

Abstract
The melanocortin receptors are involved in many physiological functions, including pigmentation, sexual function, feeding behavior, and energy homeostasis, making them potential targets to treat obesity, sexual dysfunction, etc. Understanding the basis of the ligand-receptor interactions is crucial for the design of potent and selective ligands for these receptors.
The conformational preferences of the cyclic melanocortin ligands MTII (Ac-Nle4-c[Asp5-His6-DPhe7-Arg8-Trp9-Lys10]–NH2) and SHU9119 (Ac-Nle4-c[Asp5-His6-DNal(2′)7-Arg8-Trp9-Lys10]–NH2), which show agonist and antagonist activity at the h-MC4R, respectively, were comprehensively investigated by solution NMR spectroscopy in different environments. In particular, water and water/DMSO (8:2) solutions were used as isotropic solutions and an aqueous solution of DPC (dodecylphosphocholine) micelles was used as a membrane mimetic environment. NMR-derived conformations of these two ligands were docked within h-MC4R models. NMR and docking studies revealed intriguing differences which can help explain the different activities of these two ligands.
Keywords: Melanocortin peptides, MC4 receptor, NMR spectroscopy, DPC micelle solution, Docking studies
1. Introduction
The melanocortin family contains five human receptors (h-MC1R-h-MC5R) cloned to date and stimulates the cAMP second messenger and other signal transduction pathways [1–8]. Melanocortin receptors belong to the class A superfamily of rhodopsin-like G-protein-coupled receptors (GPCRs), characterized by having seven trans-membrane α-helices (TM1-TM7) linked by three extracellular and three intracellular loops [2,5,6]. The endogeneous agonists of the MCRs, the melanocortins, are a family of peptides comprised of α-, β-, and γ-melanocyte stimulating hormones (MSH) and adreno-corticotropic hormone (ACTH). They are derived from post-translational modification of a common precursor, proopiomelanocortin (POMC) [9]. The natural melanocortins are all agonist for h-MCRs1 with exception of the h-MC2R, for which only ACTH is a full agonist [10]. Also, synthetic melanocortins have different pharmacological profiles for the five h-MCRs. For example, NDP-α-MSH [11] and MTII [12] are agonist for all h-MCRs except the h-MC2R [13]. There are also the endogeneous protein antagonists known as agouti and agouti-related protein (AGRP) [14,15]. Interaction of these effectors with MCRs results in the modulation of numerous biological functions which include among others regulation of skin pigmentation (MC1R), steroid production (MC2R), the immune response, thermoregulation, food intake, sexual function (MC3R and MC4R), and stress-induced anxiety and depression [16–21]. The MC4R subtype is regarded as a potential drug target, because it is involved in feeding and sexual behavior [14,21–24]. Mammals with a defective MC4R gene, which is expressed in the brain, are characterized by obese phenotype and increased food intake [25–27]. Pharmacological studies indicate that activation of the MC4R in rodents [24] and humans [17] modulates erectile function. Consequently, research efforts have been focused on the development of potent and MC4R-selective agonists as potential antiobesity drugs or as treatments for sexual dysfunction [28]. On the other hand, a MC4R antagonist that blocks the satiety-inducing effect of α-MSH could be helpful for treatment of anorexia or cancer cachexia [29].
A molecular understanding of MTII and SHU9119 activity at the h-MC4R may have important implications in the design of drugs. In addition, the identification of the essential amino acid residues of the h-MC4R responsible for MTII agonism and SHU9119 antagonism should be important for understanding the signalling events that regulate the melanocortin system under physiologic conditions [30,31].
Hence, we first studied the conformational preferences of the cyclic melanocortin ligands MTII, and SHU9119 (Table 1), agonist and antagonist at h-MC4R, respectively. Conformational analysis was carried out by NMR spectroscopy in water, water/DMSO solutions, and 200 mM aqueous solution of DPC as membrane mimetic environment. Then, NMR-derived structures of MTII and SHU9119 were docked within the h-MC4 receptor model, in the active and inactive state, respectively.
Table 1.
Ligand sequences.
| Peptide | Sequence |
|---|---|
| MTII | Ac-Nle4-c[Asp5-His6-DPhe7-Arg8-Trp9-Lys10]–NH2 |
| SHU9119 | Ac-Nle4-c[Asp5-His6-DNal(2′)7-Arg8-Trp9-Lys10]–NH2 |
| NDP-MSH | Ser1-Tyr2-Ser3-Nle4-Glu5-His6-DPhe7-Arg8-Trp9-Gly10 -Lys11-Pro12-Val13-Gly14 |
2. Results
2.1. Chemistry
Peptides were synthesized using the solid phase approach and standard Fmoc methodology in a manual reaction vessel [32] (Experimental Section).
The purification was achieved using a semi-preparative RP-HPLC C-18 bonded silica column (Vydac 218TP1010). The purified peptide was 98% pure as determined by analytical RP-HPLC. The correct molecular weight and composition of the peptide was confirmed by mass spectrometry and amino acid analysis (Table S1, Supporting Information).
2.2. NMR analysis
Complete 1H NMR chemical shift assignments were achieved for MTII and SHU9119 according to the Wüthrich procedure [33] via the usual systematic application of DQF-COSY [34,35], TOCSY [36], and NOESY [37] experiments (Tables S2–S9, Supporting Information) with the support of the XEASY software package [38]. The conformational preferences for the two peptides were investigated by solution NMR spectroscopy in different solvent environments. In particular, water and water/DMSO (8:2) solutions were used as isotropic solutions and 200 mM aqueous solution of DPC (dodecylphosphocholine micelles) was used as a membrane mimicking environment.
2.2.1. Water solution
NMR analysis was performed in water at pH 5 and a peptide concentration of about 2 mM. First, we analyzed the peptides at 25 °C. Both peptides showed similar NMR parameters (Tables S2–S3) as previously reported [39,40]. Almost all NMR parameters indicate structural flexibility: a) temperature effects |Δδ/ΔT| > 6 ppb/K; b) 3JHN-Hα coupling constants are all within the range 6–8 Hz; c) No standard α-helix or β-sheet structure from Ha CSI (chemical shift index) values [41] (Figure S1, Supporting Information); d) No unambiguous medium- or long-range backbone NOE connectivities were found in the ROESY or NOESY. Strong dαN(i, i+1) NOEs, which are generally observed in extended structures, appeared along almost the entire length of the peptides.
To reduce the conformational flexibility, we acquired the spectra also at 5 °C. At this temperature, most of the NMR parameters did not change significantly (Tables S4–S5), though improvements of the signal quality in the NOESY spectra were observed. Weak dαN(i, i+2) NOEs between Nle4 and His6 and between His6 and Arg8 could be observed. Also the methyl protons of the N-terminal acetyl group show a weak NOE contact with the Hα of Asp5. Medium dNN(i, i+1) NOE between DPhe7 (DNal(2′)7) and Arg8 was observed. Among the possible observable contacts, the dαN(i, i+2) NOE between Asp5 and DPhe7 could not be observed due to spectral overlap.
2.2.2. Cryoscopic solution
To further reduce the peptide conformational flexibility, NMR spectra were acquired at −10 °C in a cryomixture solution of water/DMSO 8:2. Such cryomixtures have been shown to produce physico-chemical conditions compatible with those of biological fluids [42]. Again, most of the NMR parameters did not change significantly (Tables S6–S7). Apart from the two dαN(i, i+2) NOEs observed also in water (5 °C) the NOESY spectra showed dαN(i, i+2) NOEs between Asp5 and DPhe7 (DNal(2′)7). Furthermore, dβN(i, i+3) NOEs between Asp5 and Arg8 and dNN(i, i+2) NOEs between His6 and Arg8 also were observed.
2.2.3. DPC micelles
Several NMR parameters indicate that MTII and SHU9119 are highly structured in DPC solution. In particular, 3JHN-Hα coupling constants (Tables S8–S9) and Hα CSI values (Figure S1) and many NOE signals (Tables S10–S11, Supporting Information) clearly point to a folded structure encompassing the N-terminal residues (4–7) and extended conformation of residues 8e9. Non-trivial medium range NOE interactions, among which dαN(i, i+2) 4–6, 5–7, 6–8, dNN(i, i+2) 6–8, and dβN(i, i+3) 5–8, are observed indicating that the membrane mimetic environment stabilizes intrinsic conformational tendencies of the peptide. Additional medium and long range NOE contacts were observed. In particular, various NOEs connected the Nle4 side chain with both DPhe7 (DNal(2′)7) and Trp9 aromatic moieties indicating spatial proximity of these side chains. Other interesting contacts were observed between the methyl protons of the N-terminal acetyl group and the HN of Asp5, and between amide protons of the C-terminal NH2 group and the Hδ1 and Hε1 of Trp9.
2.2.4. Structure determination
NOE distance restraints obtained for MTII and SHU9119 in DPC micelles were used as the input data for a simulated annealing structure calculation using the program DYANA [43]. The annealing procedure produced 100 conformations from which 20 structures were chosen, whose interprotonic distances best fitted the NOE derived distances, and then refined through successive steps of restrained and unrestrained EM calculations using the Insight/Discover package (Accelrys Inc, San Diego, CA).
2.2.5. Structure description
Superposition of the 10 lowest energy conformers of MTII and SHU9119 are shown in Fig. 1. Since a β-turn may be defined as four consecutive non-helical residues that have a Cα(i)–Cα(i+3) distance < 7 Å, two β-turns that involve Nle4 to DPhe7 (DNal(2′)7) and Asp5 to Arg8, can be identified. Examination of the backbone dihedral angles at the central (i+1, and i+2) residues of the turns showed that these turn structures most closely resembled type I (residue 4 to 7) and type II (residue 5 to 8) β-turns, although deviations from the standard dihedral angles of these two types of β-turn occurred (Table S12, Supporting Information). Residues 8 to 10 are in extended conformations. The side chain χ1-angles of Asp5, Arg8, Trp9 and Lys10 are also well defined, preferring trans, gauche−, trans and gauche− orientations, respectively. Side chains of Nle4 and His6 are more flexible. The DPhe7 orientation in MTII is also well defined as trans. The DNal(2′)7 orientation in SHU9119 is less defined showing an equilibrium between trans and gauche+ rotamers. These results are consistent with the measured 3JHαHβ coupling constants (Tables S8–S9) [44,45].
Fig. 1.
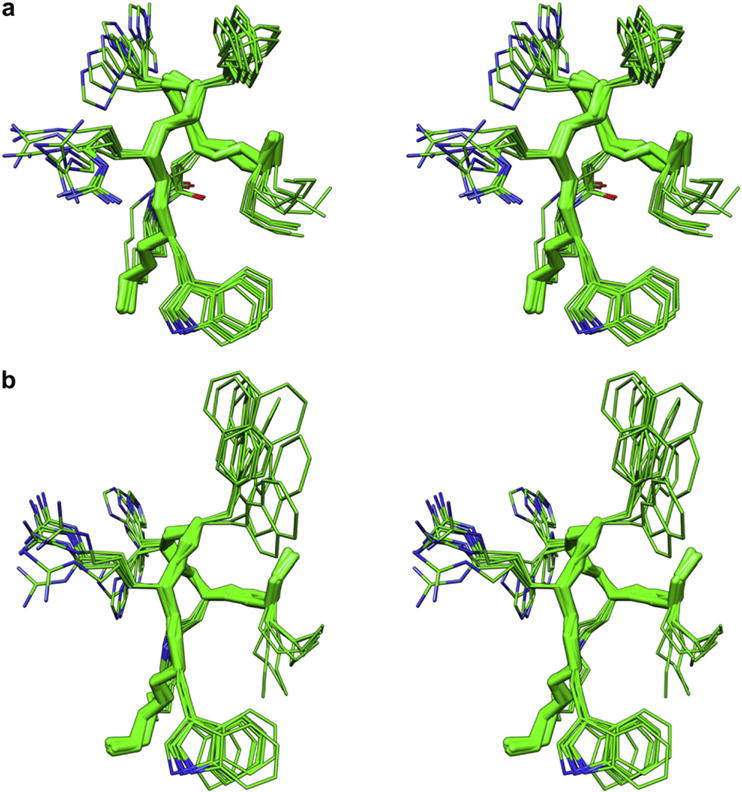
Stereoviews of the 10 lowest energy conformers of MTII (a), and SHU9119 (b). Structures were superimposed using the backbone heavy atoms of residues 5–10. Heavy atoms are shown with different colours (carbon, green; nitrogen, blue; oxygen, red). Hydrogen atoms are not shown for clarity (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The peptide surface has amphipathic nature. In fact, considering the pseudo-plane defined by the backbone atoms (green ribbon, Fig. 1) the hydrophobic residues Nle4, DPhe7 (DNal(2′)7) and Trp9 lie on one side (right in Fig. 1) while the positively charged residues His6 and Arg8 lie on the other side.
2.3. Docking studies
NMR-derived structures of MTII and SHU9119 were docked within the h-MC4R models proposed by Mosberg [46,47]. In particular, the agonist MTII was docked within an “active state” model (h-MC4Ra), while the antagonist SHU9119 was docked within an “inactive state” model (h-MC4Ri). Fig. 2 shows a snake-like diagram of the h-MC4R sequence.
Fig. 2.
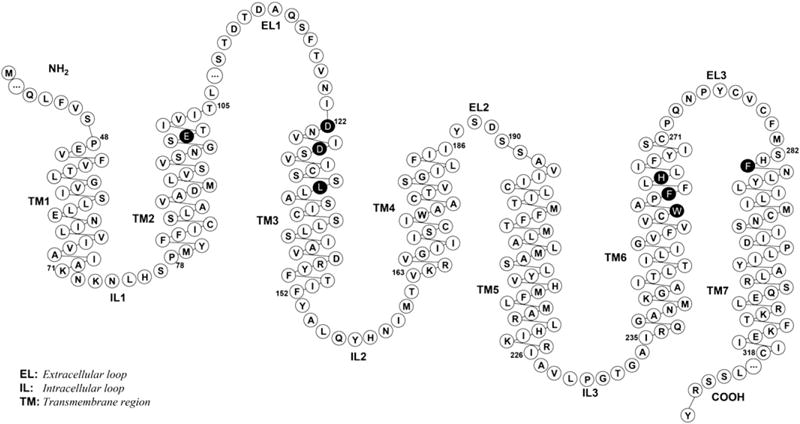
Snake-like diagram of the h-MC4R sequence. This plot was generated with the RbDe software [48]. Black residues indicate that mutation data are available. The ‘…’ indicates hidden residues (see reference [46] for the complete sequence).
Since the currently available docking programs may not work very well for peptide compounds (more than eight rotatable bonds) [49], manual docking was conducted. The NMR-derived MTII structure was placed in between the trans-membrane domains of the h-MC4Ra. The following criteria were employed to achieve meaningful docking modes: (i) The positively charged side of the amphipathic surface of the peptides should be close to the carboxylate groups of Asp122, Asp126, and Glu100, as suggested by several mutagenesis studies [46,47,50–57]; (ii) DPhe7 residue should point towards Leu133 residue as suggested by a mutagenesis study [50]; (iii) No steric clashes should occur between any atom. To assess the stability of the MTII/h-MC4Ra complex we analyzed the potential ligand/receptor interactions, energy minimization and MD simulations for 1 ns at a constant temperature of 300 K. During the MD simulation, the ligand, the EL’s, and all the receptor side chains were allowed to relax, while the TM’s and intracellular loops (ILS) backbone atoms were held frozen. The distances between the peptide and the key receptor residues were monitored along the complete 1 ns MD trajectory (Supporting Information). The mean structure of the last 0.5 ns of MD was energy minimized and used for subsequent analysis.
To inspect the variations in the ligand conformation, the rmsd with the respect to the starting structure was calculated. Interestingly, the rmsd of the MTII backbone atoms turned out to be stable throughout all of the MD simulations (0 < rmsd < 0.8 Å), indicating that the peptide settles into the receptor-binding site in a stable conformation. Also the side chain orientations are those described by NMR. In particular, the DPhe7 and Trp9 side chain prefer a trans orientation about χ1 angle (χ1 ≈ 160° for DPhe7, χ1 ≈ −163° for Trp9). As shown in Fig. 3a, the hypothetical binding site of MTII is located among TM2-TM7, and EL3. C- and N-terminal residues point towards the extracellular side. The binding mode of the peptide is determined mainly by the interactions shown in Fig. 3b and summarized in Table 2.
Fig. 3.
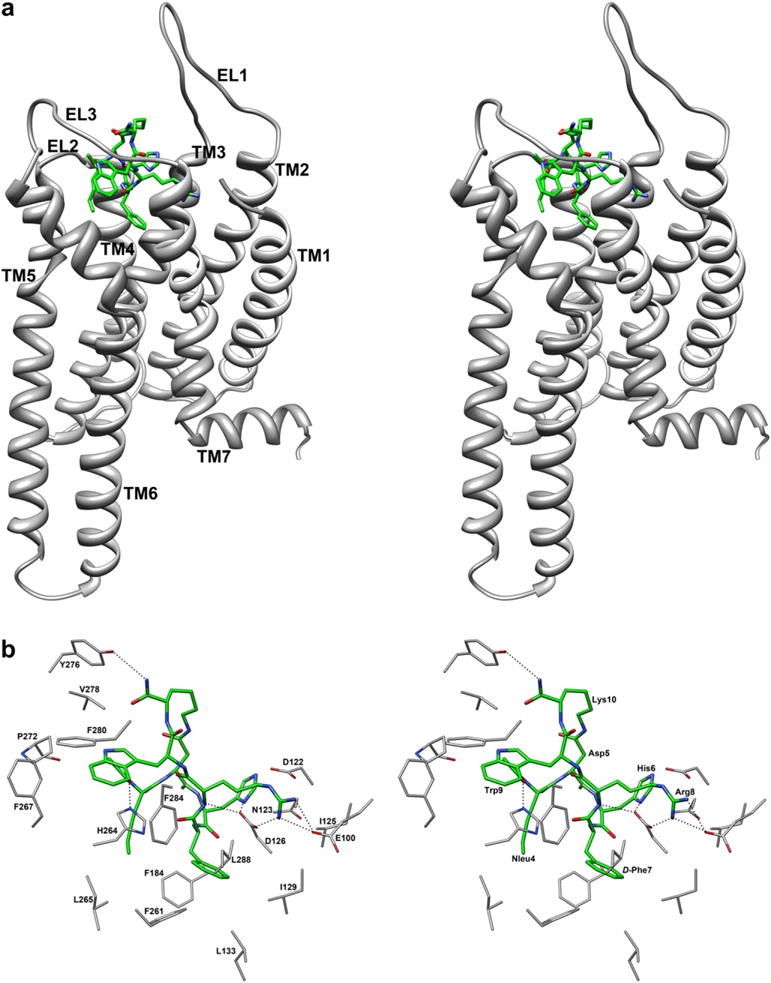
(a) Stereoview of h-MC4Ra model complexed with MTII. MTII heavy atoms are shown with different colours (carbon, green; nitrogen, blue; oxygen, red). Hydrogen atoms are not shown for clarity. Receptor backbones are represented in gray and labeled. (b) Stereoview of MTII within the binding pocket of h-MC4Ra. Hydrogen bonds are represented with dashed lines (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 2.
MTII/h-MC4Ra interactions.
| Residuea | Surrounding residue |
|---|---|
| Nle4 | Val193 (TM5), His264b (TM6), Leu265 (TM6), Tyr268 (TM6) |
| Asp5 | |
| His6 | Asp122 (TM3), Asn123(TM3), Asp126 (TM3) |
| DPhe7 | Asp126 (TM3), Ile129 (TM3), Leu133 (TM3), Phe184 (TM4), Phe261 (TM6), Phe284 (TM7), Leu288 (TM7) |
| Arg8 | Glu100 (TM2), Asp122 (TM3), Ile125 (TM3), Asp126 (TM3), Ile129(TM3) |
| Trp9 | His264 (TM6), Phe267 (TM6), Pro272 (EL3), Val278 (EL3), Phe280 (EL3), Phe284 (TM7) |
| Lys10 | Tyr276 (EL3), Val278 (EL3), Met281 (EL3) |
For sake of clarity, the residue numbers of the ligands are reported as superscript while those of the receptor are not.
Receptor residues involved in mutagenesis studies are shown in bold.
In particular, (i) a tight charge-reinforced hydrogen-bonding network involving the carboxylate groups of Glu100 and Asp126 with the protonated guanidinium group of Arg8 of MTII are established. The guanidinium group of Arg8 is also involved in an electrostatic interaction with the carboxylate group of Asp122. Such interactions, which we assume to be anchoring points of the ligand to the h-MC4Ra, remained stable during the entire MD simulation (Figure S2, Supporting Information). The oxygen atoms of the carboxylate of Asp126 form a charge-reinforced hydrogen bond with the protonated imidazole group of His6, which was not stable during the MD simulation (Figure S2). (ii) Two hydrophobic pockets, delineated by residues listed in Table 1, host the side chains of DPhe7, and Trp9 of MTII. Particularly, the side chain of DPhe7 occupies the hydrophobic pocket involving residues Ile129, Leu133, Phe184, Phe261, Phe284, while the indole system of Trp9 is surrounded by Phe280, Pro272, Phe267, Phe284 and appears to be optimally oriented for a π-stacking interaction with the imidazolic system of His264. iii) Terminal groups also contribute to the complex stabilization. The Nle4 side chain is close to Val193 and Tyr268. The acetyl group CO of Nle4 (N-terminal) engages hydrogen bonds with imidazole NHε of His264. Amide group NH2 of Lys10 (C-terminal) established a hydrogen bond with the phenolic OH of Tyr276. These H bonds are not stable during the MD production run (data not shown).
The NMR-derived SHU9119 structure was placed within the trans-membrane domains of the h-MC4Ri model, following the same criteria used for MTII (see above) to achieve meaningful binding interactions. Energy minimization and MD simulations (1 ns) were run to assess the stability of the SHU9119/h-MC4Ri complex and to analyze the potential ligand/receptor interactions. The mean structure of the last 0.5 ns of MD was extensively minimized and used for subsequent analysis. SHU9119 backbone atoms turned out to be stable throughout all of the MD simulations (0 < rmsd < 0.9 Å), indicating that the peptide settles into the receptor-binding site in a stable conformation. Also the side chain orientations are those described by NMR. In particular, Trp9 prefers a trans orientation about the χ1 angle (χ1 ≈ 175°). Finally, the DNal(2′)7 side chain adopts a gauche+ conformation (χ1 ≈ 80°).
As shown in Fig. 4a, the hypothetical binding site of SHU9119 is located among TM2-TM7, and EL3. C- and N-terminal residues point towards the extracellular side. The binding mode of the peptide is determined mainly by the interactions showed in Fig. 4b and Table 3. In particular, (i) considering Arg8 and His6, the same interactions observed in the MTII/h-MC4Ra complex are observed also for SHU9119. Again, Hbonds involving the Arg8 guanidinium group remained stable during the whole MD production run (Figure S3, Supporting Information), while those of His6 were not. (ii) Two hydrophobic pockets, involving the residues listed in Table 3, host the aromatic side chains of DNal(2′)7, and Trp9 of SHU9119. These pockets only partially overlap with those hosting the aromatic side chains of MTII. Particularly, the side chain of DNal(2′)7 occupy the hydrophobic pockets involving residues Leu133, Phe184, Cys196, Leu197, Met200, Phe261, Phe262, Leu265, while the indole group of Trp9 is surrounded by His264, Phe267, Met281, Phe284 and appears to be optimally oriented for a π-stacking interaction with the aromatic group of Tyr268. This is different from MTII, because Trp9 in MTII makes π-stacking with His264. The Nle4 side chain is close to Leu265, Tyr268, and Tyr276. The acetyl group CO of Nle4 (N-terminal) in SHU9119 isn’t involved in hydrogen-bonding. Finally, the amide group NH2 and the oxygen atom of the terminal carboxamide group of Lys10 (C-terminal) established two hydrogen bonds: with the OH of Ser116 and with the phenolic OH of Tyr268. These Hbonds are not stable during the MD production run (data not shown).
Fig. 4.
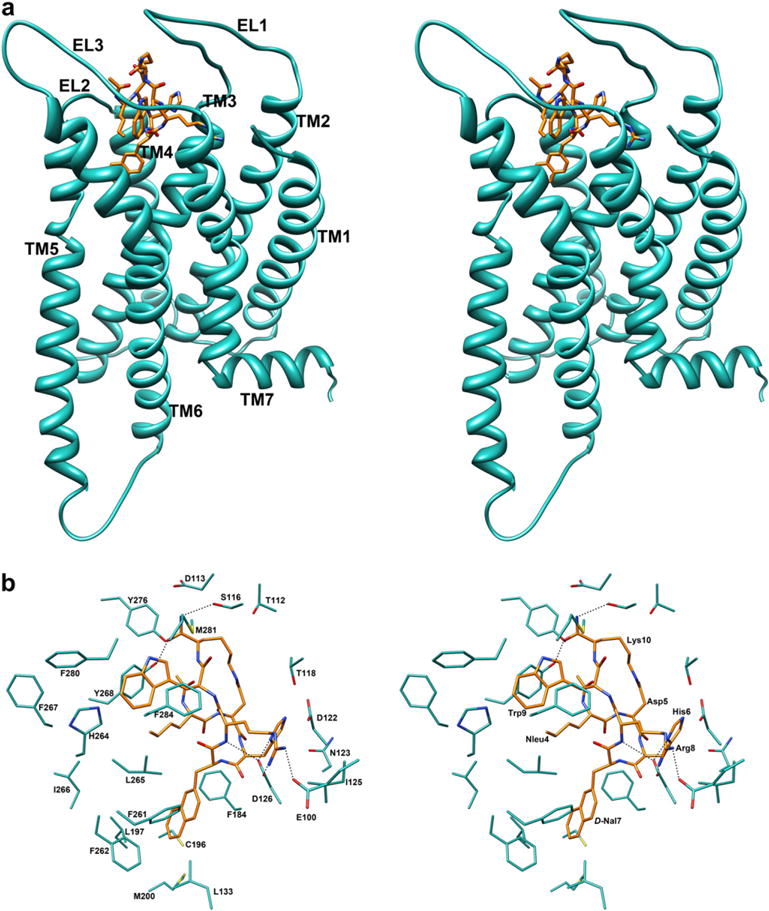
(a) Stereoview of h-MC4Ri model complexed with SHU9119. SHU9119 heavy atoms are shown with different colours (carbon, orange; nitrogen, blue; oxygen, red). Hydrogen atoms are not shown for clarity. Receptor backbones are represented in cyan and labeled. (b) Stereoview of SHU9119 within the binding pocket of h-MC4Ri. Hydrogen bonds are represented with dashed lines (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.).
Table 3.
SHU9119/h-MC4Ri Interactions.
| Residuea | Surrounding residue |
|---|---|
| Nle4 | His264b (TM6), Leu265 (TM6), Tyr268 (TM6), Tyr276(EL3) |
| Asp5 | Phe184 (TM4) |
| His6 | Thr118 (EL1), Asp122 (TM3), Asn123 (TM3), Asp126 (TM3), Phe184 (TM4) |
| DNal7 | Leu133 (TM3), Phe184 (TM4), Cys196 (TM5), Leu197 (TM5), Met200 (TM5), Phe261 (TM6), Phe262 (TM6), Leu265 (TM6) |
| Arg8 | Glu100 (TM2), Asp122 (TM3), Ile125 (TM3), Asp126 (TM3), |
| Trp9 | His264 (TM6), Leu265 (TM6), Tyr268 (TM6), Phe267 (TM6), Phe280 (EL3), Met281 (EL3), Phe284 (TM7) |
| Lys10 | Thr112 (EL1), Asp113 (EL1), Ser116 (EL1), Thr118 (EL1), Tyr268 (TM6), Tyr276 (EL3), Val278 (EL3), Met281 (EL3) |
For sake of clarity, the residue numbers of the ligands are reported as superscript while those of the receptor are not.
Receptor residues involved in mutagenesis studies are evidenced in bold.
3. Discussion
We investigated the conformational preferences of the cyclic melanocortin ligands MTII and SHU9119 by solution NMR spectroscopy in different environmental situations: water, water/DMSO (8:2) and an aqueous solution of DPC (dodecylphosphocholine). In water and water/DMSO cryoscopic mixture, NMR parameters were very similar for both peptides and indicate structural flexibility. A few NOEs, however, point to a tendency of the peptides to form a turn-helical conformation at the N-terminus (residue 4–8). The data could be indicative of a nascent helix in solution [58]. The nascent helix consists of a population of different conformations, in which a significant proportion contains backbone conformations in the α-region of (ϕ, ψ) space in the Ramachandran plot, rather than of any single defined solution conformation.
In DPC micelle solution, the peptides exhibited a higher conformational stability. The use of DPC micelles to study the conformational properties of MTII and SHU9119 is motivated on the basis of their interaction with a membrane receptor. For peptides acting as a ligand for membrane receptors (such as a GPCR), the use of membrane mimetic media, such as SDS or DPC, is suggested hypothesizing a membrane-assisted mechanism of interactions between these peptides and their receptors. According to this model, the membrane surface plays a key role to facilitate the transition of the peptide from a flexible conformation, adopted in the extracellular environment, to a more specific conformation which is recognized by the receptor [59]. NMR has proven useful to examine the structures of bioactive peptides that cross membrane barriers [60–62].
Two consecutive β-turns that involved Nle4 to DPhe7/DNal(2′)7 (distorted type I) and Asp5 to Arg8 (distorted type II) and a short extended segment along residues Trp9 and Lys10 were observed in the calculated structures of MTII and SHU9119 (Fig. 1 and Table S12). It is noteworthy that an amphiphilic molecular surface was obtained for the message sequence residues in both peptides. The main conformational difference observed in the structures of the two ligands was established in a different orientation of the DPhe7 and DNal(2′)7 side chains. DPhe7 of MTII preferred the trans rotamer, while the DNal(2′)7 side chain of SHU9119 was more flexible.
A type II β-turn structure encompassing residues 5–8 was already found by NMR analysis of MTII and SHU9119 in water solution [39,40]. This β-turn led to stacking between the aromatic rings of His6 and DPhe7 in MTII while no aromatic stacking between His6 and DNal(2′)7 was found in SHU9119. This stacking was not observed in the structures obtained in DPC micelles. Considering the β-turn encompassing residues 4–7, it has never been observed in the structure of MTII or SHU9119. Interestingly, the presence of this turn is in accordance with the results of N-methylation of MTII backbone amide bonds. In fact, N-methylation of DPhe7, which should destabilize this β-turn, caused a total loss of binding as well as adenylate cyclase activity at the h-MC4R (h-MC1R, h-MC3R and h-MC5R) [63]. N-Methylation of Arg8 caused a dramatic reduction of the binding (about 500-fold at the h-MC4R) but yielded a compound that retained full agonist activity toward all subtypes of melanocortin receptors.
To gain insight into the interaction mode of these ligands with the h-MC4R, we first undertook a docking study between MTII and h-MC4R model. Since the crystal structure of a GPCR in the active conformation has not yet been obtained, we used a h-MC4R model in the “active state” proposed by Mosberg et al. (h-MC4Ra) [46]. According to these authors, upon activation, the receptor experiences a rearrangement which involves mainly the TM6 helix. The TM6 helix shifts outward and rotates counterclockwise (viewed from the extracellular side) during activation, moving its intracellular end away from TM3 and toward TM5. As a result of this and other changes, the receptor structure tightens near its extracellular surface but opens up at the cytoplasmic side, providing a cavity for binding of the Gαs subunit. In the active state model, several side chains change their orientation among which Trp258, in accordance with earlier spectroscopic results [64]. Similar conformational changes upon activation of the MC4R were subsequently proposed also by Hogan et al. [53]. During the manuscript preparation another model of the h-MC4Ra has been published [65]. This model was based on recent crystal structures of the GPCR opsin in the ligand-free and in the G-protein-interacting conformations [66,67]. Interestingly, our h-MC4Ra model and that built by Chapman et al. are quite similar showing an rmsd of the TM’s backbone heavy atoms of 2.0 Å (Figure S4, Supporting Information).
For the MTII/h-MC4Ra complex, docking and the MD simulations (Fig. 3) indicated that: (i) the structure adequately fits the binding site and is stable during the MD trajectory; (ii) the binding site, situated in the entrance of the TM bundle on the extracellular side, is formed by TM2-TM7, and EL3 (Fig. 3a); (iii) the pharmacophore residues DPhe7, the Arg8 and Trp9 side chains establish the highest number of interactions with the receptor. In particular, Arg8 residue is involved in a charge-reinforced hydrogen-bonding network with carboxylate groups of Glu100, Asp122, and Asp126 which was stable during the MD simulations (Figure S2). In contrast, the His6 imidazole group participates only in an unstable hydrogen bond with Asp126 (Figure S2). Two wide hydrophobic pockets host the side chains of DPhe7, and Trp9 of MTII (Fig. 3b). The N- and C-terminal groups point towards the extracellular side and are involved only in limited interactions with the receptor consistent with the observation that these termini can be substituted with retention of potent binding affinity.
The proposed binding mode is in qualitative accordance with the known structure–activity relationships of MTII. In fact, substitution of DPhe7 or Trp9, which show a large number of receptor interactions, with alanine resulted in compounds with very low affinities for h-MC4R (h-MC3R and h-MC5R) [68]. The Arg8 involved in stable interactions with the receptor, its replacement with the neutral residue, alanine, led to an active analogue but with more than a 1000-fold reduced affinity at h-MC4R compared to the parent compound, in accordance with the stable interactions exhibited by this residue. In contrast, the substitution of His6 with alanine yielded a peptide with activation and binding affinity similar to MTII towards the h-MC4R (h-MC3R and h-MC5R). Therefore, the imidazole group was shown not to be essential to binding of MTII with the h-MC4R (h-MC3R and h-MC5R). A similar result was reported for the ‘core’ peptide Ac-His6-DPhe7-Arg8-Trp9-amide in which the omission of histidine resulted in the tripeptide that was only 2-fold less potent at h-MC4R than the tetrapeptide [56]. Considering N-terminal acetyl group, an analogue of MTII without the acetyl group was as potent as MTII at the h-MC4R (h-MC3R and h-MC5R) [69]. Replacement of Ac-Nle4 with Ala or Ac-Ala yielded compounds with agonist potencies at h-MC4R similar to that of MTII. The analogue without both acetyl group and norleucine was 200-fold less active at h-MC4R. Also replacement of MTII residues with proline (Pro-scan) gave similar results. Proline replacement was acceptable only at Nle4 and His6 positions yielding compounds with agonist potencies at the h-MC4R similar to that of MTII [69].
Interestingly, many residues of the receptor involved in the interaction with MTII were identified as molecular determinants of ligand binding by mutagenesis studies (Table 2) [31,47,50–56]. In particular, His264 has been demonstrated to be essential for melanocortin peptide activation of the MC4R [70]. π-stacking interaction of imidazolic nucleus of His264 and indole system of Trp9 can trigger the MC4R activation. Interestingly, this π-stacking interaction is not observed in the SHU9119/h-MC4Ri complex described below.
A docking study between SHU9119 and h-MC4Ri also was performed. The h-MC4Ri model, built by Mosberg et al. and based on the rhodopsine crystal structure, was used [47]. The main differences between inactive and active models of h-MC4R were discussed above. The obtained complex (Fig. 4) and the MD simulations indicated that SHU9119 positioning within the h-MC4Ri is similar to that observed for MTII/h-MC4Ra (Figs. 4 and 5). In particular, backbone atoms of MTII and SHU9119 are almost superimposable lying inside the TM2-TM7 bundle. Also the side chains of the positively charged residues His6 and the Arg8 show the same orientation and the Arg8 residue is involved in a charge-reinforced hydrogen-bonding network with carboxylate groups of Glu100, Asp122, and Asp126 which was stable during the MD simulations period (Figure S3).
Fig. 5.
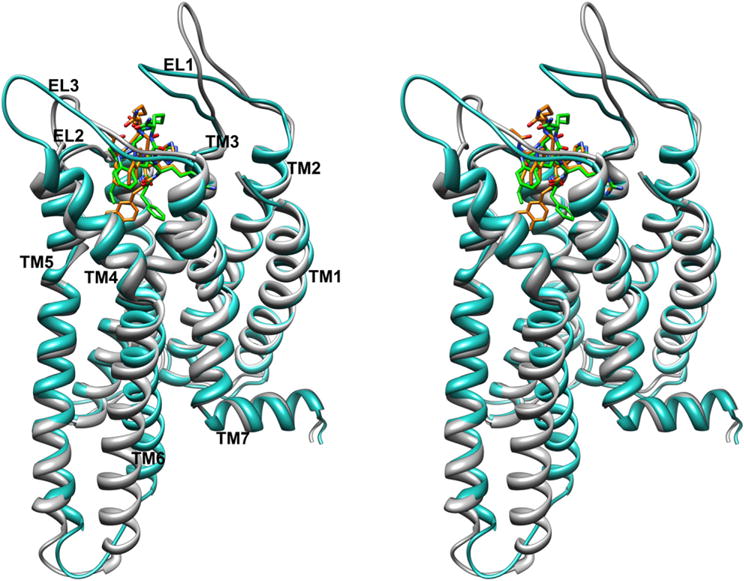
Stereoview of h-MC4R models in the active (gray) and inactive (cyan) conformations complexed with MTII (carbon, green; nitrogen, blue; oxygen, red) and SHU9119 (carbon, orange; nitrogen, blue; oxygen, red), respectively. The h-MC4R models are superimposed using the backbone heavy atoms of TM residues apart from TM6. Hydrogen atoms are not shown for clarity. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In contrast, the DNal(2′)7 and Trp9 binding pockets are quite different compared to those of the corresponding residues of MTII. These differences depend on the different orientations of the DNal(2′)7 and Trp9 side chains and on the movement of TM6 during activation [47]. In the SHU9119/h-MC4Ri complex the DNal(2′)7 prefers a gauche+ orientation due to steric interaction with Leu133, while in the MTII/h-MC4Ra complex the DPhe7 side chain could adopt a trans orientation. Furthermore, the χ2 torsion angle of Trp9 rotates from 9,7° in the MTII/h-MC4Ra to −56,0° in SHU9119/h-MC4Ri. Interestingly, the 2′-naphthalene and indole moieties of SHU9119 show many van der Waals interactions with hydrophobic residues of the TM6 helix which could stabilize the inactive state of the h-MC4R (Table 3). As a matter of fact, different groups have proposed that large aromatic side chain substitutions at the Phe7 position of α-MSH analogues can interfere with MC4R activation by interacting with receptor residues within TM6, physically hindering the conformational changes necessary to elicit full efficacy [52,57]. Our model is also supported by the observation that the DNal(2′)7 naphthalene external ring fills the same cleft as the Phe113 benzene ring of AGRP (Agouti related protein, an endogenous antagonist) in a model of AGRP/h-MC4Ri complex (Fig. 6) [47]. It can be observed in the same Fig. 6 that also the Arg8 guanidinium group of SHU9119 is perfectly overlapped with the same groups of Arg111 of AGRP.
Fig. 6.
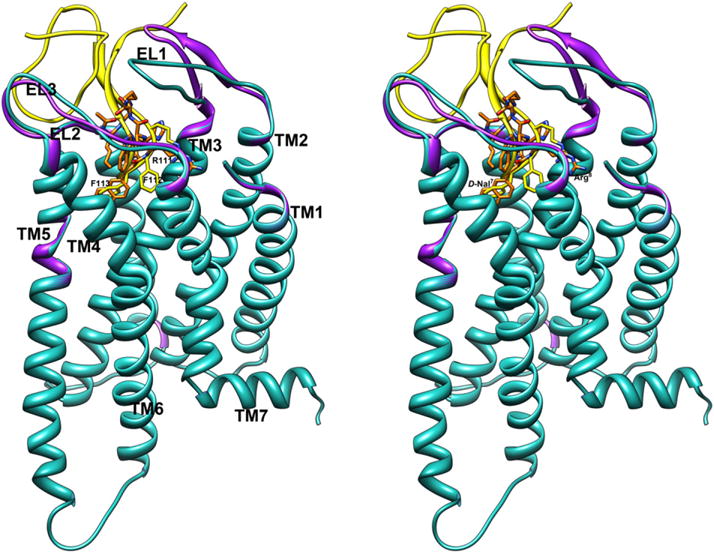
Stereoview of SHU9119/h-MC4Ri (orange-cyan) and AGRP/h-MC4Ri (gold-purple) models in the inactive conformations. On the left side, AGRP’s labels are shown; on the right side, SHU9119’s labels are shown. The h-MC4R models are superimposed using the backbone heavy atoms of TM residues. Hydrogen atoms are not shown for clarity. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Similar conformation and positioning of MTII and SHU9119 within the MC4R are not surprising since it was shown that the single substitution of Leu133 with a methionine residue in the receptor converted SHU9119 from an antagonist into an agonist at the h-MC4R [50]. Probably, according to our model, when Leu133 was replaced with methionine, which is more flexible than leucine, the hindering amino acid was removed and the DNal(2′)7 bulky aromatic side chain could be accommodated in the same cleft occupied by DPhe7 of the agonist MTII. An analogous point mutation in the h-MC3R had the same effect on SHU9119 activity [71]. Interestingly, SHU9119 behaves as an agonist at the h-MC1R and h-MC5R where a methionine or a (smaller) valine residue, respectively, occupies the position corresponding to Leu133 according to the sequence alignment reported in the reference [47].
Other groups have suggested modeled docked conformations of melanotropin peptides with the MC4R for both agonist and antagonist ligands. In particular concerning the agonists, a few models of the NDP-MSH [46,65,72], a model of α-MSH-ND (the open analogue of MTII) [73], and a model of the tetrapeptide His-DPhe-Arg-Trp [53] complexed with h-MC4R have been proposed. It is noteworthy that different ligand conformations were employed for the peptide agonist/MC4R models proposed (Table 4). As a consequence of the lack of an accepted melanocortin peptide active conformation, all the models proposed in literature, included the MTII/h-MC4R presented here, were only partially superposable.
Table 4.
Different conformations employed for the peptide agonist/MC4R models.
| Peptide | Conformation | Central residues | Reference |
|---|---|---|---|
| NDP-MSH | Type II (hairpin) | 6–7 | [46] |
| NDP-MSH | Type I′ | 6–7 | [72] |
| NDP-MSH | Type II′ | 7–8 | [65] |
| α-MSH-ND | Type I | 6–7 | [73] |
| Corea | Type II′ | 7–8 | [53] |
His-DPhe-Arg-Trp tetrapeptide core sequence.
For example, considering the NDP-MSH/h-MC4R complex proposed by Mosberg et al. [46], it is quite different from our MTII/h-MC4R model described above (Fig. 7). NDP-MSH [11] and MTII [12] differ by cyclization at Asp5-Lys10 residues (in MTII), but share the same pharmacophoric sequence His6-DPhe7-Arg8-Trp9.
Fig. 7.
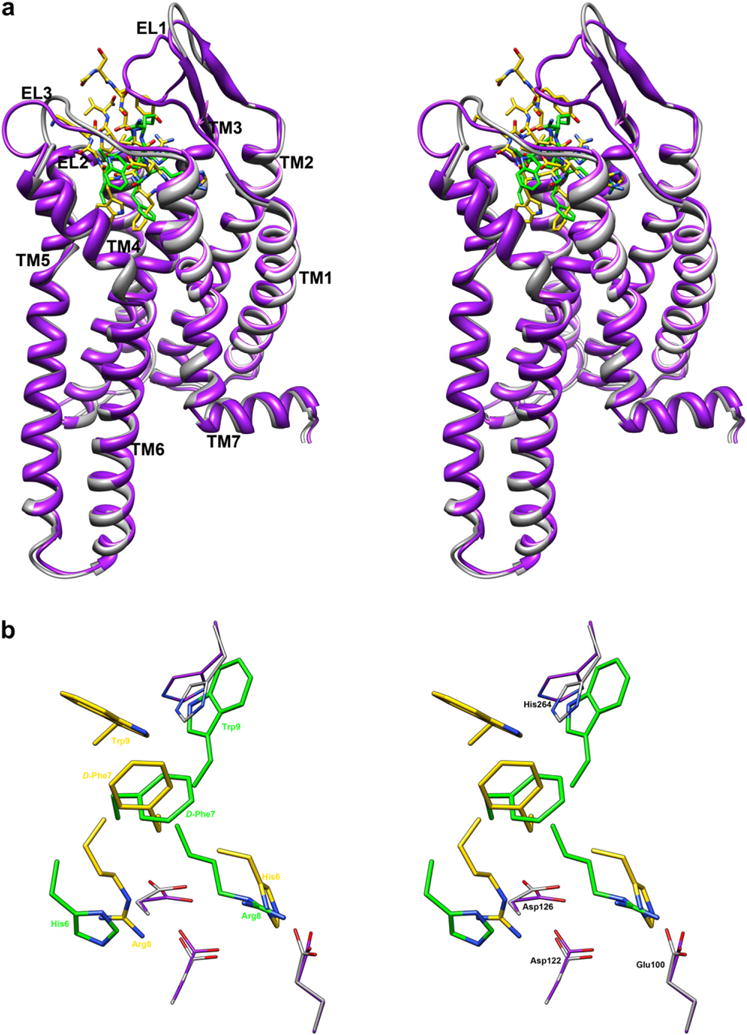
(a) Stereoview of MTII/h-MC4Ra (green-gray) and NDP-MSH/h-MC4Ra (gold-purple) models in the active conformations. (b) Bottom stereoview of MTII and NDP-MSH within the binding pocket of h-MC4Ra. Only pharmacophoric side chains of ligands (His6-Arg9) are showed for clarity. Important residues of receptors are represented. The h-MC4R models are superimposed using the backbone heavy atoms of TM residues. Hydrogen atoms are not shown for clarity. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The receptor coordinates of the two complexes are very similar; indeed, we started from the Mosberg’s h-MC4Ra coordinates, and only marginal changes of the extracellular loops could be observed after the MTII/h-MC4Ra complex optimization (Fig. 7). In contrast, ligand conformations are different considering the common tetrapeptide fragment. A β-hairpin-like structure with a distorted type II β-turn spanning His6-DPhe7 was proposed for Mosberg’s NDP-MSH, while our NMR-derived MTII structure shows two consecutive β-turns spanning residues Asp5-His6 and His6-DPhe7 (see above). Also side chain orientation of Trp9 was different in the two peptides being trans in MTII and gauche− in NDP-MSH. Hence, even if both peptides are located within the TM2-TM7 bundle at the extracellular side, their interactions with h-MC4Ra appear to be different. In particular, in the MTII/h-MC4Ra model His6 and Arg8 are swapped compared to NDP-MSH/h-MC4Ra in Mosberg’s model. In Mosberg’s model, His6 forms the most stable interactions with the Glu100, Asp122, and Asp126 negatively charged side chains, while Arg8 is more solvent exposed. Furthermore, in the NDP-MSH/h-MC4Ra model, the indole group of Trp9 roughly occupies the same position as DNal(2′)7 or Phe113 of the antagonists SHU9119 and AGRP, respectively. Finally, the DPhe7 residues are located in similar positions within the receptor.
Some differences in the SAR data of NPD-MSH and MTII were observed both in terms of binding affinity: alanine substitutions were generally better tolerated in linear NDP-MSH than in cyclic MTII [51,57,68]; and in terms of efficacy: the D-(4-Cl)Phe7 substitution in NDP-MSH substantially reduces Emax but does not appreciably affect MC4R activation by the cyclic MTII [30,57]. Different SARs could indicate different orientations of MTII and NDP-MSH (or other linear peptides) within the binding pocket thus justifying the different interactions found in the complex models. Finally, Mosberg et al. also proposed an MTII/h-MC4Ra complex model [74]. In this complex, the receptor model was the same as previously developed by the authors while the MTII structure was modeled from that of NDP-MSH. The lack of details about the interactions within this complex model does not allow any comparison with our model.
Considering the peptide antagonists, a SHU9119/h-MC4R complex model has been very recently proposed [75]. Apart from Arg8 which was close to Glu100, Asp122, and Asp126 also in this model, other side chain interactions were different from those observed in our model. Again, a different backbone conformation of the bound peptide, a type-I β-turn in that case, can explain these differences. A few AGRP derived peptides were also docked within MC4R models. The triplet peptide Arg-Phe-Phe, the smallest conserved motif of AGRP which mediates the key interactions with MC4R, was docked into the h-MC4R [76]. A bicyclic hAGRP derivative was docked into the mouse MC4R (m-MC4R) [77]. The refined averaged NMR structure of hAGRP(87–132) was docked both into a h-MC4R [47] and a m-MC4R [78] model. When considered the Arg111-Phe112-Phe113 triad, the docked structures of AGRP derivatives all maintain similar putative ligand-receptor locations, which are illustrated in Fig. 6.
In conclusion, NMR-derived MTII and SHU9119 structures show two consecutive β-turns spanning residues Asp5-His6 and His6-DPhe7 (or DNal(2′)7) with some differences in the Phe/Nal7 side chain orientation. Computational docking experiments of these structures, using three-dimensional homology molecular model of the h-MC4R, identified the main interactions between MC4 receptor and its peptide ligands. These findings may be crucial to increase our knowledge of structure–function relationships focused on the design of new potent MC4 receptor ligands.
4. Experimental section
4.1. Synthesis
Nα-Fmoc-protected amino acids, HBTU and HOBt were purchased from Inbios (Naples, Italy). Wang resin was purchased from Advanced ChemTech (Louisville, KY). Synthesis of MTII and SHU-9119 were performed by standard FMOC Strategy [32].
4.2. NMR sample preparation
99.9% 2H2O were obtained from Aldrich (Milwaukee, USA), 98% DPC-d38 was obtained from Cambridge Isotope Laboratories, Inc. (Andover, USA), [(2,2,3,3-tetradeuterio-3-(trimethylsilanyl)]propionic acid (TSP) from MSD Isotopes (Montreal, Canada).
4.3. NMR spectroscopy
The samples for NMR spectroscopy were prepared by dissolving the appropriate amount of peptide to obtain a concentration 1–2 mM in 0.55 ml of 1H2O (pH 5.5), 0.05 ml of 2H2O for water samples, 0.48 mL of 1H2O (pH 5.5), 0.12 mL of DMSOd6 for cryoscopic solution, 200 mM of DPC-d38 for micelle samples. NMR spectra were recorded on a Varian INOVA 700 MHz spectrometer equipped with a z-gradient 5 mm triple-resonance probe head. All the spectra were recorded at a temperature of 25 °C. The spectra were calibrated relative to TSP (0.00 ppm) as internal standard. One-dimensional (1D) NMR spectra were recorded in the Fourier mode with quadrature detection. Water suppression was achieved by using the double-pulsed field gradient spin-echo (DPFGSE) scheme [79]. 2D DQF-COSY [34,35], TOCSY [36], NOESY [37], and PE-COSY [80] spectra were recorded in the phase-sensitive mode using the method of States [81]. Data block sizes were 2048 addresses in t2 and 512 equidistant t1 values. Before Fourier transformation, the time domain data matrices were multiplied by shifted sin2 functions in both dimensions. A mixing time of 70 ms was used for the TOCSY experiments. NOESY experiments were run with mixing times in the range of 150–300 ms. The qualitative and quantitative analyses of DQF-COSY, TOCSY, and NOESY spectra, were obtained using the interactive program package XEASY [38]. 3JHN-Hα coupling constants were obtained from 1D 1H NMR and 2D DQF-COSY spectra. The temperature coefficients of the amide proton chemical shifts were calculated from 1D 1H NMR and 2D TOCSY experiments performed at different temperatures by means of linear regression.
4.4. Structural determinations
The NOE-based distance restraints were obtained from NOESY spectra collected with a mixing time of 200 ms. The NOE cross peaks were integrated with the XEASY program and were converted into upper distance bounds using the CALIBA program incorporated into the program package DYANA [43]. Cross peaks which overlapped more than 50% were treated as weak restraints in the DYANA calculation. For each examined peptide, an ensemble of 200 structures was generated with the simulated annealing of the program DYANA. An error-tolerant target function (tf-type = 3) was used to account for the peptide intrinsic flexibility of the peptide. The annealing procedure produced 200 conformations from which 50 structures were chosen, whose interprotonic distances best fitted NOE derived distances, and then refined through successive steps of restrained and unrestrained EM calculations using the Discover algorithm (Accelrys, San Diego, CA) and the consistent valence force field (CVFF) [82] as previously described. Coupling constants were not used in the constrained simulated annealing calculation, however, backbone and side chain conformations are in accordance with the experimental 3JHN-Hα and JHα-Hβ coupling constants, respectively. The PROMOTIF program, was used to extract details on the location and types of structural secondary motifs [83]. Graphical representation were carried out with the InsightII program (Accelrys, San Diego, CA). RMS deviation analysis between energy minimized structures were carried out with the program MOLMOL [84].
4.5. Docking procedures
The peptides MTII and SHU91119 were manually docked in the proposed binding site of the h-MC4Ra and h-MC4Ri, respectively. Employing the criteria described in the Results section, we generated 10 structures for both MTII/h-MC4Ra and SHU9119/h-MC4Ri complexes. Refinement of each structure was achieved by in vacuo energy minimization with the Discover algorithm (50 000 steps; ε = 1). The backbone atoms of the TM and IL domains of the h-MC4R were held in their position; the ligand and EL’s were free to relax. Minimization was followed by a brief MD simulation period (200 ps). After this period, many poses (7 and 8 out of the 10 poses for MTII and SHU9119, respectively) were discarded since the ligand was driven away from its starting position and lost the salt-bridge with the conserved Asp residues. The other structures (3 for MTII and 2 for SHU9119) converged to a very similar conformation (rmsd of the backbone atoms < 1 Å) and the lowest energy complex for each ligand was chosen as the starting point for subsequent 1 ns MD simulations (time step = 1 fs, T = 300 K). The backbone coordinates of the TM helices were fixed during the MD simulations because, without environmental constraints (i.e. lipid bilayer and water solution), they can move away from each other and can lose their helical structure. Fixing TM helices should still allows for sufficient spatial/conformational sampling of the docked complexes since the ligand, in the discarded poses (see above), significantly changed both the initial position and conformation, after the MD simulations. An average structure was calculated from the last 0.5 ns trajectory and energy-minimized using the steepest descent and conjugate gradient methods until a rmsd of 0.05 kcal/mol per Å was reached. All the MD trajectories were analyzed by means of the Analysis module of the InsightII package [85]. Molecular graphics images of the complexes were produced using the UCSF Chimera package. Rescoring of the ligand/receptor models according to the AutoDock4 (AD4) [86–88] scoring function was attained using a script provided within the MGLTools software package (http://mgltools.scripps.edu/).
Supplementary Material
Acknowledgments
The LC-MS and NMR spectral data were provided by Centro di Ricerca Interdipartimentale di Analisi Strumentale, Università degli Studi di Napoli “Federico II”. The assistance of the staff is gratefully appreciated. This work was supported by a grant from the Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) (PRIN 2007), and a grant from the U.S. Public Health Service National Institutes of Health DK-17420.
Abbreviations
- h-MCR
human Melanocortin Receptor
- cAMP
cyclic adenosine monophosphate
- GPCR
G-protein-coupled receptor
- MSH
melanocyte stimulating hormones
- ACTH
adreno-corticotropic hormone
- POMC
proopiomelanocortin
- AGRP
agouti-related protein
- DPC
dodecyl phosphocholine
- SAR
structure activity relationship
- NMR
nuclear magnetic resonance
- DQF-COSY
double quantum filtered correlated spectroscopy
- PE COSY
primitive exclusive correlated spectroscopy
- TOCSY
total correlated spectroscopy
- NOESY
nuclear Overhauser enhancement spectroscopy
- ROESY
rotating-frame Overhauser effect spectroscopy
- NOE
nuclear Overhauser effect
- MD
molecular dynamic
- EM
energy minimization
- 1D, 2D and 3D
one-, two- and three-dimensional
- TSP
3-(trimethylsilanyl)propionic acid
- DMSO
dimethylsulfoxide
- IL
intracellular loop
- EL
extracellular loop
- TM
trans-membrane domain
- Nal(2′)
(2′)-Naphthylalanine
- SDS
sodium dodecylsulphate
Appendix. Supplementary material
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ejmech.2011.05.038.
Footnotes
Abbreviations used for amino acids and designation of peptides follow the rules of the IUPAC-IUB Commission of Biochemical Nomenclature in J. Biol. Chem. 1972, 247, 977–983. Amino acid symbols denote L-configuration unless indicated otherwise. Additional abbreviations are also used in the article.
References
- 1.Chhajlani V, Wikberg JE. Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Lett. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- 2.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 3.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci U S A. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 5.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993;268:8246–8250. [PubMed] [Google Scholar]
- 6.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 7.Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Molecular cloning, expression, and characterization of a fifth melanocortin receptor. Biochem Biophys Res Commun. 1994;200:1214–1220. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]
- 8.Cone RD. The Melanocortin Receptors Human Press, Totowa NJ. 2000 [Google Scholar]
- 9.Pritchard LE, Turnbull AV, White A. Pro-opiomelanocortin processing in the hypothalamus: impact on melanocortin signalling and obesity. J Endocrinol. 2002;172:411–421. doi: 10.1677/joe.0.1720411. [DOI] [PubMed] [Google Scholar]
- 10.Schioth HB, Chhajlani V, Muceniece R, Klusa V, Wikberg JE. Major pharmacological distinction of the ACTH receptor from other melanocortin receptors. Life Sci. 1996;59:797–801. doi: 10.1016/0024-3205(96)00370-0. [DOI] [PubMed] [Google Scholar]
- 11.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc Natl Acad Sci U S A. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Obeidi F, Hruby VJ, Castrucci AM, Hadley ME. Design of potent linear alpha-melanotropin 4–10 analogues modified in positions 5 and 10. J Med Chem. 1989;32:174–179. doi: 10.1021/jm00121a032. [DOI] [PubMed] [Google Scholar]
- 13.Haskell-Luevano C, Nikiforovich G, Sharma SD, Yang YK, Dickinson C, Hruby VJ, Gantz I. Biological and conformational examination of stereochemical modifications using the template melanotropin peptide, Ac-Nle-c [Asp-His-Phe-Arg-Trp-Ala-Lys]-NH2, on human melanocortin receptors. J Med Chem. 1997;40:1738–1748. doi: 10.1021/jm960845e. [DOI] [PubMed] [Google Scholar]
- 14.Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, Wilkison WO, et al. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature. 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- 15.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 16.Wikberg JE. Melanocortin receptors: perspectives for novel drugs. Eur J Pharmacol. 1999;375:295–310. doi: 10.1016/s0014-2999(99)00298-8. [DOI] [PubMed] [Google Scholar]
- 17.Wessells H, Fuciarelli K, Hansen J, Hadley ME, Hruby VJ, Dorr R, Levine N. Synthetic melanotropic peptide initiates erections in men with psychogenic erectile dysfunction: double-blind, placebo controlled crossover study. J Urol. 1998;160:389–393. [PubMed] [Google Scholar]
- 18.Chaki S, Okuyama S. Involvement of melanocortin-4 receptor in anxiety and depression. Peptides. 2005;26:1952–1964. doi: 10.1016/j.peptides.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 19.Benoit SC, Schwartz MW, Lachey JL, Hagan MM, Rushing PA, Blake KA, Yagaloff KA, Kurylko G, Franco L, Danhoo W, Seeley RJ. A novel selective melanocortin-4 receptor agonist reduces food intake in rats and mice without producing aversive consequences. J Neurosci. 2000;20:3442–3448. doi: 10.1523/JNEUROSCI.20-09-03442.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xi N. The MC4 Receptor as a drug discovery target. Drugs Future. 2006;31:163–173. [Google Scholar]
- 21.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 22.Wessells H, Gralnek D, Dorr R, Hruby VJ, Hadley ME, Levine N. Effect of an alpha-melanocyte stimulating hormone analog on penile erection and sexual desire in men with organic erectile dysfunction. Urology. 2000;56:641–646. doi: 10.1016/s0090-4295(00)00680-4. [DOI] [PubMed] [Google Scholar]
- 23.Vergoni AV, Bertolini A. Role of melanocortins in the central control of feeding. Eur J Pharmacol. 2000;405:25–32. doi: 10.1016/s0014-2999(00)00538-0. [DOI] [PubMed] [Google Scholar]
- 24.Van der Ploeg LH, Martin WJ, Howard AD, Nargund RP, Austin CP, Guan X, Drisko J, Cashen D, Sebhat I, Patchett AA, Figueroa DJ, DiLella AG, Connolly BM, Weinberg DH, Tan CP, Palyha OC, Pong SS, MacNeil T, Rosenblum C, Vongs A, Tang R, Yu H, Sailer AW, Fong TM, Huang C, Tota MR, Chang RS, Stearns R, Tamvakopoulos C, Christ G, Drazen DL, Spar BD, Nelson RJ, MacIntyre DE. A role for the melanocortin 4 receptor in sexual function. Proc Natl Acad Sci U S A. 2002;99:11381–11386. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lubrano-Berthelier C, Cavazos M, Dubern B, Shapiro A, Stunff CL, Zhang S, Picart F, Govaerts C, Froguel P, Bougneres P, Clement K, Vaisse C. Molecular genetics of human obesity-associated MC4R mutations. Ann N Y Acad Sci. 2003;994:49–57. doi: 10.1111/j.1749-6632.2003.tb03161.x. [DOI] [PubMed] [Google Scholar]
- 26.Yeo GS, Lank EJ, Farooqi IS, Keogh J, Challis BG, O’Rahilly S. Mutations in the human melanocortin-4 receptor gene associated with severe familial obesity disrupts receptor function through multiple molecular mechanisms. Hum Mol Genet. 2003;12:561–574. doi: 10.1093/hmg/ddg057. [DOI] [PubMed] [Google Scholar]
- 27.Tao YX, Segaloff DL. Functional characterization of melanocortin-4 receptor mutations associated with childhood obesity. Endocrinology. 2003;144:4544–4551. doi: 10.1210/en.2003-0524. [DOI] [PubMed] [Google Scholar]
- 28.Hong Q, Bakshi RK, Dellureficio J, He S, Ye Z, Dobbelaar PH, Sebhat IK, Guo L, Liu J, Jian T, Tang R, Kalyani RN, Macneil T, Vongs A, Rosenblum CI, Weinberg DH, Peng Q, Tamvakopoulos C, Miller RR, Stearns RA, Cashen D, Martin WJ, Chen AS, Metzger JM, Chen HY, Strack AM, Fong TM, Maclntyre E, Van der Ploeg LH, Wyvratt MJ, Nargund RP. Optimization of privileged structures for selective and potent melanocortin subtype-4 receptor ligands. Bioorg Med Chem Lett. 2010;20:4483–4486. doi: 10.1016/j.bmcl.2010.06.038. [DOI] [PubMed] [Google Scholar]
- 29.Wisse BE, Schwartz MW, Cummings DE. Melanocortin signaling and anorexia in chronic disease states. Ann N Y Acad Sci. 2003;994:275–281. doi: 10.1111/j.1749-6632.2003.tb03190.x. [DOI] [PubMed] [Google Scholar]
- 30.Hruby VJ, Lu D, Sharma SD, Castrucci AL, Kesterson RA, al-Obeidi FA, Hadley ME, Cone RD. Cyclic lactam alpha-melanotropin analogues of Ac-Nle4-cyclo[Asp5, D-Phe7, Lys10] alpha-melanocyte-stimulating hormone-(4–10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 31.Chen M, Cai M, McPherson D, Hruby V, Harmon CM, Yang Y. Contribution of the transmembrane domain 6 of melanocortin-4 receptor to peptide [Pro5, DNal (2′)8]-gamma-MSH selectivity. Biochem Pharmacol. 2009;77:114–124. doi: 10.1016/j.bcp.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart JM, Yang JD. Solid Phase Peptide Synthesis. Pierce Chemical; Rockford, IL: 1984. [Google Scholar]
- 33.Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons, Inc; New York: 1986. [Google Scholar]
- 34.Piantini U, Sorensen OW, Ernst RR. Multiple quantum filters for elucidating NMR coupling network. J Am Chem Soc. 1982;104:6800–68001. [Google Scholar]
- 35.Marion D, Wuthrich K. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem Biophys Res Commun. 1983;113:967–974. doi: 10.1016/0006-291x(83)91093-8. [DOI] [PubMed] [Google Scholar]
- 36.Braunschweiler L, Ernst RR. Coherence transfer by isotropic mixing: application to proton correlation spectroscopy. J Magn Reson. 1983;53:521–528. [Google Scholar]
- 37.Jenner J, Meyer BH, Bachman P, Ernst RR. Investigation of exchange processes by two-dimensional NMR spectroscopy. J Chem Phys. 1979;71:4546–4553. [Google Scholar]
- 38.Bartels C, Xia TH, Billeter M, Guntert P, Wuthrich K. The program Xeasy for Computer-supported Nmr spectral-analysis of biological Macromolecules. J Biomol Nmr. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 39.Al-Obeidi F, O’Connor SD, Job C, Hruby VJ, Pettitt BM. NMR and quenched molecular dynamics studies of superpotent linear and cyclic alpha-melanotropins. J Pept Res. 1998;51:420–431. doi: 10.1111/j.1399-3011.1998.tb00640.x. [DOI] [PubMed] [Google Scholar]
- 40.Ying J, Kover KE, Gu X, Han G, Trivedi DB, Kavarana MJ, Hruby VJ. Solution structures of cyclic melanocortin agonists and antagonists by NMR. Biopolymers. 2003;71:696–716. doi: 10.1002/bip.10596. [DOI] [PubMed] [Google Scholar]
- 41.Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 42.Fink AL. Protein folding in cryosolvents and at subzero temperatures. Methods Enzymol. 1986;131:173–185. doi: 10.1016/0076-6879(86)31041-3. [DOI] [PubMed] [Google Scholar]
- 43.Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 44.Pachler KGR. Nuclear magnetic resonance study of some alpha-amino acidse–II. Rotational isomerism. Spectrochim Acta. 1964;20:581–587. [Google Scholar]
- 45.Cung MT, Marraud M. Conformational dependence of the vicinal proton coupling constant for the Calpha-Cbeta bond in peptides. Biopolymers. 1982;21:953–967. [Google Scholar]
- 46.Pogozheva ID, Chai BX, Lomize AL, Fong TM, Weinberg DH, Nargund RP, Mulholland MW, Gantz I, Mosberg HI. Interactions of human melanocortin 4 receptor with nonpeptide and peptide agonists. Biochemistry. 2005;44:11329–11341. doi: 10.1021/bi0501840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chai BX, Pogozheva ID, Lai YM, Li JY, Neubig RR, Mosberg HI, Gantz I. Receptor-antagonist interactions in the complexes of agouti and agouti-related protein with human melanocortin 1 and 4 receptors. Biochemistry. 2005;44:3418–3431. doi: 10.1021/bi0478704. [DOI] [PubMed] [Google Scholar]
- 48.Konvicka K, Campagne F, Weinstein H. Interactive construction of residue-based diagrams of proteins: the RbDe web service. Protein Eng. 2000;13:395–396. doi: 10.1093/protein/13.6.395. [DOI] [PubMed] [Google Scholar]
- 49.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 50.Yang Y, Chen M, Lai Y, Gantz I, Georgeson KE, Harmon CM. Molecular determinants of human melanocortin-4 receptor responsible for antagonist SHU9119 selective activity. J Biol Chem. 2002;277:20328–20335. doi: 10.1074/jbc.M201343200. [DOI] [PubMed] [Google Scholar]
- 51.Yang YK, Fong TM, Dickinson CJ, Mao C, Li JY, Tota MR, Mosley R, Van Der Ploeg LH, Gantz I. Molecular determinants of ligand binding to the human melanocortin-4 receptor. Biochemistry. 2000;39:14900–14911. doi: 10.1021/bi001684q. [DOI] [PubMed] [Google Scholar]
- 52.Haskell-Luevano C, Cone RD, Monck EK, Wan YP. Structure activity studies of the melanocortin-4 receptor by in vitro mutagenesis: identification of agouti-related protein (AGRP), melanocortin agonist and synthetic peptide antagonist interaction determinants. Biochemistry. 2001;40:6164–6179. doi: 10.1021/bi010025q. [DOI] [PubMed] [Google Scholar]
- 53.Hogan K, Peluso S, Gould S, Parsons I, Ryan D, Wu L, Visiers I. Mapping the binding site of melanocortin 4 receptor agonists: a hydrophobic pocket formed by I3.28(125), I3.32(129), and I7.42(291) is critical for receptor activation. J Med Chem. 2006;49:911–922. doi: 10.1021/jm050780s. [DOI] [PubMed] [Google Scholar]
- 54.Chen M, Cai M, Aprahamian CJ, Georgeson KE, Hruby V, Harmon CM, Yang Y. Contribution of the conserved amino acids of the melanocortin-4 receptor in [corrected] [Nle4, D-Phe7]-alpha-melanocyte-stimulating [corrected] hormone binding and signaling. J Biol Chem. 2007;282:21712–21719. doi: 10.1074/jbc.M702285200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oosterom J, Nijenhuis WA, Schaaper WM, Slootstra J, Meloen RH, Gispen WH, Burbach JP, Adan RA. Conformation of the core sequence in melanocortin peptides directs selectivity for the melanocortin MC3 and MC4 receptors. J Biol Chem. 1999;274:16853–16860. doi: 10.1074/jbc.274.24.16853. [DOI] [PubMed] [Google Scholar]
- 56.Haskell-Luevano C, Hendrata S, North C, Sawyer TK, Hadley ME, Hruby VJ, Dickinson C, Gantz I. Discovery of prototype peptidomimetic agonists at the human melanocortin receptors MC1R and MC4R. J Med Chem. 1997;40:2133–2139. doi: 10.1021/jm960840h. [DOI] [PubMed] [Google Scholar]
- 57.Fleck BA, Ling N, Chen C. Substituted NDP-MSH peptides paired with mutant melanocortin-4 receptors demonstrate the role of transmembrane 6 in receptor activation. Biochemistry. 2007;46:10473–10483. doi: 10.1021/bi700406k. [DOI] [PubMed] [Google Scholar]
- 58.Dyson HJ, Rance M, Houghten RA, Wright PE, Lerner RA. Folding of immunogenic peptide fragments of proteins in water solution. II. The nascent helix. J Mol Biol. 1988;201:201–217. doi: 10.1016/0022-2836(88)90447-0. [DOI] [PubMed] [Google Scholar]
- 59.Gysin B, Schwyzer R. Head group and structure specific interactions of enkephalins and dynorphin with liposomes: investigation by hydrophobic photolabeling. Arch Biochem Biophys. 1983;225:467–474. doi: 10.1016/0003-9861(83)90055-3. [DOI] [PubMed] [Google Scholar]
- 60.Dhanasekaran M, Palian MM, Alves I, Yeomans L, Keyari CM, Davis P, Bilsky EJ, Egleton RD, Yamamura HI, Jacobsen NE, Tollin G, Hruby VJ, Porreca F, Polt R. Glycopeptides related to beta-endorphin adopt helical amphipathic conformations in the presence of lipid bilayers. J Am Chem Soc. 2005;127:5435–5448. doi: 10.1021/ja0432158. [DOI] [PubMed] [Google Scholar]
- 61.Egleton RD, Bilsky EJ, Tollin G, Dhanaseharan M, Lowery J, Alves I, Davis P, Porreca F, I YH, Yeomans L, Keyari CM, Polt R. Biousian glycopeptides penetrate the blood-brain-barrier. Tetrahedron: Asymmetry. 2005;16:65–75. [Google Scholar]
- 62.Yamamoto T, Nair P, Jacobsen NE, Davis P, Ma SW, Navratilova E, Moye S, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. The importance of micelle-bound states for the bioactivities of bifunctional peptide derivatives for delta/mu opioid receptor agonists and neurokinin 1 receptor antagonists. J Med Chem. 2008;51:6334–6347. doi: 10.1021/jm800389v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doedens L, Opperer F, Cai M, Beck JG, Dedek M, Palmer E, Hruby VJ, Kessler H. Multiple N-methylation of MT-II backbone amide bonds leads to melanocortin receptor subtype hMC1R selectivity: pharmacological and conformational studies. J Am Chem Soc. 2010;132:8115–8128. doi: 10.1021/ja101428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin SW, Sakmar TP. Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. Biochemistry. 1996;35:11149–11159. doi: 10.1021/bi960858u. [DOI] [PubMed] [Google Scholar]
- 65.Chapman KL, Kinsella GK, Cox A, Donnelly D, Findlay JB. Interactions of the melanocortin-4 receptor with the peptide agonist NDP-MSH. J Mol Biol. 2010;401:433–450. doi: 10.1016/j.jmb.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 67.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 68.Bednarek MA, Silva MV, Arison B, MacNeil T, Kalyani RN, Huang RR, Weinberg DH. Structure-function studies on the cyclic peptide MT-II, lactam derivative of alpha-melanotropin. Peptides. 1999;20:401–409. doi: 10.1016/s0196-9781(99)00048-0. [DOI] [PubMed] [Google Scholar]
- 69.Bednarek MA, Macneil T, Kalyani RN, Tang R, Van der Ploeg LH, Weinberg DH. Analogs of MTII, lactam derivatives of alpha-melanotropin, modified at the N-terminus, and their selectivity at human melanocortin receptors 3, 4, and 5. Biochem Biophys Res Commun. 1999;261:209–213. doi: 10.1006/bbrc.1999.0981. [DOI] [PubMed] [Google Scholar]
- 70.Adan RA, Oosterom J, Toonen RF, Kraan MV, Burbach JP, Gispen WH. Molecular pharmacology of neural melanocortin receptors. Receptors and Channels. 1997;5:215–223. [PubMed] [Google Scholar]
- 71.Chen M, Aprahamian CJ, Celik A, Georgeson KE, Garvey WT, Harmon CM, Yang Y. Molecular characterization of human melanocortin-3 receptor ligand-receptor interaction. Biochemistry. 2006;45:1128–1137. doi: 10.1021/bi0521792. [DOI] [PubMed] [Google Scholar]
- 72.Cho MK, Lee CJ, Lee CH, Li SZ, Lim SK, Baik JH, Lee W. Structure and function of the potent cyclic and linear melanocortin analogues. J Struct Biol. 2005;150:300–308. doi: 10.1016/j.jsb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 73.Lim S, Li S, Lee C, Yoon C, Baik J, Lee W. Minimization of MC1R selectivity by modification of the core structure of alpha-MSH-ND. Chem Biol. 2001;8:857–870. doi: 10.1016/s1074-5521(01)00057-6. [DOI] [PubMed] [Google Scholar]
- 74.Xiang Z, Pogozheva ID, Sorenson NB, Wilczynski AM, Holder JR, Litherland SA, Millard WJ, Mosberg HI, Haskell-Luevano C. Peptide and small molecules rescue the functional activity and agonist potency of dysfunctional human melanocortin-4 receptor polymorphisms. Biochemistry. 2007;46:8273–8287. doi: 10.1021/bi7007382. [DOI] [PubMed] [Google Scholar]
- 75.Lee CJ, Yun JH, Lim SK, Lee W. Solution structures and molecular interactions of selective melanocortin receptor antagonists. Mol Cells. 2010;30:551–556. doi: 10.1007/s10059-010-0152-6. [DOI] [PubMed] [Google Scholar]
- 76.Yang X, Wang Z, Dong W, Ling L, Yang H, Chen R. Modeling and docking of the three-dimensional structure of the human melanocortin 4 receptor. J Protein Chem. 2003;22:335–344. doi: 10.1023/a:1025386022852. [DOI] [PubMed] [Google Scholar]
- 77.Wilczynski A, Wang XS, Bauzo RM, Xiang Z, Shaw AM, Millard WJ, Richards NG, Edison AS, Haskell-Luevano C. Structural characterization and pharmacology of a potent (Cys101-Cys119, Cys110-Cys117) bicyclic agouti-related protein (AGRP) melanocortin receptor antagonist. J Med Chem. 2004;47:5662–5673. doi: 10.1021/jm049620r. [DOI] [PubMed] [Google Scholar]
- 78.Wilczynski A, Wang XS, Joseph CG, Xiang Z, Bauzo RM, Scott JW, Sorensen NB, Shaw AM, Millard WJ, Richards NG, Haskell-Luevano C. Identification of putative agouti-related protein(87-132)-melanocortin-4 receptor interactions by homology molecular modeling and validation using chimeric peptide ligands. J Med Chem. 2004;47:2194–2207. doi: 10.1021/jm0303608. [DOI] [PubMed] [Google Scholar]
- 79.Hwang TL, Shaka AJ. Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J Magn Reson. 1995;112:275–279. [Google Scholar]
- 80.Mueller L. P. E: COSY, a simple alternative to E. COSY. J Magn Reson. 1987;72:191–196. [Google Scholar]
- 81.States DJ, Haberkorn RA, Ruben DJ. A two-dimensional nuclear overhauser experiment with pure absorption phase in four quadrants. J Magn Reson. 1982;48:286–292. [Google Scholar]
- 82.Maple JR, Dinur U, Hagler AT. Derivation of force fields for molecular mechanics and dynamics from ab initio energy surfaces. Proc Natl Acad Sci U S A. 1988;85:5350–5354. doi: 10.1073/pnas.85.15.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hutchinson EG, Thornton JM. PROMOTIFea program to identify and analyze structural motifs in proteins. Protein Sci. 1996;5:212–220. doi: 10.1002/pro.5560050204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 85.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. Ucsf Chimeraea visualization system for exploratory research and analysis. J Comput Chem ( 2004:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 86.Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 87.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 88.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.