

Abstract
Nanoscale particle tracking in three dimensions is crucial to directly observe dynamics of molecules and nanoparticles in living cells. Here we present a three-dimensional particle tracking method based on temporally focused two-photon excitation. Multiple particles are imaged at 30 frames/s in volume up to 180 × 180 × 100 µm3. The spatial localization precision can reach 50 nm. We demonstrate its capability of tracking fast swimming microbes at speed of ~200 µm/s. Two-photon dual-color tracking is achieved by simultaneously exciting two kinds of fluorescent beads at 800 nm to demonstrate its potential in molecular interaction studies. Our method provides a simple wide-field fluorescence imaging approach for deep multiple-particle tracking.
OCIS codes: (170.2520) Fluorescence microscopy, (170.7160) Ultrafast technology, (180.4315) Nonlinear microscopy
1. Introduction
In biological applications particle tracking is an essential method on investigating a wide range of molecular behaviors and interactions in living cells, such as protein movement and interactions, RNA transport [1–4]. Tracking single particles also reveals processes in virus infection, chemical reactions and microfluidic flow fields [5–7]. Particle tracking techniques are typically based upon optical imaging and frame-by-frame particle localization. Since most processes happen in three-dimensional (3D) space, it is desirable to obtain 3D trajectory of molecules/nanoparticles at nanometer precision. Current approaches on achieving 3D tracking can be characterized into three categories. First, a feed-back loop is used to control 3D motion of the sample stage or scanning the laser beam for lateral (x & y) and axial (z) changes [8–10]. Although this approach can achieve 1 nm resolution, it is limited to track a single particle based on the input of the feed-back loop. Second, based on super resolution imaging modalities such as photoactivated localization microscopy (PALM) [11] and stochastic optical reconstruction microscopy (STORM) [12], point spread function (PSF) engineering approach encodes the z position information into the shapes of the microscope’s PSF, such as astigmatism, double helix, Airy function, saddle-point and Tetrapod PSFs [13–18]. Currently this approach can track nanoscale emitters over axial range up to 20 µm. The third approach is based on defocused imaging to extrapolate the axial position of particles [19–24]. Unlike the Gaussian-like point spread function in the in-focus image of a single nanoscale fluorescence emitter, the intensity distribution in the defocused image has complex concentric ring structures due to Fresnel diffraction [25], and at large defocusing distance the outmost ring contains most of the energy. By measuring the center and radius of these rings the 3D position of a nanoscale emitter can be determined. This approach has achieved Ångström accuracy [23] and over 100 µm range in axial dimension [24].
All current defocused imaging for 3D tracking approaches are based on wide-field fluorescence, absorption or phase contrast microscopy. Two-photon excitation has the advantages of reduced tissue scattering, photobleaching, photodamage and phototoxicity compared with one-photon excitation [26, 27], and has been widely used in biomedical research, particularly for in vivo imaging. Developing particle tracking technique based on two-photon excitation will greatly advance our capability for tracking single molecules/nanoparticles trafficking in vivo. Most two-photon microscopic techniques use tightly focused laser beam and raster scanning to achieve efficient two-photon excitation and 2D images. Integrating particle tracking with two-photon microscopy requires sophisticated feed-back control loop for sample motion or beam steering [9, 28, 29]. Also this approach is limited to tracking one particle only. Over the last decade temporal focusing two-photon microscopy, a type of wide-field fluorescence microscopy, has achieved 2D imaging without scanning the laser beam [30, 31]. In temporal focusing setup, the spectrum of a femtosecond laser pulse is first spatially separated by a diffraction grating, then collimated by a lens, at last the microscope objective lens recombines them to a focal region. The laser pulse is broadened after the grating diffraction due to spectral-temporal relationship. Temporal focusing occurs at the focal region because the different spectral components only spatially overlap within the focal region of the objective lens; therefore, the pulse width is the shortest at the focal plane. At the focal region laser beam will not form a single spot as in the conventional microscope. Instead, due to the diffraction grating and the collimation lens, the focal region is a light sheet with a diameter from tens to hundreds micrometers depending on the focal length of lenses. Therefore, when using a CCD camera as a 2D detector, this temporal focusing two-photon microscope can obtain 2D images like a wide-field fluorescence microscope. Based on the success of this technique, several groups have adopted temporal focusing to image cellular dynamics in thick tissues and to ablate tissue [32–37]. Since it does not require scanning the laser beam, it could reach 1000 frames/second 2D imaging speed with an amplified femtosecond laser system as the light source [33]. In temporal focusing two-photon microscopy, 3D imaging can be achieved by scanning the sample in axial dimension [37, 38] or by using a spatial light modulator to change the phase front of the excitation beam for different depth of focus [39]. Applying temporal focusing for 3D particle tracking has been developed by introducing astigmatism in the excitation laser beam [40]. This method can track particles within 2 µm axial range with 10 ms temporal resolution. Compared with laser scanning two-photon tracking, the advantage of temporal focusing two-photon microscopy is its capability of tracking multiple particles simultaneously.
In this study, we present a multiple-particle tracking technique that combines temporal focusing two-photon microscopy and defocused imaging. In a temporal focusing two-photon microscope, the pulse width is at its shortest at the focal plane and this pulse broadens as it moves away from the focal plane. For instance, the pulse has a full width at half maximum (FWHM) of 84 fs in temporal domain at the focal plane, and its FWHM increases to 800 fs at the plane which is 100 µm away from the focal plane [31]. The relatively slow pulse broadening makes the axial resolution on the order of several micrometers depending on the femtosecond laser pulse width. This z-sectioning capability is not as sharp as the submicron z resolution from laser scanning two-photon microscope. Our approach utilizes this slow pulse broadening to achieve efficient two-photon excitation in a relatively thick volume (~100 µm), and moves the sample away from the focal plane for defocused imaging. Together the two-photon excited fluorescence signal from molecules/nanoparticles form defocused ring structures in the image collected by a CCD camera. By calculating the ring central position and radius we determine the 3D position (x, y, z) of molecules or nanoparticles and tracking their trajectories in time-lapse images. One challenge in this method is the low two-photon excitation efficiency while moving the sample away from focal plane. There are two focal planes in a temporal focusing two-photon microscope: the plane where excitation laser pulse reaches its shortest width (temporal focal plane), and the plane which projects onto EMCCD in focus (imaging focal plane). Usually these two focal planes overlap to meet the requirements of achieving both maximum excitation and sharpest PSF on detector simultaneously. It has been demonstrated that changing the group velocity dispersion (GVD) of the femtosecond laser pulses could lead to the displacement of the plane of the temporal focus along the optical axis of the objective lens, yielding z-scanning as a function of GVD [41, 42]. Therefore, in our setup GVD modulation is implemented to separate the temporal focal plane from the imaging focal plane. When nanoscale emitters are efficiently excited by the temporal focused laser pulse, the projected image on EMCCD is defocused.
2. Optical setup
The temporal focusing two-photon microscope setup is shown in Fig. 1. The femtosecond laser pulses come from a chirped pulse amplifier (Solstice ACE, 35 fs, 5 kHz, 6 W, Spectra-Physics, Mountain View, California, USA). A half wave plate (HWP) and a polarizing beam splitter control the power delivered into the microscope. The light has a double pass through a pair of prisms for dispersion compensation in order to achieve the shorted pulse at objective lens focal plane, then is deflected by a grating (900 lines/mm, Edmund Optics, Barrington, New Jersey, USA). Different frequency components of laser spectrum are collimated by a reflective lens (L1, f = 300 mm), then are diffracted by an acousto-optic modulator (AOM, TED8-200-50-800, Brimrose, Sparks, Maryland, USA) with 1mm × 35 mm active aperture. This AOM is driven by a 200 MHz RF wave from its driver, and this driving RF wave could be further modulated by an arbitrary function generator (AFG 3102C, Tektronix, Beaverton, Oregon, USA). The laser spectrum is diffracted by the 200 MHz acoustic wave (f0) traveling in AOM. When there is no modulation by AFG, the output electric field of the laser pulse after AOM, , equals to the input electric field, , in frequency domain. Since the laser spectrum has been spatially spread out on the AOM, each frequency, ω, corresponds to a fixed location on AOM. When AFG applies modulation function, , on the 200 MHz acoustic wave, the output electric field is the product of input electric field and this modulation function, [43, 44]. This modulation functionis synthesized by the AFG, and it can take arbitrary form to create arbitrary ultrafast laser pulses. When modulates the frequency f (centered at 200 MHz) of RF wave which takes the form of , it applies group velocity dispersion on the ultrafast laser pulses, and parameter α determines the amount of dispersion. The maximum amount of dispersion is determined by the bandwidth of this AOM, which is 50 MHz in this case that corresponds to group delay dispersion of ~104 fs2. This modulation function from AFG is synchronized to the femtosecond laser pulse at 5 kHz. After passing a pair of relay lenses (L2 and L3) and a long-pass dichroic beam splitter (FF665-Di02, Semrock, Rochester, New York, USA), the beam shoots into the microscope objective lens (LD plan-NEOFLUAR 63 × NA 0.75, Zeiss, Jena, Germany). The fluorescent signal from the sample is epi-collected by the same objective lens. A second dichroic beam splitter (FF552-Di02, Semrock, Rochester, New York, USA) separates the green and red fluorescence signals. Two EMCCD cameras (iXon Ultra 897, Andor Technology, Belfast, UK) detect the green and red fluorescence signals respectively. Two bandpass filters (FF03-543/22, and FF03-607/36, Semrock, Rochester, New York, USA) are in the detection path to select the green and red fluorescence respectively. The sample is mounted on a motorized translational stage (NPXYZ100SG, Newport, Irvine, California, USA).
Fig. 1.
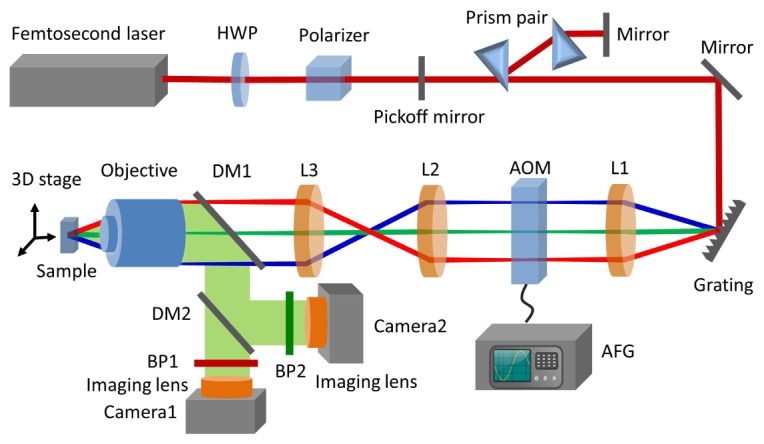
Experimental setup of the temporal focusing two-photon microscope with spatial light modulation. HWP: half-wave plate, AOM: acousto-optic modulator, AFG: arbitrary function generator, DM: dichroic mirror, L: lens, BP: bandpass filter.
3. Materials and methods
Two types of fluorescent nanospheres with diameter of 100 nm are used in the two-channel experiment. One is green sphere with the excitation peak at 505 nm and emission peak at 515 nm (FluoSpheres carboxylate-modified, F8803, Thermo Fisher Scientific, Waltham, MA, USA). The other one is red sphere with the excitation peak at 580 nm and emission peak at 605 nm (FluoSpheres carboxylate-modified, F8801, Thermo Fisher Scientific, Waltham, MA, USA). Both spheres’ initial concentrations are diluted with distilled water at the ratio of 1:1000. The sphere samples are well suspended in the solution with the help of a vortex mixer before they are prepared in the slides and cover glasses. The whole process is finished at a dark environment to preserve the optical properties of samples.
Cafeteria roenbergensis is a D-shaped biflagella single-cell marine zooplankton (~10 µm) that is widely spread throughout the world’s oceans and feeds mainly on bacteria and phytoplankton [45, 46]. As a major microzooplankton predator of marine bacteria, Cafeteria contributes to the critical pathway of nutrient recycling in the ocean and is an important link in the marine food web. Therefore, studying the grazing pattern of such protists is important to understand their roles in ecosystem [45]. Cafeteria culture is maintained in F/2 medium with E. coli bacteria as its food and together with yeast, trace metal, aspirin and vitamins. A small portion of Cafeteria is taken from a large stock, then centrifuged several times and diluted to proper concentrations based on the counts from a hemocytometer. Fluorescent nanospheres are added to the F/2 medium with Cafeteria starving for four days. It is observed that the Cafeteria may treat the spheres as food and swallow them. By recording the fluorescent signal from beads, we are able to track the fast motion of Cafeteria.
We use MATLAB curve fitting module to fit the outmost ring in defocused images to the following Gaussian distribution function:
| (1) |
where I is the pixel intensity, A is the amplitude, k is a parameter related to ring width, x0 is the ring center coordinate in x dimension, y0 is the ring center coordinate in y dimension, r0 is the ring radius. In the MATLAB curve fitting module, non-linear least squares analysis and the method of least absolute residuals are used to improve the accuracy of the fitting model. The curve fitting algorithms show the center values of x0, y0 and r0, and the intervals of each of the three fitting parameters with 95% confidence level ( ± 2σ). From the intervals given in this curve fitting results, we obtain the standard deviation of x0, y0 and r0.
4. Results
The temporal focusing two-photon microscope is first characterized with fluorescent nanospheres (100 nm). The PSF (Fig. 2(a)) of one single bead shows FWHM of 0.5 µm (Fig. 2(a)), which is at the diffraction limit. By using 1-µm diameter fluorescent microspheres and intentionally saturating the main lobe intensity in detection, we are able to observe the peripheral fringes in PSF (Fig. 2(b)). The z-sectioning capability (axial resolution) for temporal focusing two-photon microscope is on the order of several micrometers depending on several parameters such as laser spectrum bandwidth and objective lens numerical aperture. We scan the sample in axial dimension over a range of 70 µm at 2 µm step (Fig. 2(b)), and record a series of images, with the in-focus plane as the origin (z = 0 µm) and + z direction indicating sample moving towards objective lens. The in-focus image (z = 0 µm) is intentionally saturated in order to increase the brightness of defocused images. Concentric rings show up in defocused images (z ≥ 20 µm) with peak intensity in the outmost ring, and the central lobe becomes blurred. This ring structure does not present when the sample is moving away from the objective (z < 0 µm) as observed in another wide-field fluorescence imaging experiment [20]. Such asymmetric ring formation on two sides of the focal point was predicted by Fresnel diffraction simulation [25]. The normalized fluorescence intensity change in axial dimension is plotted as the red curve in Fig. 2(d) by measuring the central lobe peak intensity with unsaturated in-focus image as the peak intensity. The FWHM of this curve is about 10 µm which is a littler larger than many other experiments since the N.A. of our objective is 0.75. Implementing defocused imaging for this conventional temporal two-photon microscope is challenging, since the ring intensity in defocused images is much lower than that in in-focus image. And the dynamic range of detector is typically not suitable for such large intensity variation. To overcome this challenge we apply extra GVD on the AOM to shift the temporal focal plane away from the imaging focal plane. The amount of applied GVD determines how far the temporal focal plane can be shifted from its original position [41, 42]. Although a larger GVD will increase the tracking depth, however, it will increase the pulse width thus decrease the two-photon excitation efficiency. As an example the maximum two-photon excitation happens at the z = 30 µm plane with added GVD of 5000 fs2 (Fig. 2(c)). Although the temporal focal plane has been shifted, the imaging focal plane is still at the z = 0 µm plane as this plane is determined by the objective lens and imaging lens in front of camera only. Therefore, the z = 30 µm plane image still shows defocused rings. Pulse broadening becomes prominent when the shifting of the temporal focal plane becomes large [41], which leads to larger depth of efficient two-photon excitation as shown in Fig. 2(b). The normalized peak fluorescence intensity from the outmost ring vs. axial dimension shows a very broad peak with FWHM around 30 µm (blue curve in Fig. 2(d)). Such large FWHM in axial dimension of detected fluorescence signal facilitates large range of tracking fluorescent molecules/nanoparticles in depth.
Fig. 2.

Characterization of the temporal focusing two-photon microscope using a 100 nm fluorescent nanosphere. (a) PSF of a 100 nm bead. (b) Series of fluorescent images of nanosphere at different z positions (z = −10 to 60 µm). When this nanosphere moves closer to the objective lens ( + z), defocused rings start forming. (c) Series of fluorescent images of nanosphere at different z positions (z = −10 to 60 µm) after the temporal focal plane is shifted to z = 30 µm plane with applied GVD on AOM. (d) Maximum intensity plots along axial dimension for images in (b) and (c).
The calculated ring radius (R) increases nonlinearly over the entire range (Fig. 3(a)). The relationship of R vs. Z shows good linearity within a short distance, e.g. 20 µm (Fig. 3(b)). By scanning the nano-positioning stage at 10 nm/step in a range of 160 nm we study the detailed resolution (Fig. 3(c)). The curve in Fig. 3(c) shows linear relationship with abnormal kinks. The localization precision ultimately depends on the signal-to-noise ratio of the obtained image rings. The standard deviation (σ) of R from fitting algorithm is in the range of 1-2 nm which is comparable to a previous defocused imaging study [19]. By leaving the sample stationary and imaging for 4 minutes at 15 s interval, we calculate a time series of the 3D position of sample (Fig. 3(d)). The position in all three dimensions fluctuates in time with standard deviation in the range of 20 nm. Such fluctuation is possibly due to mechanical instability of the system. This contributes to the kinks in Fig. 3(c), since the next position’s image is taken more than 30 s later after the previous position’s image.
Fig. 3.
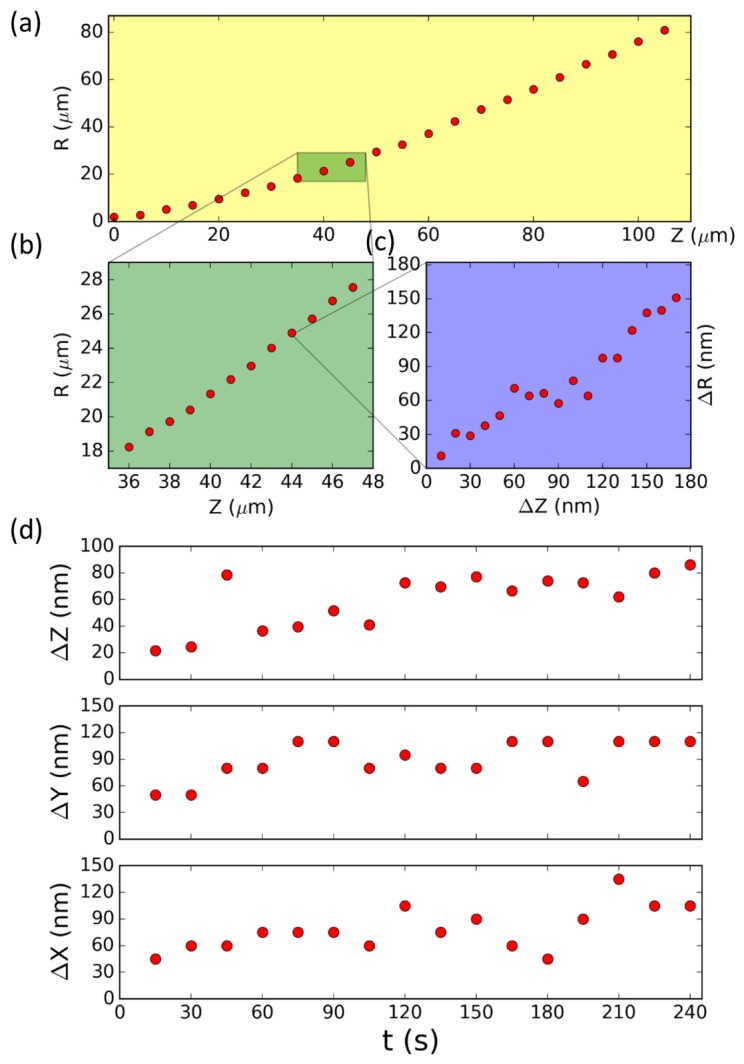
Particle 3D position calculation based on defocused images. (a) Calculated ring radius R vs. Z relationship in large axial scan range (ΔZ = 110 µm) is nonlinear. (b) The R vs. z relationship shows linearity in short scan range (ΔZ = 11 µm). (c) Within ultrashort range (ΔZ = 180 nm) the R vs. Z relationship shows overall linearity with measurement standard deviation of 2 nm. The large kinks in this curve is due to mechanical instability. (d) Mechanical stability measurement of one stationary sphere showing its 3D position (X, Y, Z) has fluctuations with standard deviation σX, σY and σZ around 20 nm.
Since our defocused imaging based particle tracking technique is capable of obtaining wide-field images, it is possible to track multiple particles simultaneously. We mix green and red nanospheres (100 nm) in solution. Both nanospheres are excited by 800 nm light, and their fluorescence is detected in the green and red channels respectively (Fig. 1, and Fig. 4(a)-4(c)). Figure 4(a) is the merged image of both channels with Fig. 4(b) and 4(c) showing green and red channels respectively. The green fluorescence signal is slightly lower than the red signal. In order to make the brightness in both channels comparable, we increased the gain in green channel with slightly higher background noise. Total 14 particles show up in both channels. Based on these defocused images, the 3D position of these particles within a volume of 180 × 180 × 100 µm3 are calculated and presented in Fig. 4(d).
Fig. 4.

Dual-color multiple-particle imaging. (a) Overlay of both green- and red- channel images that are collected simultaneously by two camera showing 8 green spheres and 6 red spheres and 8 green spheres (bar: 20 µm). Respective (b) green channel image, and (c) red channel image. (d) 3D projection of total 14 spheres in the volume of 180 × 180 × 100 µm3.
Defocused imaging based particle tracking has achieved tracking multiple bacteria at moving speed of 100 µm/s [24]. Exploring microbial motility in 3D with dynamic imaging is a powerful tool to study hydrodynamics of microorganisms, mechanics of microbial motility, and chemotaxis of microbes [47]. The swimming speed of Cafeteria is several hundred micrometers per second. We record a real-time (30 frames/s) movie of Cafeteria motion in 3D (Visualization 1 (4.3MB, AVI) , play back speed 10 frames/s) with sequential frames from the movie displayed in Fig. 5(a). Two Cafeteria are observed in this movie; the first one (right) has fast motion in all three dimensions and the second one (left) moves into the field of view and stays almost stationary within the rest time span of movie. The trajectories of both Cafeteria are plotted in Fig. 5(b). The instantaneous speeds during this period of time are plotted Fig. 5(c). The first Cafeteria has a speed in the range of 100-250 µm/s, and the second Cafeteria has an initial speed around 250 µm/s and slows down to the range of 0-50 µm/s. There have speculations on the difference of grazing patterns between fast swimming and stationary Cafeterias. Current study has only provided information on the stationary population [46]. Our method provides the capability of studying those fast swimming Cafeteria.
Fig. 5.

Dynamic tracking of microbe Cafeteria (Cro) swimming. (a) Frame by frame images (33 ms interval) of two Cafeteria with one near stationary on the left, and the other one fast swimming on the right (see Visualization 1 (4.3MB, AVI) ). (b) 3D trajectories of these two Cafeteria (black lines). (c) Instantaneous speed of two Cafeteria with color graph showing one Cafeteria is moving at high speed 100-250 µm/s, and the other one with an initial speed of over 250 µm/s and slows down to rest.
5. Discussion
In summary we present a multiple-particle tracking scheme based on temporal focusing two-photon microscopy. The current imaging rate of our approach is 30 frames/s. However, this is not the practical limit, since the camera exposure time is set at 1 ms for most experiments and it can be shorter for brighter fluorophores. The electronics of camera is currently limiting our imaging speed. At 512 × 512 pixels the frame rate limit of our camera is 56 frames/s [48]. It could be improved by reducing the number of pixels in each frame. For instance, at 64 × 64 pixels crop mode the frame rate limit is increased to 1492 frames/s. It has been reported that camera-based 3D particle tracking scheme has achieved kHz imaging rate [23]. Therefore, it is reasonable to predict that our approach can reach millisecond temporal resolution. The trade-off of achieving high imaging rate is degraded spatial resolution or reduced field of view within each image. If the camera works in binning mode, e.g. 4 × 4 pixels bin together to make 1 pixel, the pixel resolution is sacrificed. Or if the camera works in crop mode, e.g. only the central 64 × 64 pixels are used to capture light, the spatial coverage is reduced.
The spatial resolution of our approach is 2 nm in all three dimensions which is comparable to a previous study based on epifluorescence microscopy [19]. We have used a curve fitting algorithm in MATLAB to calculate the ring radius and central position. Many advanced algorithms have been developed to optimize the precision and accuracy of localization in single-particle tracking and super resolution microscopy [49]. With more careful design to improve mechanical stability we expect that this approach could achieve Ångström order resolution which had been achieved by wide-field microscopy [23]. The ultimate resolution limit is decided by the signal-to-noise ratio in images which is determined by shot noise of fluorophores and noise from camera electronics.
The advantage of tracking particles with defocused imaging is the capability of imaging and tracking multiple particles simultaneously. This capability is particularly useful for studying molecular interactions in living cells and high-throughput tracking of bacteria. When multiple particles are imaged and they are close to each other, their defocused rings can overlap, which will decrease signal-to-noise ratio, and eventually reduce the precision of determining the 3D positions of particles. To overcome this obstacle, we have implemented the dual-color imaging method where the two interacting particles could be labeled with different fluorophores and their emission is spectrally separated and detected by different CCD cameras.
Acknowledgments
We acknowledge Drs. Chuan Xiao and Supriyo Ray for insightful discussions and providing Cafeteria sample.
Funding
National Science Foundation (NSF) (1429708, 1205302). National Institute of Health (SC2GM103719).
References and links
- 1.Saxton M. J., Jacobson K., “Single-Particle Tracking: Applications to Membrane Dynamics,” Annu. Rev. Biophys. Biomol. Struct. 26(1), 373–399 (1997). 10.1146/annurev.biophys.26.1.373 [DOI] [PubMed] [Google Scholar]
- 2.Deschout H., Cella Zanacchi F., Mlodzianoski M., Diaspro A., Bewersdorf J., Hess S. T., Braeckmans K., “Precisely and accurately localizing single emitters in fluorescence microscopy,” Nat. Methods 11(3), 253–266 (2014). 10.1038/nmeth.2843 [DOI] [PubMed] [Google Scholar]
- 3.Kusumi A., Tsunoyama T. A., Hirosawa K. M., Kasai R. S., Fujiwara T. K., “Tracking single molecules at work in living cells,” Nat. Chem. Biol. 10(7), 524–532 (2014). 10.1038/nchembio.1558 [DOI] [PubMed] [Google Scholar]
- 4.Park H. Y., Lim H., Yoon Y. J., Follenzi A., Nwokafor C., Lopez-Jones M., Meng X., Singer R. H., “Visualization of Dynamics of Single Endogenous mRNA Labeled in Live Mouse,” Science 343(6169), 422–424 (2014). 10.1126/science.1239200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindken R., Rossi M., Grosse S., Westerweel J., “Micro-Particle Image Velocimetry (microPIV): recent developments, applications, and guidelines,” Lab Chip 9(17), 2551–2567 (2009). 10.1039/b906558j [DOI] [PubMed] [Google Scholar]
- 6.Wang W., Tao N., “Detection, counting, and imaging of single nanoparticles,” Anal. Chem. 86(1), 2–14 (2014). 10.1021/ac403890n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruthardt N., Lamb D. C., Bräuchle C., “Single-particle Tracking as a Quantitative Microscopy-based Approach to Unravel Cell Entry Mechanisms of Viruses and Pharmaceutical Nanoparticles,” Mol. Ther. 19(7), 1199–1211 (2011). 10.1038/mt.2011.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters I. M., de Grooth B. G., Schins J. M., Figdor C. G., Greve J., “Three dimensional single-particle tracking with nanometer resolution,” Rev. Sci. Instrum. 69(7), 2762–2766 (1998). 10.1063/1.1149012 [DOI] [Google Scholar]
- 9.Levi V., Ruan Q., Gratton E., “3-D particle tracking in a two-photon microscope: application to the study of molecular dynamics in cells,” Biophys. J. 88(4), 2919–2928 (2005). 10.1529/biophysj.104.044230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katayama Y., Burkacky O., Meyer M., Bräuchle C., Gratton E., Lamb D. C., “Real-Time Nanomicroscopy Via Three-Dimensional Single-Particle Tracking,” ChemPhysChem 10(14), 2458–2464 (2009). 10.1002/cphc.200900436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Betzig E., Patterson G. H., Sougrat R., Lindwasser O. W., Olenych S., Bonifacino J. S., Davidson M. W., Lippincott-Schwartz J., Hess H. F., “Imaging Intracellular Fluorescent Proteins at Nanometer Resolution,” Science 313(5793), 1642–1645 (2006). 10.1126/science.1127344 [DOI] [PubMed] [Google Scholar]
- 12.Rust M. J., Bates M., Zhuang X., “Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM),” Nat. Methods 3(10), 793–796 (2006). 10.1038/nmeth929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang B., Wang W., Bates M., Zhuang X., “Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy,” Science 319(5864), 810–813 (2008). 10.1126/science.1153529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavani S. R. P., Thompson M. A., Biteen J. S., Lord S. J., Liu N., Twieg R. J., Piestun R., Moerner W. E., “Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function,” Proc. Natl. Acad. Sci. U.S.A. 106(9), 2995–2999 (2009). 10.1073/pnas.0900245106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia S., Vaughan J. C., Zhuang X., “Isotropic three-dimensional super-resolution imaging with a self-bending point spread function,” Nat. Photonics 8(4), 302–306 (2014). 10.1038/nphoton.2014.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shechtman Y., Sahl S. J., Backer A. S., Moerner W. E., “Optimal Point Spread Function Design for 3D Imaging,” Phys. Rev. Lett. 113(13), 133902 (2014). 10.1103/PhysRevLett.113.133902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shechtman Y., Weiss L. E., Backer A. S., Sahl S. J., Moerner W. E., “Precise Three-Dimensional Scan-Free Multiple-Particle Tracking over Large Axial Ranges with Tetrapod Point Spread Functions,” Nano Lett. 15(6), 4194–4199 (2015). 10.1021/acs.nanolett.5b01396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spille J.-H., Kaminski T., Königshoven H.-P., Kubitscheck U., “Dynamic three-dimensional tracking of single fluorescent nanoparticles deep inside living tissue,” Opt. Express 20(18), 19697–19707 (2012). 10.1364/OE.20.019697 [DOI] [PubMed] [Google Scholar]
- 19.Speidel M., Jonás A., Florin E.-L., “Three-dimensional tracking of fluorescent nanoparticles with subnanometer precision by use of off-focus imaging,” Opt. Lett. 28(2), 69–71 (2003). 10.1364/OL.28.000069 [DOI] [PubMed] [Google Scholar]
- 20.Wu M., Roberts J. W., Buckley M., “Three-dimensional fluorescent particle tracking at micron-scale using a single camera,” Exp. Fluids 38(4), 461–465 (2005). 10.1007/s00348-004-0925-9 [DOI] [Google Scholar]
- 21.Toprak E., Balci H., Blehm B. H., Selvin P. R., “Three-Dimensional Particle Tracking Via Bifocal Imaging,” Nano Lett. 7(7), 2043–2045 (2007). 10.1021/nl0709120 [DOI] [PubMed] [Google Scholar]
- 22.Yu J., Wu C., Sahu S. P., Fernando L. P., Szymanski C., McNeill J., “Nanoscale 3D Tracking with Conjugated Polymer Nanoparticles,” J. Am. Chem. Soc. 131(51), 18410–18414 (2009). 10.1021/ja907228q [DOI] [PubMed] [Google Scholar]
- 23.Huhle A., Klaue D., Brutzer H., Daldrop P., Joo S., Otto O., Keyser U. F., Seidel R., “Camera-based three-dimensional real-time particle tracking at kHz rates and Ångström accuracy,” Nat. Commun. 6, 5885 (2015). 10.1038/ncomms6885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taute K. M., Gude S., Tans S. J., Shimizu T. S., “High-throughput 3D tracking of bacteria on a standard phase contrast microscope,” Nat. Commun. 6, 8776 (2015). 10.1038/ncomms9776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibson S. F., Lanni F., “Diffraction by a circular aperture as a model for three-dimensional optical microscopy,” J. Opt. Soc. Am. A 6(9), 1357–1367 (1989). 10.1364/JOSAA.6.001357 [DOI] [PubMed] [Google Scholar]
- 26.Denk W., Strickler J. H., Webb W. W., “Two-photon laser scanning fluorescence microscopy,” Science 248(4951), 73–76 (1990). 10.1126/science.2321027 [DOI] [PubMed] [Google Scholar]
- 27.Zipfel W. R., Williams R. M., Webb W. W., “Nonlinear magic: multiphoton microscopy in the biosciences,” Nat. Biotechnol. 21(11), 1369–1377 (2003). 10.1038/nbt899 [DOI] [PubMed] [Google Scholar]
- 28.Levi V., Ruan Q., Plutz M., Belmont A. S., Gratton E., “Chromatin Dynamics in Interphase Cells Revealed by Tracking in a Two-Photon Excitation Microscope,” Biophys. J. 89(6), 4275–4285 (2005). 10.1529/biophysj.105.066670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perillo E. P., Liu Y.-L., Huynh K., Liu C., Chou C.-K., Hung M.-C., Yeh H.-C., Dunn A. K., “Deep and high-resolution three-dimensional tracking of single particles using nonlinear and multiplexed illumination,” Nat. Commun. 6, 7874 (2015). 10.1038/ncomms8874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oron D., Tal E., Silberberg Y., “Scanningless depth-resolved microscopy,” Opt. Express 13(5), 1468–1476 (2005). 10.1364/OPEX.13.001468 [DOI] [PubMed] [Google Scholar]
- 31.Zhu G., van Howe J., Durst M., Zipfel W., Xu C., “Simultaneous spatial and temporal focusing of femtosecond pulses,” Opt. Express 13(6), 2153–2159 (2005). 10.1364/OPEX.13.002153 [DOI] [PubMed] [Google Scholar]
- 32.Therrien O. D., Aubé B., Pagès S., Koninck P. D., Côté D., “Wide-field multiphoton imaging of cellular dynamics in thick tissue by temporal focusing and patterned illumination,” Biomed. Opt. Express 2(3), 696–704 (2011). 10.1364/BOE.2.000696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng L.-C., Chang C.-Y., Lin C.-Y., Cho K.-C., Yen W.-C., Chang N.-S., Xu C., Dong C. Y., Chen S.-J., “Spatiotemporal focusing-based widefield multiphoton microscopy for fast optical sectioning,” Opt. Express 20(8), 8939–8948 (2012). 10.1364/OE.20.008939 [DOI] [PubMed] [Google Scholar]
- 34.Block E., Greco M., Vitek D., Masihzadeh O., Ammar D. A., Kahook M. Y., Mandava N., Durfee C., Squier J., “Simultaneous spatial and temporal focusing for tissue ablation,” Biomed. Opt. Express 4(6), 831–841 (2013). 10.1364/BOE.4.000831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi H., Yew E. Y. S., Hallacoglu B., Fantini S., Sheppard C. J. R., So P. T. C., “Improvement of axial resolution and contrast in temporally focused widefield two-photon microscopy with structured light illumination,” Biomed. Opt. Express 4(7), 995–1005 (2013). 10.1364/BOE.4.000995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schrödel T., Prevedel R., Aumayr K., Zimmer M., Vaziri A., “Brain-wide 3D imaging of neuronal activity in Caenorhabditis elegans with sculpted light,” Nat. Methods 10(10), 1013–1020 (2013). 10.1038/nmeth.2637 [DOI] [PubMed] [Google Scholar]
- 37.Spesyvtsev R., Rendall H. A., Dholakia K., “Wide-field three-dimensional optical imaging using temporal focusing for holographically trapped microparticles,” Opt. Lett. 40(21), 4847–4850 (2015). 10.1364/OL.40.004847 [DOI] [PubMed] [Google Scholar]
- 38.Vaziri A., Tang J., Shroff H., Shank C. V., “Multilayer three-dimensional super resolution imaging of thick biological samples,” Proc. Natl. Acad. Sci. U.S.A. 105(51), 20221–20226 (2008). 10.1073/pnas.0810636105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papagiakoumou E., Anselmi F., Bègue A., de Sars V., Glückstad J., Isacoff E. Y., Emiliani V., “Scanless two-photon excitation of channelrhodopsin-2,” Nat. Methods 7(10), 848–854 (2010). 10.1038/nmeth.1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lien C.-H., Lin C.-Y., Chen S.-J., Chien F.-C., “Dynamic particle tracking via temporal focusing multiphoton microscopy with astigmatism imaging,” Opt. Express 22(22), 27290–27299 (2014). 10.1364/OE.22.027290 [DOI] [PubMed] [Google Scholar]
- 41.Durst M. E., Zhu G., Xu C., “Simultaneous spatial and temporal focusing in nonlinear microscopy,” Opt. Commun. 281(7), 1796–1805 (2008). 10.1016/j.optcom.2007.05.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Straub A., Durst M. E., Xu C., “High speed multiphoton axial scanning through an optical fiber in a remotely scanned temporal focusing setup,” Biomed. Opt. Express 2(1), 80–88 (2011). 10.1364/BOE.2.000080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weiner A. M., “Femtosecond pulse shaping using spatial light modulators,” Rev. Sci. Instrum. 71(5), 1929–1960 (2000). 10.1063/1.1150614 [DOI] [Google Scholar]
- 44.Li C., Wagner W., Ciocca M., Warren W. S., “Multiphoton femtosecond phase-coherent two-dimensional electronic spectroscopy,” J. Chem. Phys. 126(16), 164307 (2007). 10.1063/1.2721562 [DOI] [PubMed] [Google Scholar]
- 45.Fenchel T., “Marine Plankton Food Chains,” Annu. Rev. Ecol. Syst. 19(1), 19–38 (1988). 10.1146/annurev.es.19.110188.000315 [DOI] [Google Scholar]
- 46.Boenigk J., Arndt H., “Particle handling during interception feeding by four species of heterotrophic nanoflagellates,” J. Eukaryot. Microbiol. 47(4), 350–358 (2000). 10.1111/j.1550-7408.2000.tb00060.x [DOI] [PubMed] [Google Scholar]
- 47.Son K., Brumley D. R., Stocker R., “Live from under the lens: exploring microbial motility with dynamic imaging and microfluidics,” Nat. Rev. Microbiol. 13(12), 761–775 (2015). 10.1038/nrmicro3567 [DOI] [PubMed] [Google Scholar]
- 48.“iXon Ultra 897 Specification,” http://www.andor.com/scientific-cameras/ixon-emccd-camera-series/ixon-ultra-897.
- 49.Small A., Stahlheber S., “Fluorophore localization algorithms for super-resolution microscopy,” Nat. Methods 11(3), 267–279 (2014). 10.1038/nmeth.2844 [DOI] [PubMed] [Google Scholar]