

Abstract
Purpose
We report the underlying genotype and explore possible genotypic-phenotypic correlations in a large cohort of choroideremia patients.
Methods
We studied prospectively a cohort of 79 patients diagnosed within a tertiary referral service for patients with retinal dystrophies. Phenotypic evaluation consisted of clinical examination, including visual acuity and residual retinal area by fundus autofluorescence (FAF). Genotype was established by sequencing. We also investigated whether particular genotypes were associated with more severe phenotypes by performing analysis of covariance (ANCOVA), with visual acuity and FAF as the dependent variables and age as the covariant.
Results
A total of 74 (94%) of patients in our cohort had causative mutations by sequencing, the majority of which were anticipated to be null. Of these, 35 (47%) had insertions and deletions, 13 (18%) had mutations predicted to affect splicing, and 26 (35%) had single point mutations. In the latter case, 13 of 21 (62%) pedigrees with single point mutations were C to T transitions at C-phosphate-G (CpG) dinucleotides. These mutations were spread across 5 of only 24 CpG dinucleotides in the entire CHM cDNA. Furthermore, these 5 locations are the only sites at which C to T transitions result in a stop codon. No clear evidence was found for genotype–phenotype correlation except in the instance of a patient with a large deletion involving neighbouring sequences.
Conclusions
In patients with a diagnosis of choroideremia made by a specialty service, there is a high likelihood of establishing a genetic diagnosis. The majority of causative mutations appear to be null and, therefore, may benefit from gene replacement therapy. A disproportionate number of single point mutations observed were C to T transitions, consistent with the evolutionary decay of CpG dinucleotides through methylation and subsequent deamination. Hence, the development of choroideremia in such patients may represent the unwanted consequence of human evolution; de novo mutations are predicted to arise at these sites in future generations. (ClinicalTrials.gov number, NCT01461213.)
Keywords: choroideremia, sequencing, genotyping, phenotyping
Choroideremia (OMIM 303100) is an X-linked recessive retinochoroidal dystrophy, first described by Mauthner in 1871,1 that affects approximately 1 in 50,000 males. The disease results in stereotypic changes to the ocular fundus that are characterised by centripetal loss of retinal and choroidal tissue. The functional correlates of these structural changes are early loss of scotopic vision and constriction of the visual field, both of which commence in the second decade of life. Central visual function, including visual acuity, typically is well-preserved until the fifth decade of life; however, there is variability in the rate of decline in visual function and the age at which central vision is affected. The causative gene, CHM (OMIM #300390), codes for rab-escort protein 1 (REP1),2 a 653 amino acid polypeptide crucial for intracellular trafficking within the human eye that comparative modeling has shown to be comprised of three principal domains.3 To date, almost all of the identified causative mutations appear to be null. Although the clinical phenotype can vary in terms of the age of onset and rate of decline, no evidence has been found for genotype–phenotype correlation.4,5
We describe the causative mutations in a large cohort of patients who also were examined clinically and explore potential genotype–phenotype correlations. By so doing, we further aimed to make inferences regarding the importance of particular regions of the CHM gene with respect to mutagenesis and to infer the importance of particular regions of the REP1 protein essential for normal function. Finally, we also sought to determine the prevalence of presumed null mutations and the rate of detection of mutations by sequencing. Both of these latter issues are especially important in assessing the potential suitability of gene therapy via a “gene replacement” strategy, a potential treatment that currently is under investigation by our group (ClinicalTrials.gov number, NCT01461213)6,7 and by others (ClinicalTrials.gov numbers, NCT02077361, NCT02341807).
Methods
The study was approved by the local Institute Review Board before its commencement and conformed to the tenets of the Declaration of Helsinki.
Clinical Examination Functional Assessment
Clinical examination consisted of full medical and ophthalmic history, anterior and posterior segment examination, and assessment of IOP. Visual acuity was assessed using a Bailey-Lovie style letter chart.8 For illustrative purposes, the visual acuities in the 7 patients who were count fingers (CF) or less were converted to logMAR values, as outlined by Holladay.9 A diagnosis of choroideremia, based upon clinical findings alone, was made by a senior clinician with expertise in inherited retinal disease (REM or SMD).
Structural Assessment
Fundus autofluorescence (FAF) imaging was performed with a Heidelberg Spectralis optical coherence tomography (OCT)–confocal scanning laser ophthalmoscopy (cSLO) machine with the BluePeak blue laser autofluorescence module (Heidelberg Engineering GmbH, Heidelberg, Germany) according to the manufacturer's protocols. Variations in corneal curvature were accounted for by entering the patient's keratometry values (“C-curve”). For assessing areas of autofluorescence, FAF imaging by the Spectralis with the 30° and 55° fields was performed. Fundus autofluorescence images were imported into Adobe Photoshop (Version 12; Adobe Systems, Inc., San Jose, CA, USA) and adjusted to similar levels of contrast. The area of autofluorescent zones was estimated using an automated pixel count using the Histogram function at 400% zoom, which then was converted into area measurements in mm2 using the imported scale bar.
Genotyping
DNA extraction using the Chemagic Magnetic Separation Module 1 (PerkinElmer Chemagen Technologie GmbH, Baesweiler Germany) was performed on whole blood specimens. Bidirectional sequencing was used to explore the 15 CHM gene exons and the intron/exon junctions. Amplification by polymerase chain reaction (PCR) was performed using standard conditions, with annealing of primers at 60°C. Then, 15 μL of PCR product was purified by removing Effluvia (dNTPs and primers) using Agencourt AMPure XP (Beckman Coulter Genomics, Danvers, MA, USA). The BigDye Terminator 3.1 Cycle Sequencing Kit (ABI; Life Technologies, Waltham, MA, USA) was used for sequencing, using the same primers as PCR, with the exception of exons 3 and 13, where alternative sequencing primers were used to account for repetitive sequences in the PCR products. Final purification was performed with Agencourt CleanSEQ (Beckman Coulter Genomics) and sequence analysis was performed using the 3730xl DNA Analyzer (ABI; Life Technologies). CHM exonic deletions and duplications were sought using multiplex ligation dependent probe amplification (MLPA) using a commercially available SALSA MLPA kit (P366 CHM-RP2-RPGR; MRC Holland, Amsterdam, The Netherlands). Reactions were conducted according to the manufacturer's instructions and analyzed with the 3130xl DNA Analyzer (ABI; Life Technologies).
Known nonpathogenic polymorphisms were excluded from the analysis (defined as variants described on nondisease-associated variant databases with quoted frequency data).10,11 Frameshift, nonsense, and splice site (affecting positions ±1, ±2 at the intron/exon junction) changes were considered pathogenic, as were deletions or duplications involving one or more exons. In cases of single exon deletions (determined by MLPA), genomic DNA was sequenced to exclude the occurrence of a rare single nucleotide polymorphism (SNP) at the relevant primer bindings site. In the absence of further information, remaining changes were considered to be of uncertain significance. Full details of the sequencing methods and interpretation of genetic data have been reported previously.10,11
Phenotype–Genotype Correlations
The majority of causative mutations, regardless of mechanism, were predicted to be null. We categorized mutations by mechanism into small deletions, small insertions, and insertion/deletions, large deletions (≥1 exon), splice site mutations, missense mutations, premature termination codons (i.e., point mutations resulting in a stop codon), and also grouped together patients in whom no mutation could be identified. We also identified those patients with mutations within 55 base pairs (bp) of the final exon junction as such mutations generally are not thought to result in nonsense mediated decay (NMD).12 This would, in turn, enable us to speculate on the importance of the C-terminus in REP1 functioning. Additionally, we compared mutations by mechanism. Results then were analyzed for correlation to visual function (best corrected visual acuity [BCVA]) and structure (FAF area, where available) by analysis of covariance (ANCOVA) with age as the controlled variable and using the data from right eyes in analysis.
Results
Spectrum of Genetic Mutations
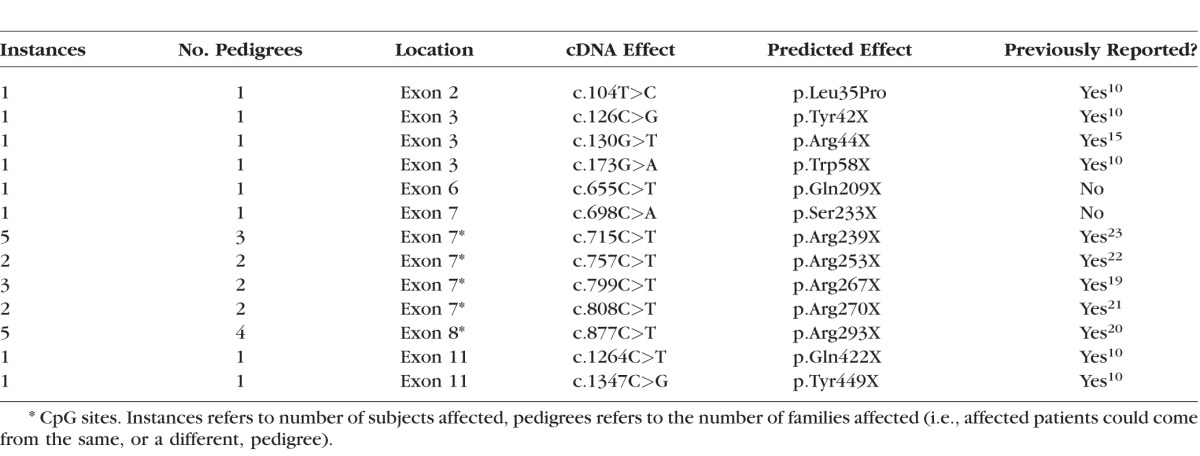
Of 79 patients who had undergone clinical examination, 74 had mutations identified (94%), making this the largest cohort of patients from a single center with identified mutations described. The mutations in our cohort are summarized in Figure 1 and Tables 1 to 5. The majority of mutations were predicted to be null. Two patients were identified in whom expression of a truncated protein was anticipated because of premature stop codons occurring downstream of 55 bp from the final exon junction. One patient was identified with a missense mutation (c.104T>C, p.Leu35Pro) known to be associated with low levels of REP1 expression.13 Mutations of uncertain significance included one patient with noncontiguous duplications of exons 1 to 2 and 9 to 12 who was subsequently demonstrated in our laboratory to have severely reduced expression of REP1 in peripheral blood by Western blotting (Edwards TL, Patricio MI, Williams J, Simunovic MP, MacLaren RE, submitted for publication, 2016). One patient was identified in whom a 6 bp (in frame) insertion at c.343 results in p.Ala115delinsValPheThr. One patient was identified with a small (in frame) deletion of 9 bp (c.872_880delAAAAGCGAA). Finally, four new variants presumed to cause aberrant splicing were identified (Table 3); one patient with a previously described c.940+3delA mutation of similarly uncertain significance was identified.10 Most small intragenic mutations were located within the region 700 to 900 (Fig. 1); one striking feature of this region is the presence of 5 CpG sites in which C to T transitions result in a premature stop codon. Mutations to 4 of these sites were reported previously by McTaggart et al.14 to be recurrent, that is, observed in multiple unrelated pedigrees. Furthermore, these are the only CpG sites (of a total of 24) in which C to T transition results in a stop codon.
Figure 1.

Number of pedigrees by cDNA location of mutations. The two mutations between 1701 and 1800 occur within 55 bp of the final exonic junction and are anticipated to result in expression of a truncated REP1 protein.
Table 1.
Summary of Major Deletions
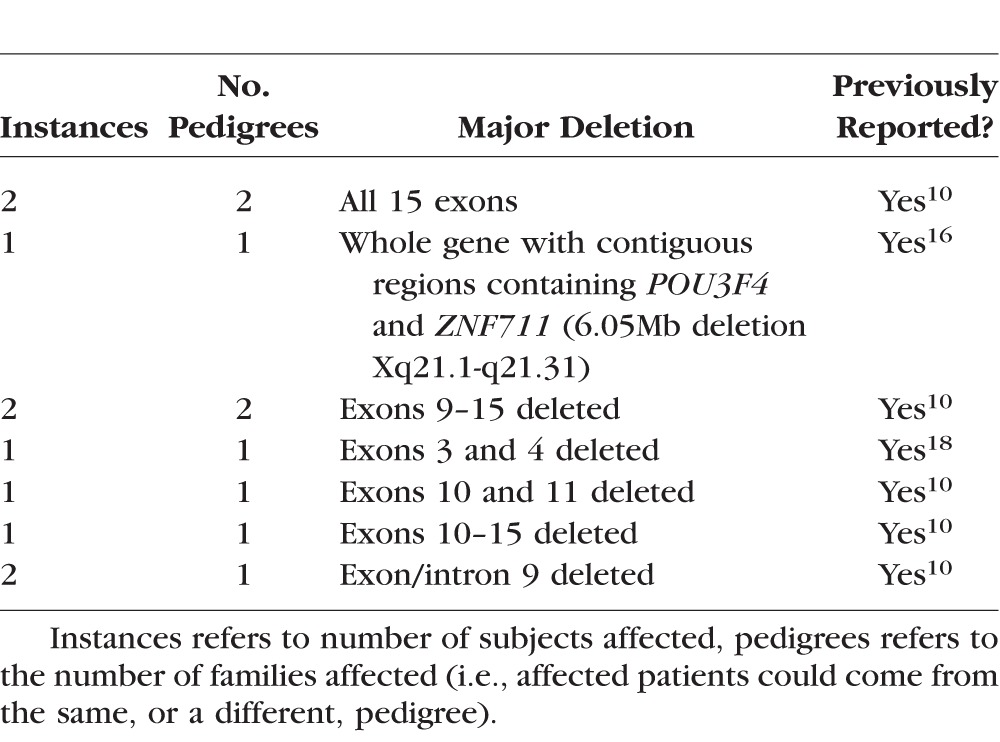
Table 5.
Insertions

Table 3.
Summary of Splice Site Mutations

Table 2.
Summary of Minor Deletions and Insertions
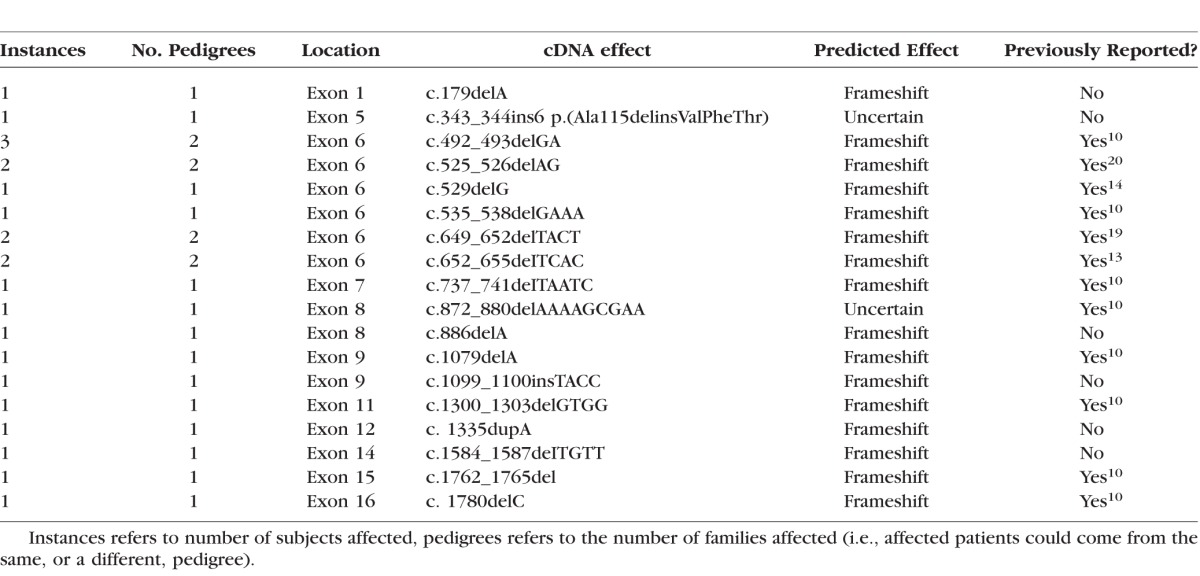
Table 4.
Point Mutations
Correlation Between Mutation and Visual Function
The majority of CHM mutations are predicted to be null, either through major deletions, or more commonly, through nonsense mutations. One mutation resulted from noncontiguous exon duplications and two occurred within 55 bp of the final intron/exon junction and would not be predicted to result in NMD. In view of the small number of subjects with such mutations, it is difficult to establish their effects on phenotype (Fig. 1). Analysis of covariance (with age as the covariant) does not suggest a significant difference between mutations involving the C-terminus and those occurring upstream of this location (P > 0.05) in terms of logMAR acuity (Fig. 2). Similarly, no correlation between genotype by category and logMAR acuity was observed (P > 0.05; Fig. 2), nor was a correlation between genotype and FAF area observed (P > 0.05; Fig. 3).
Figure 2.

Visual acuity (logMAR) versus age (years). PTC, premature termination codon; LD, large deletion (≥1 exon); SD, small deletion; MIS, missense mutation; SI, small insertion or insertion/deletion; SS, splice site mutation; NMF, no mutation found; LI, large insertion.
Figure 3.

Fundus autofluorescence area (mm2) versus age (years).
Correlation Between Mutation and Systemic Disease
One patient was identified with a “choroideremia plus” syndrome.15 This individual has a history of deafness and developmental delay, and was demonstrated to have a large deletion involving the neighbouring POU3F4 and ZNF711 genes. The former is a transcription factor implicated in X-linked congenital deafness,16 while the latter is a zinc finger protein previously described as a cause of X-linked “mental retardation.”17
Discussion
We identified a total of 48 unique mutations in 74 patients from a cohort of 79 affected males examined clinically who had been diagnosed with choroideremia. Thus, the rate of detection (94%) was higher than that reported previously using the same molecular diagnostic techniques in patients referred from a variety of centers10 (rather than from a single center with particular interest in choroideremia and inherited eye disease).
In keeping with previous reports, the majority of identified mutations were anticipated to be null.18–23 This, combined with the low rate of patients in whom sequencing failed to identify a genetic cause, reaffirms the logic of a gene-replacement strategy as a possible means of treatment, such as that currently undertaken at our center.6,7
In our cohort, there was a notable absence of disease-causing missense mutations that also would cause protein misfolding. The paucity of missense mutations in choroideremia is in contrast to the majority of human genetic diseases (which are caused predominantly by such mutations). This may be due to the role of REP1 as a chaperone protein, because the folding may be determined by the Rab27a ligand, rather than posttranslational modification of the REP1 protein itself. Since Rab27a binds equally well to REP2, which shares less than 80% sequence homology to REP1,24 it is logical to assume that a significant number of single amino acid substitutions in the REP1 sequence would be well tolerated. A preponderance of nonsense mutations has been observed similarly in mutations to other GTPase regulators, such as RPGR and KRIT1. Possible explanations (some more plausible than others) have been advanced previously for this phenomenon.4 One possibility is that some missense mutations may result in milder or different phenotypes that previously have evaded detection/recognition. With the advent of rapid sequencing, the likelihood of such occurrences is becoming increasingly remote. A further suggestion is that some missense mutations may exert a dominant negative effect and thereby disrupt the activity of REP2. Therefore, such mutations might be anticipated to be lethal in utero in males, while female carriers might remain largely asymptomatic by virtue of lyonization. The fact that REP1 is a monomer makes this possibility less likely. However, a further (and related) possibility to be considered is that mutations resulting in the expression of a protein product also might be less amenable to a gene replacement strategy because expressed mutant proteins could interfere with wild-type function. Even if this were to be the case, such mutations form the minority of cases; furthermore, overexpression may negate such hypothesized effects.
Two nonsense mutations were observed to occur within 55 bp of the final intron-exon junction and, therefore, would not be anticipated to result in NMD.12 Previous studies of the REP1 molecule suggest that the C-terminus acts as mobile shield that covers a conserved hydrophobic patch on the surface of REP1 and its presence is proposed to be essential for the maintenance of solubility.25 Furthermore, the C-terminus is essential for membrane attachment.26 The fact that C-terminus involving mutations are pathogenic further suggests that it is essential for the normal functioning of REP1 in vivo. It should be noted that some caution must be exercised in the interpretation of these findings: in particular, it has been argued that X-chromosome coded genes may be less likely to be subject to NMD than autosomes,27 and so the differential effects of the location of mutations with respect to the final intron-exon junction may be blunted.
We found no evidence for genotype–phenotype correlation by genetic mechanism with one exception: a single patient with a large mutation involving the CHM gene as well as the neighboring POU3F4 and ZNF711 genes. Mutations to POU3F4 have been identified as a cause of sensorineural hearing loss and mutations of ZNF711 have been described previously as a cause of intellectual impairment. As would be anticipated, this patient's medical history was notable for hearing impairment and developmental disability. Choroideremia with deafness and development delay (syn. Xq21 deletion syndrome) was first described by Ayazi in a single pedigree.28 Although subsequent studies localized this syndrome to Xq21.1-21.3, the first complete molecular analysis of the syndrome was performed only recently by Iossa et al.,17 who similarly identified loss of the CHM, POU3F4, and ZNF711 genes.
The most frequently encountered mutation, in terms of number of pedigrees affected, occurred at a CpG site (877 C>T, Arg293X). Furthermore, the majority of point mutations occurred at the only 5 CpG sites that would code for a stop codon as the result of a C>T transition. Such transitions are believed to occur with high frequency in evolution through inherent instability via methylation and subsequent deamination. Furthermore, these mutations were found to be recurrent, that is, they each occurred in more than one pedigree. Given the known instability of CpG sites and our observations, we hypothesized that these 5 mutations would be anticipated to be encountered with increased frequency in future generations. We also may assume that similar C>T transitions are occurring in the other CpG sites but we do not detect them, either because they are silent (i.e., if C>T is at codon position 3) or because the single amino acid change does not significantly impair REP1 function, for the reasons explained above.
In summary, therefore, we found no evidence for a correlation between different CHM mutations and phenotype in our cohort. Therefore, what factors may account for phenotypic variation among choroideremia patients? First, it may be that such correlations elude us because of the small numbers of patients with certain mutations, as outlined above. Leaving this possibility aside, it is further possible that polymorphisms in other genes whose products participate in the prenylation reactions facilitated by REP1 enable REP2 to partially compensate for the loss of the former, thereby blunting the effects of null mutations. We found clinical evidence to support the assertion that the C-terminus of REP-1 is crucial for its correct functioning, as two of our patients had mutations in this region, which are anticipated to result in an expressed, though aberrant, protein product. Furthermore, we found that 62% of the unique point mutations responsible for choroideremia in our cohort occurred at 5 CpG sites in which C>T transition results in a stop codon. Such mutations are recurrent and may represent the unwanted consequence of CpG instability that is a feature in the evolution of humans from the great apes.29,30 Our data affirmed that patients screened in specialist centers are highly likely to have an identified causative mutation revealed by sequencing (94% of patients in our sample) and confirmed that a gene replacement approach is a logical treatment strategy to be explored.
Acknowledgments
Supported by the Foundation Fighting Blindness (MPS); RANZCO Eye Foundation (MPS); the Bayer Global Ophthalmology Awards Program Fellowship (MPS), the Oxford Nuffield Foundation (TLE), the Health Innovation Challenge Fund, Royal College of Surgeons of Edinburgh; the Wellcome Trust, UK Department of Health (jointly funded by the Wellcome Trust and the Department of Health) Grant No. HICF-091984; NIHR Oxford Biomedical Research Centre, Oxford Nuffield Medical Fellowship.
Disclosure: M.P. Simunovic, None; J.K. Jolly, None; K. Xue, None; T.L. Edwards, None; M. Groppe, None; S.M. Downes, None; R.E. MacLaren, None
References
- 1. Mauthner L. Ein Fall von Choroideremia. Ber Naturw Med Vereins. 1871; 11. [Google Scholar]
- 2. Seabra MC,, Brown MS,, Goldstein JL. Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl transferase. Science. 1993; 259: 377–381. [DOI] [PubMed] [Google Scholar]
- 3. Sergeev YV,, Smaoui N,, Sui R,, et al. The functional effect of pathogenic mutations in Rab escort protein 1. Mutat Res. 2009; 665: 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Freund P,, Furgoch M,, MacDonald I. Genotype-phenotype analysis of male subjects affected by choroideremia. Invest Ophthalmol Vis Sci. 2013; 54: 1567–1567. [Google Scholar]
- 5. Freund PR,, Sergeev YV,, MacDonald IM. Analysis of a large choroideremia dataset does not suggest a preference for inclusion of certain genotypes in future trials of gene therapy. Mol Genet Genomic Med. 2016; 4: 34–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. MacLaren RE,, Groppe M,, Barnard AR,, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014; 383: 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Edwards TL,, Jolly JK,, Groppe M,, et al. Visual acuity after retinal gene therapy for choroideremia. N Engl J Med. 2016; 374: 1996–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bailey IL,, Lovie JE. New design principles for visual acuity letter charts. Am J Optom Physiol Opt. 1976; 53: 740–745. [DOI] [PubMed] [Google Scholar]
- 9. Holladay JT. Proper method for calculating average visual acuity. J Refract Surg. 1997; 13: 388–391. [DOI] [PubMed] [Google Scholar]
- 10. Ramsden SC,, O'Grady A,, Fletcher T,, et al. A clinical molecular genetic service for United Kingdom families with choroideraemia. Eur J Med Genet. 2013; 56: 432–438. [DOI] [PubMed] [Google Scholar]
- 11. Ellingford JM,, Barton S,, Bhaskar S,, et al. Molecular findings from 537 individuals with inherited retinal disease [published online ahead of print May 11 2016] J Med Genet. doi:10.1136/jmedgenet-2016-103837. [DOI] [PMC free article] [PubMed]
- 12. Nagy E,, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998; 23: 198–199. [DOI] [PubMed] [Google Scholar]
- 13. Esposito G,, De Falco F,, Tinto N,, et al. Comprehensive mutation analysis (20 families) of the choroideremia gene reveals a missense variant that prevents the binding of REP1 with Rab geranylgeranyl transferase. Hum Mutat. 2011; 32: 1460–1469. [DOI] [PubMed] [Google Scholar]
- 14. McTaggart KE,, Tran M,, Mah DY,, Lai SW,, Nesslinger NJ,, MacDonald IM. Mutational analysis of patients with the diagnosis of choroideremia. Hum Mutat. 2002; 20: 189–196. [DOI] [PubMed] [Google Scholar]
- 15. Poloschek CM,, Kloeckener-Gruissem B,, Hansen LL,, Bach M,, Berger W. Syndromic choroideremia: sublocalization of phenotypes associated with Martin-Probst deafness mental retardation syndrome. Invest Ophthalmol Vis Sci. 2008; 49: 4096–4104. [DOI] [PubMed] [Google Scholar]
- 16. Iossa S,, Costa V,, Corvino V,, et al. Phenotypic and genetic characterization of a family carrying two Xq21.1-21.3 interstitial deletions associated with syndromic hearing loss. Mol Cytogenet. 2015; 8: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tarpey PS,, Smith R,, Pleasance E,, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009; 41: 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garcia-Hoyos M,, Lorda-Sanchez I,, Gomez-Garre P,, et al. New type of mutations in three spanish families with choroideremia. Invest Ophthalmol Vis Sci. 2008; 49: 1315–1321. [DOI] [PubMed] [Google Scholar]
- 19. van den Hurk JA,, Schwartz M,, van Bokhoven H,, et al. Molecular basis of choroideremia (CHM): mutations involving the Rab escort protein-1 (REP-1) gene. Hum Mutat. 1997; 9: 110–117. [DOI] [PubMed] [Google Scholar]
- 20. van Bokhoven H,, Schwartz M,, Andreasson S,, et al. Mutation spectrum in the CHM gene of Danish and Swedish choroideremia patients. Hum Mol Genet. 1994; 3: 1047–1051. [DOI] [PubMed] [Google Scholar]
- 21. Fujiki K,, Hotta Y,, Hayakawa M,, et al. REP-1 gene mutations in Japanese patients with choroideremia. Graefes Arch Clin Exp Ophthalmol. 1999; 237: 735–740. [DOI] [PubMed] [Google Scholar]
- 22. Hayakawa M,, Fujiki K,, Hotta Y,, et al. Visual impairment and REP-1 gene mutations in Japanese choroideremia patients. Ophthalmic Genet. 1999; 20: 107–115. [DOI] [PubMed] [Google Scholar]
- 23. Francis PJ,, Fishman GA,, Trzupek KM,, MacDonald IM,, Stone EM,, Weleber RG. Stop mutations in exon 6 of the choroideremia gene, CHM associated with preservation of the electroretinogram. Arch Ophthalmol. 2005; 123: 1146–1149. [DOI] [PubMed] [Google Scholar]
- 24. Larijani B,, Hume AN,, Tarafder AK,, Seabra MC. Multiple factors contribute to inefficient prenylation of Rab27a in Rab prenylation diseases. J Biol Chem. 2003; 278: 46798–46804. [DOI] [PubMed] [Google Scholar]
- 25. Rak A,, Pylypenko O,, Niculae A,, Pyatkov K,, Goody RS,, Alexandrov K. Structure of the Rab7:REP-1 complex: insights into the mechanism of Rab prenylation and choroideremia disease. Cell. 2004; 117: 749–760. [DOI] [PubMed] [Google Scholar]
- 26. Alory C,, Balch WE. Organization of the Rab-GDI/CHM superfamily: the functional basis for choroideremia disease. Traffic. 2001; 2: 532–543. [DOI] [PubMed] [Google Scholar]
- 27. Yin S,, Deng W,, Zheng H,, Zhang Z,, Hu L,, Kong X. Evidence that the nonsense-mediated mRNA decay pathway participates in X chromosome dosage compensation in mammals. Biochem Biophys Res Commun. 2009; 383: 378–382. [DOI] [PubMed] [Google Scholar]
- 28. Ayazi S. Choroideremia, obesity, and congenital deafness. Am J Ophthalmol. 1981; 92: 63–69. [DOI] [PubMed] [Google Scholar]
- 29. Hernando-Herraez I,, Heyn H,, Fernandez-Callejo M,, et al. The interplay between DNA methylation and sequence divergence in recent human evolution. Nucleic Acids Res. 2015; 43: 8204–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hernando-Herraez I,, Garcia-Perez R,, Sharp AJ,, Marques-Bonet T. DNA methylation: insights into human evolution. PLoS Genet. 2015; 11: e1005661. [DOI] [PMC free article] [PubMed] [Google Scholar]