

ABSTRACT
A major shortcoming to plasmid-based genetic tools is the necessity of using antibiotics to ensure plasmid maintenance. While selectable markers are very powerful, their use is not always practical, such as during in vivo models of bacterial infection. During previous studies, it was noted that the uncharacterized LAC-p01 plasmid in Staphylococcus aureus USA300 isolates was stable in the absence of a known selection and therefore could serve as a platform for new genetic tools for Staphylococcus species. LAC-p01 was genetically manipulated into an Escherichia coli-S. aureus shuttle vector that remained stable for at least 100 generations without antibiotic selection. The double- and single-stranded (dso and sso) origins were identified and found to be essential for plasmid replication and maintenance, respectively. In contrast, deletion analyses revealed that none of the four LAC-p01 predicted open reading frames were necessary for stability. Subsequent to this, the shuttle vector was used as a platform to generate two plasmids. The first plasmid, pKK22, contains all genes native to the plasmid for use in S. aureus USA300 strains, while the second, pKK30, lacks the four predicted open reading frames for use in non-USA300 isolates. pKK30 was also determined to be stable in Staphylococcus epidermidis. Moreover, pKK22 was maintained for 7 days postinoculation during a murine model of S. aureus systemic infection and successfully complemented an hla mutant in a dermonecrosis model. These plasmids that eliminate the need for antibiotics during both in vitro and in vivo experiments are powerful new tools for studies of Staphylococcus.
IMPORTANCE Plasmid stability has been problematic in bacterial studies, and historically antibiotics have been used to ensure plasmid maintenance. This has been a major limitation during in vivo studies, where providing antibiotics for plasmid maintenance is difficult and has confounding effects. Here, we have utilized the naturally occurring plasmid LAC-p01 from an S. aureus USA300 strain to construct stable plasmids that obviate antibiotic usage. These newly modified plasmids retain stability over a multitude of generations in vitro and in vivo without antibiotic selection. With these plasmids, studies requiring genetic complementation, protein expression, or genetic reporter systems would not only overcome the burden of antibiotic usage but also eliminate the side effects of these antibiotics. Thus, our plasmids can be used as a powerful genetic tool for studies of Staphylococcus species.
INTRODUCTION
For decades, bacterial plasmids have been a mainstay of bacterial genetics. They are used for a variety of techniques, including the reintroduction of DNA back into cells for in trans genetic complementation, induction or control of protein expression, and the generation of both transcriptional and translational gene expression reporter systems. For all of these studies, it is essential that the entire bacterial population retains the plasmid to ensure the same behavior of each cell present in the culture to yield accurate and consistent results. To accomplish this, plasmid-encoded antibiotic resistance cassettes are used in conjunction with antibiotic supplementation to inhibit or kill any bacteria lacking the plasmid, ensuring plasmid maintenance. The use of antibiotics, however, presents certain drawbacks, and it is often believed that resistant cells are unaffected by the antibiotics present. This is certainly not the case, as it has been well documented that antibiotics lead to many changes to cells even at subinhibitory levels (1–7; reviewed in references 8 and 9). This is particularly true in the case of antibiotic resistance mechanisms that do not lead to antibiotic degradation but instead are either the result of antibiotic efflux away from the antibiotic's target or the result of a mutated protein that no longer interacts with the antibiotic. Additionally, a growing body of literature has demonstrated that these antibiotic-resistant protein variants come with a fitness or functional cost and can have adverse effects (reviewed in references 10–12). Finally, there are times when antibiotic use is difficult or ineffective, including (i) long-term experiments where antibiotics degrade spontaneously due to light or temperature sensitivity or as a result of enzymatic inactivation of the antibiotic and (ii) environments where antibiotic delivery is difficult, such as during ex vivo experiments or during animal models of infection.
As is the case with many bacteria, plasmid-based genetic tools have greatly enhanced in vitro studies of Staphylococcus aureus. Plasmids within S. aureus can replicate through either theta or rolling-circle replication (RCR). Many of the genetic tools used when studying S. aureus make use of RCR plasmids that are often derived from the naturally occurring pE194 and pT181 plasmids (13). The RCR mechanism of plasmid replication involves the binding of a Rep protein to the plasmid double-stranded origin (dso) and the generation of a single-stranded DNA intermediate that is then converted into a double-stranded DNA molecule (reviewed in reference 14). Unfortunately, much like plasmids in other bacteria, S. aureus plasmids can be inherently unstable without antibiotic selection. Indeed, this instability is a key component to existing allelic-exchange plasmids (15–18). Plasmid instability has hampered many studies of S. aureus, particularly during immune-cell interaction studies and in vivo models of infection, where antibiotic treatment is difficult or impossible. Some have suggested feeding antibiotics to animals to encourage plasmid maintenance during infection studies, but it is not clear if antibiotics will reach or maintain selective levels at all infection sites over the course of in vivo studies. Such treatment would likely change the natural microbiota of the animal and may confound results. Lastly, some antibiotics do not traverse host cell membranes or remain ineffective in eukaryotic cells and therefore do not allow for plasmid maintenance of intracellular bacteria during phagocytosis studies (19–23). Indeed, the impermeability of host cells to certain antibiotics is the rationale for using gentamicin to kill extracellular bacteria during phagocytosis experiments (23).
As an alternative to plasmid use, and to circumvent plasmid stability issues, several tools have been generated to integrate plasmids into the S. aureus chromosome at specific sites. These theoretically neutral sites include the lipase-encoding geh gene (24), phage integration sites (24), and the SaPI1 pathogenicity island (25). While integration into geh provides an easy screen, i.e., loss of lipase activity, this technique inactivates lipase, which has been implicated in virulence (26). Insertion into phage integration sites replaces the phage in strains that contain them, and some phages have been shown to play a role in virulence (27, 28). A common feature of all of these integration systems is that they generally involve integration into the chromosome of the highly mutagenized strain RN4220, since it readily accepts DNA from Escherichia coli. Once integrated into RN4220, the desired DNA fragments are then transduced into the target strain of interest. However, phage can move tens of thousands of base pairs of DNA. Any of this DNA can then be readily incorporated into the target strain genome, leading to possible unknown gene and nucleotide changes.
Here we describe the construction of plasmids based on the naturally occurring S. aureus plasmid LAC-p01 that maintain stability in Staphylococcus cells in the absence of antibiotic pressure. Furthermore, we have demonstrated that these plasmids are stable during infection in a murine model of S. aureus sepsis and dermonecrosis.
MATERIALS AND METHODS
Strains and media.
E. coli DH5α and DH5αλpir (for R6Kγ origin) (29) were utilized for plasmid construction and were grown in lysogeny broth (LB) medium supplemented with ampicillin (100 μg ml−1) or tryptic soy broth (TSB) medium with trimethoprim (Tmp) (10 μg ml−1) as needed for selection. S. aureus strains (Table 1) were grown in TSB supplemented with trimethoprim (10 μg ml−1) as needed. Reagents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Hanover Park, IL).
TABLE 1.
Strains, plasmid construction, and oligonucleotides
| Strain, plasmid, or oligonucleotide | Relevant characteristic(s) or sequencea | Source or reference |
|---|---|---|
| Strains | ||
| AH1263 | USA300 CA-MRSA strain LAC lacking LAC-p03 | 57 |
| JB24 | AH1263 hla::ΦΝΣ | 30 |
| JE2 | LAC-13C lacking LAC-p01 (contains no plasmids) | 49 |
| LAC-13C | USA300 CA-MRSA strain LAC lacking LAC-p03 (parent of JE2) | 31 |
| RN4220 | Highly transformable S. aureus | 58 |
| Plasmids | ||
| pCK8 | hla complement plasmid; PCR product (primers JB21 and JB29, AH1263 template) containing hla and its promoter in SmaI-digested pKK22 | This study |
| pCN51 | pT181cop-wt repC Ermr Ampr ColE1 oriV blaZTT | 59 |
| pJB127 | SphI/PstI-digested pCN51, treated with Klenow fragment, and self-ligated | 60 |
| pJB138 | Source of atl transcriptional terminator | 31 |
| pJB178 | Source of PsarAP1::dfrA and R6Kγ oriV | Lab stock |
| pJB1000 | EcoRI/AscI-digested PCR product (primers KK3 and KK4, pJB178 template) containing PsarAP1::dfrA in same sites of pJB127 | This study |
| pJB1001 | EcoRI/SalI-digested PCR product (primers KK5 and KK6, pJB178 template) containing R6Kγ oriV in same sites of pJB1000 | This study |
| pKK1 | PCR product (primers KK5 and KK7, pJB1001 template) into EcoRV-digested LAC-p01 | This study |
| pKK3 | PCR product (primers KK9 and KK17, pKK1 template), XhoI digested and self-ligated | This study |
| pKK5 | PCR product (primers KK13 and KK14, pKK1 template), XhoI digested and self-ligated | This study |
| pKK7 | PCR product (primers KK12 and KK18, pKK1 template), XhoI digested and self-ligated | This study |
| pKK8 | PCR product (primers KK12 and KK13, pKK1 template), XhoI digested and self-ligated | This study |
| pKK10 | PCR product (primers KK25 and KK26, pKK1 template), XhoI digested and self-ligated | This study |
| pKK13 | pKK8 digested with XhoI, treated with Klenow fragment, and self-ligated | This study |
| pKK15 | PCR product (primers KK27 and KK28, pKK1 template), XhoI digested and self-ligated | This study |
| pKK16 | BamHI-digested pKK1, treated with Klenow fragment and self-ligated | This study |
| pKK17 | PCR product (primers KK29 and KK30, pKK13 template), XhoI digested and self-ligated | This study |
| pKK18 | PCR product (primers KK31 and KK32, pJB138 template) containing the atl transcriptional terminator digested with XhoI and ligated into SalI-digested pKK16 | This study |
| pKK22 | PCR product (primers KK33 and KK34, pJB138 template) containing the atl transcriptional terminator digested with ClaI and ligated into NarI-digested pKK16 | This study |
| pKK29 | PCR product (primers KK38 and KK39, pKK22 template), self-ligated | This study |
| pKK30 | PCR product (primers KK12 and KK13, pKK29 template), XhoI digested and self-ligated | This study |
| Oligonucleotides | ||
| JB21 | ccgaattCTTGTACTGTTTGATATGGAACTCCTG | This study |
| JB29 | gcgaattctgcagTTAATTTGTCATTTCTTCTTTTTCC | 30 |
| JBKU45 | GGCAAATGTATCGAGCAAGA | This study |
| JBKU46 | TCATTTGGTAAGTGCCAAGG | This study |
| KK3 | ccgaattcGAGCTGATATTTTTGACTAAACCAAATGC | This study |
| KK4 | ttggcgcgcCTATTTCCCTTTTCTACGCACTAAATGTAAG | This study |
| KK5 | ccgtcgacGAAAGCCTGGCCACGATGC | This study |
| KK6 | ccGAATTCCCATGTCAGCCGTTAAGTGTTC | This study |
| KK7 | CAAAGGCGCCTGTCACTTTGCTTGATATATG | This study |
| KK9 | ccctcgagGTTGTTTTTTGAAACGAGTGTGAACGAG | This study |
| KK12 | ccctcgagTTACATGCAAATTAAAAATTAGCATAAAATCC | This study |
| KK13 | cctcgagAATATTTGAGGGGATTTTGAAACGAGTTTCTTC | This study |
| KK14 | cctcgagAAGACTTACTCAAATTATAAATACCTTTATTTTG | This study |
| KK17 | ccctcgagATTAAACTCCTTTGTTTTTATATG | This study |
| KK18 | ccctcgagTATATATTTTTAAGTTAAAAACCTATAT | This study |
| KK25 | ccctcgagACTCGTTCACACTCGTTTCAAAAAAC | This study |
| KK26 | ccctcgagATATATATAGCTGTAACTGTTGATATTACAGTG | This study |
| KK27 | cctcgagCATATTTTTCTTCCATCTCGTCAAAAGCATCATC | This study |
| KK28 | ccctcgagGAGCTATTAAGCCGACCATTCGACAAG | This study |
| KK29 | ccctcgagGCCAAATCTCGATATGGATATGTTCCTGAAGG | This study |
| KK30 | cctcgagCGATTTTATTTGATAATCCTTTTGACTTTTTAG | This study |
| KK31 | ggctcgagTAAATAAGCAACATGAACATAGGATCAAAAGTC | This study |
| KK32 | cactcgaGCGGCCGCTAGCCTAGGAGCTCGGTACCCGGGGATCCGAATC | This study |
| KK33 | cgcgtatcgatTAAATAAGCAACATGAACATAGGATCAAAAGTC | This study |
| KK34 | cgcgtatcgatGCGGCCGCTAGCCTAGGAGCTCGGTACCCGGGGATCCGAATC | This study |
| KK38 | P-GCCAAATCTCGATATGGATATGTTCCTGAAGG | This study |
| KK39 | P-CGATTTTATTTGATAATCCTTTTGACTTTTTAG | This study |
Antibiotic resistance abbreviations: Ampr, ampicillin resistance (bla); Ermr, erythromycin resistance; dfrA, trimethoprim resistance. CA-MRSA, community-associated, methicillin-resistant S. aureus. Oligonucleotide sequences are provided in the 5′-3′ orientation. Lowercase italics indicate nonhomologous sequences added for cloning purposes. “P” indicates the addition of a 5′ phosphate.
Molecular genetic techniques.
Restriction enzymes and T4 DNA ligase were purchased from New England BioLabs (Beverly, MA). Oligonucleotides were synthesized by IDT DNA Technologies (Coralville, IA). PCR was performed with an Applied Biosystems GeneAmp PCR system 9700 (Life Technologies, Carlsbad, CA) using KOD DNA polymerase (Novagen, Madison, WI). PCR products were purified using a ZymoResearch (Orange, CA) DNA Clean & Concentrator-5 kit or a Zymoclean gel DNA recovery kit. PCR products were sequenced by ACGT, Inc. (Wheeling, IL), to ensure that no unintended changes occurred during cloning. Sequencing results were analyzed using Vector NTI (Invitrogen, Carlsbad, CA).
Construction of pKK1 and its derivatives.
Plasmid construction details are found in Table 1. First, the Pcad promoter region was removed from pCN51 by digestion with SphI and PstI, treatment with Klenow fragment, and self-ligation of the product. The resulting plasmid, pJB127, has the pCN51 multiple-cloning site (MCS) upstream from the blaZ transcriptional terminator (blaZTT). The PsarAP1::dfrA trimethoprim resistance cassette of pJB178 was amplified by PCR, and the resulting product was digested with EcoRI and AscI, followed by ligation into the same sites of pJB127 such that the blaZ terminator serves to prevent readthrough from PsarAP1. The resulting plasmid was designated pJB1000. Next, the R6Kγ origin of replication was amplified from pJB178; the PCR product was digested with SalI and EcoRI and ligated into the same sites of pJB1000 to generate pJB1001. Finally, the R6Kγ-PsarAP1::dfrA-blaZTT module was amplified by PCR from pJB1001 and ligated into EcoRV-digested LAC-p01 to create pKK1.
All deletion versions of pKK1 were generated using PCR as previously described (16, 30, 31). For example, in order to delete open reading frame 2 (ORF2), primers were designed 5′ and 3′ to ORF2 such that PCR around the entire pKK1 plasmid would remove ORF2. The primers each contained an XhoI restriction site, and the PCR product was digested with XhoI and self-ligated to produce the new plasmid. Following ligation, the product was cleaned using the ZymoResearch Clean & Concentrator-5 kit and treated with DpnI to remove any remaining original plasmid template. All plasmids were confirmed by restriction digest and gel electrophoresis prior to electroporation into RN4220. In addition, most plasmids were completely sequenced to confirm sequences.
Using dialyzed pKK22 or pKK30 isolated from RN4220, plasmids were transferred into S. aureus JE2 or Staphylococcus epidermidis 1457 electrocompetent cells, respectively, using a Bio-Rad (Hercules, CA) GenePulser Xcell by following the manufacturer's recommended settings for S. aureus.
Plasmid copy number determination.
In order to determine the copy number of pKK22, quantitative PCR (qPCR) was used to amplify a 131-bp region overlapping PsarAP1 and the beginning of the dfrA gene. The pKK22 results were compared to those of a JE2 derivative (provided by Ken Bayles, University of Nebraska Medical Center) that contains this same cassette integrated in the chromosome. As a negative control, the JE2 wild-type strain was included in the experiment. Genomic DNA was isolated from all three strains grown to exponential phase (3 h) in TSB using a Wizard genomic DNA purification kit (Promega, Madison, WI) according to the manufacturer's instructions for Gram-positive bacteria with the slight modification of lysing bacterial pellets using 20 μg of lysostaphin. Following this, 100 pg of total genomic DNA was used to amplify the PsarAP1-dfrA region using primer pair JBKU45 and JBKU46 with FastStart essential DNA green master mix in a LightCycler 96 system (Roche).
As a second method to quantify plasmid copy number, the same strains as those described above were grown to exponential phase and standardized to the equivalent of 10 optical density units (ODs) of cells in 500 μl of Tris-EDTA buffer. Then, cells were lysed using a Fast-Prep-24 5G instrument (MP Biomedicals) according to manufacturer's recommended settings for S. aureus. Cell debris was subsequently pelleted at 21,000 × g for 5 min. Supernatants were diluted 50-fold and used for PCR as described above. For both experiments, two independent experiments were performed, each with three biological replicates.
Plasmid stability assays.
Two types of stability assays were performed. The initial assays maintained the cultures in continuous exponential phase. To achieve this, overnight cultures grown in TSB with antibiotic were diluted to an A600 of 0.1 in 12.5 ml TSB with antibiotic in 125-ml flasks. Cultures were grown at 37°C with shaking (250 rpm) until mid-exponential phase (A600 = 1.0 to 2.0) and diluted 1 × 109 in TSB without antibiotics (designated generation 0). The cultures were diluted twice daily (1 × 104 in morning and 1 × 107 in afternoon) to maintain the cells in continuous exponential phase (A600 ≤ 2.0) of growth. At each dilution, the optical density was determined and, when combined with the dilution factor, it was used to determine the number of generations that had occurred. At regular intervals, the cultures were streaked for isolated colonies on tryptic soy agar (TSA) plates and incubated overnight at 37°C, and the resulting colonies were transferred onto TSA with Tmp. Following incubation, the percentage of colonies that were Tmp resistant was determined.
A similar assay was used for noncontinuous exponential-phase testing. In the morning, the A600 was determined from overnight cultures grown in TSB with Tmp, and the cultures were diluted 1 × 104 into 12.5 ml TSB in 125-ml flasks (generation 0). The cultures were grown at 37°C with shaking (250 rpm) until late afternoon, at which point the A600 was determined again, and the cultures were diluted 1 × 107 into 12.5 ml TSB and incubated overnight at 37°C with shaking (250 rpm). This process was continued for 2 additional days. The number of generations passed was determined using the A600 taken at each dilution and factoring in the dilution factor. The morning cultures were streaked for isolated colonies on TSA and incubated overnight at 37°C, and the resulting colonies were transferred to TSA supplemented with Tmp. The percentage of resistant colonies was indicative of the presence of the plasmid and, therefore, plasmid stability. In some assays, random colonies were chosen and confirmed to have the plasmid by plasmid isolation and gel electrophoresis following restriction digestion. This also ensured that the plasmids had not undergone any structural changes during these long-term assays.
In vivo stability.
Stability assays during the murine sepsis infection model were performed using 8-week-old C57BL/6 mice (Jackson Laboratories). All mice were infected via retro-orbital injection with 4 × 107 CFU of JE2 harboring pKK22. At 3 or 7 days postinfection, mice were humanely euthanized, and kidneys were harvested. Kidney pairs were homogenized in lysing matrix H tubes (MP Biomedicals, Santa Ana, CA) containing phosphate-buffered saline (PBS) using a Fast-Prep-24 5G instrument (MP Biomedicals) according to the manufacturer's recommended settings for mouse kidneys. Subsequent homogenates were serially diluted in PBS containing 0.1% Triton X-100 and plated on TSA. Following overnight incubation, 100 colonies from each mouse were passaged onto TSA containing 10 μg ml−1 trimethoprim in order to screen for plasmid retention. Additionally, random colonies from each mouse were screened for the existence of pKK22. This was performed by isolating plasmid DNA, digesting the preparation with PstI to linearize pKK22, and viewing by DNA gel electrophoresis. These studies were conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (32). This protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Kansas Medical Center.
Mouse model of skin infection.
Animal work was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (32), and the protocol was approved by the University of New Mexico Health Sciences Center Institutional Animal Care and Use Committee. A murine model of dermonecrosis was performed as previously described (33). Briefly, 8-week-old female SKH1 mice (Charles River Laboratories, Wilmington, MA) were anesthetized with isoflurane and inoculated via subcutaneous injection in the right flank with ∼4.7 × 107 CFU of S. aureus. Mice were weighed daily, and infection sites were photographed for ImageJ analysis (34) of the dermonecrosis area. Mice were euthanized by CO2 asphyxiation on day 3 postinfection, a 2.25-cm2 area of skin surrounding the site of infection was excised and homogenized, and serial dilutions were plated on sheep blood agar to determine the number of CFU.
Resource sharing.
One goal of this project was to construct usable plasmids to be made available for the scientific community. To this end, the DH5αλpir strain and a methylation-deficient (both dam- and dcm-negative) GM2163λpir host E. coli strain as well as pKK22 and pKK30 are available through BEI Resources. In addition, plasmid maps and sequence files can be found at bosemicrolab.com.
Accession number(s).
The sequences of pKK22 (accession no. KX085042) and pKK30 (accession no. KX085043) have been deposited in GenBank.
RESULTS
Construction of pKK1.
LAC-p01 (identical to pUSA01 in sequenced strain FPR3757 [accession number NC_007790]) is found in many S. aureus USA300 isolates and has high sequence conservation among these strains (35). This 3.1-kb plasmid is predicted to encode a replication protein and four hypothetical proteins of unknown function; hence, it is commonly called the “cryptic” plasmid (Fig. 1). During other studies using a derivative of the strain LAC, we noted that the plasmid LAC-p01 was extremely stable without any known selection. Indeed, LAC-p01 remained in a strain that had undergone six rounds of targeted mutagenesis, a procedure requiring many generations of growth (data not shown). Thus, in our hands, LAC-p01 appeared to be extremely stable despite lacking any known selection. With the goal of developing LAC-p01 into a potential usable stable plasmid system, we first sought to determine if LAC-p01 was as stable as our previous studies suggested. Since LAC-p01 lacks any means of selection for tracking, and genetic manipulation of plasmids is difficult in S. aureus, we first converted LAC-p01 into a tractable plasmid. To this end, we developed a module containing three elements: (i) an R6Kγ origin of replication to allow maintenance and manipulation in E. coli, (ii) an antibiotic resistance cassette consisting of the dfrA gene under the control of the sarA P1 promoter that uniquely allows for selection on trimethoprim in both E. coli and Staphylococcus species, and (iii) the blaZ transcriptional terminator (blaZTT) to prevent transcriptional readout from the module. This entire module is 1,507 bp, which keeps the final plasmid relatively small (4.6 kb) to facilitate genetic manipulation and transfer between bacteria.
FIG 1.
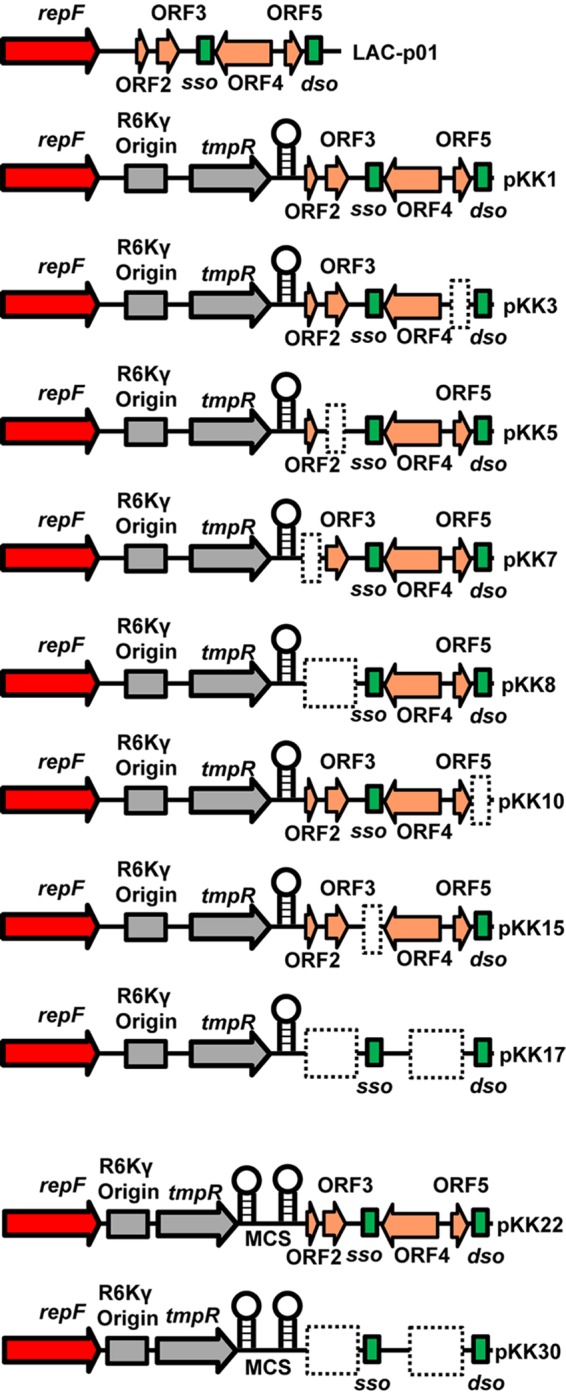
Schematic of LAC-p01 and its derivatives used in these studies. The stem-loop indicates an added transcriptional terminator, and the dashed boxes show deleted regions in each plasmid. MCS denotes the addition of a multiple-cloning site.
Relatively little is known about the architecture of LAC-p01, and this plasmid has very few unique restriction endonuclease recognition sites in nonpredicted genes. A unique EcoRV site between the gene annotated to encode the Rep protein and putative ORF2 was chosen because (i) it is predicted to be intergenic, (ii) it is far enough upstream (209 bp) of ORF2 to possibly lie upstream of any predicted promoter element, and (iii) RNA sequencing (RNA-seq) data indicated that this site was either not in a predicted expressed RNA or of minimal chance to be on the extreme 3′ end of an RNA antisense to ORF2 and ORF3 (36). Therefore, the R6Kγ-PsarAP1::dfrA-blaZTT module was cloned into the EcoRV site of LAC-p01, resulting in pKK1.
pKK1 is stable in vitro.
Since the presence of dfrA in pKK1 allows for tracking the plasmid easily, in contrast to its parent LAC-p01, we performed plasmid stability assays to determine if pKK1 was as stable as we hypothesized. In one set of experiments, RN4220 harboring pKK1 was kept in continuous exponential phase for approximately 120 generations by performing serial dilutions twice daily. As shown in Fig. S1 in the supplemental material, pKK1 was found to be 100% stable over the course of the experiment. In an alternate procedure, the cultures were allowed to enter stationary phase and then were diluted and allowed to regrow to stationary phase. This process was continued for approximately 100 generations. Again, pKK1 was found to be maintained in 100% of cells (Fig. 2A). Due to the ease of performing the second experiment, it was chosen as the standard stability test moving forward. In total, pKK1 has been checked in 4 experiments consisting of 12 independent cultures and over 2,200 colonies screened. Only 2 colonies have been identified to have lost pKK1 from a single experiment. In these assays, the cells are grown nonselectively on TSA plates prior to transfer to plates containing trimethoprim. Consequently, the number of generations calculated is a conservative estimate of the number of generations taking place since replication on the TSA plate is not considered in the calculation.
FIG 2.
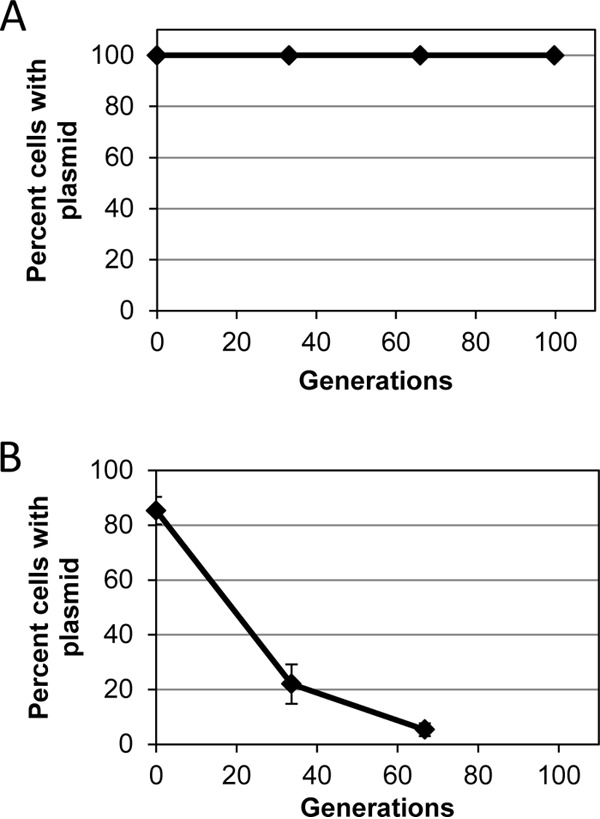
Stability of pKK1 (A) or pKK15 (Δsso) (B) in S. aureus RN4220 without antibiotic selection. Data are the mean (n = 3) with standard deviation of results from a representative experiment. For both panels, both x- and y-axis error bars are present and may be smaller than symbols. See the text for experimental details.
Identification of essential elements for pKK1 replication and plasmid copy number.
Considering the stability of pKK1, we sought to further develop this plasmid into a genetic tool by removing unnecessary DNA to find the minimal genetic elements required to retain plasmid stability. However, as mentioned above, LAC-p01 has not been characterized. Despite this, it is known that RCR plasmids normally have several critical features necessary for replication: a gene encoding a Rep protein, a double-stranded origin (dso), and typically one single-stranded origin (sso) (reviewed in reference 14).
The dso of RCR plasmids can vary greatly but consist of an initiation region containing a nic site (14, 37). We identified a region of LAC-p01 (TTTCTTCTTGTCTTG/ATACTA, bp 2951 to 2971, with a backslash indicating the nic site) with a 15/20 (38) or 15/18 match (39) to the pC194 dso initiation region and with 17/18 nucleotides (nt) identical to the pST1 nic region (39, 40). The dso should be essential for plasmid replication, and therefore deletion of this region would yield a plasmid that cannot replicate in S. aureus. Thus, a 40-bp region starting with and including the sequence indicated above was deleted in pKK1. As expected, construction of this plasmid in E. coli was successful. Consistent with the prediction that this region is essential for replication in S. aureus, we were unable to transfer this plasmid into RN4220.
RCR plasmids typically have a single sso, and previous studies have identified a canonical sequence found in the ssoA class of single-strand origins. The ssoA consists of a long (∼150-nt), single stem-loop with several bulges and contains the recombination site B (RSB, TTTATGCCGTGAAA) at the base of the stem of the hairpin and a 6-nt consensus sequence (CS-6, TAGCGT) in the terminal loop (14, 41–43). LAC-p01 contains a 13/14 bp match (TTTATGCCGaGAAA) to the canonical RSB from nt 1932 to 1945 and a perfect CS-6 (nt 1999 to 2004) between predicted ORF3 and ORF4. Based on mfold analysis (44–46), the region identified is predicted to be part of a large stem-loop (see Fig. S2 in the supplemental material) similar to the suggested structure of ssoA regions in similar RCR plasmids (42). Considering the conservation of these sites in other RCR plasmids, we predicted that nt 1930 to 2088 constitute the LAC-p01 sso. Previous studies have demonstrated that deletion of the sso in the RCR plasmid pT181 led to plasmid instability in the absence of antibiotic pressure (47). Therefore, this region was specifically deleted in pKK1. As expected, the resulting plasmid, pKK15, was found to be unstable in S. aureus in the absence of trimethoprim (Fig. 2B). Even at the initial point of transfer to TSB lacking trimethoprim, only 85% of cells were found to have pKK15. Since these cells were grown in trimethoprim until this dilution event, the loss of plasmid likely occurred following plating onto the nonselective TSA plates, resulting in a colony lacking the plasmid. Based on the results of these studies, we have identified regions of LAC-p01 consistent with known sequence and phenotypes associated with dso and sso (Fig. 1).
Often, low-copy-number plasmids are preferred for use since expression from high-copy-number plasmids can have deleterious effects. Therefore, we sought to determine the copy number of our pKK plasmids. To this end, we used qPCR for the PsarAP1-dfrA cassette of pKK22 (described below and shown in Fig. 1) and compared it to the same cassette found integrated in the S. aureus chromosome. Determining copy number can be problematic, since different methodologies provide differing results (48). Therefore, we used two methods to isolate DNA in order to determine the copy number of pKK22. The use of a genomic DNA extraction kit resulted in a copy number determination of 6 copies per chromosome, while simply using extracts from cells treated with a cell disruptor yielded a result of 10 copies per chromosome.
Putative ORFs are not required for stability.
The original LAC-p01 plasmid has been annotated to contain four predicted open reading frames (ORF2, ORF3, ORF4, and ORF5) (Fig. 1) thought to encode hypothetical proteins of unknown function. Considering the unknown function of these predicted proteins, we sought to determine if deletion of any of these predicted genes would alter plasmid stability. To this end, deletions either singly or in combination were made of ORF2, ORF3, ORF4, and ORF5. We did observe an occasional cell having lost the plasmid at the endpoint of the experiment (70 to 113 generations) (Table 2). However, even in the worst case [pKK7 (ΔORF2)], only 3 colonies out of 150 (2%) screened were found to have lost the plasmid, and no plasmid loss was observed from this same plasmid in additional experiments. In addition, this was after an overnight incubation on TSA without antibiotic prior to screening on TSA with trimethoprim, and therefore the plasmid may have been retained prior to this growth step, which does not account for the generations when growing on a plate. Even pKK17 (ΔORF2-3,4-5), which contains a deletion in all of the hypothetical ORFs, was found to be stable out to ∼100 generations without antibiotic selection. These studies demonstrate that none of the predicted open reading frames in pKK1 play a role in plasmid stability.
TABLE 2.
Stability of pKK1 derivatives at experimental endpoint
| Plasmid | Deletion | Triala | No. of generations | No. of colonies with plasmid present/total no. | % of colonies with plasmid |
|---|---|---|---|---|---|
| pKK1 | None | 1 | 99.7 | 150/150 | 100 |
| 2 | 69.8 | 148/150 | 98.7 | ||
| 3 | 113.0 | 150/150 | 100 | ||
| pKK3 | ΔORF5 | 1 | 100.7 | 150/150 | 100 |
| 2 | 100.2 | 150/150 | 100 | ||
| 3 | 99.9 | 149/150 | 99.3 | ||
| pKK5 | ΔORF3 | 1 | 101 | 149/150 | 99.3 |
| 2 | 89.5 | 148/150 | 98.7 | ||
| 3 | 99.8 | 150/150 | 100 | ||
| pKK7 | ΔORF2 | 1 | 101.7 | 150/150 | 100 |
| 2 | 103.9 | 150/150 | 100 | ||
| 3 | 103 | 147/150 | 98 | ||
| pKK8 | ΔORF2-3 | 1 | 101.2 | 149/150 | 99.3 |
| 2 | 95.0 | 150/150 | 100 | ||
| 3 | 99.8 | 150/150 | 100 | ||
| pKK17 | ΔORF2-3,4-5 | 1 | 99.4 | 150/150 | 100 |
| 2 | 101.3 | 150/150 | 100 | ||
| 3 | 101.4 | 150/150 | 100 |
Each trial consisted of 3 independent cultures and 50 colonies screened per culture.
Generation of USA300-specific and general-use plasmids.
One goal of this study was to generate plasmids that would be useful in S. aureus isolates of different lineages. LAC-p01 is natively found in many USA300 isolates. Since these plasmids are constructed from LAC-p01, it is expected that the introduction of these LAC-p01 derivatives into strains carrying the original plasmid would lead to loss of that plasmid. Indeed, this was the basis for how LAC was cured of LAC-p01 to generate JE2, which is LAC devoid of all plasmids (49). Thus, the use of pKK1 is appropriate for USA300 strains that normally harbor LAC-p01 and would render the strains isogenic. However, many S. aureus strains do not normally contain LAC-p01, and the introduction of pKK1 would introduce ORF2 to ORF5, which, while of unknown function, introduces genes not native to these S. aureus isolates. Therefore, it was important to produce two plasmids, one for use in LAC-p01-harboring strains and one for bacteria that do not contain this plasmid. In addition, while the addition of the E. coli origin and trimethoprim resistance cassette was a necessary improvement to LAC-p01, the plasmids needed modifying in order to increase their usefulness. To this end, a multiple-cloning site (MCS) flanked by the atl transcription terminator was added to pKK1 to facilitate cloning. The terminator was added so that transcripts cloned into the MCS would not extend into other parts of the plasmid. A BamHI site existing between PsarAP1 and drfA was removed to make it available in the MCS. The resulting plasmids, pKK22 (for LAC-p01-containing strains) and pKK30 (lacking ORF2, ORF3, ORF4, and ORF5; for other strains), contain an MCS flanked by the atl and blaZ transcriptional terminators (Fig. 1 and 3). These plasmids were checked to ensure that those alterations did not affect plasmid stability. As shown in Table 3, both pKK22 and pKK30 were found to be stable out to ∼100 generations without antibiotic selection. For each plasmid, only a single colony examined appeared to have lost the plasmid and only in a single experiment.
FIG 3.
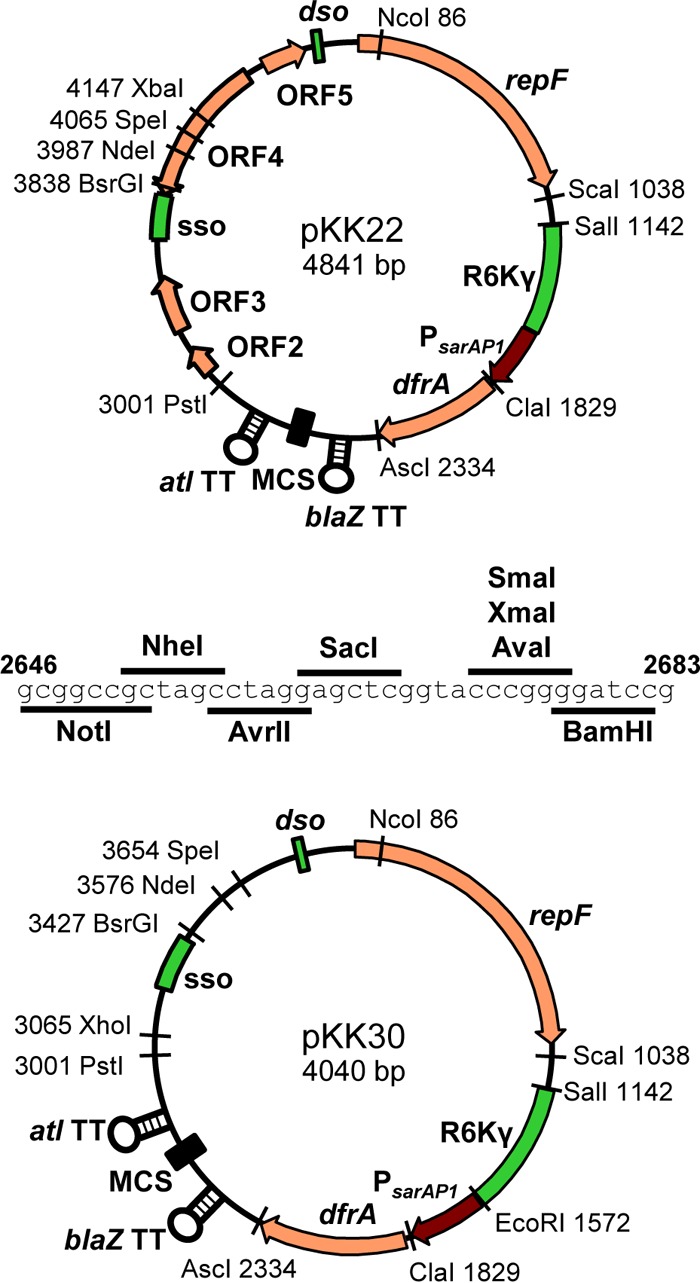
Diagrams of pKK22 and pKK30. The sequence of the multiple-cloning site (MCS) is shown, and stem-loops indicate transcriptional terminators. Numbers indicate locations of restriction endonuclease recognition sites. Note that the ClaI site is Dam methylation blocked.
TABLE 3.
In vitro stability of pKK22 and pKK30
| Plasmid | Bacterium | Triala | No. of generations | No. of colonies with plasmid present/total no. | % of colonies with plasmid |
|---|---|---|---|---|---|
| pKK22 | S. aureus | 1 | 99.6 | 150/150 | 100 |
| 2 | 99.7 | 149/150 | 99.3 | ||
| 3 | 99.9 | 150/150 | 100 | ||
| pKK30 | S. aureus | 1 | 99.9 | 150/150 | 100 |
| 2 | 100.6 | 149/150 | 99.3 | ||
| 3 | 99.4 | 150/150 | 100 | ||
| S. epidermidis | 1 | 100.2 | 150/150 | 100 | |
| 2 | 99.1 | 150/150 | 100 | ||
| 3 | 99.3 | 150/150 | 100 |
Each trial consisted of 3 independent cultures and 50 colonies screened per culture.
It would be expected that a plasmid that replicated in S. aureus would also function in other Staphylococcus species. To test this, pKK30 was transferred into S. epidermidis 1457 and tested for stability. As shown in Table 3, pKK30 was stable in S. epidermidis for at least 100 generations. This strain contains a native >10-kb plasmid and was confirmed to be present along with pKK30 at the end of a 100-generation experiment, demonstrating that there is not a compatibility issue between these two plasmids.
Prior to testing during animal models of infection, we wanted to ensure that the plasmid did not have adverse effects on the cells. To test this, the growth kinetics of JE2 (no plasmid), LAC-13C (JE2 harboring LAC-p01), and JE2 harboring pKK22 were compared. As shown in Fig. 4, we observed similar levels of growth for these strains.
FIG 4.
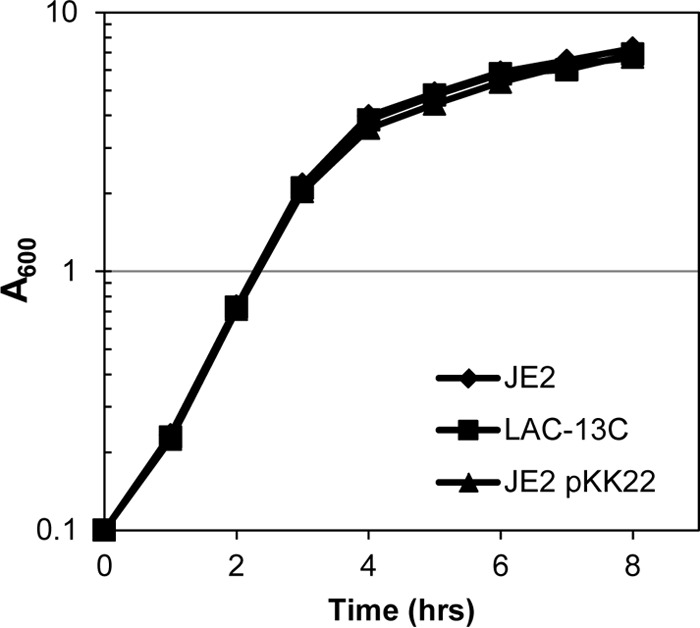
Growth of JE2 (no plasmid), LAC-13C (JE2 harboring native LAC-p01), and JE2 harboring pKK22 in TSB. Overnight cultures were diluted to an A600 of 0.1 in 50 ml of TSB in a 500-ml flask and grown at 37°C with shaking (250 rpm). Data are the mean result of three independent trials with standard deviation, although error bars are smaller than symbols.
In vivo stability of pKK22.
While developing a stable plasmid for in vitro work was important, it was the ultimate goal to generate a plasmid that would be stable within S. aureus during in vivo models of infection. To this end, pKK22 was transferred into the USA300 LAC derivative JE2 and mice were inoculated via retro-orbital injection with this strain. Following infection, bacteria were isolated from the kidneys at 3 or 7 days postinoculation and screened for trimethoprim resistance, indicative of the presence of pKK22. As shown in Table 4, all 800 colonies screened from 8 independently infected mice 3 days postinoculation maintained the plasmid. The same result was observed for 700 colonies after 7 days of infection. At each time point, random colonies were chosen and plasmid DNA was isolated and found to contain pKK22 following restriction enzyme digestion and gel electrophoresis (data not shown). This stability was observed in a second experiment in which 600 colonies from six independently infected mice were examined. As with the first experiment, random colonies from day 7 postinfection were confirmed to contain pKK22 by restriction digestion (data not shown). Importantly, no structural changes in the plasmid were observed following isolation from the host in any experiment. Furthermore, sequencing of pKK22 from randomly chosen isolates showed no difference from pKK22 prior to animal infection. Based on these results, pKK22 is a plasmid which maintains stability in the absence of antibiotics during in vitro experiments and at least over a 7-day course of in vivo infection.
TABLE 4.
In vivo stability of pKK22
| Expt | Day postinfection | No. of colonies screeneda | No. of colonies with plasmid present | % of colonies with plasmid |
|---|---|---|---|---|
| 1 | 3 | 800 | 800 | 100 |
| 7 | 700 | 700 | 100 | |
| 2 | 3 | 600 | 600 | 100 |
| 7 | 600 | 600 | 100 |
One hundred colonies from each animal were screened.
Complementation of an hla mutant.
While our data demonstrate that pKK plasmids are stable in vivo, we sought to demonstrate a utility for the pKK plasmids. To this end, we cloned the hla gene and its native promoter into pKK22. The α-hemolysin toxin encoded by hla is essential for necrosis in murine models of S. aureus skin infection (50). As such, we expected that this new construct would complement an hla mutant in vivo. Over the course of a 3-day skin infection, mice infected with an hla isogenic deletion mutant showed no dermonecrosis (Fig. 5A and B), reduced weight loss (a measure of morbidity) (Fig. 5C), and increased bacterial clearance (Fig. 5D) compared to mice infected with the wild-type isolate. As expected, infection with the hla isogenic mutant carrying hla borne on the pKK22 plasmid restored dermonecrosis, weight loss, and bacterial burden to levels consistent with those caused by the wild-type isolate (Fig. 5A to D). Along with the stability results in the murine sepsis model, these data demonstrate that pKK22 can be used in vivo without the requirement of antibiotic addition.
FIG 5.

pKK22 was used to complement an hla mutant during a murine model of dermonecrosis. AH1263 (wild type [WT]) harboring pKK22 and hla mutant JB24 harboring pKK22 or pCK8 (pKK22 with hla) were used to inoculate SKH1 mice. (A) Representative infection site images (scale bar = 5 mm). (B) Calculated area under the curve (AUC) for area of dermonecrosis (ND, not detected). (C) Percent weight loss. (D) Bacterial burden at the site of infection. Data shown are median values plus 5th to 95th percentiles. All data were taken 3 days postinfection with 4 mice per group. ns, not significant by Mann-Whitney test for nonparametric data.
DISCUSSION
The ability to provide genes for complementation or reporter constructs on plasmids during an in vivo model of infection is an important tool in studying bacterial pathogenesis. Here we described the generation of such a plasmid and have shown that it remains stable during in vitro growth in S. aureus and S. epidermidis without the need of antibiotic pressure. Moreover, the plasmid was stably maintained in S. aureus during an in vivo infection model. The construction of these plasmids represents an important step forward in the genetic tools available to the staphylococcal research community.
An important aspect of studying bacterial pathogens is the ability to provide genes for complementation of mutant strains, expression of mRNA and proteins under nonnative promoters, or expression of transcriptional and translational reporter systems. Plasmid-based systems are often a mainstay of bacterial genetics, since they are easy to produce and transfer between cells. They can also be used for overexpression or to increase the output from low-level transcripts, since they most often exist in many copies per cell. However, most plasmids are inherently lost without antibiotic pressure. To overcome this hurdle, several techniques are currently available to provide these genetic constructs at nonnative sites within the chromosome. Once integrated into the chromosome, these systems are stable without the necessity of antibiotics; however, these techniques are hampered by the necessity of moving genetic constructs into theoretically neutral sites and locations of convenience such as phage integration sites. Unfortunately, these locations are generally not neutral and lead to the inactivation or replacement of phage or genes having been shown to play a role in virulence (26–28). Furthermore, these techniques are often performed in the mutagenized strain RN4220, since it readily accepts E. coli DNA, at which point the integrated DNA is mobilized into a target strain by transduction. However, phage can move tens of thousands of base pairs of DNA, leading to unknown transferred mutations due to sequence difference between strains. The pKK plasmids generated here provide the benefit of plasmids (ease of use and transfer) and the stability often desired in chromosome cassettes (antibiotic pressure not needed).
One concern when using plasmid-based genetic systems is plasmid origin incompatibility. Indeed, it is well documented that bacteria do not maintain plasmids with identical or similar origins of replication and that this incompatibility can be used to group plasmids. Therefore, the appropriate pKK plasmid needs to be chosen depending on the strain used. Any of the pKK plasmids will replace the existing 3.1-kb plasmids LAC-p01, pUSA01, and sequence-identical plasmids found in S. aureus USA300 isolates. In this case, pKK22 should be used, as it contains all of the genes of the original plasmid. It is recommended that, following transfer, cells be plated on medium containing trimethoprim and then replated on the same medium to ensure that the cells keep pKK22 and not the native plasmid, since competition will occur between the two plasmids. Indeed, while transferring pKK22 into AH1263 (LAC strain harboring LAC-p01), we identified colonies during screening which had both plasmids. Several additional streaks on Tmp were sufficient to exclude LAC-p01 from the cells. For bacteria not containing LAC-p01, pKK30 should be chosen so as not to bring additional genes (ORF2 to ORF5) into the strain. For other bacteria, known native plasmids should be confirmed once pKK30 has been introduced. Of note, S. epidermidis 1457 contains a >10-kb plasmid, and no compatibility issues were identified with pKK30, as both were present at the end of the experiment. Regarding other commonly used S. aureus plasmids, in previous studies we have been able to stably maintain plasmids with pT181, pE194, pC194, and pE194ts origins along with the original LAC-p01 plasmid (30, 31). In addition, we have observed no compatibility issues with pLI50, which contains the pUB110 origin of replication (not shown). Thus, the pKK plasmids are compatible with the major origins commonly used for S. aureus genetic manipulation.
Plasmids used for genetic manipulation of bacteria, including S. aureus, have unpredictable stability and thus are typically used with continuous antibiotic pressure. This has been circumvented in other plasmid systems by the addition of essential genes on the plasmid (51–53). While this approach is useful, these tools generally require a mutated host strain to make the plasmid-borne gene essential, narrowing the number of strains in which the plasmids are maintained. Outside of genetic engineering of plasmids, some naturally occurring plasmids have evolved to have addiction modules, e.g., toxin-antitoxin systems, which ensure plasmid maintenance by killing daughter cells that do not receive the plasmid. In other cases, such as LAC-p01, it is uncertain as to why the plasmids have been maintained without a known selection or function. Indeed, it was expected that we would identify a stability system on LAC-p01 that would account for its remarkable stability. However, with the exception of deletion of the sso, we did not find a region of the plasmid that was necessary for plasmid stability. While only the annotated hypothetical ORFs were targeted for deletion and analysis, it is possible that a nonpredicted ORF or unknown RNA on LAC-p01 plays an unidentified role in stability.
The construction of plasmids that are retained without antibiotic selection represents an important step forward in studies of human pathogens. These plasmids eliminate the need for antibiotics during experiments, thus removing the unintentional consequences that antibiotics have on cells. In addition, stable plasmids provide a resource for studies in which antibiotic selection is difficult, such as long-term studies where antibiotics degrade or during experiments of bacterial internalization by host cells that are impermeable to many antibiotics. Finally, one of the most challenging cases where plasmid stability limits research studies is during in vivo models of infection. While some Staphylococcus plasmids may be well maintained during short-term experiments, there are few options available for long-term studies. There are several reports of plasmids that are stable (≥90%) for 60 generations in vitro for Staphylococcus carnosus (54–56) and S. aureus (54); however, this is the first report of a genetically tractable plasmid shown to be stably maintained during both long-term in vitro and in vivo experiments without antibiotic pressure. The creation of the pKK plasmids described here provides a valuable new genetic resource for studies of Staphylococcus species.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anne Dunn (University of Oklahoma) and Eric Stabb (University of Georgia) for providing DH5αλpir and GM2163λpir, Ken Bayles (University of Nebraska Medical Center) for supplying the chromosome-integrated PsarAP1-dfrA strain, and Paul Fey (University of Nebraska Medical Center) for supplying S. epidermidis 1457. We are grateful to Nicole Matocha and Michael Manley for technical assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02370-16.
REFERENCES
- 1.Blickwede M, Goethe R, Wolz C, Valentin-Weigand P, Schwarz S. 2005. Molecular basis of florfenicol-induced increase in adherence of Staphylococcus aureus strain Newman. J Antimicrob Chemother 56:315–323. doi: 10.1093/jac/dki233. [DOI] [PubMed] [Google Scholar]
- 2.Blickwede M, Wolz C, Valentin-Weigand P, Schwarz S. 2005. Influence of clindamycin on the stability of coa and fnbB transcripts and adherence properties of Staphylococcus aureus Newman. FEMS Microbiol Lett 252:73–78. doi: 10.1016/j.femsle.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 3.Brunelle BW, Bearson BL, Bearson SM. 2014. Chloramphenicol and tetracycline decrease motility and increase invasion and attachment gene expression in specific isolates of multidrug-resistant Salmonella enterica serovar Typhimurium. Front Microbiol 5:801. doi: 10.3389/fmicb.2014.00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goh EB, Yim G, Tsui W, McClure J, Surette MG, Davies J. 2002. Transcriptional modulation of bacterial gene expression by subinhibitory concentrations of antibiotics. Proc Natl Acad Sci U S A 99:17025–17030. doi: 10.1073/pnas.252607699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hung KF, Byrd JJ, Bose JL, Kaspar CW. 2006. Reduction of acid tolerance by tetracycline in Escherichia coli expressing tetA(C) is reversed by cations. Appl Environ Microbiol 72:4472–4474. doi: 10.1128/AEM.02519-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuroda H, Kuroda M, Cui L, Hiramatsu K. 2007. Subinhibitory concentrations of beta-lactam induce haemolytic activity in Staphylococcus aureus through the SaeRS two-component system. FEMS Microbiol Lett 268:98–105. doi: 10.1111/j.1574-6968.2006.00568.x. [DOI] [PubMed] [Google Scholar]
- 7.Shen L, Shi Y, Zhang D, Wei J, Surette MG, Duan K. 2008. Modulation of secreted virulence factor genes by subinhibitory concentrations of antibiotics in Pseudomonas aeruginosa. J Microbiol 46:441–447. doi: 10.1007/s12275-008-0054-x. [DOI] [PubMed] [Google Scholar]
- 8.Davies J, Spiegelman GB, Yim G. 2006. The world of subinhibitory antibiotic concentrations. Curr Opin Microbiol 9:445–453. doi: 10.1016/j.mib.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Yim G, Wang HH, Davies J. 2006. The truth about antibiotics. Int J Med Microbiol 296:163–170. doi: 10.1016/j.ijmm.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 10.Andersson DI, Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8:260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 11.Andersson DI, Levin BR. 1999. The biological cost of antibiotic resistance. Curr Opin Microbiol 2:489–493. doi: 10.1016/S1369-5274(99)00005-3. [DOI] [PubMed] [Google Scholar]
- 12.Bjorkman J, Andersson DI. 2000. The cost of antibiotic resistance from a bacterial perspective. Drug Resist Updat 3:237–245. doi: 10.1054/drup.2000.0147. [DOI] [PubMed] [Google Scholar]
- 13.Prax M, Lee CY, Bertram R. 2013. An update on the molecular genetics toolbox for staphylococci. Microbiology 159:421–435. doi: 10.1099/mic.0.061705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruiz-Maso JA, Macho NC, Bordanaba-Ruiseco L, Espinosa M, Coll M, Del Solar G. 2015. Plasmid rolling-circle replication. Microbiol Spectr 3:PLAS-0035-2014. doi: 10.1128/microbiolspec.PLAS-0035-2014. [DOI] [PubMed] [Google Scholar]
- 15.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Bose JL. 2014. Genetic manipulation of staphylococci, p 101–111. In Fey PD. (ed), Staphylococcus epidermidis: methods and protocols, vol 1106 Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 17.Bose JL, Fey PD, Bayles KW. 2013. Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl Environ Microbiol 79:2218–2224. doi: 10.1128/AEM.00136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sau S, Sun J, Lee CY. 1997. Molecular characterization and transcriptional analysis of type 8 capsule genes in Staphylococcus aureus. J Bacteriol 179:1614–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maurin M, Raoult D. 1993. Antibiotic penetration within eukaryotic cells, p 24–33. In Raoult D. (ed), Antimicrobial agents and intracellular pathogens. CRC Press, Boca Raton, FL. [Google Scholar]
- 20.Solberg CO, Hellum KB. 1978. Protection of phagocytosed bacteria against antimicrobial agents. Scand J Infect Dis Suppl 14:246–250. [PubMed] [Google Scholar]
- 21.Tulkens PM. 1991. Intracellular distribution and activity of antibiotics. Eur J Clin Microbiol Infect Dis 10:100–106. doi: 10.1007/BF01964420. [DOI] [PubMed] [Google Scholar]
- 22.van den Broek PJ. 1991. Activity of antibiotics against microorganisms ingested by mononuclear phagocytes. Eur J Clin Microbiol Infect Dis 10:114–118. doi: 10.1007/BF01964422. [DOI] [PubMed] [Google Scholar]
- 23.Vaudaux P, Waldvogel FA. 1979. Gentamicin antibacterial activity in the presence of human polymorphonuclear leukocytes. Antimicrob Agents Chemother 16:743–749. doi: 10.1128/AAC.16.6.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luong TT, Lee CY. 2007. Improved single-copy integration vectors for Staphylococcus aureus. J Microbiol Methods 70:186–190. doi: 10.1016/j.mimet.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Yoong P, Ram G, Torres VJ, Novick RP. 2014. Single-copy vectors for integration at the SaPI1 attachment site for Staphylococcus aureus. Plasmid 76:1–7. doi: 10.1016/j.plasmid.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu C, Xiong N, Zhang Y, Rayner S, Chen S. 2012. Functional characterization of lipase in the pathogenesis of Staphylococcus aureus. Biochem Biophys Res Commun 419:617–620. doi: 10.1016/j.bbrc.2012.02.057. [DOI] [PubMed] [Google Scholar]
- 27.Bae T, Baba T, Hiramatsu K, Schneewind O. 2006. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol Microbiol 62:1035–1047. doi: 10.1111/j.1365-2958.2006.05441.x. [DOI] [PubMed] [Google Scholar]
- 28.Novick RP, Schlievert P, Ruzin A. 2001. Pathogenicity and resistance islands of staphylococci. Microbes Infect 3:585–594. doi: 10.1016/S1286-4579(01)01414-9. [DOI] [PubMed] [Google Scholar]
- 29.Dunn AK, Martin MO, Stabb EV. 2005. Characterization of pES213, a small mobilizable plasmid from Vibrio fischeri. Plasmid 54:114–134. doi: 10.1016/j.plasmid.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Bose JL, Daly SM, Hall PR, Bayles KW. 2014. Identification of the Staphylococcus aureus vfrAB operon, a novel virulence factor regulatory locus. Infect Immun 82:1813–1822. doi: 10.1128/IAI.01655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bose JL, Lehman MK, Fey PD, Bayles KW. 2012. Contribution of the Staphylococcus aureus Atl AM and GL murein hydrolase activities in cell division, autolysis, and biofilm formation. PLoS One 7:e42244. doi: 10.1371/journal.pone.0042244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 33.Malachowa N, Kobayashi SD, Braughton KR, DeLeo FR. 2013. Mouse model of Staphylococcus aureus skin infection. Methods Mol Biol 1031:109–116. doi: 10.1007/978-1-62703-481-4_14. [DOI] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kennedy AD, Porcella SF, Martens C, Whitney AR, Braughton KR, Chen L, Craig CT, Tenover FC, Kreiswirth BN, Musser JM, DeLeo FR. 2010. Complete nucleotide sequence analysis of plasmids in strains of Staphylococcus aureus clone USA300 reveals a high level of identity among isolates with closely related core genome sequences. J Clin Microbiol 48:4504–4511. doi: 10.1128/JCM.01050-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carroll RK, Weiss A, Broach WH, Wiemels RE, Mogen AB, Rice KC, Shaw LN. 2016. Genome-wide annotation, identification, and global transcriptomic analysis of regulatory or small RNA gene expression in Staphylococcus aureus. mBio 7:e01990–15. doi: 10.1128/mBio.01990-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan SA. 2005. Plasmid rolling-circle replication: highlights of two decades of research. Plasmid 53:126–136. doi: 10.1016/j.plasmid.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 38.del Solar G, Giraldo R, Ruiz-Echevarria MJ, Espinosa M, Diaz-Orejas R. 1998. Replication and control of circular bacterial plasmids. Microbiol Mol Biol Rev 62:434–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marsin S, Forterre P. 1998. A rolling circle replication initiator protein with a nucleotidyl-transferase activity encoded by the plasmid pGT5 from the hyperthermophilic archaeon Pyrococcus abyssi. Mol Microbiol 27:1183–1192. doi: 10.1046/j.1365-2958.1998.00759.x. [DOI] [PubMed] [Google Scholar]
- 40.Hashiba H, Takiguchi R, Joho K, Aoyama K, Hirota T. 1993. Identification of the replication region of Streptococcus thermophilus no. 29 plasmid pST1. Biosci Biotechnol Biochem 57:1646–1649. doi: 10.1271/bbb.57.1646. [DOI] [PubMed] [Google Scholar]
- 41.del Solar GH, Puyet A, Espinosa M. 1987. Initiation signals for the conversion of single stranded to double stranded DNA forms in the streptococcal plasmid pLS1. Nucleic Acids Res 15:5561–5580. doi: 10.1093/nar/15.14.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kramer MG, Khan SA, Espinosa M. 1997. Plasmid rolling circle replication: identification of the RNA polymerase-directed primer RNA and requirement for DNA polymerase I for lagging strand synthesis. EMBO J 16:5784–5795. doi: 10.1093/emboj/16.18.5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novick RP, Projan SJ, Rosenblum W, Edelman I. 1984. Staphylococcal plasmid cointegrates are formed by host- and phage-mediated general rec systems that act on short regions of homology. Mol Gen Genet 195:374–377. doi: 10.1007/BF00332777. [DOI] [PubMed] [Google Scholar]
- 44.Waugh A, Gendron P, Altman R, Brown JW, Case D, Gautheret D, Harvey SC, Leontis N, Westbrook J, Westhof E, Zuker M, Major F. 2002. RNAML: a standard syntax for exchanging RNA information. RNA 8:707–717. doi: 10.1017/S1355838202028017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuker M, Jacobson AB. 1998. Using reliability information to annotate RNA secondary structures. RNA 4:669–679. doi: 10.1017/S1355838298980116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gruss AD, Ross HF, Novick RP. 1987. Functional analysis of a palindromic sequence required for normal replication of several staphylococcal plasmids. Proc Natl Acad Sci U S A 84:2165–2169. doi: 10.1073/pnas.84.8.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skulj M, Okrslar V, Jalen S, Jevsevar S, Slanc P, Strukelj B, Menart V. 2008. Improved determination of plasmid copy number using quantitative real-time PCR for monitoring fermentation processes. Microb Cell Fact 7:6. doi: 10.1186/1475-2859-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of non-essential Staphylococcus aureus genes. mBio 4:e00537-12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR. 2010. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis 202:1050–1058. doi: 10.1086/656043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galan JE, Nakayama K, Curtiss R III. 1990. Cloning and characterization of the asd gene of Salmonella typhimurium: use in stable maintenance of recombinant plasmids in Salmonella vaccine strains. Gene 94:29–35. doi: 10.1016/0378-1119(90)90464-3. [DOI] [PubMed] [Google Scholar]
- 52.Hagg P, de Pohl JW, Abdulkarim F, Isaksson LA. 2004. A host/plasmid system that is not dependent on antibiotics and antibiotic resistance genes for stable plasmid maintenance in Escherichia coli. J Biotechnol 111:17–30. doi: 10.1016/j.jbiotec.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 53.Skogman SG, Nilsson J. 1984. Temperature-dependent retention of a tryptophan-operon-bearing plasmid in Escherichia coli. Gene 31:117–122. doi: 10.1016/0378-1119(84)90201-4. [DOI] [PubMed] [Google Scholar]
- 54.Keller G, Schleifer KH, Gotz F. 1983. Construction and characterization of plasmid vectors for cloning in Staphylococcus aureus and Staphylococcus carnosus. Plasmid 10:270–278. doi: 10.1016/0147-619X(83)90041-0. [DOI] [PubMed] [Google Scholar]
- 55.Kreutz B, Gotz F. 1984. Construction of Staphylococcus plasmid vector pCA43 conferring resistance to chloramphenicol, arsenate, arsenite and antimony. Gene 31:301–304. doi: 10.1016/0378-1119(84)90226-9. [DOI] [PubMed] [Google Scholar]
- 56.Wieland KP, Wieland B, Gotz F. 1995. A promoter-screening plasmid and xylose-inducible, glucose-repressible expression vectors for Staphylococcus carnosus. Gene 158:91–96. doi: 10.1016/0378-1119(95)00137-U. [DOI] [PubMed] [Google Scholar]
- 57.Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5:e10146. doi: 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 59.Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lehman MK, Bose JL, Sharma-Kuinkel BK, Moormeier DE, Endres JL, Sadykov MR, Biswas I, Bayles KW. 2015. Identification of the amino acids essential for LytSR-mediated signal transduction in Staphylococcus aureus and their roles in biofilm-specific gene expression. Mol Microbiol 95:723–737. doi: 10.1111/mmi.12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.