

Abstract
Purpose.
Monosomy 3 (M3) or the presence of a specific RNA expression profile, known as class 2, is strongly associated with death from uveal melanoma (UM). Given the important role of epigenetic processes in cancer development and progression, we compared the transcriptional profiles of a selection of epigenetic regulators between primary UM with a good and a bad prognosis.
Methods.
Transcriptional levels of 59 epigenetic regulator genes were measured by quantitative PCR (qPCR) in 20 UM, 12 with monosomy of chromosome 3 (M3) and 8 with disomy of chromosome 3 (D3). Validation was performed in an independent cohort. Expression levels were compared to clinicopathological characteristics, including class type. Bisulfite sequencing was used to evaluate the role of DNA methylation in gene silencing.
Results.
In the first set of tumors, general downregulation of transcription of the genes encoding epigenetic regulatory enzymes was seen in association with M3. The 10 genes with the highest differential expression between M3 and D3 were selected and were analyzed in a second set of tumors. In the validation set, significantly lower levels of KAT2B (P = 0.008), HDAC11 (P = 0.009), KMT1C (P = 0.05), KDM4B (P = 0.003), KDM6B (P = 0.04), and BMI-1 (P = 0.001) transcripts were found in tumors with M3/class 2. Methylation of C-phosphate-G (CpG) residues was not observed on the putative regulatory regions of KAT2B, KDM4B, or KDM6B.
Conclusions.
Expression levels of a number of histone-modifying genes and polycomb family members are significantly lower in uveal melanoma with monosomy 3/class 2, supporting a general dysregulation of epigenetic modifiers in UM with a bad prognosis.
Keywords: uvea, melanoma, epigenetics, gene expression, oncology
Epigenetic processes are known to play an important role in cancer development and progression. Several epigenetic regulator genes are significantly less expressed in monosomy 3/class 2 uveal melanoma. This indicates a general dysregulation in bad-prognosis uveal melanoma.
Introduction
Uveal melanoma (UM) is the most common primary intraocular tumor in adults, with 5.1 cases per million annually in the United States.1,2 The mean age at diagnosis is 58 years,3 with most cases occurring in patients between the ages of 50 and 80.1 Uveal melanoma frequently leads to liver metastases, for which hardly any treatment has been found to be effective. Survival at 5 years ranges from 69% to 82%, falling to 57% to 62% at 10 years,4 despite often successful treatment of the primary tumor. However, many clinicopathological and genetic predictive factors have been identified, allowing early recognition of individuals at risk. In order to improve survival, adjuvant treatments should be developed and provided to high-risk individuals to prevent the outgrowth of metastases.
The most prominent genetic prognostic indicator is the chromosome 3 status. Monosomy of chromosome 3 (M3) is a frequent event in UM,5 contributing to a poor prognosis, with one report indicating a 55% 10-year mortality for those with M3 compared to 0% for patients with disomy of chromosome 3 (D3).6 This has triggered great interest in potential tumor suppressor genes that may be located on this chromosome. The most significant discovery so far has been the identification of a mutation of the BRCA-associated protein-1 (BAP1) gene on 3p21.1 on the remaining chromosome 3 in metastasizing tumors.7 There are also chromosomal alterations that predict a good prognosis, such as gain of chromosome 6p.8
The fact that M3 and gain of 6p are virtually exclusive to metastasizing and nonmetastasizing tumors, respectively, suggests that this is the level at which tumors diverge along two distinct pathways,9,10 and that loss of M3, BAP1 in particular, produces a highly aggressive tumor with a distinctive transcriptome, while an as yet unknown regulator on chromosome 6 prevents the development of this pattern. Uveal melanomas can be separated into two prognostically relevant groups based on a panel of differentially expressed genes: one with a low (class 1) and the other with a high (class 2) likelihood of metastatic spread.11–13
In the past decade, the important role of epigenetic mechanisms has begun to become apparent in a broad array of pathologies, and new treatments may focus on interfering in disease-associated epigenetic regulation. Epigenetics is the study of mitotically heritable changes in gene function that do not entail a change in gene sequence.14 Epigenetic modifications allow dynamic changes in chromatin structure. A number of epigenetic mechanisms that control gene transcription have been elucidated, most importantly DNA methylation and histone modification, which together define the epigenome.
Posttranslational modifications of histones by histone-modifying enzymes (HMEs) lead to changes in the state of chromatin compaction, which facilitates DNA-based processes such as transcription, replication, recombination, and repair. These modifications are carried out by “writers,” and many are potentially reversible by “erasers.” The net result is known as the histone code.15,16
Major regulators of transcription are the polycomb repressive complexes (PRC) 1 and 2, which are “readers” of the histone code responsible for transcriptional repression. Polycomb repressive complex 2 is responsible for initiation of gene repression, while reader PRC1 monoubiquitinates histone 2A at K119, leading to maintenance of gene repression. The Polycomb group (PcG) proteins are particularly important in regulating lineage choices by governing expression of genes involved in differentiation and development, such as the HOX genes (reviewed in Ref. 17). Global changes in cellular patterns of histone modification are associated with clinical outcome in many malignancies, for example, in prostate,18 lung,19,20 gastric,21 breast, ovarian, and pancreatic cancer.22 This supports evidence that aberrant histone modifications are critical in tumor genesis and implies that functional consequences of alterations in expression, activity, or specificity of HMEs have significant effects on outcome in cancer.
Little is known about the role of epigenetics in the pathology of UM. There are, however, a number of examples of loci silenced by promoter methylation, including pINK4a,23 RASSF1A,24 TIMP3,25 RASEF,26 and EFS,27 most of which encode genes with a role in cell cycle regulation. When UMs were clustered according to the global DNA methylome, they divided into the same two classes as when clustered according to their gene expression profile,10 suggesting an epigenetic contribution to the underlying molecular pathology that produces this transcriptome. Histone modifications may also play a role in UM development and progression. In UM, KMT6 (EZH2), a member of PRC2, is associated with repression of expression of the major histocompatibility complex (MHC) class II transactivator (CIITA), the master regulator of MHC-II genes,28 which is frequently silenced in tumor cells by epigenetic mechanisms.29,30 BAP1 is another HME, the loss of which is strongly implicated in the development of aggressive UM. It encodes a deubiquitinating enzyme,7 and BAP1 silencing or deletion is associated with hyperubiquitination of histone 2A; depletion of BAP1 in UM cell lines resulted in loss of melanocytic differentiation and a shift in gene expression to a class 2 profile.31
We hypothesize that epigenetic regulators in UM contribute to increased malignancy; we investigated changes in the expression of genes that play an important role in DNA methylation and histone modification and studied the expression of components of the PRCs, which mainly catalyze histone lysine modifications and are involved in DNA methylation. First, we measured the gene expression profile of 52 epigenetic modifier enzymes of the DNA methyltransferase (DNMT), lysine methyltransferase (KMT), lysine demethylase (KDM), lysine acetyltransferase (KAT), histone deacetylase (HDAC), and sirtuin (SIRT) groups, as well as seven PcG proteins of PRC1 and PRC2, and compared their expression between D3 and M3 UM.
Thereafter, we analyzed whether the most important differentially expressed genes were associated with loss of one chromosome 3 or the presence of a class 2 gene expression profile in a second set of UM. Subsequently, we looked for associations with clinicopathological prognostic factors and any relations with survival (as expected because of their relation to monosomy of chromosome 3/a class 2 gene expression profile). Finally, we investigated the mechanisms of altered transcription in some of the differentially expressed genes.
Materials and Methods
Study Population and Tumor Characteristics
We used tumor samples collected from 20 patients at the Leiden University Medical Center, Leiden, The Netherlands. For a validation study, we used a second independent set of 19 tumor samples. Uveal melanoma–containing eyes were removed by enucleation, and tumor material was snap frozen and stored at −80°C. Clinicopathological data were obtained from medical files and histology reports and collected in a database. Patient and tumor characteristics are described in Table 1. Each sample underwent conventional histopathological evaluation. Tumor, node, and metastasis (TNM) classification of the tumors occurred using the seventh edition of the American Joint Committee on Cancer (AJCC) staging manual.32 Follow-up information on date and cause of death of patients was retrieved from the Integral Cancer Center West, which checks and registers the survival status of patients on a yearly basis. The median follow-up time was 61.5 months (range, 2–169 months). Follow-up time is determined as the time period from enucleation until moment of death or last date when follow-up was checked. We do not have missing information on follow-up.
Table 1.
Overview of the Characteristics of the Patients Included in the Test and the Validation Sets
|
Clinicopathological Parameters |
Total |
Tumor Set |
|||
|
n |
Test Set,
n= 20 |
% |
Validation Set, n = 19 |
% |
|
| Mean age at diagnosis, y (SD) | 39 | 56.9 (±17.5) | 59.0 (±14.3) | ||
| Sex | |||||
| Male | 21 | 12 | 60 | 9 | 47 |
| Female | 18 | 8 | 40 | 10 | 53 |
| Eye | |||||
| Right | 22 | 10 | 50 | 12 | 63 |
| Left | 17 | 10 | 50 | 7 | 37 |
| Tumor diameter, mm (SD) | 39 | 13.5 (±4.7) | 13.1 (±3.0) | ||
| Tumor prominence, mm (SD) | 39 | 7.8 (±2.6) | 6.8 (±3.0) | ||
| Histopathologic cell type | |||||
| Spindle | 15 | 8 | 40 | 7 | 37 |
| Mixed/epithelioid | 24 | 12 | 60 | 12 | 63 |
| Ciliary body involvement | |||||
| No | 27 | 15 | 75 | 12 | 63 |
| Yes | 12 | 5 | 25 | 7 | 37 |
| Scleral ingrowth | |||||
| None | 5 | 2 | 10 | 3 | 16 |
| Superficial | 21 | 9 | 45 | 12 | 63 |
| Deep | 7 | 5 | 25 | 2 | 10.5 |
| Extrascleral | 6 | 4 | 20 | 2 | 10.5 |
| TNM stage | |||||
| Stage I | 4 | 0 | 0 | 4 | 21 |
| Stage IIA | 11 | 8 | 40 | 3 | 16 |
| Stage IIB | 13 | 7 | 35 | 6 | 32 |
| Stage IIIA | 10 | 4 | 20 | 6 | 32 |
| Stage IIIB | 0 | 0 | 0 | 0 | 0 |
| Stage IIIC | 1 | 1 | 5 | 0 | 0 |
| Stage IV | 0 | 0 | 0 | 0 | 0 |
| Metastasis | |||||
| No | 16 | 6 | 30 | 10 | 53 |
| Yes | 23 | 14 | 70 | 9 | 47 |
| Chromosome 3 status | |||||
| Disomy | 16 | 8 | 40 | 8 | 42 |
| Monosomy | 23 | 12 | 60 | 11 | 58 |
| Class | |||||
| 1 | ND | ND | ND | 10 | 53 |
| 2 | ND | ND | ND | 9 | 47 |
, number of tumors; SD, standard deviation; ND, not determined.
Tumor Material
DNA and RNA were isolated from 25 sections of 20 μm from fresh-frozen tissue that was collected in Eppendorf tubes. DNA was isolated using the QIamp DNA Mini Kit, and RNA was isolated using the RNeasy Mini Kit (both from Qiagen, Venlo, The Netherlands). Both were isolated according to the instructions of the manufacturer (vacuum protocol). Concentrations were measured using a Nanodrop (NanoDrop Products, Wilmington, DE, USA). Chromosome status was determined by single nucleotide polymorphism (SNP) analysis with the Affymetrix 250K_NSP microarray chip (Affymetrix, Santa Clara, CA, USA). After RNA isolation, cDNA was synthesized from RNA samples (1 μg) using SuperScript III (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol. RNA obtained from the frozen material from the tumors in the validation set was tested in the 15-gene classification assay as described by Onken et al.13 and sent to the department of Ophthalmology and Visual Sciences of Washington University School of Medicine (St. Louis, MO, USA) for class assignment. Data on chromosome 3 status and class were concordant in 17 out of the 19 cases in which both features were determined (the validation set). There were two cases with monosomy 3 that were discordantly classified as class 1. According to Dutch national regulations, human material remaining after pathological examination is available for use in research. The Tenets of the Declaration of Helsinki were followed.
Quantitative PCR Analysis
Quantitative PCR (qPCR) was performed on a Bio-Rad MyiQ cycler with SYBR Green supermix (Bio-Rad Laboratories, Hercules, CA, USA) and with primers to amplify the products of the 52 epigenetic modifier enzymes and seven PcG proteins listed in Table 2. Primer sequences will be published elsewhere but are available upon request. The PCR program used was 95°C for 3 minutes, followed by 50 cycles (30 seconds 95°C; 30 seconds Tann; 30 seconds 72°C) (where Tann is the specific annealing temperature of the individual primer pair), and was concluded by melting curve analysis. For each reaction, 50 ng cDNA and 5 pmol forward and reverse primer were used. The PCR data are shown as relative expression (2ΔCt), normalized against the geometric mean Ct value of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), RNA polymerase II (RPII), hypoxanthine-guanine phosphoribosyltransferase (HPRT), and β-glucuronidase (GUS). Normalization of expression of the geometric mean of multiple household genes was used to account for variation of household gene expression between samples.33 The Ct values of the genes that showed the highest differential expression are presented in Supplementary Table S1.
Table 2.
Epigenetic Gene Groups Categorized According to Mode of Action
|
Target |
DNA |
Histones |
|||||
| Modification |
Methylation |
Acetylation |
Deacetylation |
Methylation |
Demethylation |
Methylation |
|
| Function |
Writer |
Writer |
Eraser |
Writer |
Eraser |
Writer | Reader |
| (PRC2) |
(PRC1) |
||||||
| Enzymes | DNMT1 | KAT2A | Class I | KMT1A | KDM1 | EED | PHC1 |
| DNMT3a | KAT2B | HDAC1 | KMT1B | KDM2A | SUZ12 | RING1 | |
| DNMT3b | KAT3A | HDAC2 | KMT1C | KDM3A | JARID2 | BMI-1 | |
| KAT3B | HDAC3 | KMT1D | KDM3B | KMT6 | |||
| HDAC8 | KMT2A | KDM4A | |||||
| Class II A | KMT2B | KDM4B | |||||
| HDAC4 | KMT2C | KDM4C | |||||
| HDAC5 | KMT2D | KDM5A | |||||
| HDAC7 | KMT2E | KDM5B | |||||
| HDAC9 | KMT2F | KDM5C | |||||
| Class II B | KMT2G | KDM6A | |||||
| HDAC6 | KMT5A | KDM6B | |||||
| HDAC10 | KMT5B | ||||||
| Class III | KMT5C | ||||||
| SIRT1 | KMT7 | ||||||
| SIRT2 | |||||||
| SIRT3 | |||||||
| SIRT4 | |||||||
| SIRT5 | |||||||
| SIRT6 | |||||||
| SIRT7 | |||||||
| Class IV | |||||||
| HDAC11 | |||||||
DNA methyltransferases are responsible for DNA methylation. Polycomb repressive complexes 1 and 2 are responsible for initiation and maintenance of gene repression, respectively. The modifiers can also be classified according to function: acetyltransferases, methyltransferases, and PRC2 complex “write” the histone code, while deacetylases and demethylases “erase” it. An additional functional category of epigenetic modifiers comprises “readers” of the histone code, such as PRC1.
Bisulfite Sequencing
Bisulfite sequencing was used to analyze DNA methylation patterns. Genomic DNA was isolated from the two highest- and two lowest-expressing tumor samples for each of the genes of interest. DNA was treated with bisulfite EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA) according to the manufacturer's instructions. Speculative promoter regions of KDM4B, KDM6B, and KAT2B were located by detecting regions 5′ to the transcriptional start site with dense CpG islands and DNase I activity using UCSC Genome Browser Version hg19 (release date February 2009; UCSC Genome Informatics Group, Santa Cruz, CA, USA). Primers were designed to amplify this region using BiSearch Primer Design Tool (BiSearch Web Server, available in the public domain at http://bisearch.enzim.hu/), which generates primers to amplify anticipated bisulfite-converted CpG islands (CPIs; defined as a DNA sequence with GC content > 50% and observed-to-expected CpG ratio of >0.634). Sequences were amplified (AccuPrime Taq DNA Polymerase System; Invitrogen) according to the manufacturer's protocol. Optimal PCR conditions were no added MgCl2, Ta of 55°C, and 40 cycles for each of the primer pairs listed. The PCR products were purified (NucleoSpin Extract II kit; Macherey-Nagel, Inc., Bethlehem, PA, USA) and cloned into pGEMTeasy plasmid (Promega, Madison, WI, USA) according to the manufacturer's protocol. Twelve colonies containing a successfully ligated vector for each tumor sample were selected based on blue-white screening, and DNA was isolated (PureYield Plasmid Miniprep System; Promega) after overnight culture at 37°C in Luria Broth (LB) medium containing ampicillin in a shaking incubator. Ligation of the correct insert was confirmed using restriction enzyme digestion with EcoRI or NotI. As not all clones contained an insert of the correct size, results of at least two individual clones that had been successfully ligated were sequenced using the vector's M13 primer sites at the Leiden Genome Technology Center. Sequences were analyzed for CpG island methylation using the online tool QUantification tool for Methylation Analysis.35 For bisulfite sequencing of the three analyzed genes, the following primer sequences, 5′–3′, were used: for KDM4B, for the region spanning −63177 to −62839 (relative to CDS coding sequence) F: 5′TGGTATTTTGTAAATTGGGG, R: 5′CCRACTCTCTATTCTCATTAAAAAAA; for KDM6B, for the region spanning −3940 to −3552 F: GGAGATAAGATGAGTAGATAT, R: 5′AAATAAAACRATCCAAAACCCTC, and for KAT2B; for the region spanning −442 to −241 F: 5′AAAAGAGGTYGTGGGGGGTTTTTTA, R: 5′CCAAAAAAAAAAAAACTAACRAC (R: nucleotide A/G. Y: nucleotide C/T).
Statistics
Expression levels of each of the individual 59 genes were compared between M3 and D3 tumors using the Mann-Whitney U test, which compares median gene transcript levels. This statistical test was also used for comparison of gene expression levels to different clinicopathological parameters (sex, chromosome status, class, cell type, TNM stage). The relative expression levels of transcripts are presented as median (range). To correct for multiple testing, a Bonferroni correction was applied. The possible correlation with largest basal tumor diameter was analyzed using the Spearman correlation test. To investigate the association with the parameter “death due to metastases,” a univariate Cox regression analysis was conducted by dividing the genes into two groups using the median expression level. All tests were performed using the statistical software package SPSS 20.0.0 (IBM SPSS Statistics for Windows, version 20.0.0; IBM Corp., Armonk, NY, USA). Differences were considered to be significant if P < 0.05 after correction for multiple testing.
Results
Expression Patterns of Epigenetic Regulator Genes in M3 Versus D3 Tumors
We tested the expression levels of epigenetic regulator genes that can be categorized according to function (Table 2), depending on whether they methylate DNA, modify the histone code by “writing” or “erasing” it, or are part of PRC1 or PRC2. We determined with qPCR the expression of 59 genes involved in epigenetic regulation in 20 UM samples and compared their expression between M3 and D3 tumors. The set consisted of 12 M3 and 8 D3 UM of varying diameter and cell type, obtained following enucleation without any prior treatment. The results were validated in another set that included 19 UM samples. As we did several analyses with diverse groups of genes in different sets of tumors, a flowchart of analyses is presented for clarification (Fig. 1).
Figure 1.
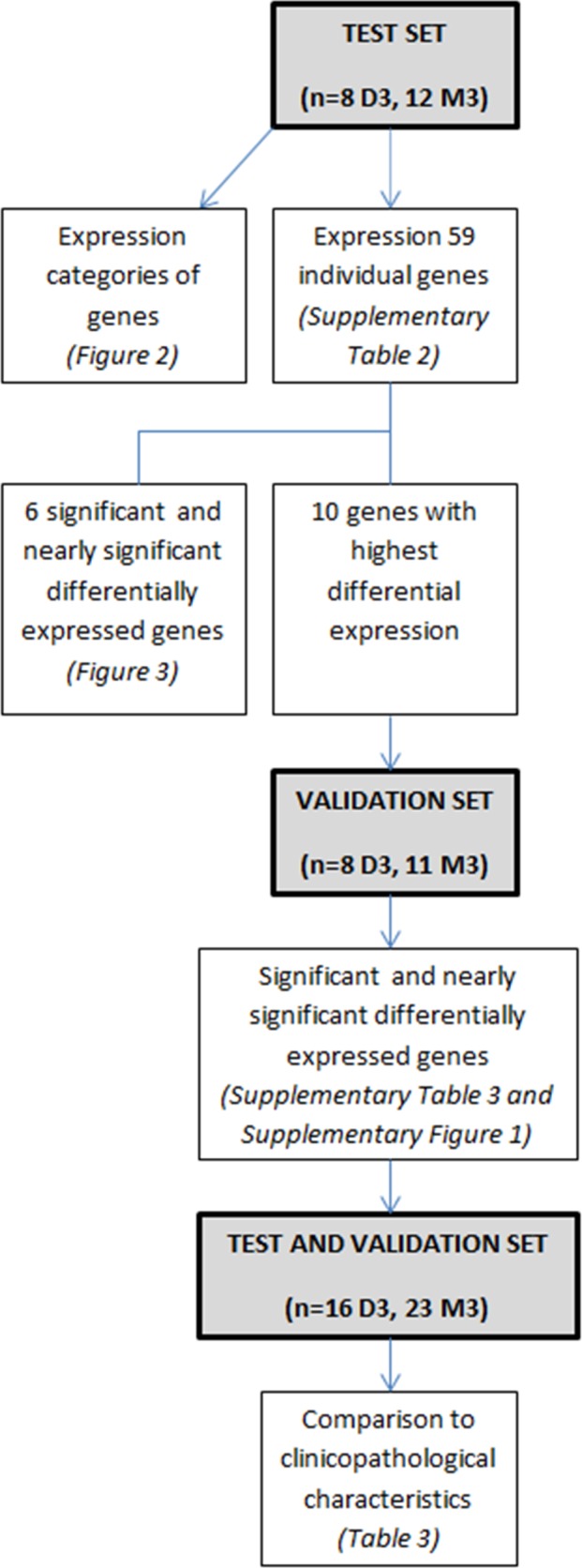
Flowchart of the different analyses in consecutive order. Gray boxes represent the sets of tumors used, with the number of D3 and M3 cases between brackets.
Transcription Profiles of Subcategories of Epigenetic Modifiers
Expression of the different functional categories varied widely between tumors, but overall, a trend was noted for a lower expression of the epigenetic gene-modifier subcategories in M3 tumors and a higher and more variable gene expression in D3 tumors. After Bonferroni correction to correct for the number of analyzed categories, M3 tumors showed a significantly higher expression of the KAT (P < 0.02), SIRT (P = 0.034), KMT (P = 0.009), and PRC2 (P = 0.007) gene categories than D3 tumors (Fig. 2).
Figure 2.

Box plot comparing expression of different modifier groups with the mean normalized expression of four household genes, between D3 (n = 8) and M3 (n = 12) tumor samples in the test set. Quantitative PCR was performed on each of 20 UM samples to amplify 59 genes, along with four household genes for each individual test gene. The mean of the Ct values for four household genes was compared with the Ct value of the test gene. Here, 59 genes have been categorized into eight modifier groups for analysis. Significant differences in expression (P < 0.05) of the modifier groups KAT, SIRT, KMT, and PRC2 between D3 and M3 tumors were found.
Transcriptional Profiles of Individual Genes
When looking at individual epigenetic modifier genes, the expression profiles of over 50% of the genes tested were found to be significantly different between M3 and D3 tumors (P < 0.05) (Supplementary Table S2). Following Bonferroni correction for the number of genes analyzed in the test set, differential expression of three genes remained significant: KAT2B (P = 0.017), JARID2 (P = 0.017), and BMI-1 (P = 0.017). Three showed an almost significant difference: SIRT5 (P = 0.059), HDAC11 (P = 0.059), and KDM6B (P = 0.059) (Fig. 3). The aforementioned trend of broader spread of expression values in D3 tumors was observed again.
Figure 3.

Box plot comparing expression of individual genes with the mean normalized expression of four household genes, between D3 (n = 8) and M3 (n = 12) tumor samples. Quantitative PCR was performed on a test set of 20 UM samples to amplify 59 genes. The mean of the Ct values for four household genes was compared with the Ct value of the test gene. Here, six genes for which (nearly) significant differential expression between D3 and M3 tumors was found are represented.
Validation of Differential Expression of Gene Expression
Several epigenetic modifier genes had shown an (almost) significant differential expression after correction in the first set. To validate these results, expression of the 10 genes with the highest differential expression in the test set was determined by qPCR in a second independent validation set, which was made up of 19 UMs, containing 8 D3 tumors and 11 M3 tumors. The significance in difference of transcriptional profile in relation to the chromosome 3 status was maintained for KAT2B (P = 0.008) and BMI-1 (P = 0.001), and was now also significant for HDAC11 (P = 0.009), KDM4B (P = 0.003), KDM6B (P = 0.038), and KMT1C (P = 0.05) with an almost significant difference in JARID2 expression (P = 0.083). The difference between D3 and M3 tumors was not reproducible for SIRT5 (Supplementary Table S3; Supplementary Fig. S1).
Validation of Selection: A Comparison of Epigenetic Gene Expression and Other Clinicopathological Prognostic Factors
The basis of the study was to determine whether high-risk tumors differed in their expression of epigenetic regulators, and for this goal we had selected tumors with and without M3. We selected the genes that had shown an (almost) significant differential expression between D3 and M3 tumors in the validation set and, using the tumors of the test as well as the validation set, we compared expression levels with other clinical and pathological variables and death due to metastases. Expression levels of the epigenetic regulators were not associated with sex (data not shown) or largest basal diameter (LBD). A low expression of KMT1C (P = 0.014) was associated with mixed/epithelioid cell type, while a low expression of KAT2B (P = 0.061) as well as HDAC11 (P = 0.053) was almost significantly associated with a mixed/epithelioid cell type. Low expression levels of almost all regulators were associated with a higher TNM stage (Table 3). We were able to compare gene expression levels of 19 tumors with the class 1 or 2 status: Tumors with a class 2 gene expression profile showed a decreased expression of KAT2B (P = 0.004), HDAC11 (P = 0.003), KMT1C (P = 0.05), KDM4B (P = 0.001), KDM6B (P = 0.014), and BMI-1 (P = 0.001).
Table 3.
A Comparison of Clinicopathological Parameters in All 39 Analyzed Cases With Expression Levels of Genes That Showed the Highest Differential Expression Between M3 and D3 Tumors in the Test Set and/or the Validation Set
|
Clinicopathological Parameters |
Epigenetic Regulator |
|||||||
|
KAT2B |
SIRT5 |
HDAC11 |
KMT1C |
KDM4B |
KDM6B |
JARID2 |
BMI-1 |
|
| LBD, n = 39 | ||||||||
| Correlation coefficient | −0.244 | −0.204 | −0.267 | −0.148 | −0.162 | −0.22 | −0.174 | −0.193 |
| P value* | 0.13 | 0.21 | 0.1 | 0.37 | 0.32 | 0.18 | 0.29 | 0.24 |
| Histological subtype, n = 39 | ||||||||
| Spindle, n = 15 | ||||||||
| Median | 19.5 | 9.7 | 8.1 | 76.7 | 18.9 | 4.1 | 33.4 | 23.7 |
| (range) | (7.5–53.8) | (4.2–23.1) | (1.7–22.2) | (26.0–142.8) | (9.5–32.3) | (1.0–23.9) | (11.5–134) | (6.3–79.4) |
| Mixed/epithelioid, n = 24 | ||||||||
| Median | 11.8 | 9.2 | 5.7 | 37.4 | 15 | 2.6 | 28.3 | 15.7 |
| (range) | (3.7–43.7) | (3.5–26.4) | (0.2–19.1) | (18.6–122) | (7.5–45.9) | (0.6–15.3) | (3.7–115.2) | (7.4–77.6) |
| P value† | 0.061 | 0.4 | 0.053 | 0.014 | 0.18 | 0.1 | 0.14 | 0.11 |
| TNM 7 stage, n = 39 | ||||||||
| I-IIB, n = 28 | ||||||||
| Median | 18.8 | 11.2 | 6.9 | 65.5 | 18.9 | 3.8 | 33.7 | 23.6 |
| (range) | (3.7–53.8) | (4.2–23.1) | (0.2–22.2) | (18.6–143) | (7.5–45.9) | (0.6–23.9) | (11.9–134) | (6.3–79.4) |
| IIIA-IIIC, n = 11 | ||||||||
| Median | 8.8 | 5.4 | 4.0 | 31.4 | 10.6 | 1.6 | 16.1 | 15.7 |
| (range) | (4.4–20.7) | (3.5–26.4) | (0.2–19.0) | (23.5–92.2) | (8.8–37.7) | (0.8–4.5) | (3.7–101) | (7.4–54.8) |
| P value† | 0.008 | 0.016 | 0.19 | 0.039 | 0.011 | 0.01 | 0.006 | 0.19 |
| Chromosome 3 status, n = 39 | ||||||||
| Disomy 3, n = 16 | ||||||||
| Median | 22 | 13.6 | 10.7 | 77.4 | 21.7 | 4.6 | 61.6 | 30 |
| (range) | (9.0–53.8) | (4.2–26.4) | (2.9–22.2) | (32.1–143) | (16.4–45.9) | (2.1–23.9) | (12.6–134.3) | (23.1–79.4) |
| Monosomy 3, n = 23 | ||||||||
| Median | 10.5 | 7.3 | 2.7 | 31.5 | 12.2 | 2.3 | 24.2 | 12.9 |
| (range) | (3.7–20.7) | (3.5–23.1) | (0.2–12.6) | (18.6–115) | (7.5–32.3) | (0.6–4.3) | (3.7–91.9) | (6.3–34.7) |
| P value† | <0.001 | 0.003 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| Class, n = 19 | ||||||||
| Class 1, n = 10 | ||||||||
| Median | 20.4 | 13.1 | 14.1 | 46.5 | 20.5 | 4.3 | 45.7 | 25.9 |
| (range) | (9.0–53.8) | (4.2–26.4) | (2.0–22.2) | (26.0–92.2) | (11.6–37.7) | (1.4–5.8) | (12.6–115) | (10.3–54.8) |
| Class 2, n = 9 | ||||||||
| Median | 7.6 | 9.1 | 6.1 | 29.4 | 10.2 | 2.5 | 24.2 | 12.9 |
| (range) | (3.7–20.7) | (3.5–23.1) | (1.7–7.3) | (18.6–98.3) | (7.5–16.9) | (1.4–4.3) | (3.7–91.9) | (6.3–16.7) |
| P value† | 0.004 | 0.25 | 0.003 | 0.05 | 0.001 | 0.014 | 0.09 | 0.001 |
| Death due to metastases, n = 39 | ||||||||
| B value | 1.067 | 0.771 | 1.524 | 0.411 | 1.636 | 1.435 | 1.123 | 2.206 |
| HR | 2.907 | 2.162 | 4.593 | 1.508 | 5.132 | 4.2 | 3.075 | 9.076 |
| (95% CI for HR) | (1.165–7.254) | (0.900–5.194) | (1.742–12.108) | (0.631–3.606) | (1.856–14.188) | (1.646–10.717) | (1.228–7.696) | (2.938–28.039) |
| P value‡ | 0.022 | 0.09 | 0.002 | 0.36 | 0.002 | 0.003 | 0.016 | <0.001 |
A comparison with class was possible only in the validation group (n = 19). Note that Ct values of KAT2B were normalized against the Ct values of two household genes due to limited material. CI, confidence interval; HR, hazard ratio.
Spearman correlation analysis.
Mann-Whitney U test.
Univariate Cox regression analysis.
Univariate Cox regression analysis showed an association between low expression levels of KAT2B (P = 0.022), HDAC11 (P = 0.002), KDM4B (P = 0.002), KDM6B (P = 0.003), JARID2 (P = 0.016), and BMI-1 (P < 0.001) and death due to metastases (Table 3).
Methylation of Regulatory Regions of Differentially Expressed Genes
To investigate the cause of KDM4B, KDM6B, and KAT2B downregulation in M3 tumors and evaluate the contribution of epigenetic mechanisms, the methylation status of putative regulatory regions of each gene was assessed using bisulfite sequencing (Fig. 4). For each gene, the two highest- and the two lowest-expressing tumor samples were selected for analysis. The number of clones successfully sequenced for each tumor sample is provided in Supplementary Table S4. For the three genes investigated, the selected putative regulatory regions appear largely unmethylated in the tumor samples selected for analysis, regardless of the transcript levels of the genes investigated. This indicates that DNA methylation does not contribute to the altered expression levels of KDM4B, KDM6B, and KAT2B between the high- and low-expressing tumor groups.
Figure 4.

Methylation analysis of putative regulatory regions of genes significantly differentially expressed in M3 versus D3 tumors. Each CpG dinucleotide is represented by a circle. Results of at least two individual clones are illustrated as a pie chart. Unmethylated CpG residues are indicated by white circles. Percentage of the circle colored black represents the percentage of clones that were methylated at a particular CpG dinucleotide. The length of the line between each circle indicates relative distance between CpG residues in the genome. Numbers are used to identify tumor samples. For each gene, the two lowest-expressing (upper rows) and highest-expressing (lower rows) tumor samples were analyzed. CpG residues are numbered according to number of residues in the region amplified (upper rows) and according to base-pair position in region amplified (lower rows).
Discussion
A lot is already known about epigenetics and its role in cancer initiation and progression. The enzymes that epigenetically modify DNA or histones are promising targets for therapy because, unlike mutations in DNA or gains and losses of DNA, epigenetic modifications are potentially reversible. Although there is accumulating evidence supporting the contribution of epigenetics to the pathogenesis of UM, only a few epigenetic regulators have been studied in this malignancy. Across all analyses—when epigenetic regulator genes were analyzed collectively, grouped to function, or individually—the present study demonstrates a striking pattern of decreased expression of genes involved in epigenetic regulation in M3/class 2 tumors compared to D3/class 1 tumors. Additionally, a much broader range of gene expression levels was present in D3 tumors. When looking at all tumors together, we show a statistically significant association between M3/class 2 in UM and downregulation of expression of KAT2B, SIRT5, HDAC11, KMT1C, KDM4B, KDM6B, and BMI-1.
The dysregulation of genes involved in creating, removing, and reading epigenetic marks in M3 tumors reflects previous findings regarding other functional categories of genes, including genes involved in HLA protein expression,28–30 G-protein coupled signaling, calcium-response pathways, cell adhesion marker expression, and retinoic acid response pathways.36 Discovery of distinct gene expression patterns associated with M3 tumors11 paved the way for division of tumors into two prognostic classes based on gene expression profiling.12 Although a small proportion of M3 tumors will not have the class 2 gene expression profile that is associated with poor prognosis,12 these two features characterize the majority of metastasizing UMs. In our own experience, loss of one chromosome 3 and the class 2 gene expression pattern are highly correlated phenomena, as is also observed in the cases studied here.37 The association between the loss of many different epigenetic regulators and loss of one chromosome 3 may also be functionally related, as many of the regulators play a role in maintaining genomic integrity.
Associations between cancer and dysregulation of the specific HMEs found to be differentially expressed in this study have been described previously. In particular, dysregulation of the lysine acetyl transferase KAT2B (P300/CBP-associated factor; PCAF) is of interest as it is localized on chromosome 3p24.3, the loss of which chromosome is a very important prognostic factor in UM. Deletion of 3p is a frequent change in many cancers, such as esophageal squamous cell carcinoma and renal carcinoma, and KAT2B has been identified as a candidate tumor suppressor gene (TSG).38
Lysine demethylase 6B (JMJD3) demethylates the repressive histone mark H3K27me3/2.39 It is also a strong candidate for a TSG, and low expression levels have been demonstrated in malignancy, such as glioblastoma.40 On the other hand, overexpression of KDM6B leads to cell cycle arrest.41 Lysine residues K9 and K27 on H3 are the specific substrates for KMT1C, which shows methyltransferase activity for these amino acids in vitro.42 The main effect of this histone methylation modification is transcriptional repression. In our study, we found a downregulation of KMT1C expression in prognostically bad tumors, which is in contrast to overexpression levels found in other tumors.43
Upregulation of HDAC11 has previously been linked to tumor development. Histone deacetylases deacetylate both histone and nonhistone substrates, for example, transcription factors such as p53, nuclear factor κB (NFκB), and hypoxia-inducible transcription factor 1-α (HIF1α). They are involved in many cellular processes such as development, angiogenesis, proliferation, and apoptosis44 and are associated with numerous malignancies.45 HDAC11 is the sole member of the class IV HDAC group and is among the top 1% to 2% of genes overexpressed in cancers such as breast, hepatocellular, and renal pelvis urothelial carcinoma.46 The decreased expression of HDAC11 in M3 UM is most likely explained by its localization on chromosome 3p25.1-3p25.2.
Sirtuin 5 is another HDAC whose expression was downregulated in M3 tumors. However, the function of SIRT5 is not completely understood, and there are limited studies on the role of SIRT5 in cancer development and progression.47 A recent study showed downregulation of SIRT5 and other members of the sirtuin family in head and neck squamous cell carcinoma.48 We do not know which functional consequences this downregulation may have. As a loss of sirtuin expression has been associated with aging, and aging is associated with inflammation, it is interesting to speculate that similarly, loss of sirtuins may play a role in the inflammation associated with M3 tumors.49
The PcG proteins are essential for maintaining cells in an undifferentiated state; the repressive H3K27me3 mark is created by the PRC2 component KMT6 and recruits PRC1. Polycomb repressive complex 1 ubiquitinates H2A, resulting in silencing of genes that determine cell fate. It is possible that the downregulation of BAP1, a member of the polycomb repressive deubiquitinase (PR-DUB) complex that deubiquitinates H2A, in M3 tumors may be a key contributor to the class 2 transcriptome. It was expected that key PcG proteins would be more highly expressed in M3 tumors. However, in this study, expression of BMI-1 and JARID2 was significantly downregulated in M3 tumors. Lysine methyltransferase 6, which has previously been shown to have a role in MHC class II expression in UM,28 was not differentially expressed. Although these results do not support a stem-like epigenetic landscape in M3 tumors as may have been predicted, in these tumors expression of other HMEs involved in differentiation, such as KDM6B, was downregulated. In differentiating cells, KDM6B and KDM7A collaborate to remove the repressive H3K27me3 mark at the bivalent promoters of their developmental target genes, and may also facilitate release of “poised” RNA polymerase II and subsequent transcription.50 Downregulation of expression of KDM6B has been implicated in glioblastoma development by inhibition of the differentiation of glioblastoma stem cells.40
Epigenetic dysregulation may have a role in the development of monosomy 3 in UM, instead of being a secondary phenomenon. Genomic instability is a feature of metastasizing UMs.10 Epigenetic marks are important for maintenance of genetic stability; alterations in particular marks, for example, hypomethylation in subtelomeric, telomeric, or pericentric regions, are associated with chromosome instability,51 and global DNA hypomethylation is associated with aneuploidy and rearrangements of chromosomes.52 Lysine demethylase 4B has a suppressive effect on genomic instability53 and showed decreased expression in M3 UM in this study. Lysine demethylase 4B has recently been identified as a DNA damage response protein.54
Methylation of Regulatory Regions
Promoter methylation has been previously described as a mechanism for KAT2B downregulation in esophageal squamous cell carcinoma.55 This, along with the general dysregulation of epigenetic markers that we observed, prompted us to investigate the methylation status of putative CpG-rich regulatory regions of several of the genes consistently differentially expressed between D3 and M3 tumors. Bisulfite sequencing indicated that selected regions for KDM4B, KDM6B, and KAT2B were unmethylated. One limitation of our approach is the sequencing of only one putative CpG island for each gene. Previous studies have shown that CpG islands at various points along a gene and its promoter can be differentially methylated in the same gene.56 Additionally, only two high- and two low-expressing tumor samples were sequenced for each gene, and the number of clones sequenced varied from 2 to 10 due to varying success in ligation of the correct insert. Despite these drawbacks, these results suggest that DNA methylation most likely does not account for the differential expression of the selected genes in the tumor samples analyzed.
Future Experiments
While our data indicate that in a comparison of M3 and D3 tumors, mRNA expression levels of epigenetic modifiers differ globally, the implications at a functional level can only be speculated upon. To determine whether dysregulation of epigenetic modifiers has a detectable effect on the genome, future experiments should focus on comparing the global methylation of the genome and profiles of histone modification between tumors with good and poor prognosis. Analyzing global histone modification profiles using immunohistochemical staining specific for particular histone marks has been shown to be predictive of outcome in other cancers.18 The mark H3K27me3 is of particular interest given its link to KDM6B and BAP1. Chromatin immunoprecipation (ChIP) sequencing may detect specific local changes as a result of dysregulated epigenetic modifiers, yielding functional models of the consequences of epigenetic dysregulation for prognosis in UM. Additionally, small interfering RNA (siRNA)–mediated knockdown of expression of genes of interest may yield information regarding their roles in tumor development or progression.
The complexity of epigenetic regulation means that biological and clinical consequences of combinatorial variations in expression profiles of epigenetic genes must also be considered and investigated. Considering the diverse range of functions each epigenetic regulator has in cellular processes such as differentiation, proliferation, and transcription, future research on the functional consequences of altered expression of each of these genes may yield therapeutic targets and expose crucial pathological mechanisms underlying the pathways that produce two tumor types with such polarized clinical outcomes.
Acknowledgments
We thank Mieke Versluis for helping with the tissue samples.
Supported by the Eye Cancer Foundation (MD).
Disclosure: N. Herlihy, None; M. Dogrusöz, None; T.H. van Essen, None; J.W. Harbour, None; P.A. van der Velden, None; M.C.J.A. van Eggermond, None; G.W. Haasnoot, None; P.J. van den Elsen, None; M.J. Jager, None
Footnotes
NH and MD are joint first authors.
PJvdE and MJJ are joint senior authors.
References
- 1. McLaughlin CC, Wu XC, Jemal A, Martin HJ, Roche LM, Chen VW. Incidence of noncutaneous melanomas in the U.S. Cancer. 2005; 103: 1000–1007. [DOI] [PubMed] [Google Scholar]
- 2. Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011; 118: 1881–1885. [DOI] [PubMed] [Google Scholar]
- 3. Shields CL, Furuta M, Thangappan A, et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch Ophthalmol. 2009; 127: 989–998. [DOI] [PubMed] [Google Scholar]
- 4. Jovanovic P, Mihajlovic M, Djordjevic-Jocic J, Vlajkovic S, Cekic S, Stefanovic V. Ocular melanoma: an overview of the current status. Int J Clin Exp Pathol. 2013; 6: 1230–1244. [PMC free article] [PubMed] [Google Scholar]
- 5. Scholes AG, Damato BE, Nunn J, Hiscott P, Grierson I, Field JK. Monosomy 3 in uveal melanoma: correlation with clinical and histologic predictors of survival. Invest Ophthalmol Vis Sci. 2003; 44: 1008–1011. [DOI] [PubMed] [Google Scholar]
- 6. Damato B, Dopierala JA, Coupland SE. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin Cancer Res. 2010; 16: 6083–6092. [DOI] [PubMed] [Google Scholar]
- 7. Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010; 330: 1410–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Damato B, Dopierala J, Klaasen A, van Dijk M, Sibbring J, Coupland SE. Multiplex ligation-dependent probe amplification of uveal melanoma: correlation with metastatic death. Invest Ophthalmol Vis Sci. 2009; 50: 3048–3055. [DOI] [PubMed] [Google Scholar]
- 9. Parrella P, Sidransky D, Merbs SL. Allelotype of posterior uveal melanoma: implications for a bifurcated tumor progression pathway. Cancer Res. 1999; 59: 3032–3037. [PubMed] [Google Scholar]
- 10. Landreville S, Agapova OA, Harbour JW. Emerging insights into the molecular pathogenesis of uveal melanoma. Future Oncol. 2008; 4: 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tschentscher F, Hüsing J, Hölter T, et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003; 63: 2578–2584. [PubMed] [Google Scholar]
- 12. Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004; 64: 7205–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Onken MD, Worley LA, Tuscan MD, Harbour JW. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010; 12: 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003; 33: 245–254. [DOI] [PubMed] [Google Scholar]
- 15. Jenuwein T, Allis CD. Translating the histone code. Science. 2001; 293: 1074–1080. [DOI] [PubMed] [Google Scholar]
- 16. Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003; 983: 84–100. [DOI] [PubMed] [Google Scholar]
- 17. Schuettengruber B, Cavalli G. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development. 2009; 136: p3531–p3542. [DOI] [PubMed] [Google Scholar]
- 18. Seligson DB, Horvath S, Shi T, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005; 435: 1262–1266. [DOI] [PubMed] [Google Scholar]
- 19. Barlesi F, Giaccone G, Gallegos-Ruiz MI, et al. Global histone modifications predict prognosis of resected non small-cell lung cancer. J Clin Oncol. 2007; 25: 4358–4364. [DOI] [PubMed] [Google Scholar]
- 20. Song JS, Kim YS, Kim DK, Park SI, Jang SJ. Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathol Int. 2012; 62: 182–190. [DOI] [PubMed] [Google Scholar]
- 21. Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, Jang SJ. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol. 2008; 15: 1968–1976. [DOI] [PubMed] [Google Scholar]
- 22. Wei Y, Xia W, Zhang Z, et al. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol Carcinog. 2008; 47: 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van der Velden PA, Metzelaar-Blok JAW, Bergman W, et al. Promoter hypermethylation: a common cause of reduced p16INK4a expression in uveal melanoma. Cancer Res. 2001; 61: 5303–5306. [PubMed] [Google Scholar]
- 24. Maat W, van der Velden PA, Out-Luiting C, et al. Epigenetic inactivation of RASSF1a in uveal melanoma. Invest Ophthalmol Vis Sci. 2007; 48: 486–490. [DOI] [PubMed] [Google Scholar]
- 25. Van der Velden PA, Zuidervaart W, Hurks MH, et al. Expression profiling reveals that methylation of TIMP3 is involved in uveal melanoma development. Int J Cancer. 2003; 106: 472–479. [DOI] [PubMed] [Google Scholar]
- 26. Maat W, Beiboer SH, Jager MJ, Luyten GP, Gruis NA, van der Velden PA. Epigenetic regulation identifies RASEF as a tumor-suppressor gene in uveal melanoma. Invest Ophthalmol Vis Sci. 2008; 49: 1291–1298. [DOI] [PubMed] [Google Scholar]
- 27. Neuman LC, Weinhäusel A, Thomas S, Horsthemke B, Lohmann DR, Zeschnigk M. EFS shows biallelic methylation in uveal melanoma with poor prognosis as well as tissue-specific methylation. BMC Cancer. 2011; 11: 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holling TM, Bergevoet MW, Wilson L, et al. A role for EZH2 in silencing of IFN-gamma inducible MHC2TA transcription in uveal melanoma. J Immunol. 2007; 179: 5317–5325. [DOI] [PubMed] [Google Scholar]
- 29. Van der Stoep N, Biesta P, Quinten E, Van den Elsen PJ. Lack of IFNγ-mediated induction of the Class II Transactivator (CIITA) through promoter methylation is predominantly found in developmental tumor cell lines. Int J Cancer. 2002; 97: 501–507. [DOI] [PubMed] [Google Scholar]
- 30. Eggermond MCJA, Boom DR, Klous P, et al. Epigenetic regulation of CIITA expression in human T cells. Biochem Biopharm. 2011; 82: 1430–1437. [DOI] [PubMed] [Google Scholar]
- 31. Landreville S, Agapova OA, Matatall KA, et al. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin Cancer Res. 2012; 18: 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Finger PT. The 7th edition AJCC staging system for eye cancer: an international language for ophthalmic oncology. Arch Pathol Lab Med. 2009; 133: 1197–1198. [DOI] [PubMed] [Google Scholar]
- 33. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002; 3: research0034.1–research0034.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002; 99: 3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008; 36: W170–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCannel TA, Burgess BL, Rao NP, Nelson SF, Straatsma BR. Identification of candidate tumor oncogenes by integrative molecular analysis of choroidal melanoma fine-needle aspiration biopsy specimens. Arch Ophthalmol. 2010; 128: 1170–1177. [DOI] [PubMed] [Google Scholar]
- 37. Van Essen TH, van Pelt SI, Versluis M, et al. Prognostic parameters in uveal melanoma and their association with BAP1 expression. Br J Ophthalmol. 2014; 98: 1738–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qin YR, Fu L, Sham PC, et al. Single-nucleotide polymorphism-mass array reveals commonly deleted regions at 3p22 and 3p14.2 associate with poor clinical outcome in esophageal squamous cell carcinoma. Int J Cancer. 2008; 123: 826–830. [DOI] [PubMed] [Google Scholar]
- 39. Xiang Y, Zhu Z, Han G, Lin H, Xu L, Chen CD. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007; 17: 850–857. [DOI] [PubMed] [Google Scholar]
- 40. Ene CI, Edwards L, Riddick G, et al. Histone demethylase Jumonji D3 (JMJD3) as a tumor suppressor by regulating p53 protein nuclear stabilization. PLoS One. 2012; 7: e51407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Terashima M, Ishimura A, Yoshida M, Suzuki Y, Sugano S, Suzuki T. The tumor suppressor Rb and its related Rbl2 genes are regulated by Utx histone demethylase. Biochem Biophys Res Commun. 2010; 399: 238–244. [DOI] [PubMed] [Google Scholar]
- 42. Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem. 2001; 276: 25309–25317. [DOI] [PubMed] [Google Scholar]
- 43. Huang J, Dorsey J, Chuikov S, et al. G9a and Glp methylate lysine 373 in the tumor suppressor P53. J Biol Chem. 2010; 285: 9636–9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009; 10: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007; 26: 5420–5432. [DOI] [PubMed] [Google Scholar]
- 46. Deubzer HE, Schier MC, Oehme I, et al. HDAC11 is a novel drug target in carcinomas. Int J Cancer. 2013; 132: 2200–2208. [DOI] [PubMed] [Google Scholar]
- 47. Shih J, Donmez G. Mitochondrial sirtuins as therapeutic targets for age-related disorders. Genes Cancer. 2013; 4: 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lai CC, Lin PM, Lin SF, et al. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumour Biol. 2013; 34: 1847–1854. [DOI] [PubMed] [Google Scholar]
- 49. Maat W, Ly LV, Jordanova ES, de Wolff-Rouendaal D, Schalij-Delfos NE, Jager MJ. Monosomy of chromosome 3 and an inflammatory phenotype occur together in uveal melanoma. Invest Ophthalmol Vis Sci. 2008; 49: 505–510. [DOI] [PubMed] [Google Scholar]
- 50. Chen S, Ma J, Wu F, et al. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev. 2012; 26: 1364–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herrera LA, Prada D, Andonegui MA, Dueñas-González A. The epigenetic origin of aneuploidy. Curr Genomics. 2008; 9: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tuck-Muller CM, Narayan A, Tsien F, et al. DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet Cell Genet. 2000; 89: 121–128. [DOI] [PubMed] [Google Scholar]
- 53. Whetstine JR, Nottke A, Lan F, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006; 125: 467–481. [DOI] [PubMed] [Google Scholar]
- 54. Young LC, McDonald DW, Hendzel MJ. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following γ-irradiation. J Biol Chem. 2013; 288: 1376–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu C, Qin YR, Xie D, et al. Characterization of tumor suppressive function of P300/CBP-associated factor at frequently deleted region 3p24 in esophageal squamous cell carcinoma. Oncogene. 2009; 28: 2821–2828. [DOI] [PubMed] [Google Scholar]
- 56. Wierda RJ, Kuipers HF, van Eggermond MC, et al. Epigenetic control of CCR5 transcript levels in immune cells and modulation by small molecules inhibitors. J Cell Mol Med. 2012; 16: 1866–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]