testing (P < 0.001). Comparison of P7C3-S243–treated mice and vehicle-treated mice showed no significant differences in baseline or provocative IOP values. These results demonstrated that abnormal RGC physiology observed 5 weeks after blast exposure leads to progressive visual dysfunction, which can be observed using provocative PERG. Furthermore, treatment with P7C3-S243 prevents the induced decline in PERG function that is revealed using provocative testing.
Provocative PERG was recorded in sham-injury mice treated with P7C3-S243 (Sham+P7C3-S243), vehicle-treated sham-injury mice (Sham+Veh), blast-injury mice (“TBI”) treated with P7C3-S243 (TBI+ P7C3-S243), and blast-injury mice treated with vehicle (TBI+Veh) 4 weeks after injury (Fig. 10C). There was no significant difference in provocative PERG amplitude among Sham+ P7C3-S243 (24.18 ± 3.23 μV, n = 5), Sham+Veh (23.19 ± 3.27 μV, n = 4), and TBI+ P7C3-S243 groups (19.34 ± 1.76 μV, n = 5), but there was a significant decrease in the provocative PERG amplitude of TBI+Veh-treated mice (12.74 ± 2.66 μV, n = 5, P < 0.05, ANOVA). Thus, P7C3-S243 exerted a protective effect against the drop in provocative PERG amplitudes seen after TBI, indicating its ability to foster protection at this early time point of damage that ultimately leads to chronic loss of function.
Chronic Neutral Position PERG is Preserved After TBI by Treatment With P7C3-S243
Standard (neutral position) PERG was analyzed 16 weeks after blast exposure to determine whether daily treatment with P7C3-S243 could prevent chronic neutral position PERG deficits otherwise observed at this post-blast interval (Fig. 11). The PERG amplitudes of TBI+P7C3-S243 and TBI+Veh mice were compared to historical PERG amplitudes from aged-matched sham mice (as shown in Fig. 1). The average PERG amplitude of TBI+P7C3-S243–treated mice (22.7 ± 2.35 μV, n = 5) was not significantly different from that of sham-blasted mice (24.78 ± 1.45 μV, n = 12, P < 0.05, ANOVA with Dunnett's multiple comparison test), but the PERG amplitude of TBI + Veh mice (19.92 ± 0.80 μV, n = 9) was significantly lower than sham-blasted mice (P < 0.05, ANOVA with Dunnett's multiple comparison test). Thus, treatment with the neuroprotective agent P7C3-S243 effectively preserved visual function 16 weeks after blast injury in mice.
Figure 11.

Analysis of the efficacy of P7C3-S243 to prevent chronic neutral position PERG dysfunction. Analysis of mice exposed to TBI and treated with P7C3-S243 showed no significant difference compared to PERG amplitudes from sham-injured control mice. There was a significant decrease in vehicle-treated mice compared to PERG amplitudes from control mice. *P < 0.05, ANOVA with Dunnett's multiple comparison test.
Discussion
To our knowledge, this is the first report to describe two important findings related to visual dysfunction after TBI. First, we have identified a latent period of temporary recovery during which time in vivo RGC function under nonstressed conditions returns to baseline levels. At this time, abnormalities are revealed by identical testing conducted under conditions of RGC stress, and by in vitro measures of individual RGC physiology. Second, we showed that treatment after TBI with P7C3-S243 prevents acquisition of visual deficits, pointing towards future new therapeutic options for patients.
We have demonstrated that RGCs are highly susceptible to blast-mediated TBI, with a temporal bimodal decline in function as evaluated by neutral position PERG. The latent period, when no significant RGC functional deficits can be observed by neutral position PERG testing, begins approximately 2 weeks after injury and continues until 16 weeks after injury. By 16 weeks after injury, neutral position PERG deficits return. Although the neutral position PERG response returns to baseline levels during the latent period after blast exposure (4 weeks post blast), we have identified ongoing abnormalities of neuronal structure and function that remain undetected by standard clinical testing, yet lead to chronic visual dysfunction. For example, multielectrode recordings show that after blast exposure many ON and OFF RGCs have abnormal responsiveness to light during this temporary recovery latent period, including increased response duration to full-field flashes and increased spontaneous activity when compared to retinas of mice not exposed to blast injury. This measure is only obtainable in vitro in acutely dissected retina, however, and, thus, cannot be implemented clinically. Here, we showed that in vivo provocative PERG testing correlates with in vitro MEA deficits, thus providing a novel and effective way to detect subclinical disease and monitor its progression in living patients. This can serve to identify patients destined to develop permanent visual dysfunction at an earlier time point than is detected otherwise in routine clinical eye exam. The hope is that this earlier treatment in patients would increase the likelihood of therapeutic protection in these patients from chronic visual damage.
During the chronic post-TBI period, when neutral-position PERG deficits reappear, we also observed a concomitant loss of RGCs. In addition, we also have observed significant dendritic retraction of RGCs between 4 and 16 weeks after blast-injury. Taken together, these findings suggested that functional and structural visual decrements observed during the chronic post-TBI period are likely permanent, resulting from loss of cells or from dendritic rearrangement that renders RGCs anatomically disadvantaged. Conversely, functional abnormalities detectable during the latent period appear to be reversible, as we demonstrated by treatment with the neuroprotective compound P7C3-S243.
One feature that distinguishes our model from other models of traumatic ocular injury, or the injuries observed in some contact sports, is the lack of retinal detachment that is not observed in our model. This may be due to the fact that our model delivers a diffuse blast wave to the entire head, which is not directed exclusively to the eye. The lack of retinal detachment or commotio retinae also suggests our model recapitulates mild TBI, and lacks the intense sharp, shearing forces necessary for damage to outer nuclear layer neurons.
One current challenge for our healthcare system is to develop objective clinical testing procedures to identify individuals with increased susceptibility to chronic damage to the visual system and brain after TBI. We have shown here that provocative PERG testing elucidates blast-mediated visual deficits before permanent RGC damage/dysfunction. This testing paradigm has been validated in mice24 and humans32 susceptible to development of glaucoma, a disease affecting primarily RGCs. Corresponding abnormalities in the provoked posture-related decrements in PERG also were detected at a post-injury interval (4 weeks), similar to the onset of abnormal in vitro RGC MEA physiology. While a majority of cells had abnormal RGC spontaneous and light-evoked activity, a smaller proportion of all RGCs were lost after blast-exposure. We hypothesized that MEA-recorded RGC responses return to baseline levels in the months following TBI due to loss of RGCs. Perhaps their abnormal activity following damage is a form of “agonal” physiology preceding their death. By contrast, less severely injured RGCs likely recover normal physiologic behavior with time. In cases of repeated trauma or more severe injury, the ability of the nervous system to compensate in this manner could become compromised, leading to progressive deterioration in function. Thus, early intervention designed to preserve normal functioning of RGCs could help mitigate long-term visual impairments after TBI, and also may be applicable to other CNS regions.
While significant visual deficits have been observed thus far in veterans with TBI, the possibility exists that other visual deficits are under-reported or overshadowed by other clinical ailments. For example, one recent report33 suggests that visual symptoms of blast-exposed individuals resolve within 5 to 6 years following injury. Alternatively, it is possible that many veterans who ultimately will suffer chronic visual decrements due to TBI may currently be in the latent phase of the disease process. It also is entirely likely that many veterans with visual disturbances have not reported to medical care.
Also of important concern to healthcare providers is the lack of treatment options for patients suffering from the effects of blast-mediated TBI. In this study, we have demonstrated that treatment with the neuroprotective compound P7C3-S243 prevents RGC dysfunction measured by provocative PERG testing. Animals treated with vehicle, by contrast, exhibited significant RGC dysfunction. While we still do not understand the molecular mechanisms responsible for neuronal dysfunction following TBI, we do know that P7C3-S243 functions by augmenting activity of NAMPT, the rate-limiting enzyme in NAD salvage, after injury.22 Here, we treated immediately after blast-mediated TBI with P7C3-S243, and then continuously for the duration of the experiment. In future experiments we will determine the critical treatment window within which P7C3-S243 must be administered to prevent visual deficits following TBI.
Conclusions
Efficient observation and analysis of individual RGC physiology in vitro using multi-electrode recording of the intact retina revealed that the period of active RGC injury and dysfunction leading to chronic visual loss occurs much earlier than previously anticipated, approximately 4 weeks following blast exposure. Provocative PERG testing confirmed that RGCs are correspondingly more susceptible to disruption of normal physiology by stressful conditions, such as a transient, postural-induced rise in IOP during this latent period following blast injury. Furthermore, we show that permanent RGC dysfunction can be prevented by administration of the neuroprotective compound P7C3-S243. Thus, we have identified a time window for therapeutic intervention and a novel means to detect this period noninvasively, and also shown that treatment with P7C3-S243 protects RGCs from functional degeneration at acute and chronic time points.
The degenerative processes we have observed in this study include an ictal insult to the RGC followed by a temporal bimodal decline of in vivo RGC function. During the latent period we observed overt changes in the physiology of RGCs, which may underlie subclinical, difficult to quantify visual decrements that ultimately lead to decreased quality of life.34 At later time points after injury, we have observed chronic PERG deficits concomitant with atypical dendritic fields, which may leave RGCs unable to respond correctly to visual input. It is likely that other forms of optic neuropathy, such as glaucoma, follow a similar degenerative process,35–37 and, thus, also might be amenable to treatment with the P7C3 class of neuroprotective agents.
Acknowledgments
Disclosure: L.M. Dutca, None; S.F. Stasheff, None; A. Hedberg-Buenz, None; D.S. Rudd, None; N. Batra, None; F.R. Blodi, None; M.S. Yorek, None; T. Yin, None; M. Shankar, None; J.A. Herlein, None; J. Naidoo, None; L. Morlock, None; N. Williams, None; R.H. Kardon, None; M.G. Anderson, None; A.A. Pieper, P; M.M. Harper, None
Supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Rehabilitation Research and Development Center for Prevention and Treatment of Visual Loss, a Rehabilitation Research and Development Career Development Award (MMH), an RR&D Merit Study Award (1I01RX000427-01), and funds from the University of Iowa Carver College of Medicine (AAP). The authors alone are responsible for the content and writing of the paper.
Footnotes
LMD and SFS contributed equally to the work presented here and should therefore be regarded as equivalent authors.
References
- 1. SOTA State of the Art IX: Traumatic Brain Injury Research. 2008. [Google Scholar]
- 2. Dougherty AL, MacGregor AJ, Han PP, Heltemes KJ, Galarneau MR. Visual dysfunction following blast-related traumatic brain injury from the battlefield. Brain Inj. 2011; 25: 8–13. [DOI] [PubMed] [Google Scholar]
- 3. Lew HL, Poole JH, Vanderploeg RD, et al. Program development and defining characteristics of returning military in a VA Polytrauma Network Site. J Rehabil Res Dev. 2007; 44: 1027–1034. [PubMed] [Google Scholar]
- 4. Cockerham GC, Rice TA, Hewes EH, et al. Closed-eye ocular injuries in the Iraq and Afghanistan wars. N Engl J Med. 2011; 364: 2172–2173. [DOI] [PubMed] [Google Scholar]
- 5. Cockerham GC, Goodrich GL, Weichel ED, et al. Eye and visual function in traumatic brain injury. J Rehabil Res Dev. 2009; 46: 811–818. [DOI] [PubMed] [Google Scholar]
- 6. Lemke S, Cockerham GC, Glynn-Milley C, Cockerham KP. Visual quality of life in veterans with blast-induced traumatic brain injury. JAMA Ophthalmol. 2013; 131: 1602–1609. [DOI] [PubMed] [Google Scholar]
- 7. Goldstein LE, Fisher AM, Tagge CA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012; 4: 134ra160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mohan K, Kecova H, Hernandez-Merino E, Kardon RH, Harper MM. Retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury. Invest Ophthalmol Vis Sci. 2013; 54: 3440–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yin T, Britt J, De Jesus-Cortes H, et al. P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Reports. 2014; 8: 1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luo X, Frishman LJ. Retinal pathway origins of the pattern electroretinogram (PERG). Invest Ophthalmol Vis Sci. 2011; 52: 8571–8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miura G, Wang MH, Ivers KM, Frishman LJ. Retinal pathway origins of the pattern ERG of the mouse. Exp Eye Res. 2009; 89: 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Viswanathan S, Frishman LJ, Robson JG. The uniform field and pattern ERG in macaques with experimental glaucoma: removal of spiking activity. Invest Ophthalmol Vis Sci. 2000; 41: 2797–2810. [PubMed] [Google Scholar]
- 13. Pieper AA, Xie S, Capota E, et al. Discovery of a proneurogenic, neuroprotective chemical. Cell. 2010; 142: 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. MacMillan KS, Naidoo J, Liang J, et al. Development of proneurogenic, neuroprotective small molecules. J Am Chem Soc. 2011; 133: 1428–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Naidoo J, De Jesus-Cortes H, Huntington P, et al. Discovery of a neuroprotective chemical, (S)-N-(3-(3,6-dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(-)-P7C3-S243], with improved druglike properties. J Med Chem. 2014; 57: 3746–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pieper AA, McKnight SL, Ready JM. P7C3 and an unbiased approach to drug discovery for neurodegenerative diseases. Chem Soc Rev. 2014; 43: 6716–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walker AK, Rivera PD, Wang Q, et al. The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis [published online ahead of print April 22, 2014]. Mol Psychiatry. doi:10.103/mp.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Jesus-Cortes H, Xu P, Drawbridge J, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc Natl Acad Sci U S A. 2012; 109: 17010–17015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tesla R, Wolf HP, Xu P, et al. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2012; 109: 17016–17021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kemp SW, Szynkaruk M, Stanoulis KN, et al. Pharmacologic rescue of motor and sensory function by the neuroprotective compound p7c3 following neonatal nerve injury. Neuroscience. 2014; 284C: 202–216. [DOI] [PubMed] [Google Scholar]
- 21. Blaya MO, Bramlett HM, Naidoo J, Pieper AA, Dietrich WD. Neuroprotective efficacy of a proneurogenic compound after traumatic brain injury. J Neurotrauma. 2014; 31: 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang G, Han T, Nijhawan D, et al. The P7C3 class of neuroprotective chemicals functions by activating the rate-limiting enzyme in NAD salvage. Cell. 2014; 158: 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mohan K, Harper MM, Kecova H, et al. Characterization of structure and function of the mouse retina using pattern electroretinography, pupil light reflex, and optical coherence tomography. Vet Ophthalmol. 2012; 15 (suppl 2): 94–104. [DOI] [PubMed] [Google Scholar]
- 24. Nagaraju M, Saleh M, Porciatti V. IOP-dependent retinal ganglion cell dysfunction in glaucomatous DBA/2J mice. Invest Ophthalmol Vis Sci. 2007; 48: 4573–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Connolly SE, Hores TA, Smith LE, D'Amore PA. Characterization of vascular development in the mouse retina. Microvasc. Res. 1988; 36: 275–290. [DOI] [PubMed] [Google Scholar]
- 26. Thompson S, Blodi FR, Lee S, et al. Photoreceptor cells with profound structural deficits can support useful vision in mice. Invest Ophthalmol Vis Sci. 2014; 55: 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chichilnisky EJ. A simple white noise analysis of neuronal light responses. Network: Comput Neural Syst. 2001; 12: 199–213. [PubMed] [Google Scholar]
- 28. Maunsell JH, Gibson JR. Visual response latencies in striate cortex of the macaque monkey. J Neurophysiol. 1992; 68: 1332–1344. [DOI] [PubMed] [Google Scholar]
- 29. Reid RC, Victor JD, Shapley RM. The use of m-sequences in the analysis of visual neurons: linear receptive field properties. Vis Neurosci. 1997; 14: 1015–1027. [DOI] [PubMed] [Google Scholar]
- 30. Ventura LM, Golubev I, Lee W, et al. Head-down posture induces PERG alterations in early glaucoma. J Glaucoma. 2013; 22: 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nagaraju M, Saleh M, Porciatti V. IOP-dependent retinal ganglion cell dysfunction in glaucomatous DBA/2J mice. Invest Ophthalmol Vis Sci. 2007; 48: 4573–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ventura LM, Golubev I, Lee W, et al. Head-down posture induces PERG alterations in early glaucoma. J Glaucoma. 2013; 22: 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Magone MT, Kwon E, Shin SY. Chronic visual dysfunction after blast-induced mild traumatic brain injury. J Rehab Res Develop. 2014; 51: 71–80. [DOI] [PubMed] [Google Scholar]
- 34. Lemke S, Cockerham GC, Glynn-Milley C, Cockerham KP. Visual quality of life in veterans with blast-induced traumatic brain injury. JAMA Ophthalmol. 2013; 131: 1602–1609. [DOI] [PubMed] [Google Scholar]
- 35. Della Santina L, Inman DM, Lupien CB, Horner PJ, Wong RO. Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J Neurosci. 2013; 33: 17444–17457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Williams PA, Howell GR, Barbay JM, et al. Retinal ganglion cell dendritic atrophy in DBA/2J glaucoma. PLoS One. 2013; 8: e72282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feng L, Zhao Y, Yoshida M, et al. Sustained ocular hypertension induces dendritic degeneration of mouse retinal ganglion cells that depends on cell type and location. Invest Ophthalmol Vis Sci. 2013; 54: 1106–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]