

Abstract
First principles prediction of the structure, thermodynamics and solubility of organic molecular crystals, which play a central role in chemical, material, pharmaceutical and engineering sciences, challenges both potential energy functions and sampling methodologies. Here we calculate absolute crystal deposition thermodynamics using a novel dual force field approach whose goal is to maintain the accuracy of advanced multipole force fields (e.g. the polarizable AMOEBA model) while performing more than 95% of the sampling in an inexpensive fixed charge (FC) force field (e.g. OPLS-AA). Absolute crystal sublimation/deposition phase transition free energies were determined using an alchemical path that grows the crystalline state from a vapor reference state based on sampling with the OPLS-AA force field, followed by dual force field thermodynamic corrections to change between FC and AMOEBA resolutions at both end states (we denote the three step path as AMOEBA/FC). Importantly, whereas the phase transition requires on the order of 200 nsec of sampling per compound, only 5 nsec of sampling was needed for the dual force field thermodynamic corrections to reach a mean statistical uncertainty of 0.05 kcal/mol. For five organic compounds, the mean unsigned error between direct use of AMOEBA and the AMOEBA/FC dual force field path was only 0.2 kcal/mol and not statistically significant. Compared to experimental deposition thermodynamics, the mean unsigned error for AMOEBA/FC (1.4 kcal/mol) was more than a factor of two smaller than uncorrected OPLS-AA (3.2 kcal/mol). Overall, the dual force field thermodynamic corrections reduced condensed phase sampling in the expensive force field by a factor of 40, and may prove useful for protein stability or binding thermodynamics in the future.
Graphical Abstract
Introduction
Organic molecular crystals play a central role in chemical, material, pharmaceutical and engineering sciences.1–4 First principles prediction of their structure5–9, thermodynamics10, 11 and solubility12, 13 is a challenge for both potential energy functions and sampling methodologies.14, 15 For example, it has been shown that classical force fields based on fixed atomic partial charges (FC), such as Amber16, CHARMM17 and OPLS-AA18, lack the accuracy needed to correctly rank the relative stability of alternative polymorphs.19, 20 To achieve sufficient accuracy for crystal structure prediction, atomic multipole expansions can be used to systematically reproduce the electrostatic potential outside the van der Waals surface of a rigid molecule, as defined by electronic structure calculations.21, 22 However, molecular charge distributions are sensitive to both conformation and the electric field across the molecule, due in part to electronic polarization.23 Thus, to apply the configurational sampling algorithms needed to quantify thermodynamics, multipolar force fields must address transferability of their multipole moments as a function of molecular conformation, while also consistently treating both intra- and intermolecular polarization.23, 24 A few examples of multipolar energy functions include AMOEBA (Atomic Multipole Optimized Energetics for Biomolecular Applications)24–26, GMM (Gaussian Multipolar Model)27, 28, SIBFA (Sum of Interactions Between Fragments Ab Initio Computed)29, 30 and NEMO (Non-empirical Molecular Orbital)31, which are described in recent reviews32–34. The accuracy and transferability improvements of AMOEBA relative to FC force fields have been demonstrated in the context of water25, 35, ion solvation36, the properties of small organic molecules24, 26, 36–38 and for protein energetics.34, 39
The increased domain of applicability of advanced multipolar force fields, however, comes at a price of greater computational expense relative to FC force fields by a factor of 5–10 or more for energy and force evaluations. To ameliorate the expense of sampling advanced potential energy functions, previous work to reweight from molecular mechanics (MM) sampling has been explored in the context of determining thermodynamics for quantum mechanical (QM) or QM/MM potential energy surfaces.40–49 Recent work includes the dual-topology alchemical Hamiltonian replica exchange method (DTA-HREM)50–52, non-Boltzmann Bennett (NBB) reweighting53–55, and the multistate Bennett acceptance ratio (MBAR)56, 57 approaches. The emergence of increasingly sophisticated polarizable atomic multipole force fields for organic molecules and proteins further motivates approaches that either reweight FC trajectories or define a path that smoothly connects FC states to those defined by a more advanced multipolar force field.
Perhaps the simplest approach to computing the free energy difference between FC and more advanced force fields, such as AMOEBA, is direct reweighting via the Zwanzig relationship
| Equation 1 |
where kB is Boltzmann’s constant, T is temperature in degrees Kelvin and the angle brackets denote an ensemble average.58 This approach evaluates the potential energy of the expensive model only at intermediate samples. The reverse perturbation offers no efficiency improvement due to requiring direct sampling of the more expensive ensemble
| Equation 2 |
However, convergence of reweighting as shown in Eqs. 1 or 2 may fail due to lack of phase space overlap between force fields, which arises due to differences in bonded terms (i.e. equilibrium bond distances or bond angles) and/or intermolecular contact distances (i.e. due to the balance of van der Waals and Coulombic interactions)59, 60. Phase space overlap between resolutions can be improved by coordinating their design and parameterization60–62.
Here we explore an approach that performs more than 95% of the sampling using an inexpensive fixed charge force field, followed by the addition of two rigorous corrections to recover thermodynamics consistent with the more advanced force field. The method mitigates nontrivial differences between force fields by defining a dual force field (DFF) potential that enables explicit sampling of the transition between resolutions
| Equation 3 |
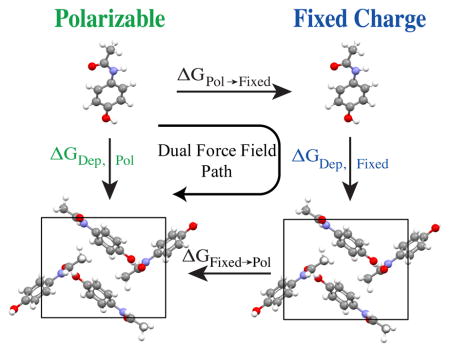
where UDFF defines a smooth transition between the fixed charge energy function at λ =0 and the polarizable AMOEBA energy function at λ =1. Calculation of free energy differences due to force field resolution changes at both vapor and crystalline end states permits the computationally demanding sublimation/deposition phase transition to be sampled with the inexpensive force field (Figure 1). The approach has similarities to “dual topology” style potential energy functions, which smoothly interpolate between chemical functional groups63.
Figure 1.

This diagram summarizes the thermodynamic cycle for computing absolute deposition free energy at the accuracy of the polarizable AMOEBA force field, but with an efficiency approaching the fixed atomic partial charge OPLS-AA force field. The vertical sublimation/deposition phase transition steps each require ~200 nsec of sampling while the horizontal steps to change resolution converge in only 5 nsec. This equates to a factor of 40 reduction in the amount of condensed phase AMOEBA sampling required.
Using five organic compounds, we show that the DFF approach defines a thermodynamic path that is more efficient than direct simulation in AMOEBA, but maintains its agreement with experiment in the context of calculating absolute sublimation/deposition phase transitions. OPLS-AA18, 64 and AMOEBA24–26 were used as the fixed charge (i.e. cheap) and polarizable multipole (i.e. expensive) force fields, respectively, as implemented in the open source Force Field X (FFX) software package (http://ffx.biochem.uiowa.edu)65–67 version 1.0.0-beta. Overall, the DFF interpolation between resolutions exhibits rapid convergence relative to the phase transition and allows decreased condensed phase sampling in the expensive potential by a factor of 40 for a thermodynamic path characterized by Growth of the Asymmetric Unit into a Crystal via alCHemy (GAUCHE) and described previously14, 15. Thus, polarizable atomic multipole AMOEBA thermodynamics are reproduced with an expense approaching that of fixed charge models.
Methods
Lattice Potential Energies
We analyzed five molecules from a prior study on deposition thermodynamics.15 These compounds are shown in Figure 2 and include acetanilide,68 paracetamol (polymorph I),69 methyl paraben (polymorph II),70 ethyl paraben,71 and phenacetin.72 Experimental lattice parameters and the space group for each compound are given in Table 1, along with asymmetric unit composition and unit cell volume in Table 2. The deposition free energy values from the prior study, computed using AMOEBA directly, will be compared to the three step DFF thermodynamic path that combines FC phase transitions and DFF corrections. Analogous to previous work15, the five molecules were optimized in the crystalline and vapor states using both the AMOEBA and OPLS-AA force fields within the FFX program. Space group and lattice parameters were constrained to their experimental values (Table 1) for both AMOEBA and OPLS-AA optimizations. AMOEBA parameters were obtained from Poltype38, while OPLS-AA 2005 parameters were obtained from Schrödinger73. To calculate lattice energies, the minimized vapor energy was subtracted from the minimized crystal energy on a per molecule basis.
Figure 2.

The structure of each organic compound.
Table 1.
Compounds studied and their associated CSD reference codes, space groups and unit cell parameters. Roman numerals following paracetamol and methyl paraben correspond to polymorph.
| Compound | CSD Code | Space Group | a | b | c | α | β | γ |
|---|---|---|---|---|---|---|---|---|
| Acetanilide | ACANIL | Pbca | 19.64 | 9.48 | 7.98 | 90 | 90.0 | 90 |
| Paracetamol I | HXACAN01 | P21/c | 11.72 | 9.40 | 12.93 | 90 | 147.0 | 90 |
| Methyl Paraben II | CEBGOF03 | P21/c | 4.82 | 14.63 | 10.24 | 90 | 99.8 | 90 |
| Ethyl Paraben | FEGLEI | P21/c | 13.76 | 13.18 | 11.58 | 90 | 125.5 | 90 |
| Phenacetin | PYRAZB10 | P21/c | 13.25 | 9.65 | 7.81 | 90 | 104.9 | 90 |
Table 2.
Molecular weight, number of molecules per asymmetric unit (AU), unit cell (UC) volume, number of unit cell molecules, volume per molecule and experimental temperature for each crystal studied.
| Compound | Mol. Weight (g/mol) | AU Molecules | UC Vol. (Å3) | Z | Vol./Z (Å3) | Temp. (K) |
|---|---|---|---|---|---|---|
| Acetanilide | 135.16 | 1 | 1486.1 | 8 | 185.8 | 297 |
| Paracetamol I | 151.16 | 1 | 776.3 | 4 | 194.1 | 297 |
| Methyl Paraben II | 152.15 | 1 | 711.3 | 4 | 177.8 | 100 |
| Ethly Paraben | 166.17 | 2 | 1710.2 | 8 | 213.8 | 297 |
| Phenacetin | 179.22 | 1 | 965.0 | 4 | 241.3 | 297 |
| Equation 4 |
A van der Waals cutoff of 12.0 Å was used, with a multiplicative switch tapering the interaction energy to zero starting at 10.8 Å, which is consistent with parameterization of the AMOEBA model for water, organic molecules and proteins24, 25, 39. Polarizable electrostatic evaluations were conducted using a smooth74 particle-mesh Ewald75 (PME) algorithm for multipoles76 which maintained the self-consistent field with a convergence criterion of 10−5 RMS Debye and supported space group symmetry.65 For this work, PME parameters included a real-space cutoff of 9 Å, a mesh density of 2.0 grid points per Å, eighth order B-splines, and an Ewald Parameter of 0.42. The molecules were minimized to a tight RMS gradient convergence criterion of 10−4 kcal/mol/Å.
Deposition Free Energy and Dual Force Field Corrections
The DFF method, as employed in this work, adds a free energy correction composed of two terms to the deposition free energy calculated using the inexpensive OPLS-AA force field to recover direct AMOEBA thermodynamics, at approximately OPLS-AA efficiency. Consistent with our earlier work on these systems15, the NVT ensemble was sampled at 298 degrees Kelvin using stochastic dynamics. For each of the three simulation legs, five independent trajectories were collected beginning from different random velocity vectors. The first simulation leg calculates the free energy to change from AMOEBA resolution into OPLS-AA resolution in vapor . Next, the inexpensive potential UFC(X) is sampled to determine the FC deposition/sublimation phase transition free energy . Finally, the last simulation leg calculates the free energy to convert back from OPLS-AA resolution to AMOEBA resolution in the crystal state . Summing the three simulation legs yields the AMOEBA/FC deposition free energy
| Equation 5 |
Orthogonal Space Sampling of the Thermodynamic Paths
The Orthogonal Space Random Walk (OSRW) method builds up a time-dependent bias by depositing two-dimensional Gaussian-shaped repulsive potentials as a function of the state variable λ and the derivative of the potential energy with respect to λ (Fλ = ∂U/∂λ)77, 78. The total potential energy is then given by
| Equation 6 |
where gm(λ, Fλ) is the sum of the repulsive potentials (i.e. hills) centered at states given by [λ(ti), Fλ(ti)]14:
| Equation 7 |
The additional biasing dimension promotes crossing of hidden barriers relative to the simpler one-dimensional bias of original metadynamics approaches.77, 78 We note that the gradient of Um (i.e. the partial derivatives with respect to all atomic coordinates), which is needed to integrate equations of motion during OSRW dynamics, requires partial derivatives of the target function UDFF(λ, X) that include ∂UDFF(λ, X)/∂λ, ∂2UDFF(λ, X)/∂λ2 and ∂2UDFF(λ, X)/∂λ ∂X. These are given by
| Equation 8 |
| Equation 9 |
and
| Equation 10 |
The first two results above (Equations 8 and 9) are the 1st and 2nd partial derivatives of the dual force field potential energy (Equation 3) with respect to λ, while the last result (Equation 10) is equivalent to the difference in the partial derivative for each force field with respect to atomic coordinate X. As shown in Figure 3, the ensemble average thermodynamic force 〈 ∂U/∂λ 〉 is smooth and well-behaved as resolutions between the OPLS-AA and AMOEBA force fields are sampled (i.e. for 0 ≤ λ ≤ 1). However, if this had not been the case (i.e. for force fields that are more dramatically different), higher powers of λ could be explored (i.e. in Equation 3 apply the substitution λ → λ2).
Figure 3.

The ensemble average thermodynamic force 〈∂U/∂λ〉 along the depositon path (Panel A) and dual force field paths (Panel B) are shown for paracetamol (Par) and acetanilide (Ace).
Simulations with Non-Crystallographic Symmetry
In cases where more than one molecule is present in the asymmetric unit (i.e. non-crystallographic symmetry), intermolecular interactions are smoothly turned off as the simulation transitions from the crystalline state to the vapor state. This allows each molecule to be independent in the vapor state (i.e. they can pass through each other). For example, due to non-crystallographic symmetry in the ethyl paraben crystal (Table 2), two molecules were simulated and computed deposition values normalized by a factor of two.
Results
Lattice Potential Energies
Displayed in Table 3 is the lattice energy for each compound using both OPLS-AA and AMOEBA force fields. The mean absolute difference for OPLS-AA relative to AMOEBA of 3.6 kcal/mol is significant relative to the goal of achieving chemical accuracy (i.e. ~1.0 kcal/mol). In particular, the three amide containing compounds (acetanilide, paracetamol and phenacetin) show increased crystalline stability of 4.0 to 6.6 kcal/mol under OPLS-AA relative to AMOEBA. The differences for the ester containing methyl and ethyl paraben compounds of 0.5 and 1.2 kcal/mol, respectively, are more modest.
Table 3.
OPLS-AA and AMOEBA lattice potential energies for each compound (kcal/mol).
| Compound | OPLS-AA
|
AMOEBA
|
Diff. | ||||
|---|---|---|---|---|---|---|---|
| Crystal | Vacuum | Lattice | Crystal | Vacuum | Lattice | ||
| Acetanilide | −28.73 | −2.01 | −26.72 | −38.05 | −15.32 | −22.73 | 3.99 |
| Paracetamol | −44.45 | −10.12 | −34.32 | −41.04 | −13.35 | −27.70 | 6.63 |
| Methyl Paraben | −23.58 | −0.13 | −23.45 | −30.77 | −8.48 | −22.29 | 1.16 |
| Ethyl Paraben | −24.33 | 0.78 | −25.11 | −28.56 | −3.98 | −24.58 | 0.53 |
| Phenacetin | −37.49 | −5.70 | −31.80 | −36.05 | −9.92 | −26.13 | 5.67 |
|
| |||||||
| Mean | −28.28 | −24.68 | 3.60 | ||||
Deposition Free Energy and Dual Force Field Corrections
As shown in Figure 1 above, the accelerated thermodynamic pathway consists of three steps. First, the transition to the cheap force field is completed in the vapor state , followed by the deposition/sublimation phase transition for the asymmetric unit using the cheap force field , and finally the transition back to the expensive force field in the crystalline state . Convergence of the independent deposition/sublimation phase transition trajectories for the asymmetric unit in both the expensive (i.e. AMOEBA) and cheap (i.e. OPLS-AA) force fields is presented in Figure 4 for both acetanilide and paracetamol. Convergence of the independent DFF transition trajectories in vapor and crystalline phases is shown in Figure 5 for both acetanilide and paracetamol. Free energy differences were computed by collecting the ensemble average thermodynamic force 〈 ∂U/∂λ 〉 for 200 equally sized bins along the λ parameterized thermodynamic path, followed by numerical thermodynamic integration. Simulation legs were considered to be converged once the mean free energy difference for the five independent trajectories changed by less than 0.1 kcal/mol for the last ¼ of the trajectory.
Figure 4.

Convergence of the deposition free energy is shown for alchemical simulations of the asymmetric unit of acetanilide (Panels A & B) and paracetamol (Panels C & D). Panels A & C use the OPLS-AA force field while Panels B & D use the AMOEBA force fields.
Figure 5.

Convergence of the free energy change for acetanilide (Panels A & B) and paracetamol (Panels C & D) dual force field simulations are shown. In Panels A & C, the AMOEBA force field is transformed into the OPLS-AA force field in vapor. In Panels B & D, the OPLS-AA force field is transformed into the AMOEBA force field in the crystalline environment.
The total DFF corrections in Table 4 follow the trend of the lattice potential energy differences shown in Table 3, albeit slightly smaller in magnitude in all cases. After 5 nsec of sampling for both of the dual force field legs, the mean standard deviation of the free energy difference had fallen to 0.05 kcal/mol (Table 4). The direct deposition/sublimation phase transitions for the asymmetric unit using OPLS-AA and AMOEBA15 are shown in Table 5. The differences between AMOEBA and OPLS-AA follow the trends seen in Table 3 for lattice potential energy differences, with OPLS-AA showing greater stabilization than AMOEBA. For example, the mean unsigned error of 3.1 kcal/mol is only slightly smaller than the mean lattice potential energy difference of 3.6 kcal/mol. The standard deviation for the phase transition free energy after 200 nsec of sampling was 0.41 kcal/mol for AMOEBA and 0.27 kcal/mol for OPLS-AA (Table 5). The DFF corrections from Table 4 were added to the pure OPLS-AA deposition values in Table 5 to yield corrected values denoted .
Table 4.
DFF thermodynamic corrections for each compound in vapor and crystalline phases after 5 nsec of sampling (kcal/mol). Each value is the mean of 5 trials ± the standard deviation.
| Compound |
|
|
Total | ||
|---|---|---|---|---|---|
| Acetanilide | 14.00±0.02 | −10.12±0.02 | 3.87±0.03 | ||
| Paracetamol | 2.72 ±0.03 | 2.42±0.02 | 5.14±0.04 | ||
| Methyl Paraben | 8.48±0.01 | −7.57±0.02 | 0.91±0.02 | ||
| Ethyl Paraben | 4.32±0.01 | −4.14±0.03 | 0.18±0.03 | ||
| Phenacetin | 3.66±0.03 | 1.16±0.14 | 4.81±0.15 |
Table 5.
Shown are asymmetric unit absolute deposition free energy values for AMOEBA ( ), OPLS-AA ( ) and OPLS-AA corrected to AMOEBA ( ). In the latter case, the total DFF corrections (Table 4) have been added the OPLS-AA deposition values (kcal/mol).
| Compound |
|
|
|
|||||
|---|---|---|---|---|---|---|---|---|
| Deposition | UE | Deposition | UE | |||||
| Acetanilide | −12.42±0.29 | −16.06±0.12 | 3.64 | −12.19±0.12 | 0.23 | |||
| Paracetamol | −13.98±0.57 | −19.43±0.29 | 5.45 | −14.30±0.29 | 0.32 | |||
| Methyl Paraben | −9.81±0.29 | −10.80±0.16 | 0.99 | −9.89±0.16 | 0.08 | |||
| Ethyl Paraben | −10.31±0.71 | −10.86±0.37 | 0.55 | −10.68±0.37 | 0.37 | |||
| Phenacetin | −14.47±0.21 | −19.10±0.39 | 4.63 | −14.29±0.40 | 0.18 | |||
|
| ||||||||
| Mean | −12.20±0.41 | −15.25±0.27 | 3.05 | −12.27±0.27 | 0.23 | |||
We note that the statistical uncertainty for the DFF AMOEBA/FC deposition is almost completely due to the OPLS-AA sublimation/deposition phase transition ( ) step of the thermodynamic cycle, and not from the DFF corrections. To further reduce the statistical uncertainty, this analysis suggests the focus should be on the OPLS-AA deposition/sublimation phase transition, which is further considered in the Conclusions below. Comparison of pure AMOEBA to the DFF AMOEBA/FC results show a mean unsigned error of only 0.2 kcal/mol, indicating successful application of the DFF thermodynamic path (Figure 1 and Eq. 5). Overall, the DFF method reduced the amount of condensed phase sampling in the more expensive AMOEBA force field by a factor of 40 (i.e. 200 nsec per trial per compound was reduced to only 5 nsec). Although the current version of FFX (1.0.0-beta) does not include optimized code for FC electrostatics, the wall clock time saved using OPLS-AA relative to AMOEBA was a factor of ~2 in this work due to elimination of the self-consistent field calculation (i.e. fixed multipole interactions with zero dipole and quadrupole components are computed for OPLS-AA). In the future, it is reasonable to expect the speed-up of the DFF AMOEBA/FC approach should reach a factor of ~5, based on codes that implement relatively optimized code paths for both fixed partial charge (i.e. OPLS-AA) and polarizable atomic multipole (i.e. AMOEBA) force fields such as TINKER79.
Absolute Deposition Thermodynamics vs. Experiment
The AMOEBA and AMOEBA/FC asymmetric unit deposition free energy values given in Table 5 can be compared to experiment after addition of 1) an ideal gas correction to account for compressing a 1 molar vapor into the volume of the crystal and 2) a correction to account for removal of the perfect symmetry constraint applied during the sublimation/deposition phase transition simulations. This later correction, a part of the GAUCHE path, has been described previously.15 As shown in Table 6, both AMOEBA and AMOEBA/FC approaches produce absolute deposition free energy values that compare favorably to experiment, with mean unsigned errors of 1.6 and 1.4 kcal/mol, respectively. For all compounds, the difference between AMOEBA and the accelerated AMOEBA/FC DFF result is not significant based on Student’s t-test. While OPLS-AA and AMOEBA deposition thermodynamics are clearly different for some crystals, the results from the AMOEBA/FC dual force field path are not distinguishable from the direct AMOEBA path (Figure 6).
Table 6.
Shown are absolute deposition free energy values for AMOEBA and AMOEBA/FC after including ideal gas and GAUCHE corrections (kcal/mol). The unsigned errors (UE) are relative to experiment.
| Compound | Expt. | Ideal Gas Correction | GAUCHE Correction |
|
UE |
|
UE | ||
|---|---|---|---|---|---|---|---|---|---|
| Acetanilide | −11.57 | 1.30 | −0.25±0.24 | −11.37±0.38 | 0.20 | −11.14±0.27 | 0.43 | ||
| Paracetamol | −16.23 | 1.27 | −1.67±0.18 | −14.38±0.60 | 1.85 | −14.70±0.34 | 1.53 | ||
| Methyl Paraben | −11.98 | 1.32 | −1.74±0.14 | −10.23±0.32 | 1.75 | −10.31±0.21 | 1.67 | ||
| Ethyl Paraben | −12.27 | 1.21 | −1.19±0.18 | −10.29±0.73 | 1.98 | −10.66±0.41 | 1.61 | ||
| Phenacetin | −14.39 | 1.14 | −3.08±0.36 | −16.41±0.42 | 2.02 | −16.23±0.54 | 1.84 | ||
|
| |||||||||
| Mean | 1.56 | 1.42 | |||||||
Figure 6.

Absolute crystal deposition free energy values from FC (OPLS-AA), AMOEBA/FC dual force field approach and direct AMOEBA calculations are compared to experiment.
Decomposition into Enthalpic and Entropic Contributions
Crystal structure prediction and the ranking of polymorphs is often based on direct use of potential energy rather than thermodynamic stability (i.e. free energy)80. Although efficient, methods that neglect entropic contributions are unable to describe changes in polymorph stability as a function of temperature. To overcome this common approximation, the GAUCHE procedure was developed to efficiently calculate absolute deposition free energy15. Insights into the origin of crystal stability differences can sometimes be obtained by decomposing free energy differences into enthalpic and entropic contributions using the relationships
| Equation 11 |
and
| Equation 12 |
where temperature (T) is 298 degrees Kelvin for the current work and the NVT ensemble was sampled using stochastic dynamics with experimental unit cell parameters (Table 1). The importance of entropic contributions is shown by comparing acetanilide to methyl and ethyl paraben in Table 7; while methyl and ethyl paraben have lower enthalpy of deposition under AMOEBA, acetanilide’s lesser entropic penalty results in a more favorable deposition free energy.
Table 7.
The decomposition of AMOEBA absolute deposition free energy values for each compound into enthalpic and entropic contributions at 298 degrees Kelvin.
| Compound |
|
|
|
|||
|---|---|---|---|---|---|---|
| Acetanilide | −11.37±0.38 | −20.81±0.15 | 9.44±0.40 | |||
| Paracetamol | −14.38±0.60 | −25.93±0.18 | 11.55±0.63 | |||
| Methyl Paraben | −10.23±0.32 | −21.19±0.23 | 10.96±0.39 | |||
| Ethyl Paraben | −10.29±0.73 | −23.30±0.24 | 13.01±0.77 | |||
| Phenacetin | −16.41±0.42 | −22.96±0.28 | 6.55±0.50 | |||
|
| ||||||
| Mean | −12.54 | −22.84 | 10.30 | |||
Conclusions
The DFF approach combines the strengths of both advanced polarizable atomic multipole force fields and efficient fixed partial charge models for organic crystal thermodynamics, while mitigating their primary weaknesses (i.e. FC accuracy limitations and the increased computational cost of AMOEBA). The AMOEBA/FC thermodynamic path was both accurate and cost effective for acetanilide, phenacetin, methyl parben, ethyl paraben and paracetamol crystals with a MUE of 1.4 kcal/mol relative to experiment, which is substantially less than the OPLS-AA MUE of 3.0 kcal/mol. Furthermore, the AMOEBA/OPLS-AA DFF method was statistically indistinguishable from using AMOEBA directly, with a MUE of only 0.2 kcal/mol relative to AMOEBA. Finally, the DFF protocol enabled sampling a path between energy functions with large inherent differences in both their bonded (i.e. equilibrium bond and angle values) and non-bonded functional forms (i.e. van der Waals, permanent electrostatics and explicit polarization). This serves to overcome limitations in reweighting procedures (e.g. the Zwanzig relationship) that require significant phase space overlap.
In future work, we plan to incorporate transition-tempering into the orthogonal space sampling algorithm (i.e. transition-tempered OSRW) to further reduce statistical uncertainty81, especially for the sublimation/deposition phase transition step. We also plan to replace the three discrete simulations that form the thermodynamic cycle described in Figure 1 with a single simulation that “on-the-fly” turns AMOEBA into OPLS-AA at the beginning of the thermodynamic path (i.e. in vacuum) and then back into AMOEBA at the end (i.e. in the crystalline state). This will serve to avoid any discrepancy in the optimal unit cell parameters or coordinates between force field resolutions for NPT ensembles. We also plan to explore the domain of applicability of DFF thermodynamic paths for applications beyond crystal thermodynamics, including acceleration of small molecule solvation thermodynamics82, protein/ligand binding83 and protein folding stability.84
Acknowledgments
We thank Dr. Ed Harder (Schrödinger, LLC) for providing the OPLS-AA parameters used in this work. All computations were performed on The University of Iowa Neon cluster with support and guidance from Glenn Johnson and Ben Rogers. NSF Award CHE-1404147 and NIH Award R01 DC002842 from NIDCD supported MJS. JML acknowledges an NIH fellowship from Award T32 GM008365 and a University of Iowa Presidential Fellowship. IJN was partially supported by fellowships from The University of Iowa Center for Research by Undergraduates. The authors declare that no competing interests exist.
References
- 1.Lommerse JPM, Motherwell WDS, Ammon HL, Dunitz JD, Gavezzotti A, Hofmann DWM, Leusen FJJ, Mooij WTM, Price SL, Schweizer B, Schmidt MU, van Eijck BP, Verwer P, Williams DE. A test of crystal structure prediction of small organic molecules. Acta Crystallographica Section B. 2000;56:697–714. doi: 10.1107/s0108768100004584. [DOI] [PubMed] [Google Scholar]
- 2.Price SL. The computational prediction of pharmaceutical crystal structures and polymorphism. Adv Drug Deliver Rev. 2004;56:301–319. doi: 10.1016/j.addr.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 3.Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM. The Chemistry and Applications of Metal-Organic Frameworks. Science. 2013;341 doi: 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]
- 4.Desiraju GR. Crystal Engineering: From Molecule to Crystal. J Am Chem Soc. 2013;135:9952–9967. doi: 10.1021/ja403264c. [DOI] [PubMed] [Google Scholar]
- 5.Price SL. From crystal structure prediction to polymorph prediction: interpreting the crystal energy landscape. Phys Chem Chem Phys. 2008;10:1996–2009. doi: 10.1039/b719351c. [DOI] [PubMed] [Google Scholar]
- 6.Bardwell DA, Adjiman CS, Arnautova YA, Bartashevich E, Boerrigter SXM, Braun DE, Cruz-Cabeza AJ, Day GM, Della Valle RG, Desiraju GR, van Eijck BP, Facelli JC, Ferraro MB, Grillo D, Habgood M, Hofmann DWM, Hofmann F, Jose KVJ, Karamertzanis PG, Kazantsev AV, Kendrick J, Kuleshova LN, Leusen FJJ, Maleev AV, Misquitta AJ, Mohamed S, Needs RJ, Neumann MA, Nikylov D, Orendt AM, Pal R, Pantelides CC, Pickard CJ, Price LS, Price SL, Scheraga HA, van de Streek J, Thakur TS, Tiwari S, Venuti E, Zhitkov IK. Towards crystal structure prediction of complex organic compounds - a report on the fifth blind test. Acta Crystallogr B. 2011;67:535–551. doi: 10.1107/S0108768111042868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price SL. Predicting crystal structures of organic compounds. Chem Soc Rev. 2014;43:2098–2111. doi: 10.1039/c3cs60279f. [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Hu W, Usvyat D, Matthews D, Schütz M, Chan GK-L. Ab initio determination of the crystalline benzene lattice energy to sub-kilojoule/mole accuracy. Science. 2014;345:640–643. doi: 10.1126/science.1254419. [DOI] [PubMed] [Google Scholar]
- 9.Thompson HPG, Day GM. Which conformations make stable crystal structures? Mapping crystalline molecular geometries to the conformational energy landscape. Chemical Science. 2014;5:3173–3182. [Google Scholar]
- 10.Hylton RK, Tizzard GJ, Threlfall TL, Ellis AL, Coles SJ, Seaton CC, Schulze E, Lorenz H, Seidel-Morgenstern A, Stein M, Price SL. Are the Crystal Structures of Enantiopure and Racemic Mandelic Acids Determined by Kinetics or Thermodynamics? J Am Chem Soc. 2015;137:11095–11104. doi: 10.1021/jacs.5b05938. [DOI] [PubMed] [Google Scholar]
- 11.Heit YN, Nanda KD, Beran GJO. Predicting finite-temperature properties of crystalline carbon dioxide from first principles with quantitative accuracy. Chemical Science. 2016;7:246–255. doi: 10.1039/c5sc03014e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliver Rev. 2002;54:355–366. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 13.Chu KA, Yalkowsky SH. Predicting Aqueous Solubility: The Role of Crystallinity. Current Drug Metabolism. 2009;10:1184–1191. doi: 10.2174/138920009790820110. [DOI] [PubMed] [Google Scholar]
- 14.Schnieders MJ, Baltrusaitis J, Shi Y, Chattree G, Zheng L, Yang W, Ren P. The structure, thermodynamics, and solubility of organic crystals from simulation with a polarizable force field. J Chem Theory Comput. 2012;8:1721–1736. doi: 10.1021/ct300035u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park J, Nessler I, McClain B, Macikenas D, Baltrusaitis J, Schnieders MJ. Absolute Organic Crystal Thermodynamics: Growth of the Asymmetric Unit into a Crystal via Alchemy. J Chem Theory Comput. 2014;10:2781–2791. doi: 10.1021/ct500180m. [DOI] [PubMed] [Google Scholar]
- 16.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1996;118:2309. [Google Scholar]
- 17.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 18.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 19.Price SL. Applications of realistic electrostatic modeling to molecules in complexes, solids and proteins. Journal of the Chemical Society, Faraday Transactions. 1996;92:2997–3008. [Google Scholar]
- 20.Lommerse JPM, Motherwell WDS, Ammon HL, Dunitz JD, Gavezzotti A, Hofmann DWM, Leusen FJJ, Mooij WTM, Price SL, Schweizer B, Schmidt MU, van Eijck BP, Verwer P, Williams DE. A test of crystal structure prediction of small organic molecules. Acta Crystallogr B. 2000;56:697–714. doi: 10.1107/s0108768100004584. [DOI] [PubMed] [Google Scholar]
- 21.Stone AJ, Alderton M. Distributed multipole analysis - methods and applications. Mol Phys. 1985;56:1047–1064. [Google Scholar]
- 22.Stone AJ. Distributed multipole analysis: Stability for large basis sets. J Chem Theory Comput. 2005;1:1128–1132. doi: 10.1021/ct050190+. [DOI] [PubMed] [Google Scholar]
- 23.Ren P, Ponder JW. Consistent treatment of inter- and intramolecular polarization in molecular mechanics calculations. J Comput Chem. 2002;23:1497–1506. doi: 10.1002/jcc.10127. [DOI] [PubMed] [Google Scholar]
- 24.Ren P, Wu C, Ponder JW. Polarizable atomic multipole-based molecular mechanics for organic molecules. J Chem Theory Comput. 2011;7:3143–3161. doi: 10.1021/ct200304d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren P, Ponder JW. Polarizable atomic multipole water model for molecular mechanics simulation. J Phys Chem B. 2003;107:5933–5947. [Google Scholar]
- 26.Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, Schnieders MJ, Haque I, Mobley DL, Lambrecht DS, DiStasio RA, Head-Gordon M, Clark GNI, Johnson ME, Head-Gordon T. Current status of the AMOEBA polarizable force field. The Journal of Physical Chemistry B. 2010;114:2549–2564. doi: 10.1021/jp910674d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piquemal JP, Cisneros GA, Reinhardt P, Gresh N, Darden TA. Towards a force field based on density fitting. J Chem Phys. 2006;124:104101. doi: 10.1063/1.2173256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elking DM, Cisneros GA, Piquemal JP, Darden TA, Pedersen LG. Gaussian Multipole Model (GMM) J Chem Theory Comput. 2009;6:190–202. doi: 10.1021/ct900348b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gresh N. Development, validation, and applications of anisotropic polarizable molecular mechanics to study ligand and drug-receptor interactions. Curr Pharm Des. 2006;12:2121–2158. doi: 10.2174/138161206777585256. [DOI] [PubMed] [Google Scholar]
- 30.Gresh N, Cisneros GA, Darden TA, Piquemal JP. Anisotropic, polarizable molecular mechanics studies of inter- and intramoecular interactions and ligand-macromolecule complexes. A bottom-up strategy. J Chem Theory Comput. 2007;3:1960–1986. doi: 10.1021/ct700134r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holt A, Bostrom J, Karlstrom G, Lindh R. A NEMO Potential that Includes the Dipole-Quadrupole and Quadrupole-Quadrupole Polarizability. J Comput Chem. 2010;31:1583–1591. doi: 10.1002/jcc.21502. [DOI] [PubMed] [Google Scholar]
- 32.Ponder JW, Case DA. Adv Protein Chem. Academic Press; 2003. Force fields for protein simulations; pp. 27–85. [DOI] [PubMed] [Google Scholar]
- 33.Ren P, Chun J, Thomas DG, Schnieders MJ, Marucho M, Zhang J, Baker NA. Biomolecular electrostatics and solvation: a computational perspective. Q Rev Biophys. 2012;45:427–491. doi: 10.1017/S003358351200011X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Y, Ren P, Schnieders M, Piquemal J-P. Reviews in Computational Chemistry. Vol. 28. John Wiley & Sons, Inc; 2015. Polarizable Force Fields for Biomolecular Modeling; pp. 51–86. [Google Scholar]
- 35.Ren P, Ponder JW. Temperature and pressure dependence of the AMOEBA water model. J Phys Chem B. 2004;108:13427–13437. [Google Scholar]
- 36.Grossfield A, Ren PY, Ponder JW. Ion solvation thermodynamics from simulation with a polarizable force field. J Am Chem Soc. 2003;125:15671–15682. doi: 10.1021/ja037005r. [DOI] [PubMed] [Google Scholar]
- 37.Shi Y, Wu C, Ponder JW, Ren P. Multipole electrostatics in hydration free energy calculations. J Comput Chem. 2011;32:967–977. doi: 10.1002/jcc.21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu JC, Chattree G, Ren P. Automation of AMOEBA polarizable force field parameterization for small molecules. Theor Chem Acc. 2012;131:1–11. doi: 10.1007/s00214-012-1138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Y, Xia Z, Zhang J, Best R, Wu C, Ponder JW, Ren P. Polarizable atomic multipole-based AMOEBA force field for proteins. J Chem Theory Comput. 2013;9:4046–4063. doi: 10.1021/ct4003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao J, Xia X. A priori evaluation of aqueous polarization effects through Monte Carlo QM-MM simulations. Science. 1992;258:631–635. doi: 10.1126/science.1411573. [DOI] [PubMed] [Google Scholar]
- 41.Luzhkov V, Warshel A. Microscopic models for quantum mechanical calculations of chemical processes in solutions: LD/AMPAC and SCAAS/AMPAC calculations of solvation energies. J Comput Chem. 1992;13:199–213. [Google Scholar]
- 42.Wesolowski T, Warshel A. Ab Initio Free Energy Perturbation Calculations of Solvation Free Energy Using the Frozen Density Functional Approach. The Journal of Physical Chemistry. 1994;98:5183–5187. [Google Scholar]
- 43.Stanton RV, Little LR, Merz KM., Jr Quantum Free Energy Perturbation Study within a PM3/MM Coupled Potential. The Journal of Physical Chemistry. 1995;99:483–486. [Google Scholar]
- 44.Li G, Zhang X, Cui Q. Free Energy Perturbation Calculations with Combined QM/MM Potentials Complications, Simplifications, and Applications to Redox Potential Calculations. The Journal of Physical Chemistry B. 2003;107:8643–8653. [Google Scholar]
- 45.Li G, Cui Q. pKa Calculations with QM/MM Free Energy Perturbations. The Journal of Physical Chemistry B. 2003;107:14521–14528. [Google Scholar]
- 46.Olsson MHM, Hong G, Warshel A. Frozen Density Functional Free Energy Simulations of Redox Proteins: Computational Studies of the Reduction Potential of Plastocyanin and Rusticyanin. J Am Chem Soc. 2003;125:5025–5039. doi: 10.1021/ja0212157. [DOI] [PubMed] [Google Scholar]
- 47.Hu H, Yang W. Dual-topology/dual-coordinate free-energy simulation using QM/MM force field. The Journal of Chemical Physics. 2005;123:041102. doi: 10.1063/1.1990113. [DOI] [PubMed] [Google Scholar]
- 48.Riccardi D, Schaefer P, Cui Q. pKa Calculations in Solution and Proteins with QM/MM Free Energy Perturbation Simulations: A Quantitative Test of QM/MM Protocols. The Journal of Physical Chemistry B. 2005;109:17715–17733. doi: 10.1021/jp0517192. [DOI] [PubMed] [Google Scholar]
- 49.Woods CJ, Manby FR, Mulholland AJ. An efficient method for the calculation of quantum mechanics/molecular mechanics free energies. The Journal of Chemical Physics. 2008;128:014109. doi: 10.1063/1.2805379. [DOI] [PubMed] [Google Scholar]
- 50.Li H, Yang W. Sampling enhancement for the quantum mechanical potential based molecular dynamics simulations: A general algorithm and its extension for free energy calculation on rugged energy surface. The Journal of Chemical Physics. 2007;126:114104. doi: 10.1063/1.2710790. [DOI] [PubMed] [Google Scholar]
- 51.Min D, Zheng L, Harris W, Chen M, Lv C, Yang W. Practically Efficient QM/MM Alchemical Free Energy Simulations: The Orthogonal Space Random Walk Strategy. J Chem Theory Comput. 2010;6:2253–2266. doi: 10.1021/ct100033s. [DOI] [PubMed] [Google Scholar]
- 52.Yang W, Cui Q, Min D, Li H. Chapter 4 - QM/MM Alchemical Free Energy Simulations: Challenges and Recent Developments. In: Ralph AW, editor. Annu Rep Comput Chem. Elsevier; 2010. pp. 51–62. [Google Scholar]
- 53.Konig G, Hudson PS, Boresch S, Woodcock HL. Multiscale Free Energy Simulations: An Efficient Method for Connecting Classical MD Simulations to QM or QM/MM Free Energies Using Non-Boltzmann Bennett Reweighting Schemes. J Chem Theory Comput. 2014;10:1406–1419. doi: 10.1021/ct401118k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konig G, Pickard FC, Mei Y, Brooks BR. Predicting hydration free energies with a hybrid QM/MM approach: an evaluation of implicit and explicit solvation models in SAMPL4. J Comput Aided Mol Des. 2014;28:245–257. doi: 10.1007/s10822-014-9708-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.König G, Mei Y, Pickard FC, Simmonett AC, Miller BT, Herbert JM, Woodcock HL, Brooks BR, Shao Y. Computation of Hydration Free Energies Using the Multiple Environment Single System Quantum Mechanical/Molecular Mechanical Method. J Chem Theory Comput. 2016;12:332–344. doi: 10.1021/acs.jctc.5b00874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dybeck EC, König G, Brooks BR, Shirts MR. Comparison of Methods To Reweight from Classical Molecular Simulations to QM/MM Potentials. J Chem Theory Comput. 2016;12:1466–1480. doi: 10.1021/acs.jctc.5b01188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys. 2008;129:10. doi: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zwanzig RW. High-temperature equation of state by a perturbation method. I Nonpolar gases. The Journal of Chemical Physics. 1954;22:1420–1426. [Google Scholar]
- 59.Lyman E, Ytreberg FM, Zuckerman DM. Resolution exchange simulation. Phys Rev Lett. 2006;96:4. doi: 10.1103/PhysRevLett.96.028105. [DOI] [PubMed] [Google Scholar]
- 60.Dybeck EC, Schieber NP, Shirts MR. Effects of a More Accurate Polarizable Hamiltonian on Polymorph Free Energies Computed Efficiently by Reweighting Point-Charge Potentials. J Chem Theory Comput. 2016 doi: 10.1021/acs.jctc.6b00397. [DOI] [PubMed] [Google Scholar]
- 61.Akin-Ojo O, Song Y, Wang F. Developing ab initio quality force fields from condensed phase quantum-mechanics/molecular-mechanics calculations through the adaptive force matching method. The Journal of Chemical Physics. 2008;129:064108. doi: 10.1063/1.2965882. [DOI] [PubMed] [Google Scholar]
- 62.Spiriti J, Zuckerman DM. Tunable Coarse Graining for Monte Carlo Simulations of Proteins via Smoothed Energy Tables: Direct and Exchange Simulations. J Chem Theory Comput. 2014;10:5161–5177. doi: 10.1021/ct500622z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kollman P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem Rev. 1993;93:2395–2417. [Google Scholar]
- 64.Jorgensen WL, Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 65.Schnieders MJ, Fenn TD, Pande VS. Polarizable atomic multipole X-ray refinement: Particle mesh Ewald electrostatics for macromolecular crystals. J Chem Theory Comput. 2011;7:1141–1156. doi: 10.1021/ct100506d. [DOI] [PubMed] [Google Scholar]
- 66.Fenn TD, Schnieders MJ. Polarizable atomic multipole X-ray refinement: Weighting schemes for macromolecular diffraction. Acta Crystallogr D. 2011;67:957–965. doi: 10.1107/S0907444911039060. [DOI] [PubMed] [Google Scholar]
- 67.LuCore Stephen D, Litman Jacob M, Powers Kyle T, Gao S, Lynn Ava M, Tollefson William TA, Fenn Timothy D, Washington MT, Schnieders Michael J. Dead-End Elimination with a Polarizable Force Field Repacks PCNA Structures. Biophys J. 2015;109:816–826. doi: 10.1016/j.bpj.2015.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown C. Further refinement of the crystal structure of acetanilide. Acta Crystallogr. 1966;21:442–445. [Google Scholar]
- 69.Haisa M, Kashino S, Kawai R, Maeda H. The Monoclinic Form of p-Hydroxyacetanilide. Acta Crystallographica Section B. 1976;32:1283–1285. [Google Scholar]
- 70.Nath NK, Aggarwal H, Nangia A. Crystal structure of methyl paraben polymorph II. Cryst Growth Des. 2011;11:967–971. [Google Scholar]
- 71.Lin X. The structure of ethyl paraben. Chin J Struct Chem. 1986;5:281. [Google Scholar]
- 72.Patel U, Patel TC, Singh TP. Structure of phenacetin, C10H13NO2. Acta Crystallographica Section C. 1983;39:1445–1447. [Google Scholar]
- 73.Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM. Integrated Modeling Program, Applied Chemical Theory (IMPACT) J Comput Chem. 2005;26:1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle-mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 75.Darden T, York D, Pedersen L. Particle-mesh Ewald - An n log(n) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 76.Sagui C, Pedersen LG, Darden TA. Towards an accurate representation of electrostatics in classical force fields: Efficient implementation of multipolar interactions in biomolecular simulations. J Chem Phys. 2004;120:73–87. doi: 10.1063/1.1630791. [DOI] [PubMed] [Google Scholar]
- 77.Zheng L, Chen M, Yang W. Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proc Natl Acad Sci USA. 2008;105:20227–20232. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee S, Chen M, Yang W, Richards NGJ. Sampling Long Time Scale Protein Motions: OSRW Simulation of Active Site Loop Conformational Free Energies in Formyl-CoA:Oxalate CoA Transferase. J Am Chem Soc. 2010;132:7252–7253. doi: 10.1021/ja101446u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ponder JW. TINKER: Software Tools for Molecular Design. TINKER: Software Tools for Molecular Design; Saint Louis, MO: 2015. 7.0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bardwell DA, Adjiman CS, Arnautova YA, Bartashevich E, Boerrigter SXM, Braun DE, Cruz-Cabeza AJ, Day GM, Della Valle RG, Desiraju GR, van Eijck BP, Facelli JC, Ferraro MB, Grillo D, Habgood M, Hofmann DWM, Hofmann F, Jose KVJ, Karamertzanis PG, Kazantsev AV, Kendrick J, Kuleshova LN, Leusen FJJ, Maleev AV, Misquitta AJ, Mohamed S, Needs RJ, Neumann MA, Nikylov D, Orendt AM, Pal R, Pantelides CC, Pickard CJ, Price LS, Price SL, Scheraga HA, van de Streek J, Thakur TS, Tiwari S, Venuti E, Zhitkov IK. Towards crystal structure prediction of complex organic compounds - a report on the fifth blind test. Acta Crystallographica Section B-Structural Science Crystal Engineering and Materials. 2011;67:535–551. doi: 10.1107/S0108768111042868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dama JF, Rotskoff G, Parrinello M, Voth GA. Transition-Tempered Metadynamics: Robust, Convergent Metadynamics via On-the-Fly Transition Barrier Estimation. J Chem Theory Comput. 2014;10:3626–3633. doi: 10.1021/ct500441q. [DOI] [PubMed] [Google Scholar]
- 82.Mobley DL, Guthrie JP. FreeSolv: a database of experimental and calculated hydration free energies, with input files. J Comput Aided Mol Des. 2014;28:711–720. doi: 10.1007/s10822-014-9747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jiao D, Golubkov PA, Darden TA, Ren P. Calculation of protein-ligand binding free energy by using a polarizable potential. Proc Natl Acad Sci USA. 2008;105:6290–6295. doi: 10.1073/pnas.0711686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dill KA, MacCallum JL. The Protein-Folding Problem, 50 Years On. Science. 2012;338:1042–1046. doi: 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]