

Abstract
Targeting Bruton tyrosine kinase (BTK) by ibrutinib is an effective treatment for patients with relapsed/refractory mantle cell lymphoma (MCL). However, both primary and acquired resistance to ibrutinib have developed in a significant number of these patients. A combinatory strategy targeting multiple oncogenic pathways is critical to enhance the efficacy of ibrutinib. Here, we focus on the BCL2 anti-apoptotic pathway. In a tissue microarray of 62 MCL samples, BCL2 expression positively correlated with BTK expression. Increased levels of BCL2 were shown to be due to a defect in protein degradation because of no or little expression of the E3 ubiquitin ligase FBXO10, as well as transcriptional upregulation through BTK-mediated canonical nuclear factor-κB activation. RNA-seq analysis confirmed that a set of anti-apoptotic genes (for example, BCL2, BCL-XL and DAD1) was downregulated by BTK short hairpin RNA. The downregulated genes also included those that are critical for B-cell growth and proliferation, such as BCL6, MYC, PIK3CA and BAFF-R. Targeting BCL2 by the specific inhibitor ABT-199 synergized with ibrutinib in inhibiting growth of both ibrutinib-sensitive and -resistant cancer cells in vitro and in vivo. These results suggest co-targeting of BTK and BCL2 as a new therapeutic strategy in MCL, especially for patients with primary resistance to ibrutinib.
INTRODUCTION
Mantle cell lymphoma (MCL), a B-cell non-Hodgkin lymphoma, remains incurable with the current treatment modalities.1,2 The B-cell antigen receptor (BCR) signaling pathway is essential for normal B-cell development, proliferation and differentiation and has an important role in the pathogenesis of several types of non-Hodgkin lymphoma, including MCL.3–6 An antigen-driven process of MCL has been suggested by sequence analysis of the immunoglobulin variable region genes that revealed a biased repertoire and stereotyped usage in >40% of cases.7 That MCL cells acquire BCR signaling for survival is supported by the observation that pharmacological inhibition of downstream signaling triggered cell apoptosis.8–12 Bruton tyrosine kinase (BTK), a key component of the early BCR signaling pathway, has emerged as a promising therapeutic target. Ibrutinib, a specific inhibitor that binds covalently to the active site of BTK at cysteine 481, has been approved for the treatment of MCL. In a recent Phase II study, treatment for relapsed or refractory MCL with ibrutinib alone achieved a response rate of 68%.2 Although this response rate is striking, approximately one-third of patients show primary resistance, and acquired resistance with a C481S mutation in BTK can also develop.11 To overcome primary and acquired resistance to BTK inhibition, a combinatory strategy that targets multiple pathways is needed.
BCR crosslinking causes BTK and other tyrosine kinases to interact with the inner leaflet of the plasma membrane to constitute the BCR signalosome, which subsequently activates many downstream pathways, including nuclear factor (NF)-κB.13,14 BCR-mediated NF-κB activation is through the canonical NF-κB pathway in which the Ikappa B kinase (IKK) complex (IKKα, IKKβ, regulatory IKKγ subunit) phosphorylates IκBα for proteasomal degradation, leading to nuclear translocation of the heterodimeric NF-κB transcription factors, predominantly the p50/p65 dimer.15,16 The CARD11, MALT1 and BCL10 (CBM) signaling complex, the intermediate between the BTK and IKK complex, is essential for NF-κB activation.16–19 Somatic mutations or chromosomal translocations impacting the CBM pathway contribute to pathogenesis of several lymphoma subtypes.16,18,20 Although genetic alterations in the canonical NF-κB pathway are rare in MCL, recent studies have revealed that the noncanonical NF-κB pathway is pathogenetically altered.10,21,22 The noncanonical NF-κB pathway targets activation of the p52/RelB NF-κB complex via processing of p100, the precursor of p52 and a RelB-specific inhibitor.23 The p100 phosphorylation leading to its processing is mediated by NF-κB-inducing kinase (NIK).23 The basal level of NIK is low because of constant degradation by a TRAF2/3-cIAP destruction complex but the signal-induced noncanonical NF-κB signaling from a subset of TNF receptor family members (for example, BAFF receptor) prevents such a degradation and mediates nuclear translocation of the p52/RelB dimer.23 The evidence of the BAFF/BAFF receptor signaling pathway in the pathogenesis of B-cell lymphomas has been well documented.24–29 However, whether BTK is required for this alternative NF-κB signaling in malignant B cells remains undefined.
BCL2 and its family member genes (for example, BCL-XL and MCL1) are targets of NF-κB signaling and are upregulated in cancer cells.30–32 In B-cell lymphomas, BCL2 is deregulated through other mechanisms as well, including chromosomal translocations,33 genomic amplification and gene mutations.34,35 Recently, we have demonstrated posttranslational regulation of BCL2 in human lymphomas.36 BCL2 protein can be targeted for proteasomal degradation by the E3 ubiquitin ligase FBXO10. The FBXO10 gene is infrequently mutated in diffuse large B-cell lymphoma (DLBCL) but its expression is reduced in the majority of cases. FBXO10 functions as a potential tumor suppressor because many types of human lymphoma cells are sensitive to FBXO10 overexpression, with the most sensitive one being MCL.36
To gain new insights into the molecular pathogenesis of MCL, we analyzed expression of and association among FBXO10, BCL2 and BTK in 62 MCL samples, and functionally dissected their role in regulation of cell proliferation, survival and apoptosis in MCL cell lines. These functional analyses together with the results from the MCL xenografts provided the mechanistic rationale for a combination of the BCL2 and BTK small molecule inhibitors, ABT-199 and ibrutinib, in the treatment of MCL.
RESULTS
BCL2 is overexpressed while FBXO10 is rarely expressed in MCL Our previous study established FBXO10 as the E3 ubiquitin ligase that targets BCL2 for proteasomal degradation in DLBCL cell lines.36 FBXO10 expression was reduced in the majority of DLBCL patient samples despite elevated levels of FBXO10 expression in germinal center B cells that the disease is derived from.36 However, it is unclear whether this is a general regulation mechanism that applies to other subtypes such as MCL. To address this question, we performed immunohistochemical analysis for expression of BCL2 and FBXO10 in a tissue microarray that contained 62 MCL cases. Five BCL2-negative Burkitt lymphoma samples were used as staining controls and six lymph nodes served as normal B-cell controls. All these MCL cases had the typical chromosomal translocation t(11:14) involving cyclin D1 with the morphological characteristics of MCL, as described previously.37 We scored protein expression in triplicate cores for each case using Inform advanced image analysis software (PerkinElmer, Waltham, MA, USA) and found high levels of BCL2 expression but no or low FBXO10 expression in MCL cases (Figure 1a, Supplementary Figure 1a). The average of FBXO10 expression in these MCL cases was significantly lower than that of normal B cells (Supplementary Figure 1b). In comparison with tissue surrounding the tumor, malignant cells from 85% (53/62) of cases were BCL2 positive, whereas FBXO10 expression was detected in only 8% (5/62) of cases.
Figure 1.
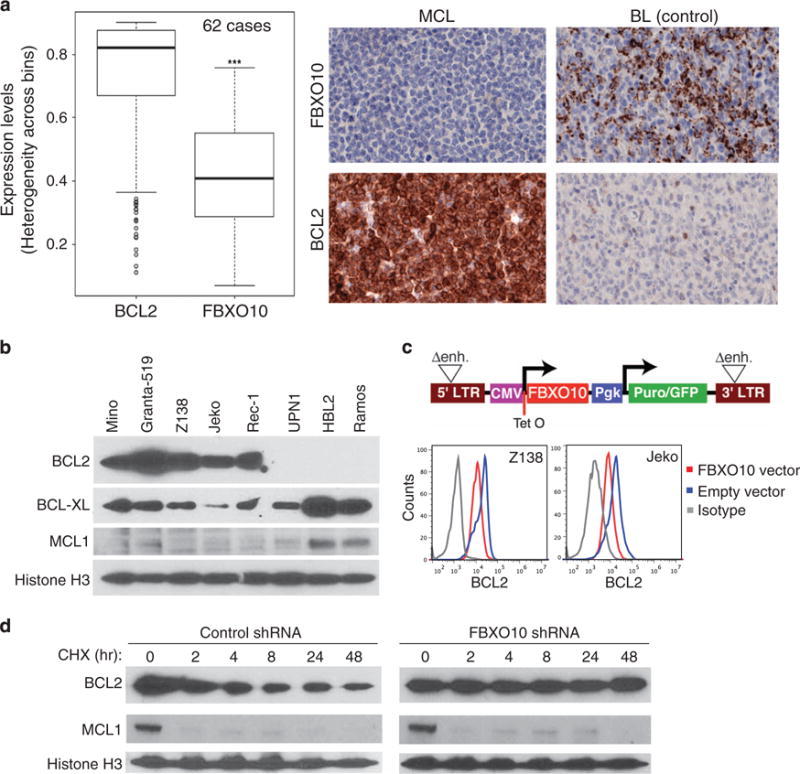
High levels of BCL2 expression and defects in FBXO10-mediated proteasomal degradation in MCL cell lines. (a) A tissue microarray (TMA) of 62 cases was used for immunohistochemical staining for BCL2 and FBXO10 expression and analyzed with Inform advanced image analysis software (PerkinElmer) (***P<0.001). Representative staining images from a MCL patient are shown. Infiltrating lymphocytes of a Burkitt lymphoma (BL) served as a positive control for FBXO10 staining and malignant cells as a negative control for BCL2 staining. (b) Immunoblotting assay for expression of BCL2, BCL-XL and MCL1. Histone H3 served as a loading control. Note genomic deletion of BCL2 in UPN-1 and BCL2 amplification in Granta-519 and Z138 (ref. 34) BL cell line Ramos served as a negative control for BCL2. (c) Structure of the inducible GFP retroviral vector. Two tetracycline repressor binding sites (Tet operators) were inserted into the CMV promoter, which drives FBXO10 expression. LTR, long terminal repeat; puror/GFP, fusion of puromycin resistance and green fluorescent protein gene; Pgk, phosphoglycerate kinase promoter; Δenh., deletion of LTR promoter sequences. Intracellular BCL2 flow cytometric analysis of two MCL cell lines after 2 days of FBXO10 expression. (d) FBXO10 silencing by shRNA prolongs BCL2 half-life. Two days after shRNA induction with 20 ng/ml of doxycycline, Z138 cells were treated with 20 μg/ml of cycloheximide and analyzed by immunoblotting for indicated proteins.
Based on this initial expression analysis, we hypothesized that a defect in FBXO10-mediated proteasomal degradation contributes to high BCL2 expression in MCL. To test this hypothesis, we genetically manipulated FBXO10 expression in MCL cell lines. We screened seven MCL cell lines and found five of them to have high levels of BCL2 expression but much lower expression of BCL-XL and MCL1 compared with the remaining two cell lines and Ramos, a Burkitt lymphoma cell line (Figure 1b). We used the inducible retroviral system to overexpress FBXO10 along with green fluorescent protein (GFP) as a marker for transduced cells (Figure 1c). Consistent with the results in DLBCL,36 flow cytometric analysis revealed that overexpression of FBXO10 resulted in reduced BCL2 expression in two representative MCL cell lines (Figure 1c). To test whether reduced levels of BCL2 expression is due to increased protein turnover by FBXO10, we silenced endogenous FBXO10 in the Z138 cell line by the same small hairpin RNA as used for our previous study.36 Indeed, after blocking protein synthesis by cycloheximide, the amount of BCL2 protein was sustained for up to 48 h in these FBXO10 short hairpin RNA (shRNA)-expressing cells but not in the control shRNA cells (Figure 1d). In contrast, non-shRNA-targeted MCL1 was turned over rapidly in both conditions. Taken together, the results indicate that BCL2 is the predominantly expressed anti-apoptotic protein in MCL, because of a specific defect in its proteasomal degradation.
BCL2 is essential for the survival of MCL cells and can be targeted by ABT-199
To examine whether BCL2 has an essential role in cell survival in MCL, we first used the selective BCL2 inhibitor ABT-199(ref. 38) and tested cell viability in seven MCL cell lines. The Burkitt lymphoma cell line Ramos served as a negative control because of no BCL2 expression despite abundant BCL-XL expression in this cell line (Figure 1b). The Trypan blue exclusion viability assay revealed a dose-dependent toxic effect of ABT-199 on all five BCL2-expressing MCL cell lines, whereas the BCL2-negative MCL cell lines, UPN-1 and HBL2, and the control cell line Ramos did not respond to the drug (Figure 2a). Flow cytometric analysis demonstrated that the toxicity was due to apoptotic cell death triggered by ABT-199 (Figure 2b). The results were consistent with previous in vitro cell viability assays for the same MCL cell lines and primary cancer cells.39 To rule out the possibility of off-target toxicity by ABT-199, we analyzed cell viability upon knockdown of endogenous BCL2 by shRNA. Two different BCL2 shRNAs were retrovirally transduced along with the marker GFP into lymphoma cells; the percentage of GFP+ cells was calculated by flow cytometric analysis during 12 days of observation (Figure 2c). As expected, both BCL2 shRNAs induced cell death in all BCL2-expressing cell lines but not in BCL2-negative cell lines (Figure 2c). The on-target effect of ABT-199 was confirmed by a rescue experiment, which demonstrated that the overexpression of BCL2 complementary DNA lacking the 3′-UTR reversed toxicity by the shRNA that targets the 3′-UTR of BCL2 (Supplementary Figure 2).
Figure 2.
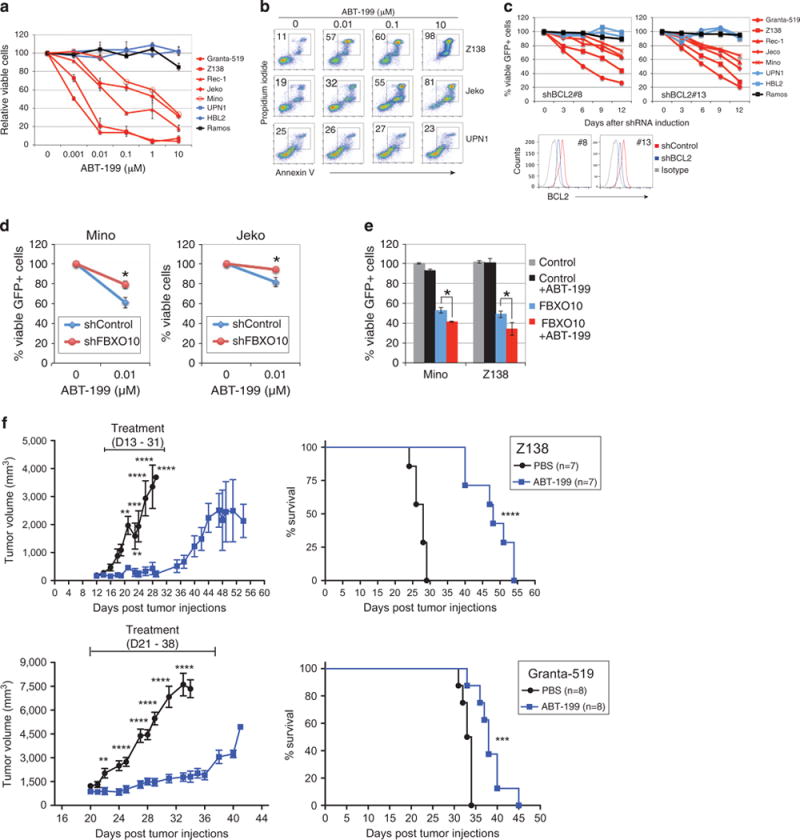
Targeting BCL2-dependent MCL by ABT-199. (a) Quantitative measurement of ABT-199-mediated toxicity by Trypan blue dye exclusion cell viability assay in the indicated cell lines after 3 days of treatment. Error bars represent mean ± s.d. of triplicates. (b) Flow cytometric analysis of apoptotic cell death by propidium iodide and annexin V co-staining after 3 days of treatment. (c) BCL2 knockdown by shRNAs is toxic to BCL2-expressing MCL cells. Histograms show reduced BCL2 expression by two shRNAs. The percentage of viable GFP+ shBCL2 expressing cells was normalized to that of the control shRNA for each time point. (d) Silencing endogenous FBXO10 attenuates ABT-199-mediated cell killing. Three days after shRNA induction, cells were treated with ABT-199 for 72 h before flow cytometric analysis. Error bars represent mean ± s.d. of triplicates (*P<0.05). (e) Ectopic expression of FBXO10 increases ABT-199-induced toxicity. The cells were pretreated with 0.01 μM ABT-199 for 2 days and then induced for FBXO10 expression for an additional day before flow cytometric analysis. Error bars represent mean ± s.d. of triplicates (*P<0.05). (f) ABT-199 inhibits xenograft tumor growth. Z138 and Granta-519 cells were established as a subcutaneous tumor (average 172 mm3 for Z138, and 864 mm3 for Granta-519) in immunodeficient mice, and then treated daily for 18 days with ABT-199 (100 mg/kg) or phosphate-buffered saline/dimethylsulfoxide control by intraperitoneal injection. Tumor progression was monitored by measuring tumor volumes (left panel) or overall survival (right panel). Error bars show the s.e.m. of seven or eight mice per group (**P<0.01, ***P<0.001, ****P<0.0001).
As FBXO10 knockdown by shRNA prolongs BCL2 half-life, we next tested whether the FBXO10 shRNA can counteract ABT-199 toxicity. We chose two MCL cell lines Mino and Jeko because both express relatively high levels of FBXO10.36 Indeed, expression of the FBXO10 shRNA prevented these two cell lines from ABT-199-mediated cell death (Figure 2d). Conversely, ectopic expression of FBXO10 synergized with ABT-199 in cell killing (Figure 2e).
Furthermore, we evaluated the ability of ABT-199 to suppress tumor growth in vivo in MCL xenografts established in immunocompromised mice. We initially subcutaneously implanted the representative cell line Z138 in the mice and observed that these cells reached an average volume of 172 mm3 after 13 days of injection. The mice bearing the Z138 tumor were then treated with ABT-199 intraperitoneally for 18 consecutive days at 100 mg per kg of body weight, an optimized dose used in a recent study.38 The results showed that ABT-199 caused complete tumor growth inhibition during the period of treatment and delayed tumor growth after ceasing treatment (Figure 2f, left top panel). Within 18 days of treatment, we killed all mice in the phosphate-buffered saline control group because tumors grew to large sizes (20 mm in any dimension) or the mice became very sick, whereas all mice with ABT-199 treatment survived and were relatively healthy (Figure 2f, right top panel). In addition, we obtained similar results from Granta-519 xenografts (Figure 2f, bottom panels). Thus, this xenograft study together with in vitro functional analyses reinforces the therapeutic potential of ABT-199.
BCR/BTK signaling in regulation of cell survival and BCL2 expression in MCL
Several recent studies have demonstrated that MCL cells acquire BTK activity for their survival and proliferation.8–10 Indeed, the oncogenic role of BTK in MCL is further supported by our biochemical and functional analyses. We found that BTK is constitutively activated in all eight MCL cell lines examined and the specific inhibitor ibrutinib blocked BTK phosphorylation/activation in these cancer cells (Figure 3a). We performed the Trypan blue viability assay and found five out of eight cell lines were sensitive to ibrutinib treatment by undergoing apoptotic cell death (Figures 3b and c), with the most sensitive ones being BCR dependent. This is, in general, in agreement with a recent study,10 but we noticed that two BCR-independent cell lines Z138 and Maver-1 had a mild sensitivity to ibrutinib. This unexpected finding prompted us to further evaluate the function of BTK in MCL by using a BTK shRNA whose target specificity was confirmed previously.15 Consistently, we observed a toxic effect of the shRNA on those MCL cell lines that were sensitive to ibrutinib (Figure 3d).
Figure 3.
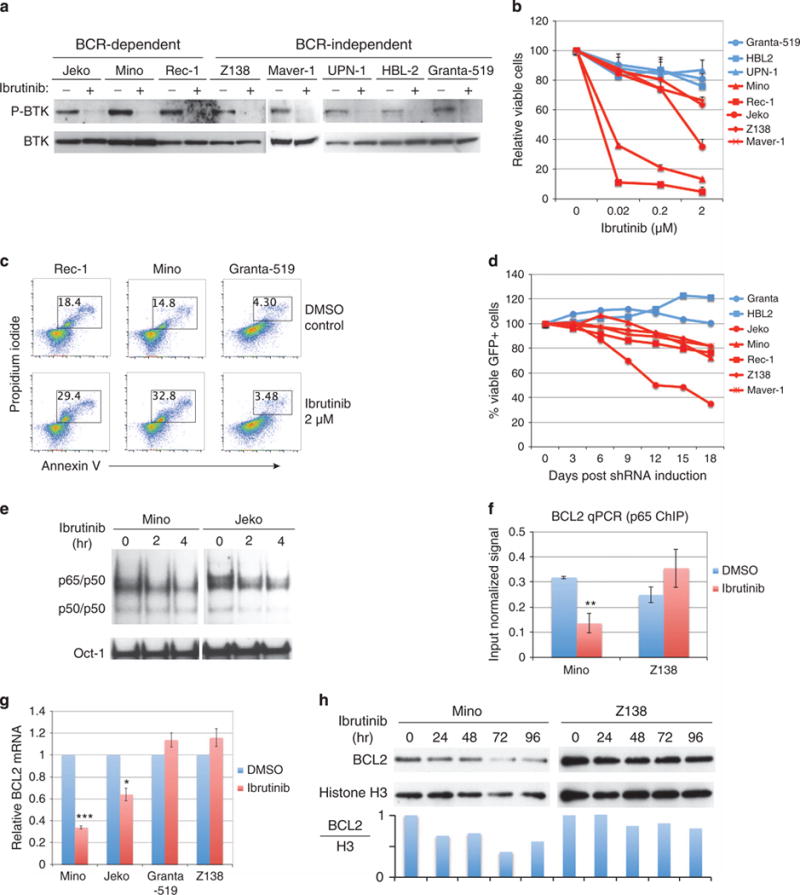
BTK-mediated canonical NF-κB activation and targeting BTK by ibrutinib in MCL. (a) Immunoblotting analysis of BTK and p-BTK in MCL cell lines. Cells were treated with 10 μM ibrutinib or dimethylsulfoxide control for 15 min. (b) Quantitative measurement of ibrutinib-mediated toxicity by Trypan blue dye exclusion viability assay in the indicated cell lines after 6 days of treatment. Error bars represent mean ± s.d. (n = 3). (c) Flow cytometric analysis of apoptotic cell death by propidium iodide and annexin V co-staining after 6 days of ibrutinib treatment. (d) Flow cytometric analysis of BTK shRNA expressing cells. The percentage of viable GFP+ shBTK expressing cells was normalized to that of the control shRNA for each time point. Data are representative of three independent experiments. (e) Electrophoretic mobility shift assay (EMSA) for NF-κB activity in Mino and Jeko cells when treated with 10 μM ibrutinib. Oct-1 served as a loading control. (f) Chromatin immuneprecipitation (ChIP) assay with anti-p65 antibody in Mino and Z138 cells after 6 h of treatment with 2 μM ibrutinib. The occupancy of p65 on BCL2 promoter region was determined by standard qPCR. IgG antibody served as a negative control and the signal from IgG ChIP was negligible. (g) Measurement of BCL2 mRNA by qPCR in the indicated MCL cell lines after 24 h of treatment with 2 μM ibrutinib. Error bars represent mean ± s.d. of triplicates (*P<0.05, ***P<0.001). (h) Measurement of BCL2 protein in Mino and Z138 cells after treatment with 10 μM ibrutinib for the indicated times.
Given that BCL2 is a target gene of NF-κB and BCR/BTK signaling contributes to high NF-κB activity in MCL, we asked whether BCL2 is upregulated through this transcriptional mechanism. First, we examined BTK-dependent NF-κB activation by electrophoretic mobility shift assay. We used Mino and Jeko as two representative BCR-dependent cell lines and found that BTK treatment indeed reduced constitutive NF-κB (p65 and p50) activity in both cell lines (Figure 3e). Chromatin immunoprecipitation assay confirmed BCL2 as an NF-κB target gene given that p65 enrichment on the BCL2 promoter region was significantly reduced when Mino cells were treated with ibrutinib (Figure 3f). Z138 is a negative control as the essential BTK downstream gene CARD11 is barely expressed in this cell line.10 BTK-dependent BCL2 transcription also reflected a reduction in both BCL2 mRNA (Figure 3g) and protein in cancer cells when treated with ibrutinib (Figure 3h).
To test whether reduced BCL2 transcription represents a mechanism accounting for apoptotic cell death triggered by ibrutinib, we expressed BCL2 complementary DNA by the above retroviral GFP vector in two sensitive cell lines Jeko and Mino and, indeed, observed elevated BCL2 protein in these cells (Supplementary Figure 3). We monitored viable GFP-positive cells in the absence or presence of various concentrations of ibrutinib by flow cytometry and found that the cells with BCL2 overexpression were more resistant to the drug treatment than the empty vector control (Supplementary Figure 3). Altogether, the data indicate that BCR/BTK signaling promotes the survival of MCL cells through transcriptional upregulation of BCL2.
Co-targeting of BTK and BCL2 in MCL
The transcriptional regulation of BCL2 mediated by BCR/BTK signaling prompted us to investigate whether there is a positive correlation in gene expression between BTK and BCL2 in primary cancer cells. To test this possibility, we performed immunohistochemical analysis using BTK antibodies on the same tissue microarray samples described for BCL2 staining. Indeed, we detected high levels of BTK expression in the vast majority of MCL (Figure 4a), which was strongly correlated with BCL2 expression (P<0.0001).
Figure 4.
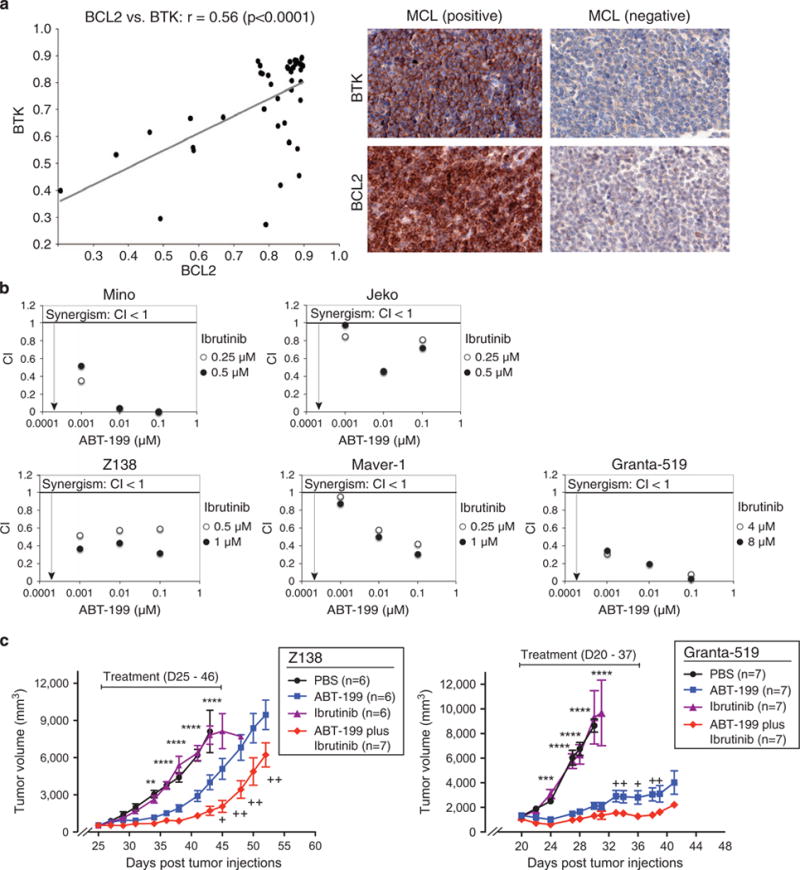
Positive correlation between BCL2 and BTK and co-targeting MCL by ABT-199 and ibrutinib. (a) Shown are a strong positive correlation between BCL2 and BTK in 62 MCL cases analyzed (left plot) and examples of immunohistochemical staining of BCL2 and BTK in positive vs negative cases (right panels). ‘r’ denotes nonparametric Spearman’s rank correlation coefficient. (b) Synergism between ABT-199 and ibrutinib in cell killing in Mino and Z138 cells. Cells were pretreated with ibrutinib for 3 days, and then exposed to ABT-199 and ibrutinib either alone or in combination for additional 3 days before Trypan blue dye exclusion viability assay. Error bars represent mean ± s.d. of triplicates. Combination index (CI) was calculated with CompuSyn software (Compusyn, Inc., Paramus, NJ, USA). (c) ABT-199 enhances the efficacy of ibrutinib in inhibiting xenograft tumor growth. Z138 cells were established as a subcutaneous tumor (average 172 mm3) in immunodeficient mice, and then treated daily for 21 days with either ABT-199 (30 mg/kg) or ibrutinib (12 mg/kg) or in combination by intraperitoneal injection (left panel). Mice with the Granta-519 tumor (average 864 mm3) were treated daily for 18 days with either ABT-199 (30 mg/kg) or ibrutinib (25 mg/kg) or in combination by intraperitoneal injection (right panel). Tumor progression was monitored by measuring tumor volumes. Error bars show the s.e.m. of six or seven mice (**P<0.01, ***P<0.001, ****P<0.0001 between phosphate-buffered saline/dimethylsulfoxide control and single ABT-199 group; +P<0.05, ++P<0.01 between single ABT-199 and ABT-199 and ibrutinib combination group).
The above data have demonstrated both transcriptional (BTK dependent) and posttranslational (reduced proteasomal degradation) mechanisms for BCL2 upregulation in MCL. Elevated BCL2, however, can also result from other mechanisms, such as amplification. Indeed, this genomic alteration is present in some MCL cell lines, including Granta-519 and Z138 used for this study.34 As a result of this BCL2 amplification, inhibition of BTK alone by ibrutinib may be less effective at generally triggering apoptosis in MCL cells. Instead, a combination of ibrutinib and ABT-199 could be more effective. We tested this hypothesis using the three BCR-independent cell lines Z138, Granta-519 and Marver-1. We also used the two BCR-dependent cell lines Mino and Jeko as positive controls because synergistic toxicity of ibrutinib and ABT-199 in BCR-dependent MCL cells was reported recently.39,40 As shown in Figure 4b, ABT-199 synergized with ibrutinib in cell killing in all the five cell lines tested. The synergistic toxicity was also observed either in cells that expressed shBTK in the presence of ABT-199 (Supplementary Figure 4a) or in cells that expressed shBCL2 in the presence of ibrutinib (Supplementary Figure 4b).
We next used the above Z138 xenograft murine model to test antitumor effect by a combination of ibrutinib and ABT-199. We reduced ABT-199 to 30 mg/kg from the above optimal dosage of 100 mg/kg in order to observe possible synergistic effects with ibrutinib. The dosage for ibrutinib was 12 mg/kg as suggested in a previous study.41 As expected, co-treatment of ABT-199 and ibrutinib remarkably inhibited tumor growth when compared with the ABT-199 alone group (Figure 4c, left panel). Interestingly, a dose of ibrutinib at 12 mg/kg had limited effects on tumor inhibition, which is distinct from DLBCL xenografts.41 This was consistent with our recent combinatorial screening study, which revealed a strong synergism between ibrutinib and ABT-199 in DLBCL.42 In chronic lymphocytic leukemia xenograft models, an optimal dose of ibrutinib was 25 mg/kg.43,44 It is likely that 12 mg/kg of ibrutinib is a suboptimal dose for the Z138 xenografts, based on these studies and our observations.
Further, we performed xenograft analysis on synergism between ibrutinib and ABT-199 in the Granta-519 cell line. Granta-519 cells have high levels of BCL2 expression (Figure 1b) and are insensitive to BTK inhibition by both ibrutinib and shRNA (Figures 3b and d). Therefore, Granta-519 can be considered a model cell line for the study of primary resistance to ibrutinib. We subcutaneously implanted Granta-519 cells (4 million per mouse) in the mice and started treatment when these cells reached an average volume of 864 mm3. The mice were treated intraperitoneally with ABT-199 (30 mg/kg), or ibrutinib (25 mg/kg), or both, for 18 consecutive days. The results showed that combination treatment caused nearly complete tumor growth inhibition during the period of treatment and delayed tumor growth after ceasing treatment (Figure 4c, right panel) when compared with the ABT-199 alone group. Consistent with in vitro analyses, ibrutinib alone had no effect on inhibition of tumor growth, the same as the phosphate-buffered saline control. Thus, both our in vitro Trypan blue toxicity assay and in vivo xenograft analysis demonstrate that co-targeting of BCL2 and BTK is a promising therapeutic strategy for MCL cases where BTK inhibition fails to reduce BCL2 expression.
Noncanonical NF-κB signaling pathway and its association with BTK in MCL
A recent study demonstrated no BCR-mediated NF-κB activation in the Z138 cell line because of almost no CARD11 expression, whereas the noncanonical NF-κB pathway is constitutively activated because of an inactivating mutation of the negative regulator gene TRAF2 (Supplementary Figure 5).10 These findings together with our observations indicating the sensitivity of Z138 cells to ibrutinib treatment prompted us to explore whether there is a link between BTK activity and the noncanonical NF-κB pathway. We started to analyze Z138 cells for their expression of CD40 and BAFF receptor (BAFF-R), two major receptors responsible for activation of the noncanonical NF-κB pathway. CD40 expression was low in Z138 cells when compared with naive B cells, DLBCL cells and some other MCL cell lines, but BAFF-R was highly expressed (Supplementary Figure 6, Figure 5a). BAFF-R expression was also detected in all other seven MCL cell lines (Figure 5a). The BAFF-R signaling pathway is functioning because its stimulation by BAFF elevated p100 to p52 processing in Mino and Jeko cells (Figure 5b). Z138 has constitutive p100 to p52 processing because of TRAF2 deficiency, which was not further increased by BAFF treatment. Similarly, the Maver-1 cell line that harbors a biallelic TRAF3 deletion and constitutive noncanonical NF-κB activation10 also did not further increase p100 to p52 processing in response to BAFF treatment.
Figure 5.
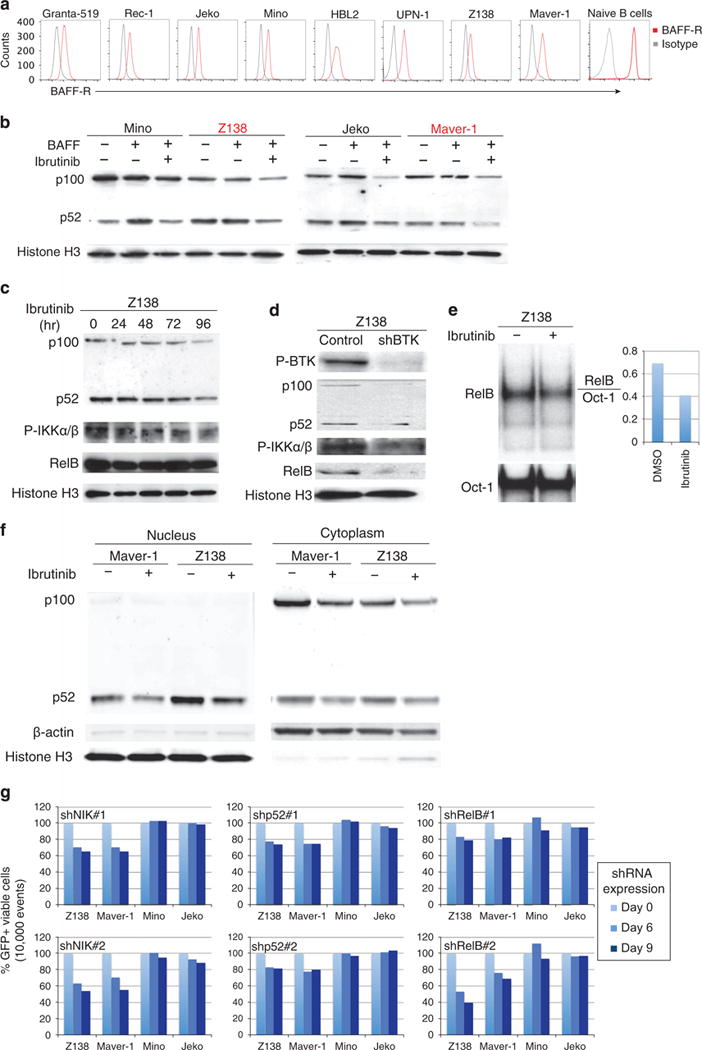
BTK-mediated noncanonical NF-κB activation in MCL. (a) Flow cytometric analysis of BAFF receptor in the indicated cell lines and naïve B cells. (b) Ibrutinib blocks BAFF-induced noncanonical p100 processing to p52 in Mino and Jeko cells. Z138 and Maver-1 cells served as controls because TRAF2 or TRAF3 mutations by which this pathway is deregulated. Cells were pretreated with 10 μM ibrutinib or dimethylsulfoxide (DMSO) control for 48 h and then stimulated with 100 ng/ml of BAFF for 24 h before immunoblotting analysis for p100 and p52 expression. (c, d) BTK inhibition by ibrutinib or BTK shRNA reduces p100 and p52 levels in Z138 cells. Cells were treated with 10 μM ibrutinib at the indicated time (c) or 4 days after shBTK induction (d) before immunoblotting analysis for expression of p100, p52, phospho-IKKs, and RelB. Histone H3 served as a loading control. (e) Electrophoretic mobility shift assay (EMSA) with anti-RelB antibody in Z138 cells treated with 10 μM ibrutinib or DMSO control for 24 h. Oct-1 served as a loading control (left panel). RelB and Oct-1 bands were quantified using the NIH Image J software (NIH, Bethesda, MD, USA) (right panel). (f) BTK inhibition reduces nuclear p52 protein in Maver-1 and Z138 cells. Cells were treated with 10 μM ibrutinib for 72 h, nuclear and cytoplasmic fractions were extracted as described in Materials and methods section before immunoblotting for p100, p52, β-actin (cytoplasm) and histone H3 (nucleus). (g) Flow cytometric analysis of cells expressing NIK, p52 or RelB shRNAs. The percentage of viable GFP+ shRNA-expressing cells was normalized to that of the control shRNA for each time point. Data are representative of three independent experiments.
A biochemical study on BTK-deficient murine B cells revealed that BTK is downstream of BAFF-R signaling for NF-κB activation.45 BTK inhibition by ibrutinib reduced BAFF-mediated p100 to p52 processing in Mino and Jeko cells. Similarly, ibrutinib also reduced the levels of both p100 and p52 in Z138 and Maver-1 cells (Figure 5b). We next treated Z138 cells with ibrutinib and observed a time-dependent reduction in p100 and 52 levels, which was accompanied by reductions in phosphorylated IKKα/β levels (Figure 5c). More marked changes in p100, p52 and phosphorylated IKKα/β, as well as RelB, were observed in Z138 cells with BTK knocked down (Figure 5d). Reduced RelB and p52 nuclear translocation because of BTK inhibition was confirmed by electrophoretic mobility shift assay with anti-RelB antibody (Figure 5e) and by immunoblotting for measuring nuclear p52 protein (Figure 5f), respectively.
To further determine whether BTK-dependent noncanonical NF-κB activation is required for cancer cell survival, we used those shRNAs that were used in a recent study to knockdown NIK, p52 and RelB.10 We obtained an expected result demonstrating that all these shRNAs selectively killed Z138 and Marver-1 cells but not the control cell lines Mino and Jeko in which the noncanonical NF-κB pathway is not active (Figure 5g).
Given that BTK can mediate both canonical and noncanonical NF-κB activation pathways, we thought to determine whether BTK targets a similar set of genes. To this end, we knocked down the expression of BTK by shRNA (shBTK) in Z138 cells (BCR-independent MCL) as well as in Mino cells (BCR-dependent MCL) and performed the whole genome transcriptome analysis in these cell lines by RNA-sequencing (RNA-seq). As expected, we found that a significant number of genes were upregulated and downregulated in either Mino or Z138 cells. Surprisingly, the set of genes that differentially expressed in Mino cells is strikingly different from that of Z138 cells (Figure 6a). Further comparison of downregulated genes in Mino and Z138 cells revealed only about 10% (58 genes) of overlapped genes (Figure 6b). Similarly, a small set of common upregulated genes was identified in the two cell lines upon BTK inhibition (Figure 6b), suggesting that BTK-mediated regulation of gene expression in BCR-independent MCL cells is distinct from those in BCR-dependent cells.
Figure 6.
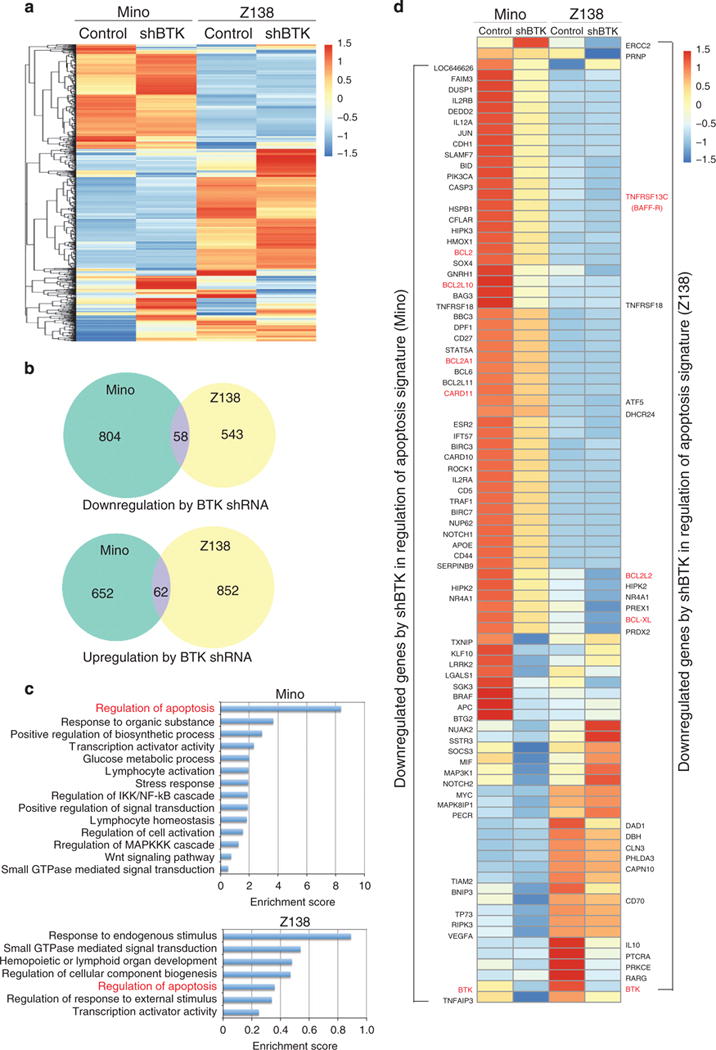
RNA-seq analysis reveals distinct sets of genes regulated by shBTK between Mino and Z138 cells. (a) A heatmap of differentially expressed genes in untreated (control) and shBTK-treated (shRTK) Mino and Z138 cells. The gene expression (RPKM) values for each gene were normalized to the standard normal distribution to generate Z-scores. The color bar indicates expression levels from low in blue to high in red. (b) Venn diagrams showing the overlap of downregulated genes (upper panel) and upregulated genes (bottom panel) between Mino and Z138 cells. (c) Enrichment for ontological categories. (d) A heatmap shows distinct apoptosis-associated genes downregulated by shBTK in Mino and Z138 cells. The complete gene lists for a to d are provided in Supplementary Tables 1c–1f.
We next focused on these downregulated genes and performed signature enrichment analysis using the DAVID gene ontology tool.46 We observed that the common enriched signature in both Mino and Z138 cell lines was associated with regulation of apoptosis, with a greater enrichment score in Mino cells than that in Z138 cells (Figure 6c). As expected, among 70 genes in Mino cells, many regulate B-cell proliferation, differentiation and apoptosis, such as BCL2, MYC, BCL6, MYC, PIK3CA and TNFAIP3 (Figure 6d). Although only 23 genes in Z138 cells were enriched in the regulation of the apoptosis pathway, some of them are well-defined cell survival genes, including BCL-XL, BCL2L2 (BCL-W), DAD1 and TNFRSF13C (BAFF-R) (Figure 6d). Notably, many pro-apoptotic genes, such as FAS, FOXO3B, BCLAF1, CASP1 and CASP4, are upregulated by BTK shRNA in Z138 cells (Supplementary Table 1). Combined, these data demonstrate that BTK mediates noncanonical NF-κB activation in MCL, and provide a molecular mechanism by which BCR-independent MCL cell lines such as Z138 respond to ibrutinib treatment.
DISCUSSION
The anti-apoptotic gene BCL2, initially identified in follicular lymphoma with the t(14;18) chromosomal translocations,33,47 is overexpressed in many types of non-Hodgkin lymphoma, including MCL.31,48–50 As there are no such chromosomal translocations in MCL, how BCL2 is upregulated in this cancer type remains elusive. In this study, we for the first time demonstrate two distinct mechanisms underlying BCL2 overexpression in MCL, one at the posttranslational level and the other at the transcriptional level. By analyzing a tissue microarray of 62 cases, we have found no expression of the E3 ubiquitin ligase FBXO10 in ~ 90% of cases. Further functional and biochemical analyses have confirmed that FBXO10 targets BCL2 for proteasomal degradation in MCL. Also, we have discovered a strong positive correlation in gene expression between BCL2 and BTK, an essential component of the BCR signaling complex. BTK activity in MCL promotes BCL2 transcription through the canonical NF-κB pathway.
The role of BTK in B-cell development, proliferation, differentiation and apoptosis has been well studied since the discovery of its mutation two decades ago that causes X-linked immunodeficiency in mice and X-linked agammaglobulinemia in humans.51 The research effort has improved understanding of how BTK mediates BCR signaling in the canonical NF-κB pathway, but little is known about the role of BTK in regulating noncanonical NF-κB signaling. To our knowledge, there is only one study in BTK-deficient murine B cells suggesting that BAFF-mediated NF-κB activation requires BTK.45 This is an important research area given that the noncanonical pathway is constitutively active because of genetic alterations or activated upon CD40 or BAFF receptor engagement in many types of human lymphomas, and NF-κB activation through these mechanisms promotes cancer cell survival and proliferation.20 Indeed, knockdown of genes in the noncanonical pathway, such as NIK, p52 or RelB, or a specific NIK inhibitor induces apoptotic cell death of MCL cell lines. We here provide compelling evidence that BTK is required for either constitutive or BAFF-mediated noncanonical NF-κB activation in some of the MCL cell lines analyzed. Future studies are required to determine the underlying molecular mechanisms.
Involvement of BTK in the noncanonical NF-κB pathway forms the molecular basis of synergism between ibrutinib and ABT-199 in cell killing, especially in BCR-independent cell lines including Z138 and Maver-1. These two cell lines have unique features in NF-κB: impaired BCR signaling-mediated canonical NF-κB activation because of low CARD11 expression but increased noncanonical NF-κB activation because of TRAF2 or TRAF3 mutation.10 BTK-mediated NF-κB activation is essential for the survival of cancer cells given their sensitivity to ibrutinib or BTK shRNA. However, BTK inhibition by ibrutinib does not reduce BCL2 expression in these cells. High levels of BCL2 protein in Z138 cells are likely due to genomic amplification plus defects in FBXO10-mediated degradation.34 Therefore, blocking of BCL2 anti-apoptotic pathway by ABT-199 is necessary to enhance the efficacy of ibrutinib. A study using siRNA or an antisense oligonucleotide against BCL2 has supported our findings about BCL2-mediated resistance to apoptosis in Z138 cells and revealed that silencing of BCL2 is associated with reduced NF-κB activity, transient loss of p53, and a decrease in cyclin D1.52 Our RNA-seq analysis has revealed that BTK activity in Z138 cells is required for expression of an array of genes that regulate cell growth, proliferation, differentiation and apoptosis, such as BCL-XL, DAD1, HIPK2, TNFRSF18 and TNFRSF13C (BAFF-R) (Figure 6, Supplementary Table 1). As both BCL2 and BTK are indispensable for cancer cell survival and function in distinct survival/signaling pathways, it is not surprising that a combination of ibrutinib and ABT-199 produces synergistic antitumor effects, as shown in our in vitro functional analysis as well as by the Z138 xenograft model.
The synergism between ibrutinib and ABT-199 has also been observed in BCR signaling-dependent cell lines such as Mino and Jeko. Similarly, this group of cell lines has a defect in BCL2 proteasomal degradation because of low levels of FBXO10 expression.36 An additional mechanism for BCL2 upregulation is increased gene transcription by active BCR/BTK signaling through canonical NF-κB activity (Figure 3f), and this is further demonstrated by our RNA-seq study (Figure 6d). The RNA-seq analysis also provides new insights into the molecular pathogenesis of BTK in MCL. In addition to regulation of apoptosis, BTK activity is involved in a diversity of biological processes, such as transcriptional regulation, response to organic substance, positive regulation of biosynthetic process, and B-cell homeostasis and activation (Figures 6c and d). These findings elucidate the molecular mechanisms underlying synergistic co-targeting of BCL2 and BTK in MCL. Given the high expression levels of BTK and BCL2 and the failure of BCL2 protein degradation in the vast majority of cases, combination treatment by ibrutinib and ABT-199 would be expected to be more effective than the single drugs against MCL.
The third group of MCL cell lines, including Granta-519, HBL2 and UPN-1, is independent of BCR signaling possibly due to reduced or lack of BCR gene expression.10 This molecular feature is similar to that observed in Z138 and Maver-1 cell lines. However, unlike Z138 cells that are dependent on noncanonical NF-κB signaling for survival, these three cell lines are all resistant to ibrutinib treatment. The mechanisms of BTK resistance are unknown despite the fact that BTK is phosphorylated and that ibrutinib can block BTK phosphorylation in these cells. In this group, the anti-apoptotic pathway is often deregulated, leading to high expression levels of BCL2 and BCL-XL (Figure 2a). In the case of Granta-519 cells, genomic amplification renders BCL2 expression to desensitize cancer cells to ibrutinib treatment but an addition of ABT-199 can restore sensitivity of cancer cells to ibrutinib (Figures 4b and c). This suggests that targeting of BCL2 or other family members is a potential therapeutic strategy to overcome primary resistance to ibrutinib. In this treatment setting, the first generation inhibitor Navitoclax targeting BCL2, BCL-XL and BCL-W may be more effective.53
In summary, this study elucidated mechanisms of BCL2 overexpression and association of this survival pathway with the BCR/BTK signaling pathway in MCL, and revealed the role of BTK in noncanonical NF-κB activation, which also promotes cancer cell survival. Our data provided a mechanistic rationale for co-targeting of these two oncogenic pathways by their specific inhibitors ABT-199 and ibrutinib as a new therapeutic strategy in a pre-clinical setting. Most importantly, the findings indicate that this combination has a synergistic effect on the killing of both BCR-dependent and -independent MCL cells. As these two signaling pathways are deregulated in DLBCL and follicular lymphoma,6,35,42,54,55 co-targeting by ibrutinib and ABT-199 may provide an attractive therapeutic strategy for these additional lymphoma patients as well.
MATERIALS AND METHODS
Patient samples
A tissue microarray of 62 cases of MCL was obtained from our previous study,37 with approval from the University of Wisconsin—Madison institutional review boards (UW protocol M-2008-1011 and MCRF protocol SHA 10109).
Xenografts
Male and female NOD-Prkdcscid-IL-2Rγ −/− (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, abbreviated as NSG) breeder pairs were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and bred in our animal care facility at the Wisconsin Institutes of Medical Research, University of Wisconsin Madison. NSG mice were bred under specific pathogen-free conditions in sterile ventilated racks. All of the animal protocols were approved by the Institutional Animal Care and Use Committees. See more details in Supplementary Materials and Methods.
RNA-seq
Control shRNA and shBTK in Mino and Z138 cells were induced with 20 ng/ml of doxycycline for 2 days. Total RNA from cells was extracted using RNeasy plus mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. RNA-seq libraries were prepared using the Illumina TruSeq stranded mRNA LT Sample Preparation Kit (San Diego, CA, USA). See more details in Supplementary Materials and Methods.
Cell culture, doxycycline-inducible system and retroviral transduction, plasmid construction, immunohistochemical staining and quantification, cell viability assay, apoptosis analysis, flow cytometry, quantitative RT–PCR (qPCR), electrophoretic mobility shift assay, immunoblotting assay, chromatin immunoprecipitation assay and statistical analysis are described in Supplementary Materials and Methods.
Acknowledgments
We thank Dr Louis Staudt for providing the reagents, Dr Sameer Mathur for providing peripheral blood mononuclear cells, Dr Wei Huang for helping with analysis of immunohistochemical data, Drs Debra Bloom and Peiman Hematti for providing CD40, BAFF and BAFF receptor antibody, and Kirsti Walker for monitoring xenografted mice. This work was supported by the UW-Madison Start-up funds, KL2 Scholar Award (UL1TR0000427 and KL2TR000428), and NIH R01 CA187299 to LR, the MACC fund and the National Institutes of Health/National Cancer Institute (K08 CA174750) to CMC, the UW-Madison Start-up funds and USDA National Institute of Food and Agriculture (Hatch 1002874) to XZ, and the UW Forward Lymphoma Fund. We thank the University of Wisconsin Carbone Cancer Center (UWCCC) for the funds to complete this project. This work was also supported in part by NIH/NCI P30 CA014520- UW Comprehensive Cancer Center Support.
Footnotes
ACCESSION CODES
All sequencing data were deposited into GEO with the accession number GSE80563.
AUTHOR CONTRIBUTIONS
LR designed research; LR, CMC, DTY, SM and BSK conceived and supervised the project; YL, MNB, LR, JW, KMG, LL, SQ, NP and SM. W-D performed research. LL, XZ and JCE analyzed data; CJT contributed reagents and intellectual input; LR, YL and MNB wrote the paper.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest. 2012;122:3416–3423. doi: 10.1172/JCI61272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiorazzi N, Hatzi K, Albesiano E. B-cell chronic lymphocytic leukemia, a clonal disease of B lymphocytes with receptors that vary in specificity for (auto)antigens. Ann N Y Acad Sci. 2005;1062:1–12. doi: 10.1196/annals.1358.002. [DOI] [PubMed] [Google Scholar]
- 4.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5:251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- 5.Kenkre VP, Kahl BS. The future of B-cell lymphoma therapy: the B-cell receptor and its downstream pathways. Curr Hematol Malig Rep. 2012;7:216–220. doi: 10.1007/s11899-012-0127-0. [DOI] [PubMed] [Google Scholar]
- 6.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12:229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hadzidimitriou A, Agathangelidis A, Darzentas N, Murray F, Delfau-Larue MH, Pedersen LB, et al. Is there a role for antigen selection in mantle cell lymphoma? Immunogenetic support from a series of 807 cases. Blood. 2011;118:3088–3095. doi: 10.1182/blood-2011-03-343434. [DOI] [PubMed] [Google Scholar]
- 8.Rinaldi A, Kwee I, Taborelli M, Largo C, Uccella S, Martin V, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol. 2006;132:303–316. doi: 10.1111/j.1365-2141.2005.05883.x. [DOI] [PubMed] [Google Scholar]
- 9.Cinar M, Hamedani F, Mo Z, Cinar B, Amin HM, Alkan S. Bruton tyrosine kinase is commonly overexpressed in mantle cell lymphoma and its attenuation by Ibrutinib induces apoptosis. Leuk Res. 2013;37:1271–1277. doi: 10.1016/j.leukres.2013.07.028. [DOI] [PubMed] [Google Scholar]
- 10.Rahal R, Frick M, Romero R, Korn JM, Kridel R, Chan FC, et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20:87–92. doi: 10.1038/nm.3435. [DOI] [PubMed] [Google Scholar]
- 11.Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4:1022–1035. doi: 10.1158/2159-8290.CD-14-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma J, Lu P, Guo A, Cheng S, Zong H, Martin P, et al. Characterization of ibrutinib-sensitive and -resistant mantle lymphoma cells. Br J Haematol. 2014;166:849–861. doi: 10.1111/bjh.12974. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15:48–58. doi: 10.1016/S1470-2045(13)70513-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14:219–232. doi: 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- 15.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee MH, Mabb AM, Gill GB, Yeh ET, Miyamoto S. NF-kappaB induction of the SUMO protease SENP2: a negative feedback loop to attenuate cell survival response to genotoxic stress. Mol Cell. 2011;43:180–191. doi: 10.1016/j.molcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 18.Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111:3701–3713. doi: 10.1182/blood-2007-09-111948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hailfinger S, Lenz G, Ngo V, Posvitz-Fejfar A, Rebeaud F, Guzzardi M, et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2009;106:19946–19951. doi: 10.1073/pnas.0907511106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110:18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Jima D, Moffitt AB, Liu Q, Czader M, Hsi ED, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123:2988–2996. doi: 10.1182/blood-2013-07-517177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun SC. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Briones J, Timmerman JM, Hilbert DM, Levy R. BLyS and BLyS receptor expression in non-Hodgkin’s lymphoma. Exp Hematol. 2002;30:135–141. doi: 10.1016/s0301-472x(01)00774-3. [DOI] [PubMed] [Google Scholar]
- 25.Novak AJ, Grote DM, Stenson M, Ziesmer SC, Witzig TE, Habermann TM, et al. Expression of BLyS and its receptors in B-cell non-Hodgkin lymphoma: correlation with disease activity and patient outcome. Blood. 2004;104:2247–2253. doi: 10.1182/blood-2004-02-0762. [DOI] [PubMed] [Google Scholar]
- 26.Abe M, Kido S, Hiasa M, Nakano A, Oda A, Amou H, et al. BAFF and APRIL as osteoclast-derived survival factors for myeloma cells: a rationale for TACI-Fc treatment in patients with multiple myeloma. Leukemia. 2006;20:1313–1315. doi: 10.1038/sj.leu.2404228. [DOI] [PubMed] [Google Scholar]
- 27.Wada K, Maeda K, Tajima K, Kato T, Kobata T, Yamakawa M. Expression of BAFF-R and TACI in reactive lymphoid tissues and B-cell lymphomas. Histopathology. 2009;54:221–232. doi: 10.1111/j.1365-2559.2008.03203.x. [DOI] [PubMed] [Google Scholar]
- 28.Takahata H, Ohara N, Ichimura K, Tanaka T, Sato Y, Morito T, et al. BAFF-R is expressed on B-cell lymphomas depending on their origin, and is related to proliferation index of nodal diffuse large B-cell lymphomas. J Clin Exp Hematop. 2010;50:121–127. doi: 10.3960/jslrt.50.121. [DOI] [PubMed] [Google Scholar]
- 29.Paterson JC, Tedoldi S, Craxton A, Jones M, Hansmann ML, Collins G, et al. The differential expression of LCK and BAFF-receptor and their role in apoptosis in human lymphomas. Haematologica. 2006;91:772–780. [PubMed] [Google Scholar]
- 30.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–3683. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tracey L, Perez-Rosado A, Artiga MJ, Camacho FI, Rodriguez A, Martinez N, et al. Expression of the NF-kappaB targets BCL2 and BIRC5/survivin characterizes small B-cell and aggressive B-cell lymphomas, respectively. J Pathol. 2005;206:123–134. doi: 10.1002/path.1768. [DOI] [PubMed] [Google Scholar]
- 32.Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012;12:121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 33.Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 34.de Leeuw RJ, Davies JJ, Rosenwald A, Bebb G, Gascoyne RD, Dyer MJ, et al. Comprehensive whole genome array CGH profiling of mantle cell lymphoma model genomes. Hum Mol Genet. 2004;13:1827–1837. doi: 10.1093/hmg/ddh195. [DOI] [PubMed] [Google Scholar]
- 35.Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012;26:1383–1390. doi: 10.1038/leu.2011.378. [DOI] [PubMed] [Google Scholar]
- 36.Chiorazzi M, Rui L, Yang Y, Ceribelli M, Tishbi N, Maurer CW, et al. Related F-box proteins control cell death in Caenorhabditis elegans and human lymphoma. Proc Natl Acad Sci USA. 2013;110:3943–3948. doi: 10.1073/pnas.1217271110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oberley MJ, Rajguru SA, Zhang C, Kim K, Shaw GR, Grindle KM, et al. Immunohistochemical evaluation of MYC expression in mantle cell lymphoma. Histopathology. 2013;63:499–508. doi: 10.1111/his.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 39.Chiron D, Dousset C, Brosseau C, Touzeau C, Maiga S, Moreau P, et al. Biological rational for sequential targeting of Bruton tyrosine kinase and Bcl-2 to overcome CD40-induced ABT-199 resistance in mantle cell lymphoma. Oncotarget. 2015;6:8750–8759. doi: 10.18632/oncotarget.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao X, Bodo J, Sun D, Durkin L, Lin J, Smith MR, et al. Combination of ibrutinib with ABT-199: synergistic effects on proliferation inhibition and apoptosis in mantle cell lymphoma cells through perturbation of BTK, AKT and BCL2 pathways. Br J Haematol. 2015;168:765–768. doi: 10.1111/bjh.13149. [DOI] [PubMed] [Google Scholar]
- 41.Yang Y, Shaffer AL, 3rd, Emre NC, Ceribelli M, Zhang M, Wright G, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell. 2012;21:723–737. doi: 10.1016/j.ccr.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci USA. 2014;111:2349–2354. doi: 10.1073/pnas.1311846111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–1189. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herman SE, Sun X, McAuley EM, Hsieh MM, Pittaluga S, Raffeld M, et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia. 2013;27:2311–2321. doi: 10.1038/leu.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shinners NP, Carlesso G, Castro I, Hoek KL, Corn RA, Woodland RT, et al. Bruton’s tyrosine kinase mediates NF-kappa B activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J Immunol. 2007;179:3872–3880. doi: 10.4049/jimmunol.179.6.3872. [DOI] [PubMed] [Google Scholar]
- 46.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 48.Iqbal J, Neppalli VT, Wright G, Dave BJ, Horsman DE, Rosenwald A, et al. BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol. 2006;24:961–968. doi: 10.1200/JCO.2005.03.4264. [DOI] [PubMed] [Google Scholar]
- 49.Knezevich S, Ludkovski O, Salski C, Lestou V, Chhanabhai M, Lam W, et al. Concurrent translocation of BCL2 and MYC with a single immunoglobulin locus in high-grade B-cell lymphomas. Leukemia. 2005;19:659–663. doi: 10.1038/sj.leu.2403661. [DOI] [PubMed] [Google Scholar]
- 50.Nagy B, Lundan T, Larramendy ML, Aalto Y, Zhu Y, Niini T, et al. Abnormal expression of apoptosis-related genes in haematological malignancies: overexpression of MYC is poor prognostic sign in mantle cell lymphoma. Br J Haematol. 2003;120:434–441. doi: 10.1046/j.1365-2141.2003.04121.x. [DOI] [PubMed] [Google Scholar]
- 51.Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 52.Tucker CA, Kapanen AI, Chikh G, Hoffman BG, Kyle AH, Wilson IM, et al. Silencing Bcl-2 in models of mantle cell lymphoma is associated with decreases in cyclin D1, nuclear factor-kappaB, p53, bax, and p27 levels. Mol Cancer Ther. 2008;7:749–758. doi: 10.1158/1535-7163.MCT-07-0302. [DOI] [PubMed] [Google Scholar]
- 53.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 54.Freedman A. Follicular lymphoma: 2014 update on diagnosis and management. Am J Hematol. 2014;89:429–436. doi: 10.1002/ajh.23674. [DOI] [PubMed] [Google Scholar]
- 55.Hiddemann W, Cheson BD. How we manage follicular lymphoma. Leukemia. 2014;28:1388–1395. doi: 10.1038/leu.2014.91. [DOI] [PubMed] [Google Scholar]