

ABSTRACT
Ethidium monoazide and propidium monoazide (EMA and PMA) have been used in combination with PCR for more than a decade to facilitate the discrimination of live and dead bacteria (LD discrimination). These methods, however, require many laborious procedures, including the use of a darkroom. Here, we demonstrate an innovative use of palladium compounds involving lower limits of detection and quantification of targeted live cells, fewer laborious procedures, lower costs, and potentially higher-throughput analysis than the use of EMA and PMA. We have also recently reported platinum compounds for LD discrimination, but platinum compounds carry costs that are 3 times higher because of the requirement for much larger amounts for LD discrimination than palladium compounds. Palladium compounds can penetrate dead (compromised) but not live bacteria and can be chelated primarily by chromosomal DNA and cell wall transmembrane proteins, with small amounts of DNA-binding proteins in vivo. The new mechanism for palladium compounds is obviously different from that of platinum compounds, which primarily target DNA. Combining palladium compounds with PCR (Pd-PCR) in water resulted in discrimination between live and dead Enterobacteriaceae bacteria that was much clearer than that seen with the PMA method. Pd-PCR correlated with reference plating or with the currently used PMA-PCR method for pasteurized milk, based on EN ISO 16140:2003 validation. Pd-PCR enabled us to specifically detect and assay viable Enterobacteriaceae cells at concentrations of 5 to 10 CFU/ml in milk while following U.S./EU regulations after a 4.5-h process in a typical laboratory exposed to natural or electric light, as specified by U.S./EU regulations.
IMPORTANCE Ethidium monoazide and propidium monoazide (EMA and PMA) facilitate the discrimination of live and dead bacteria (LD discrimination). These methods, however, require many laborious procedures, including the use of a darkroom. Here, we demonstrate an innovative use of palladium compounds involving fewer laborious procedures, lower costs, and potentially higher-throughput analysis than the use of EMA and PMA. We have also recently reported platinum compounds for LD discrimination, but platinum compounds carry costs that are 3 times higher because of the requirement for much larger amounts for LD discrimination than palladium compounds, which have also a novel reaction mechanism different from that of platinum compounds. In view of testing cost, palladium compounds are also very useful here compared with platinum compounds. Ultimately, the innovative Pd-PCR method may be also substituted for the currently used reference plating methods.
INTRODUCTION
The PCR is a widely available tool that is used to detect bacteria and viruses in food and in environmental and clinical samples. However, PCR cannot distinguish live from dead bacteria. During reverse transcription-PCR targeting mRNA, a high concentration of contaminating dead bacteria (4 log10 to 7 log10 cells/ml) will trigger a false-positive result owing to the presence of residual mRNA (1, 2). Although DNA can be deactivated in vitro by cross-linking psoralen/psoralen derivatives following elaborate exposure to UV A (3), the selective penetration of dead bacteria is an important issue.
DNA cross-linking agents, specifically, ethidium monoazide and propidium monoazide (EMA and PMA, respectively), have been used for more than a decade to address the inability of conventional PCR to distinguish live bacteria from dead bacteria (4–6). EMA and PMA molecules generally permeate only dead bacteria (compromised cells) after a brief exposure time, and these agents specifically intercalate into dead bacterial DNA, followed by cross-linking and direct DNA cleavage upon exposure to visible light (7). Live or dead bacterial suspensions must be kept on ice to prevent EMA and PMA molecules from permeating live cells whose cell walls and inner membranes have become physically injured by the gradually increasing temperature present during irradiation with a strong halogen lamp. Performing a series of EMA and PMA experimental measures is very demanding in terms of more-rapid food, environmental, and clinical tests (4–7). EMA and PMA solutions must be prepared in a darkroom owing to their high reactivity to visible light, which leads to laborious testing and significant disturbance for higher-throughput analysis. Additionally, EMA and PMA reagents are expensive for analytical testing, as demonstrated by the costs of PMA (267 dollars/mg) and EMA (52 dollars/mg) compared with the costs of the reagents used in the present study: dichloro(η-cycloocta-1,5,-diene)palladium(II) (0.11 dollars/mg), bis(benzonitrile)dichloropalladium(II) (0.08 dollars/mg), diamminedichloropalladium(II) (0.25 dollars/mg), and palladium(II)acetate (0.05 dollars/mg). The average reagent cost for these 4 palladium (Pd) compounds is 0.12 dollars/mg, which corresponds to approximately 30% of the average cost of the 5 previously reported platinum (Pt) compounds used to distinguish live from dead bacteria (8). In view of economic factors, Pd compounds are more convenient for use than Pt compounds.
We present an innovative use of Pd compounds that facilitates clearer discrimination between live and dead bacteria (here referred to as LD discrimination) without the need for special laboratory equipment or darkrooms, which are typically required for PMA and EMA methods. We demonstrate that the use of Pd compounds also contributes to less-laborious test procedures, lower costs, and potentially higher-throughput procedures than the PMA or EMA methods for LD discrimination. These advantages are primarily because Pd compounds can simply be added to live and dead bacterial suspensions in typical laboratories that are equipped with natural and/or electric lights, similarly to Pt compounds (8).
Furthermore, for possible practical use, we propose that Pd compounds could be superior to PMA (currently used in DNA elongation technology) in terms of improved accuracy, sensitivity, and specificity relative to the reference plating method (ISO, 2003), specifically when examining relative detection levels and linearity and detection and quantification limits (limits of detection [LOD] and limits of quantification [LOQ]) for live Escherichia coli cells in pasteurized milk, which can be contaminated with many PCR inhibitors and palladium inhibitors (9–11).
Concerning the reaction mechanism for Pd compounds, we were originally interested in palladium and other platinum group metals that could be chelated in mammalian cells by ligands such as cysteine or methionine amino acid residues and metallothionein as well as DNA bases (12–17). We were also very interested in the interaction between palladium compounds and bovine serum albumin (BSA) in view of the effects of Pd compounds on proteins and enzymes (18). Thus, we evaluated whether treatment with Pd compounds followed by PCR could improve LD discrimination, and then we elucidated the reaction mechanism of Pd compounds for LD discrimination.
MATERIALS AND METHODS
Bacterial strains, growth conditions, and preparation of inocula.
Two bacterial strains (Cronobacter sakazakii ATCC 29554 and E. coli JCM1649) were incubated in brain heart infusion (BHI) broth (Eiken Chemical Co., Ltd., Tokyo, Japan) at 37°C for 16 h prior to use. To obtain live cell suspensions, single-strain cultures were centrifuged at 3,000 × g for 10 min at 4°C. After removal of the supernatant, the same volume of sterile distilled water (SW) was added. Viable cell counts were obtained by plating 0.1 ml of an appropriate dilution on standard plate count agar (SPC; Kanto Chemical Co., Inc., Tokyo, Japan). Unless otherwise specified, the centrifugation was typically performed at 3,000 × g for 5 min at 4°C.
Heat-killed bacterial suspensions were prepared as follows: the pellet obtained from a 1-ml BHI culture was resuspended in 1 ml of SW and boiled in a water bath for 3 min, immediately cooled, and then coplated on SPC (Kanto Chemical) to verify the presence or absence of colonies. The live cells and heat-killed suspensions were serially diluted in SW for the LD discrimination assay. Independently, E. coli was added into pasteurized milk to evaluate viable cell counts using violet red bile agar (VRBA; Becton Dickinson, Sparks, MD, USA), as discussed later in this text.
Preparation and exposure method for Pd or Pt compounds.
Dichloro(ŋ-cycloocta-1,5-diene)palladium(II) and bis(benzonitrile)dichloropalladium(II) (Sigma, St. Louis, MO, USA) were diluted to 50 mM with dimethyl sulfoxide (DMSO) and stored at −20°C until use. Diamminedichloropalladium(II) and palladium(II)acetate (Sigma) were dissolved to 10 mM concentrations in DMSO followed by storage at −20°C until use. The frozen Pd compound solutions were thawed and diluted to appropriate levels with physiological saline solution. The Pd compound solutions (10 μl) were added to the lids of microtubes filled with bacterial suspensions (90 μl), with the exception that a 970-μl bacterial suspension was used for milk samples. This procedure was performed in a typical laboratory room, regardless of exposure to natural and/or electric visible light. Then, bacterial suspensions were incubated at 37°C for 30 min in a water bath.
For comparison of the reaction mechanisms of Pd and Pt compounds, which are mentioned below, tetrakis(triphenylphosphine)platinum(0) (Sigma) solution was prepared following the protocol of a previous paper (8).
To directly evaluate the degree of inhibition of real-time quantitative PCR (qPCR) by Pd compounds in vitro, the aforementioned Pd compounds, appropriately diluted, were added to a purified C. sakazakii DNA solution with SW (90 μl; 45 ng) at a 10-fold dilution. The exposure times and temperatures used in the subsequent experiments mimicked these conditions. Following the addition of 3 M sodium acetate solution (10 μl) and cooled ethanol (250 μl), purified DNA was dissolved in 20 μl of SW. All isolated DNA solutions were adjusted to the lowest concentration among them, and a 5-μl aliquot was used for qPCR, as described in a later section.
Preparation and exposure method for a PMA agent in current use.
As a control compound for LD discrimination, 20 mM PMA (Biotium Inc., Hayward, CA, USA) was diluted in SW in the dark to concentrations of 100, 1,000, 2,500, 5,000, and 10,000 μM. The diluted PMA solutions (10 μl) were added to E. coli or C. sakazakii suspensions (generally, 90 μl was used, with the exception that a 970-μl suspension was used for milk samples) under a safelight. The bacterial suspensions were uniformly mixed, and the tubes were kept at 5°C for 10 min (typical treatment conditions) under a safelight, followed by visible-light irradiation (5 min) (19, 20).
Bacterial inoculation into pasteurized milk.
As described above, the pellet obtained from a centrifuged live or heat-killed E. coli suspension was resuspended and serially diluted (under the detection limit for live total bacteria and Enterobacteriaceae cells by plating methods) in commercial pasteurized milk purchased from a local grocery store (here referred to simply as milk).
Milk pretreatment (12-ml milk scale) followed by direct-qPCR.
To obtain the lowest possible detection level of live targeted Enterobacteriaceae cells in milk, 12 ml of milk inoculated with live or dead E. coli cells with or without a 2.5-h enrichment (short-time enrichment) was centrifuged, followed by removal of the supernatant. Ten milliliters of phosphate-buffered saline (PBS) supplemented with 30 μl of savinase (protease from Bacillus sp.; Sigma) (≥16 U/g) was added to the obtained pellet to decompose the milk proteins, such as micellar casein. Then, the suspension was shaken using an RM-2M Intelli-Mixer (ELMI Ltd., Riga, Latvia) at 37°C for 10 min. The supernatant was removed by centrifugation. In total, 980 μl of SW was added, followed by the addition of 1,250 μM or 2,500 μM dichloro(ŋ-cycloocta-1,5-diene)palladium(II) [referred to here as Cl2(ŋ-cycloocta-1,5-diene)Pd] (20 μl).
For the control PMA treatment, 970 μl of SW was added to the aforementioned milk pellets, followed by the addition of a 5,000 or 10,000 μM PMA solution (30 μl). A total of 30 μl of SW was added to the sample (970 μl) without any added Pd compound or PMA solution. After exposure to the Pd compound or PMA solution, 10 ml of PBS was added to the samples, which were then centrifuged, and the supernatant was removed. The pellet was quantitatively transferred to a new PCR tube with 100 μl of SW, followed by centrifugation. After washing was performed once with 100 μl of SW, the pellet (corresponding to 5 μl of qPCR template) was added to 20 μl of direct-qPCR master mix (described later). Noninoculated milk or commercial anonymized milk samples (12 ml) were subjected to the same treatment procedure.
Differential DNA extraction procedures for dead E. coli cells following exposure to Pd or Pt compounds for the elucidation of reaction mechanisms.
First, to prepare heat-killed E. coli cells with cell wall transmembrane proteins (CW_TMP) or without the proteins, a live E. coli suspension (1.8 × 109 CFU/ml) was boiled for 3 min in SW or 1% Brij58 (Sigma) solution followed by immediate chilling. After centrifugation, supernatants were removed and pellets added with 1 ml of SW. Then, 10 μl of 50 mM Cl2(ŋ-cycloocta-1,5-diene)Pd solution, 125 μl of 4 mM tetrakis(triphenylphosphine)platinum(0), or no reagent was added to the aforementioned 1 ml of dead bacterial suspension with or without CW_TMP; the solutions were then left at 37°C for 1 h. The solutions were centrifuged, the supernatants were removed, pellets were washed with SW (1 ml), and DNA extraction methods (described later in the text) were performed on the pellets to elucidate the reaction mechanisms for Pd compounds compared with Pt compounds.
A lysis method involving glass beads followed by typical incubation with sodium lauryl sulfate (SDS)-supplemented protease and peptidase (SAV/PPR/SDS), phenol-chloroform/isoamyl alcohol (P/C/I) extraction, and ethanol precipitation was performed as follows. Glass beads (300 mg; diameter of 1 mm) were added to break bacterial cells and were subjected to vigorous vortex mixing for 2 min. Next, 500 μl of 10 mM Tris-HCl (pH 8.0) was added and the reaction mixture was gently shaken. Twenty microliters of savinase (≥16 U/g), 20 μl of 4% peptidase R (Amano Enzyme Inc., Nagoya, Japan), and 5 μl of 10% SDS solution were added to digest various proteins, including CW_TMP and DNA-binding proteins (DBP), and the samples were incubated at 50°C for 14 h in a Brock incubator. Thereafter, typical P/C/I extraction was performed followed by ethanol precipitation (21). Finally, a 5-μl aliquot was taken from 20 μl of the obtained DNA solution and was used for real-time PCR (qPCR). Regarding SDS treatment followed by P/C/I extraction and ethanol precipitation, the same procedure was performed except that savinase and peptidase treatments were excluded from the aforementioned DNA extraction procedure.
Independently, to obtain a highly purified DNA solution from C. sakazakii that was not modified with any Pd compounds, bacterial DNA from a 1-ml culture was purified using a commercial DNA purification kit (QuickGene SP kit DNA tissue SP-DT; Fujifilm Corp., Tokyo, Japan) to directly evaluate PCR inhibition by three types of Pd compounds in vitro.
Detailed evaluation of the reaction mechanism of a Pd compound compared with a typical fixation agent.
To elucidate the detailed reaction mechanism of a Pd compound, pellets obtained from 1 ml of E. coli boiled in SW (2.8 × 109 cells/ml) with CW_TMP were exposed to 1,000 μM Cl2(ŋ-cycloocta-1,5-diene)Pd at 37°C for 1 h or the commercial fixation agent Mildform 10NM (Wako, Osaka, Japan), whose main component is HCHO, together with methanol at 4°C overnight. After centrifugation followed by one wash performed with SW, 300 mg of glass beads was added and subjected to vigorous vortex mixing for 2 min. Next, 500 μl of 10 mM Tris-HCl (pH 8.0) was added, the reaction mixture was gently shaken, and centrifugation was performed to obtain the pellet and supernatant to elucidate the reaction mechanism of the Pd compound. Five hundred microliters of 10 mM Tris-HCl (pH 8.0) was added to the obtained pellet. Then, as mentioned above, the same three DNA extraction procedures were carried out on the pellet and supernatant obtained by centrifugation.
Direct-qPCR without the need for DNA extraction from bacteria (live and dead cells) and with or without Pd compounds for comparison with the currently used PMA method.
As described above, some bacteria (live and dead cells) treated with Pd compounds (or with none) followed by centrifugation underwent three types of DNA extraction, while other samples were directly subjected to qPCR without laborious DNA extraction (direct-qPCR) for comparison with the typical PMA treatment for routine use. Direct-qPCR master mix was used to facilitate PCR elongation following direct addition to bacterial cells (22–24). Specifically, the direct-qPCR master mix consisted of the following: 0.5 μl of Taq DNA polymerase (with standard Taq buffer [5 U/μl]; New England BioLabs, Japan, Inc., Tokyo, Japan); 2.5 μl of 10× standard Taq reaction buffer (New England BioLabs); 2.0 μl of a 2 mM deoxynucleoside triphosphate (dNTP) mixture (TaKaRa-Bio, Ohtsu, Japan); 0.75 μl of 10 μM forward primer (5′-GTTGTAAAGCACTTTCAGTGGTGAGGAAGG-3′) and 10 μM reverse primer (5′-GCCTCAAGGGCACAACCTCCAAG-3′); 5 μl of 2× SYBR green (Invitrogen, CA, USA) (10,000× stock); 2.5 μl of mixed reagent solution stored at −20°C before use (8.3% Brij 58 from Sigma; 1.9% bovine serum albumin from Sigma; 10 mM trisodium citrate dehydrate from Kanto-Kagaku, Tokyo, Japan; 30 mM MgCl2 from Nakalai-Tesque, Kyoto, Japan; 100 μg/ml lysozyme from egg white purchased from Wako Pure Chemicals Industries, Ltd., Osaka, Japan); 5 μl of PCR template (bacterial cells or isolated DNA); and 6.75 μl of SW. The forward and reverse primers, which target a specific region of the 16S rRNA gene in Enterobacteriaceae cells, produced an amplicon of 424 bp (25). The qPCR (including direct-qPCR) thermal cycle profile was 1 cycle of 95°C for 20 s; 50 cycles of 95°C for 5 s and 60°C for 45 s; and 1 cycle of 95°C for 3 min. Incidentally, the threshold cycle (CT) value in direct-qPCR presents the first PCR cycle at which the fluorescence value stemming from direct-qPCR amplification is above the threshold, and consequently, the CT value decreases in reverse proportion to the increasing number of bacterial cells in the qPCR tube. To measure the melting point of the obtained amplicon, the PCR tube was cooled to 65°C and the temperature was increased at a rate of 0.1°C/10 s to a final temperature of 95°C. The fluorescence-temperature curve was recorded.
RESULTS
Distinguishing between live and dead bacteria using Pd-direct-qPCR compared with the currently used PMA-direct-qPCR method.
The chemical structures of the Pd compounds used in this study are presented in Fig. 1A. Four types of Pd compounds were found to clearly distinguish live from dead bacteria (C. sakazakii and E. coli) in water by direct real-time PCR without DNA isolation (direct-qPCR) (Table 1), thus completely eliminating the need for laborious DNA purification in routine use.
FIG 1.

Listed Pd compounds and the qPCR elongation of purified C. sakazakii chromosomal DNA exposed to Pd compounds in vitro. (A) Chemical structures for Pd compounds. (B) Effects of Pd compounds on purified C. sakazakii DNA in vitro. The qPCR procedure (50 cycles) was performed in duplicate, and the CT values are presented as means ± standard deviations (SD) in the bar graph (n = 2). The light-gray (close to white), gray, and black bars terminating at a CT value of 50 on the y axis indicate that no qPCR amplification occurred above that level.
TABLE 1.
Pd-direct-qPCR for discriminating between live and dead bacteriaa
| Agent concn (μM) |
CT |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
C. sakazakii (1.0 × 107 CFU or cells/ml) |
E. coli (1.0 × 107CFU or cells/ml) |
|||||||||
| Dichloro(ŋ-cycloocta-1,5-diene) palladium(II) |
Bis(benzonitrile) dichloropalladium(II) |
Diamminedichloropalladium(II) |
Palladium(II) acetate |
Palladium(II) acetate |
||||||
| Live | Dead | Live | Dead | Live | Dead | Live | Dead | Live | Dead | |
| 0 | 18.3 ± 0.78 | 19.8 ± 0.42 | 18.4 ± 0.59 | 18.1 ± 0.69 | 19.3 ± 0.63 | 19.0 ± 0.84 | 18.9 ± 0.61 | 19.9 ± 0.86 | 18.1 ± 0.29 | 20.4 ± 0.33 |
| 1 | 20.4 ± 0.21 | ND × 2 | 19.4 ± 0.13 | ND × 2 | ||||||
| 10 | 20.5 ± 0.88 | ND × 2 | 19.7 ± 0.97 | ND × 2 | ||||||
| 25 | 18.6 ± 0.35 | ND × 2 | ||||||||
C. sakazakii and E. coli cells were suspended in sterile distilled water. Dead cells were prepared by boiling in sterile distilled water for 3 min. qPCR (50 cycles) was performed in duplicate, and the CT values are presented as means ± SD (n = 2). ND × 2, no-amplification results were obtained in duplicate.
At a concentration of each Pd compound that completely suppressed PCR of dead bacteria, the delta threshold cycle (ΔCT) value, i.e., the CT value of the Pd compound-treated live cells minus the CT value of the untreated live cells, was found to be 0.3 to 2.1. In contrast, for the typical PMA agent, the corresponding ΔCT values for live cells were 7.5 to 7.6 (Table 2). Thus, the four Pd compounds were superior to the currently used PMA agent for clear LD discrimination in water (Table 1 and Table 2).
TABLE 2.
PMA-direct-qPCR for discriminating between live and dead bacteriaa
| PMA concn (μM) |
CT |
|||
|---|---|---|---|---|
|
C. sakazakii (1.0 × 107CFU or cells/ml) |
E. coli (1.0 × 107 CFU or cells/ml) |
|||
| Live | Dead | Live | Dead | |
| 0 | 19.2 ± 0.24 | 18.9 ± 0.28 | 19.4 ± 0.41 | 19.0 ± 0.35 |
| 10 | 19.7 ± 0.31 | 24.9 ± 0.51 | 19.9 ± 0.38 | 25.5 ± 0.59 |
| 100 | 21.8 ± 0.54 | 39.7 ± 0.77 | 23.1 ± 0.72 | 38.4 ± 1.11 |
| 250 | 26.8 ± 0.79 | ND × 2 | 26.9 ± 0.78 | ND × 2 |
C. sakazakii and E. coli cells were suspended in sterile distilled water. Dead cells were prepared by boiling in sterile distilled water for 3 min. qPCR (50 cycles) was performed in duplicate, and the CT values are presented as means ± SD (n = 2). ND × 2, no-amplification results were obtained in duplicate.
Evaluation of the degree of inhibition of qPCR in vitro by Pd compounds.
The inhibition of qPCR with purified C. sakazakii DNA following exposure to three types of Pd compounds in vitro is presented in Fig. 1B. These 3 Pd compounds completely inhibited qPCR at a concentration of 25 μM, implying that Pd compounds could be chelated with bases of C. sakazakii DNA in vitro.
Specific detection of live E. coli in milk by Pd-direct-qPCR using a large (12-ml) milk volume.
Among the Pd compounds evaluated for use in a specific assay with live E. coli in milk, Cl2(ŋ-cycloocta-1,5-diene)Pd was most effective for this application. Figure 2A presents the results of LD discrimination of E. coli in a large-scale (12-ml) milk sample, performed in duplicate, which potentially could result in a much lower detection level than that determined using a 1-ml milk sample when Cl2(ŋ-cycloocta-1,5-diene)Pd treatments were followed by qPCR without DNA extraction (direct-qPCR). As a control, typical PMA solutions were also used in duplicate to discriminate between live and dead E. coli in 12 ml of milk (Fig. 2B). To accurately estimate the limits of detection and limits of quantification (LOD and LOQ) for live E. coli cells in milk following EN ISO 16140:2003 validation, a comparison of Pd-direct-qPCR with PMA-direct-qPCR using 12 ml of milk with artificially inoculated live E. coli was performed in 6 replicates of eight different milk samples, including real milk samples (Fig. 2C). Furthermore, Fig. 2D presents the estimation of LOD and LOQ for Pd-direct-qPCR and PMA-direct-qPCR using 12 ml of milk with artificially inoculated live E. coli (to estimate the standard deviations of the results from the use of unspiked milk for both methods) in 6 replicates at 4 different inoculations.
FIG 2.

Distinguishing between live and dead E. coli bacteria in pasteurized milk by qPCR following Pd compound treatment compared with the currently used PMA treatment. (A) The qPCR results for Pd compound treatment without DNA isolation (direct-qPCR): large-volume (12-ml) milk samples were exposed to 25 or 50 μM Cl2(ŋ-cycloocta-1,5-diene)Pd(II). (B) The qPCR results for PMA treatment without DNA isolation (direct-qPCR): large-volume (12-ml) milk samples were subjected to 150 or 300 μM PMA. (C) Pd-direct-qPCR and PMA-direct-qPCR using milk with artificially inoculated live E. coli and noninoculated 12-ml milk samples. Both methods were performed with 6 replicates for 8 different inoculations and for noninoculated milk (milk blank). (D) Linearity and variance for Pd-direct-qPCR and PMA-direct-qPCR using 12-ml milk samples with artificially inoculated live E. coli. Six CT value replicates for the currently used and alternative methods were plotted for four different levels of live E. coli (lower and higher levels).
First, CT values of 31.2 ± 0.63 were obtained for noninoculated, untreated milk (Fig. 2A). A 50 μM Cl2(ŋ-cycloocta-1,5-diene)Pd treatment, but not a 25 μM treatment, completely inhibited PCR amplification from both noninoculated milk and milk that was heavily spiked with dead E. coli (1.1 × 105 and 1.1 × 106 cells/ml); these milk samples were estimated to be potentially contaminated at the maximum level (Fig. 2A). With the application of Cl2(ŋ-cycloocta-1,5-diene)Pd treatment (50 μM) in direct-qPCR, the CT values of the live E. coli cells decreased in inverse proportion to the increased levels of live E. coli exogenously added to the milk. The linearity of the assay for the 50 μM Pd-treated live E. coli cells was obtained over a range of 1.1 × 103 to 1.1 × 106 CFU/ml (Fig. 2A).
For the currently used PMA treatment, the same positive results were obtained for noninoculated, untreated milk (Fig. 2B). A 300 μM PMA treatment, but not a 150 μM treatment, completely suppressed PCR amplification from noninoculated milk and milk heavily spiked with dead E. coli (1.1 × 105 and 1.1 × 106 cells/ml). For PMA treatment (300 μM) followed by direct-qPCR (PMA-direct-qPCR), the CT values of the newly spiked live E. coli cells decreased in inverse proportion to the cell number of live E. coli. The linearity of the assay for live E. coli was obtained over a range of 1.1 × 103 to 1.1 × 106 CFU/ml (Fig. 2B). Regarding the relative detection levels following EN ISO 16140:2003 validation in both Pd-direct-qPCR and PMA-direct-qPCR at a concentration of 2.54 log10 CFU/ml of live E. coli, the former gave 5 positive results for 6 replicates, but the latter gave 3 positive results for 6 replicates (Fig. 2C). The CT values of the Pd-treated or PMA-treated live E. coli cells decreased in inverse proportion to the cell number of spiked live E. coli (Fig. 2C). The relative detection level(s) with live E. coli in milk following EN ISO 16140:2003 validation was calculated in a range between 2.5 and 3.2 log10 CFU/ml (Pd-direct-qPCR and PMA-direct-qPCR) (Fig. 2C). In both Pd-direct-qPCR and PMA-direct-qPCR, analysis of variance (ANOVA) using a one-way layout (a factor of the live E. coli level) was performed on 6 replicates of data in a range between 3.15 and 6.15 log10 CFU/ml (lower and higher levels) to estimate the variance (square of standard deviation) of unspiked milk data (Fig. 2D). Then, the limits of detection and limits of quantification (LOD and LOQ) were calculated following EN ISO 16140:2003 validation and are also presented in Fig. 2D. As presented in Fig. 2D, a correlation coefficient (R2) value of 0.953 and a slope value of −3.445 for the regression line for Pd-direct-qPCR were obtained compared with R2 = 0.937 and a slope of −4.271 for the regression line for the currently used PMA-direct-qPCR. The LOD and LOQ of live E. coli in milk were estimated by Pd-direct-qPCR to be 0.76 and 2.3 log10 CFU/ml, respectively, and the associated detection and quantification limits for PMA-direct-qPCR were estimated to be 0.97 and 2.9 log10 CFU/ml, respectively; thus, Pd-direct-qPCR was superior to PMA-direct-qPCR regarding the LOD and LOQ (Fig. 2D).
Results of analysis of the correlation between Pd-direct-qPCR and the reference plating/PMA-direct-qPCR method.
Owing to the difficulty in obtaining domestic pasteurized milk contaminated with live Enterobacteriaceae cells, direct-qPCR following Cl2(ŋ-cycloocta-1,5-diene)Pd (50 μM) treatment (Pd-direct-qPCR), direct-qPCR without Pd or PMA compound treatments, direct-qPCR following PMA (300 μM) treatment (PMA-direct-qPCR), and the typical plating method (VRBA) were performed using 20 anonymized pasteurized milk samples (estimated dead bacterium levels of less than 3 log10 cells/ml, i.e., 2 log10 cells/ml, as shown in Fig. 2A and B) and a total of 80 milk samples artificially contaminated with live or dead E. coli cells without enrichment. The accuracy, sensitivity, and specificity of Pd-direct-qPCR or PMA-direct-qPCR relative to the reference plating method following EN ISO 16140:2003 validation were determined (Table 3). Similarly, the experimental procedures were performed using 80 corresponding milk samples subjected to 2.5-h enrichment to specifically increase the levels of low doses of spiked live E. coli cells (Table 3); the results of the comparisons of all tested methods are summarized in Table 3 and Table 4 , following EN ISO 16140:2003 validation. The results obtained with samples that underwent direct-qPCR without Pd or PMA compound treatments in noninoculated and inoculated milk samples (2.5-h enrichment) spiked with dead E. coli cells were positive, while the results obtained with Pd-direct-qPCR, PMA-direct-qPCR, and reference culture method (VRBA) samples were all negative (Fig. 3). In contrast, when the methods were applied to 36 live-E. coli-inoculated, 2.5-h-enrichment milk samples, the results obtained with both Pd- and PMA-direct-qPCR were positive (Fig. 3). The Pd-direct-qPCR method correlated well with the reference plating method (VRBA) in all 2.5-h-enrichment milk samples (Fig. 3). Additionally, the Pd-direct-qPCR results correlated well with the PMA-direct-qPCR results in all 2.5-h-enrichment milk samples (Fig. 3).
TABLE 3.
Accuracy, sensitivity, and specificity of milk data determined with Pd-direct-qPCR or PMA-direct-qPCR relative to the reference method of plating following EN ISO 16140:2003 validationa
| Method | Dairy matrix | PA | NA | ND | PD | Sum | % accuracyb | N+c | % sensitivityd | N−e | % specificityf |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pd-direct-qPCR | No-enrichment milk samples | 26 | 44 | 10 | 0 | 80 | 87.5 | 36 | 72.2 | 44 | 100.0 |
| 2.5-h-enrichment milk samples | 36 | 44 | 0 | 0 | 80 | 100.0 | 36 | 100.0 | 44 | 100.0 | |
| PMA-direct-qPCR | No-enrichment milk samples | 25 | 44 | 11 | 0 | 80 | 86.3 | 36 | 69.4 | 44 | 100.0 |
| 2.5-h-enrichment milk samples | 36 | 44 | 0 | 0 | 80 | 100.0 | 36 | 100.0 | 44 | 100.0 |
PA, number of samples showing positive agreement; PD, positive deviation; number of samples showing NA, negative agreement; ND, negative deviation; Sum, total numbers of samples.
% accuracy data were calculated as follows: 100 × (PA + NA)/Sum.
N+ data were calculated as follows: PA + ND.
% sensitivity data were calculated as follows: 100 × PA/N+.
N− data were calculated as follows: NA + PD.
% specificity data were calculated as follows: 100 × NA/N−.
TABLE 4.
Accuracy, sensitivity, and specificity of milk data determined with Pd-direct-qPCR relative to the currently used PMA-direct-qPCR method in DNA elongation following EN ISO 16140:2003 validationa
| Dairy matrix | PA | NA | ND | PD | Sum | % accuracyb | N+c | % sensitivityd | N−e | % specificityf |
|---|---|---|---|---|---|---|---|---|---|---|
| No-enrichment milk samples | 25 | 54 | 0 | 1 | 80 | 98.8 | 25 | 100.0 | 55 | 98.2 |
| 2.5-h-enrichment milk samples | 36 | 44 | 0 | 0 | 80 | 100.0 | 36 | 100.0 | 44 | 100.0 |
PA, number of samples showing positive agreement; PD, positive deviation; number of samples showing NA, negative agreement; ND, negative deviation; Sum, total numbers of samples.
% accuracy data were calculated as follows: 100 × (PA + NA)/Sum.
N+ data were calculated as follows: PA + ND.
% sensitivity data were calculated as follows: 100 × PA/N+.
N− data were calculated as follows: NA + PD.
% specificity data were calculated as follows: 100 × NA/N−.
FIG 3.

Analysis of correlation between Pd-direct-qPCR and the reference plating or currently used PMA-direct-qPCR methods using 12-ml milk samples with 2.5-h enrichment. Counts obtained by the plating method, VRBA plate count; Pd-direct-qPCR, 50 μM Cl2(ŋ-cycloocta-1,5-diene)Pd(II)-qPCR without DNA extraction; PMA-direct-qPCR, 300 μM PMA-qPCR without DNA extraction. Twenty commercially pasteurized (live bacterium-free and anonymized) milk samples and 60 milk samples with live or dead E. coli artificially added were used.
Effects of Pd compound treatment with or without cell wall transmembrane proteins of heat-killed E. coli.
Table 5 shows the influence of the Pd compound Cl2(ŋ-cycloocta-1,5-diene)Pd on heat-killed E. coli cells with or without CW_TMP compared with the Pt compound tetrakis(triphenylphosphine)platinum(0) and no exposure to Pd and Pt compounds. When DNA extraction using SAV/PPR/SDS plus P/C/I was performed on glass bead-lysed dead E. coli cells with CW_TMP following Pd treatment, the amount of DNA recovered in the upper water layer during P/C/I extraction (127.0 ± 6.36 ng/μl) drastically increased by 2 logarithmic orders compared with recovery from 2 other DNA extractions that lacked protease and peptidase digestion (E. coli cells were boiled in SW; Table 5). In contrast, as with the cells that were exposed to Pt compound or with no exposure to any agent, the amounts of DNA that transferred in the water layer following SAV/PPR/SDS plus P/C/I (48.4 ± 3.87 ng/μl for Pt; 153.1 ± 9.72 ng/μl for no treatment) increased by 1 logarithmic order (Pt) or only a little (no treatment [labeled “NT” in Table 5]) compared with the 2 other DNA extractions. In relation to the relevant qPCR elongation results for the Pd compound, the CT values determined with SAV/PPR/SDS plus P/C/I (29.3 ± 0.37) were significantly lower than those determined with 2 other DNA extractions that lacked protease and peptidase digestion (38.3 ± 0.55 and no elongation for E. coli boiled in SW) (Table 5). However, with respect to the Pt compound, newly added protease and peptidase digestion during the DNA extraction process had little influence on the CT value determined by qPCR, which was not similar to the experimental results determined with the Pd compound.
TABLE 5.
DNA recovery and qPCR with different DNA extractions from heat-killed E. coli with or without transmembrane proteins exposed to the Pd or Pt compounda
| DNA extraction using glass beads (pellet + supernatant) | Treatment for LD discrimination |
E. coli boiled in sterile water (1.8 × 109 cells/ml) |
E. coli boiled in 1% Brij58 (1.8 × 109 cells/ml)b |
||||
|---|---|---|---|---|---|---|---|
| OD260/OD280 | DNA concn (ng/μl) | CT by qPCR (cycle) (5 ng/qPCR) | OD260/OD280 | DNA concn (ng/μl) | CT by qPCR (cycle) (5 ng/qPCR) | ||
| SAV/PPR/SDS + P/C/I | Pd (1,000 μM) | 1.98 ± 0.03 | 127.0 ± 6.36 | 29.3 ± 0.37 | 1.98 ± 0.04 | 45.9 ± 4.76 | 39.5 ± 0.35 |
| Pt (1,000 μM) | 1.85 ± 0.04 | 48.4 ± 3.87 | 38.6 ± 0.54 | 2.00 ± 0.03 | 27.0 ± 2.21 | 40.1 ± 0.51 | |
| NT (no treatment) | 1.96 ± 0.03 | 153.1 ± 9.72 | 20.5 ± 0.43 | 2.01 ± 0.03 | 91.7 ± 3.67 | 22.9 ± 0.38 | |
| SDS + P/C/I | Pd (1,000 μM) | 1.77 ± 0.03 | 4.5 ± 0.42 | 38.3 ± 0.55 | 1.75 ± 0.01 | 2.8 ± 0.39 | ND |
| Pt (1,000 μM) | 1.70 ± 0.04 | 3.9 ± 0.43 | ND | 1.93 ± 0.03 | 3.2 ± 0.33 | ND | |
| NT (no treatment) | 1.92 ± 0.03 | 118.1 ± 3.85 | 21.3 ± 0.62 | 1.99 ± 0.01 | 60.3 ± 2.14 | 22.1 ± 0.58 | |
| P/C/I alone | Pd (1,000 μM) | 1.71 ± 0.03 | 2.9 ± 0.38 | ND | 1.67 ± 0.04 | 2.5 ± 0.34 | ND |
| Pt (1,000 μM) | 1.64 ± 0.06 | 2.3 ± 0.32 | ND | 1.61 ± 0.03 | 2.3 ± 0.33 | ND | |
| NT (no treatment) | 1.91 ± 0.03 | 77.1 ± 3.43 | 23.2 ± 0.73 | 1.92 ± 0.01 | 30.2 ± 0.38 | 23.7 ± 0.66 | |
Glass beads (pellet + supernatant): 10 mM Tris-HCl was added to all amounts of E. coli cells decomposed by glass beads, and their suspensions were supplied for DNA extraction. OD260, optical density at 260 nm. OD280, optical density at 280 nm. SAV/PPR/SDS: savinase, peptidase R, and SDS were added. P/C/I: DNA extraction by phenol, chloroform, and isoamyl alcohol. Pd: Cl2(ŋ-cycloocta-1,5-diene)Pd(II). Pt: tetrakis(triphenylphosphine)platinum(0). ND, no qPCR elongation of the target gene.
Boiling in 1% Brij58 to remove membrane-spanning protein for E. coli cells was performed.
In contrast, examining E. coli that lacked CW_TMP for the Pd compound (E. coli boiled in 1% Brij58; Table 5), the increase in the amount of DNA obtained by performing protease and peptidase digestion was limited to 1 logarithmic order compared with the amounts obtained from 2 other DNA extractions lacking savinase and peptidase R digestion. Regarding SAV/PPR/SDS plus PCI for the Pd compound, the influence of protease and peptidase on DNA recovery in the water layer following P/C/I extraction was greater for heat-killed E. coli cells with CW_TMP than for associated cells that lacked CW_TMP, which implied a reaction between the Pd compound and CW_TMP (Table 5).
DNA recovery from the pellet or supernatant obtained from heat-killed E. coli with cell wall transmembrane proteins exposed to the Pd or fixation agents.
Using the results from three different DNA extraction methods, Table 6 presents data representing DNA recovery in the water layer and qPCR elongation for the pellet or supernatant obtained via glass bead decomposition of heat-killed E. coli with CW_TMP that was subjected to exposure to the Pd compound or the fixation agent Mildform 10NM. Regarding the pellets, the DNA amounts (105.7 ± 4.41 and 132.3 ± 6.62 ng/μl) recovered by SAV/PPR/SDS plus P/C/I for fixation and the Pd compound, respectively, obviously increased by more than 1 logarithmic order compared with 2 other DNA extractions that lacked savinase and peptidase R digestion, as presented in Table 6. Additionally, pellet DNA recovery amounts (4.1 ± 0.42 to 12.0 ± 0.51 ng/μl) due to SDS plus P/C/I or P/C/I alone following fixation or Pd compound exposure were significantly lower by 1 or 2 logs than the recovery levels (108.5 ± 3.47 and 188.1 ± 11.13 ng/μl) seen following no exposure (no treatment [NT] data in Table 6). A series of experimental results implied that the Pd compound molecules could exert an influence on some proteins (CW_TMP) that were recovered in the pellet, similarly to the reaction mechanism of the fixation agent (HCHO), which involves covalent bonding. Regarding the CT value of qPCR elongation for the pellet, the addition of protease and peptidase during DNA extraction following exposure to the fixation agent or the Pd compound facilitated qPCR elongation as evident from the CT values of 31.1 ± 0.41 and 27.3 ± 0.38 in Table 6, compared with the CT values from two other DNA extractions that lacked savinase and peptidase R digestion.
TABLE 6.
DNA recovery and qPCR using pellet or supernatant obtained by glass bead decomposition of heat-killed E. coli with transmembrane proteins in the cell wall exposed to the Pd or fixation agenta
| Source | DNA extraction | Treatment for LD discriminationd |
E. coli boiled in distilled sterile water (2.8 × 109 cells/ml) |
||
|---|---|---|---|---|---|
| OD260/OD280 | DNA concn (ng/μl) | CT by qPCR (cycle) (5 ng/qPCR) | |||
| Pelletb | SAV/PPR/SDS + P/C/I | Fixation | 1.57 ± 0.06 | 105.7 ± 4.41 | 31.1 ± 0.41 |
| Pd (1,000 μM) | 1.64 ± 0.06 | 132.3 ± 6.62 | 27.3 ± 0.38 | ||
| NT (no treatment) | 1.88 ± 0.04 | 217.5 ± 8.95 | 20.1 ± 0.64 | ||
| SDS + P/C/I | Fixation | 1.61 ± 0.04 | 12.0 ± 0.51 | ND | |
| Pd (1,000 μM) | 1.62 ± 0.02 | 8.4 ± 0.42 | 38.1 ± 0.68 | ||
| NT (no treatment) | 1.63 ± 0.04 | 188.1 ± 11.13 | 20.3 ± 0.65 | ||
| P/C/I alone | Fixation | 1.55 ± 0.03 | 8.4 ± 0.54 | ND | |
| Pd (1,000 μM) | 1.52 ± 0.01 | 4.1 ± 0.42 | ND | ||
| NT (no treatment) | 1.61 ± 0.01 | 108.5 ± 3.47 | 21.8 ± 0.58 | ||
| Supernatantc | SAV/PPR/SDS + P/C/I | Fixation | 1.71 ± 0.09 | 10.5 ± 0.62 | 37.1 ± 0.48 |
| Pd (1,000 μM) | 1.74 ± 0.07 | 6.1 ± 0.55 | 35.5 ± 0.63 | ||
| NT (no treatment) | 1.66 ± 0.04 | 61.8 ± 4.35 | 25.7 ± 0.54 | ||
| SDS + P/C/I | Fixation | 1.53 ± 0.06 | 3.4 ± 0.41 | ND | |
| Pd (1,000 μM) | 1.68 ± 0.05 | 4.2 ± 0.56 | 40.8 ± 0.71 | ||
| NT (no treatment) | 1.60 ± 0.08 | 55.3 ± 3.34 | 25.9 ± 0.55 | ||
| P/C/I alone | Fixation | 1.48 ± 0.04 | 2.2 ± 0.36 | ND | |
| Pd (1,000 μM) | 1.62 ± 0.03 | 3.6 ± 0.34 | ND | ||
| NT (no treatment) | 1.56 ± 0.04 | 35.6 ± 2.84 | 24.6 ± 0.74 | ||
Heat-killed E. coli cells were decomposed with glass beads. SAV/PPR/SDS: savinase, peptidase R, and SDS were added. P/C/I: DNA extraction by phenol, chloroform, and isoamyl alcohol. Pd: Cl2(ŋ-cycloocta-1,5-diene)Pd(II). ND, no qPCR elongation of the target.
A 10 mM concentration of Tris-HCl was added to decomposed E. coli fragments followed by centrifugation to produce the pellet. In detail, the pellet obtained by the centrifugation was supplied for DNA extraction.
The supernatant obtained by the centrifugation of decomposed E. coli fragments was applied for DNA extraction.
Mildform 10NM was used to cross-link chromosomal DNA with transmembrane proteins in E. coli cells for fixation.
With regard to the supernatants, the addition of savinase and peptidase R during DNA extraction following exposure to the fixation agent or Pd compound promoted a small increase in DNA recovery in the water layer after P/C/I extraction compared to the recovery from 2 other DNA extractions that lacked protease and peptidase digestion, as presented in Table 6. Similarly, the addition of savinase and peptidase R during DNA extraction following exposure to the fixation agent or Pd compound led to a small decrease in the CT value compared with the CT values of 2 other DNA extractions (Table 6). Additionally, with respect to the supernatant following no exposure to either of the agents, as presented for NT (no treatment) in Table 6, DNA recovery and qPCR elongation were independent of the different DNA extraction procedures, and their values were consequently almost the same or were slightly differentiated.
DISCUSSION
As Pd compounds are not sensitive to visible light, unlike platinum compounds (Pt compound) (8), the Pd-direct-qPCR method (the alternative method) does not require any special laboratory equipment or a darkroom, which are required for the PMA method (5, 19). Pd compounds have the potential to allow significantly less laborious test procedures, lower costs, and possibly higher-throughput experiments than the PMA or EMA methods because the Pd-compound-based method does not require complicated analytical systems, as presented in the relevant sections of Materials and Methods. Furthermore, Pd compounds as well as Pt compounds are next-generation LD discrimination agents. Compared with Pt compounds that have been reported recently (8), as presented in Table 1 and Table 2, most Pd compounds allow the same clear LD discrimination as the Pt compounds but at much lower concentrations (8). Consequently, in view of testing costs, a large number of Pd compounds are superior to Pt compounds.
In terms of analytical factors, the four tested Pd compounds showed better LD discrimination of Enterobacteriaceae cells (C. sakazakii and E. coli) in water than the typically used PMA agent (Table 1 and Table 2). The small CT value increases for live cells treated with the four Pd compounds (Fig. 1A) indicate that significant penetration of Pd compounds into live cells does not occur, although PCR elongation in dead cells is completely suppressed (Table 1). In contrast, a significant increase in the CT value for PMA-treated live cells (significant penetration into live cells) is induced (Table 2). Thus, Pd-direct-qPCR could be substituted for PMA-direct-qPCR as a DNA elongation technology. However, it is crucial for Pd-direct-qPCR to be applicable for practical use across a broad range of testing.
Therefore, pasteurized milk was examined, and a test of Enterobacteriaceae bacteria was performed. Pasteurized milk contains granulocytes, many genera of dead bacteria, lipids, proteins, and sugars; in brief, pasteurized milk contains a number of PCR inhibitors (26). Milk proteins, enzymes, and nucleic acids inside or outside cow cells are also likely to disturb the function of Pd compounds (12–18).
Enterobacteriaceae bacterial testing is a worldwide concern in the food hygiene field (22). Enterobacteriaceae bacteria also serve as indicators of environmental pollution and can induce bacteremia (20). Thus, Enterobacteriaceae bacteria in milk have been targeted as a topic of broad concern.
To meet the requirements of U.S. and EU regulations regarding the detection of low levels of live coliform, we wanted to specifically detect and accurately assay live Enterobacteriaceae cells in milk at the lowest possible bacterial concentrations; simultaneously, however, Pd compounds should not allow the PCR amplification of potentially contaminating dead Enterobacteriaceae cells (5 log10 cells/ml in milk) (22). We have studied many Pd compounds, including those listed in Fig. 1A. Cl2(η-cycloocta-1,5,-diene)Pd provided the best LD discrimination for Enterobacteriaceae cells in milk, and the second best LD discrimination was obtained with bis(benzonitrile)dichloropalladium(II). According to U.S./EU regulations, live coliforms (Enterobacteriaceae-like bacteria) should not be present at a concentration of more than 5 to 10 CFU/ml, although the corresponding value in the Japanese Sanitary Act (JP) is 1 CFU/2.22 ml (22, 27). Thus, a large (12-ml) milk volume was sampled to perform testing that meets the requirements of not only the U.S./EU regulations but also the JP regulations regarding the detection of low levels of live coliforms (Fig. 2A). Because the relative detection level of Pd-treated live E. coli cells in milk is 2.5 to 3.2 log10 CFU/ml, a short period of enrichment for sampled milk is indispensable to minimally meet the requirements of current U.S./EU regulations (Fig. 2C). Thus, we compared Pd-direct-qPCR with PMA-direct-qPCR (the currently used method for DNA elongation) and with the current plating method (VRBA; the reference method) using 80 12-ml samples of commercially pasteurized (anonymized) milk and milk artificially contaminated with live or dead E. coli following 2.5 h of preenrichment (Fig. 3).
A strong correlation was observed between the results from Pd-direct-qPCR and the plating method (VRBA) with 2.5-h preenrichment of milk samples (Fig. 3). A complete or nearly complete correlation was observed between the Pd-direct-qPCR and PMA-direct-qPCR results with or without 2.5-h enrichment (Fig. 3 and Table 4). Pd-direct-qPCR is slightly superior to PMA-direct-qPCR with and without 2.5-h enrichment with respect to accuracy, sensitivity, and specificity relative to the reference plating method (Table 3).
Consequently, Pd-direct-qPCR could be substituted for PMA-direct-qPCR given its less laborious testing procedures, lower cost, possibly higher throughput, analytical parameters, and relative accuracy. Indeed, Pd-direct-qPCR allows a significant shortening of the enrichment time for typical plating (VRBA), but if lower LOD and LOQ for Pd-direct-qPCR than those reported in this study can be achieved, the short period of enrichment (2.5 h) used in this study will not be necessary, enabling Pd-direct-qPCR to ultimately be substituted for the plating (VRBA) method. The estimated total testing time to satisfy the U.S./EU regulations is less than 5 h (approximately 4.5 h), comprising a 2.5-h enrichment step, 1.25 h for a series of Pd-compound treatment procedures, and 0.75 h for direct-qPCR. This time requirement is shorter by approximately 13.5 to 19.5 h than the time required for the reference plating method.
Next, we elucidated the reaction mechanism for Pd compounds. First, as shown in Fig. 1B, Pd compounds could be chelated with purified bacterial chromosomal DNA despite the fact that bacterial cell wall transmembrane proteins (CW_TMP) and DNA-binding proteins (DBP) do not exist in vitro. This indicates that Pd compounds could be chelated with DNA, similarly to Pt compounds (8). However, in general, effective concentrations of Pd compounds for clear LD discrimination are lower than those of Pt compounds (8). These decreased concentrations of Pd compounds for LD discrimination imply that Pd compounds could also possess another reaction mechanism in addition to that of Pt compounds.
Metallothioneins contain cysteine at a rate of approximately 33% and exist in both mammalian and bacterial cells. These proteins help to mitigate exposure to heavy metals through chelation (28, 29). Additionally, palladium reportedly interacts with proteins, enzymes, and peptides through terminal amine groups, carboxylate groups, histidine imidazole groups, methionine and cysteine thiol groups, arginine, and lysine (13, 14, 30, 31–36). Furthermore, the Pd metal that chelated with the 2Cl groups in the original agent (complex form) is reported to be negatively charged.
Therefore, although we have already confirmed that Pd compounds can be chelated with DNA, we hypothesized two additional possible Pd compound-specific reaction schemes.
One potential scheme is based on adsorption or chelation to positively charged basic DBP (rich in arginine and lysine residues and containing histidine residues) in dead cells. Another potential scheme is based on the chelation of Pd compounds to CW_TMP, similarly to the results seen with the fixation agent Mildform 10MN (main component, HCHO), as indicated in Tables 5 and 6.
Negatively charged chromosomal DNA is strongly bound by positively charged basic DBP under neutral pH conditions via an electrostatic effect. Therefore, as additional information, the strong binding between DNA and basic proteins (DBP) is not easily destroyed through the physical decomposition of dead cells performed using glass beads (Tables 5 and 6).
Figure 4 also implies that the light-brown Pd compound Cl2(ŋ-cycloocta-1,5-diene)Pd, but not the yellow Pt compound, could react with CW_TMP of heat-killed E. coli, like the fixation agent Mildform 10NM. The main component of the fixation agent, formaldehyde (HCHO), cross-links to amino or imino groups of proteins as well as nucleic acids (bases); consequently, the generation of various fixed DNA-protein complexes is mediated by formaldehyde (37). Similarly, considering Fig. 4 and previous reports (17, 18, 30, 31–36), it is conceivable that the components to which the Pd compound molecules (negatively charged in neutral pH) could be chelated and/or adsorbed are CW_TMP, positively charged DBP in neutral pH, and DNA bases in E. coli cells. In contrast, as shown in Fig. 1B, Pd compounds could also be chelated with DNA bases (covalent bonding) in vitro. The potential complexes comprising CW_TMP (mainly conjugated with E. coli cell walls)-Pd-CW_TMP, CW_TMP-Pd-DNA, DBP-Pd-DBP, and DBP-Pd-DNA, which form when E. coli is boiled in SW, are listed, regardless of chelation and/or adsorption (in particular, to positively charged DBP for adsorption) (Table 5). In terms of the complexes containing DNA, if CW_TMP-Pd-DNA or DBP-Pd-DNA were produced by mediating the adsorption of Pd metal via electrostatic effects, CW_TMP and DBP should be detached from Pd metal during successive exposures to SDS plus P/C/I or P/C/I alone during the DNA extraction procedure (E. coli boiled in SW; Table 5). Above all, the majority of hydrophilic Pd-DNA (DNA that chelates Pd compounds) is most likely to be recovered in the water layer for its 2 aforementioned DNA extractions (E. coli boiled in SW; Table 5). However, in terms of SDS plus P/C/I or P/C/I alone following exposure to the Pd compound, the recovery rates are lower by more than 1 logarithmic order than those seen with no exposure to the Pd compound (Table 5). Therefore, we discarded the potential reaction mechanism involving the absorption of Pd compounds on CW_TMP and DBP. In contrast, if CW_TMP-Pd-DNA or DBP-Pd-DNA were formed via the chelation of Pd metals with CW_TMP or DBP (covalent bonding), CW_TMP-Pd-DNA or DBP-Pd-DNA complexes could be recovered in the medium layer during exposure to SDS plus P/C/I or P/C/I alone in the DNA extraction process, owing to the transfer of denatured CW_TMP and DBP to the medium layer. This might trigger the poor recovery of DNA conjugated with Pd compounds (CW_TMP-Pd-DNA or DBP-Pd-DNA) in the upper water layer. In fact, data presented in Table 5 (E. coli boiled in SW) support this hypothesis in view of DNA recovery. Furthermore, as for E. coli boiled in distilled water (DW) (Table 5), DNA extraction with SAV/PPR/SDS plus P/C/I following exposure to the Pd compound led to the same recovery rates as those seen with NT (no Pd compound exposure), as presented in Table 5. A large portion of the proteins in CW_TMP-Pd-DNA or DBP-Pd-DNA could be digested via protease and peptidase hydrolysis; consequently, protein-free Pd-DNA molecules could be recovered at a high recovery rate in the water layer during successive P/C/I processes (Table 5). Additionally, if a number of CW_TMP-Pd-CW_TMP or DBP-Pd-DBP were produced, many Pd compound molecules might not be effectively chelated with DNA bases in dead E. coli cells. This incomplete chelation is thought to trigger incomplete suppression with qPCR for dead E. coli cells that have been exposed to the Pd compound.
FIG 4.
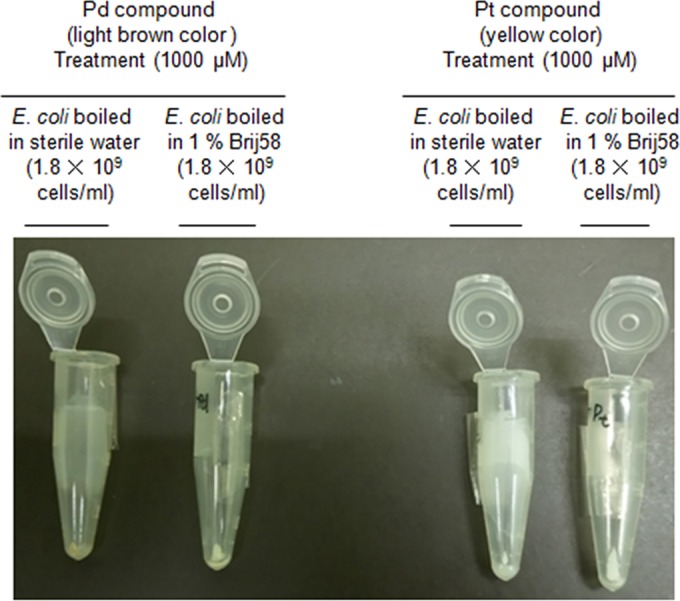
Different color images for pellets stemming from E. coli cells heat-killed in sterile water or 1% Briji 58 that were subjected to treatment with Pd or Pt compounds.
From the viewpoint of qPCR inhibition by the Pd compound for E. coli boiled in DW (Table 5), the CT values (29.3 ± 0.37) for the DNA extraction of SAV/PPR/SDS plus P/C/I were lower by approximately 9 to 14 cycles than those for DNA extractions involving P/C/I alone and SDS plus P/C/I. In other words, the DNA recovered in the upper water layer for SDS plus P/C/I or P/C/I alone could have been primarily comprised of Pd-DNA alone without any CW_TMP and DBP, thus indicating that the Pd compound molecules were more effectively chelated with DNA bases in E. coli cells than with CW_TMP-Pd-DNA and DBP-Pd-DNA. However, DNA transferred into the upper water layer following extraction involving SAV/PPR/SDS plus P/C/I DNA mainly consisted of DNA (specifically, Pd-DNA) that was cut from the aforementioned CW_TMP-Pd-DNA or DBP-Pd-DNA. Therefore, in terms of Pd-DNA derived from CW_TMP-Pd-DNA or DBP-Pd-DNA, it is conceivable that many Pd compound molecules were originally chelated with CW_TMP and DBP proteins as well; consequently, the rate of chelation of the Pd compound by its targeted gene in the DNA from E. coli cells might significantly decrease.
As E. coli boiled in 1% Brij58 largely lacks CW_TMP, the rate of Pd compound penetration into dead cells is thought to be obviously higher than that into E. coli cells boiled in SW because of circumvention of the disturbance by CW_TMP. Thus, PCR elongation for DNA extraction performed with SDS plus P/C/I and P/C/I alone could be completely suppressed, and the CT values (39.5 ± 0.35) for SAV/PPR/SDS plus P/C/I could be 10 CT units higher than the CT values for SAV/PPR/SDS plus P/C/I for E. coli boiled in DW (Table 5). In contrast, regardless of the boiling method (in other words, with or without CW_TMP), the CT values (including no amplification) for E. coli cells that were exposed to the Pt compound tetrakis(triphenylphosphine)platinum are almost the same in the three DNA extraction procedures. This experimental fact indicates that the presence or absence of CW_TMP might have little influence on the Pt compound (Table 5).
Figure 4 also indicates the significant difference between the Pd and Pt compounds concerning the reaction mechanism involving the walls of dead E. coli cells. This differentiation implies that CW_TMP could have a greater influence on the Pd compound than on the Pt compound.
Regarding CW_TMP and DBP, it is still unknown which proteins would have a greater influence on the Pd compound. To elucidate the detailed reaction mechanism of the Pd compound, dead E. coli cells boiled in DW (with CW_TMP) that were exposed to the fixation agent (HCHO, the main component in Mildform 10NM) or the Pd compound, including unexposed cells, were decomposed with glass beads followed by separation into the pellet or supernatant by successive centrifugations (Table 6). The CW_TMP-HCHO-DNA complex should be largely transferred to the pellet by the just-mentioned centrifugation step because CW_TMP can be physically inserted in solid form into E. coli cell walls. In contrast, the DBP-HCHO-DNA complex should be mainly recovered in the supernatant given that its main components comprise positively charged DBP and hydrophilic DNA, which are lacking cell walls of solid form. HCHO-DNA stemming from CW_TMP-HCHO-DNA is not recovered in the upper water layer by P/C/I DNA extraction until SAV/PPR/SDS treatment is complete. For the DBP-HCHO-DNA complex, the highest recovery rate stemmed from the DNA extraction of SAV/PPR/SDS plus P/C/I, and the recovered DNA concentration was 10.5 ± 0.62 ng/μl, which is lower by 10-fold than the relevant values (105.7 ± 4.41 ng/μl) determined for the pellet (CW_TMP-HCHO-DNA) (Table 6). Still, considering that the recovered DNA concentration (217.5 ± 8.95 ng/μl) determined for SAV/PPR/SDS plus P/C/I following NT (no treatment) of the pellet significantly differed from the value for the supernatant (61.8 ± 4.35 ng/μl) by 3.5-fold, the amount of DNA cut from CW_TMP-HCHO-DNA by protease together with peptidase is estimated to have been greater than the amount cut from DBP-HCHO-DNA by at least approximately 3-fold (Table 6). Similarly, the concentration of DNA (132.3 ± 6.62 ng/μl) cut from CW_TMP-Pd-DNA (see the data corresponding to SAV/PPR/SDS plus P/C/I and the pellet following Pd compound treatment in Table 6) by both enzymes was higher by approximately 22-fold than the associated value for the supernatant (6.1 ± 0.55 ng/μl) (DNA cut from the DBP-Pd-DNA complex by enzymes). Therefore, it is conceivable that the Pd compound could be chelated with more cell wall transmembrane proteins of dead E. coli than DNA-binding proteins. In particular, according to Table 6 data, the concentration of DNA cut in the pellet following exposure to the Pd compound greatly increased from 8.4 ± 0.42 to 132.3 ± 6.62 with the addition of protease and peptidase during DNA extraction. In contrast, regarding supernatants exposed to the Pd compound, the relevant DNA concentration increased slightly from 4.2 ± 0.56 to 6.1 ± 0.55 with the addition of both enzymes during DNA extraction. This result demonstrates that Pd compound molecules could have a greater influence on cell wall transmembrane proteins than DNA-binding proteins.
To summarize the Pd compound reaction mechanism, Pd compounds (soft Lewis acids) could be chelated with DNA bases (soft or medium Lewis bases) when there were only free DNA and Pd compounds in vitro (Fig. 1B). However, when chromosomal DNA, transmembrane proteins, and DNA-binding proteins were present in vivo, the Pd compound molecules that penetrated dead cells could be primarily chelated by DNA and transmembrane proteins and partially chelated with DNA-binding proteins.
In conclusion, in terms of the practical use of Pd compounds, Pd-direct-qPCR eliminates the associated laborious procedures and darkroom required for typical PMA-qPCR (5, 19). Pd-direct-qPCR also lowers costs compared with the recently reported Pt-direct-qPCR method in terms of reagent consumption (8) and potentially increases the throughput of experiments to levels higher than those achieved with PMA-qPCR (5, 19). In assessing the analytical parameters described in the EN ISO 16140:2003 validation, Pd-direct-qPCR could be superior to the currently used PMA-qPCR method for practical use in pasteurized milk and water samples. Concerning the practical use of Pd compounds in protein-rich pasteurized milk, centrifugation of pasteurized milk followed by the removal of supernatant and resuspension of milk pellets with sterile water is necessary. This process accounts for the greatest possible level of removal of Pd compound inhibitors, as presented in this study. Thus, our novel method represents a potential substitute for the currently used PMA-qPCR method. In summary, our method is very promising as a rapid test for food hygiene (in the milk category) and environmental (in the water category) Enterobacteriaceae tests. If the LOD and LOQ for Pd-direct-qPCR are reduced, this technology may ultimately replace the current culture method used worldwide in a matter of years.
Funding Statement
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
REFERENCES
- 1.Vaitilingom M, Gendre F, Brignon P. 1998. Direct detection of viable bacteria, molds, and yeasts by reverse transcriptase PCR in contaminated milk samples after heat treatment. Appl Environ Microbiol 64:1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheridan GEC, Masters CI, Shallcross JA, Mackey BM. 1998. Detection of mRNA by reverse transcription-PCR as an indicator of viability in Escherichia coli cells. Appl Environ Microbiol 64:1313–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cimino GD, Metchette KC, Tessman JW, Hearst JE, Issacs ST. 1991. Post-PCR sterilization: a method to control carryover contamination for the polymerase chain reaction. Nucleic Acids Res 19:99–107. doi: 10.1093/nar/19.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rudi K, Moen B, Drømtorp SM, Holck AL. 2005. Use of ethidium monoazide and PCR in combination for quantification of viable and dead cells in complex samples. Appl Environ Microbiol 71:1018–1024. doi: 10.1128/AEM.71.2.1018-1024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nocker A, Cheung C-Y, Camper AK. 2006. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J Microbiol Methods 67:310–320. doi: 10.1016/j.mimet.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 6.Soejima T, Iida K, Qin T, Taniai H, Seki M, Yoshida S. 2008. Method to detect only live bacteria during PCR amplification. J Clin Microbiol 46:2305–2313. doi: 10.1128/JCM.02171-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soejima T, Iida K, Qin T, Taniai H, Seki M, Yoshida S. 2007. Photoactivated ethidiummonoazide directly cleaves bacterial DNA and is applied to PCR for discrimination of live and dead bacteria. Microbiol Immunol 51:763–775. doi: 10.1111/j.1348-0421.2007.tb03966.x. [DOI] [PubMed] [Google Scholar]
- 8.Soejima T, Minami J-I, Xiao J-Z, Abe F. 2016. Innovative use of platinum compounds to selectively detect live microorganisms by polymerase chain reaction. Biotechnol Bioeng 113:301–310. doi: 10.1002/bit.25711. [DOI] [PubMed] [Google Scholar]
- 9.Anonymous. 2003. Microbiology of food and animal feeding stuffs. Protocol for the validation of alternative methods (ISO 16140: 2003). International Organization for Standardization, Geneva, Switzerland. [Google Scholar]
- 10.Jacobs W, Rijksinstituut voor Volksgezondheid en Milieu Ministerie van Volksgezondheid, Welzijn en Sport. 2010. MicroVal validation of a Campylobacter enumeration medium. EU-RL Campylobacter Workshop 2010. http://www.sva.se/globalassets/redesign2011/pdf/om_sva/nrl/crl/workshop-2010/2/microval-validation-of-a-campylobacter-enumeration-medium-method-comparison-study-and-interlaboratory-study-results.pdf.
- 11.Jacobs-Reitsma W, MicroVal Expert Laboratory (National Institute for Public Health and Environment Ministry of Health, Welfare and Sport). 15 May 2012. MicroVal validation of alternative methods. http://www.google.co.jp/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&ved=0CCMQFjAA&url=http%3A%2F%2Fwww.eurlsalmonella.eu%2Fdsresource%3Ftype%3Dpdf%26objectid%3Drivmp%3A181852%26versionid%3D%26subobjectname%3D&ei=8SGjVdvuO8i10ASw4IjYCg&usg=AFQjCNEiwgVWMgdTDb-KnGbSv2AY9kmTAQ&bvm=bv.97653015,d.dGo.
- 12.Rosenberg B, Van Camp L, Krigas T. 1965. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg B, VanCamp L, Trosko JE, Mansour VE. 1969. Platinum compounds: a new class of potent antitumour agents. Nature 222:385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 14.Gagnon ZE, Pate lA. 2007. Induction of metallothionein in chick embryos as a mechanism of tolerance to platinum group metal exposure. J Environ Sci Health A Tox Hazard Subst Environ Eng 42:381–387. doi: 10.1080/10934520601144691. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Q, Zhong W, Xing B, Tang W, Chen Y. 1998. Binding properties and stoichiometries of a palladium(II) complex to metallothioneins in vivo and in vitro. J Inorg Biochem 72:195–200. doi: 10.1016/S0162-0134(98)10080-6. [DOI] [PubMed] [Google Scholar]
- 16.Lovejoy KS, Todd RC, Zhang S, McCormick MS, D'Aquino JA, Reardon JT, Sancar A, Giacomini KM, Lippard SJ. 2008. cis-Diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor agent: uptake, structure, function, and prospects. Proc Natl Acad Sci U S A 105:8902–8907. doi: 10.1073/pnas.0803441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serrano FA, Matsuo AL, Monteforte PT, Bechara A, Smaili SS, Santa DP, Rodrigues T, Pereira FV, Silva LS, Machado J Jr, Santos EL, Pesquero JB, Martins RM, Travassos LR, Caires AC, Rodrigues EG. 2011. A cyclopalladated complex interacts with mitochondrial membrane thiol-groups and induces the apoptotic intrinsic pathway in murine and cisplatin-resistant human tumor cells. BMC Cancer 11:296. doi: 10.1186/1471-2407-11-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karami K, Hosseini-Kharat M, Sadeghi-Aliabadi H, Lipkowski J, Mirian M. 2014. In vitro cytotoxicity studies of palladacyclic complexes containing the symmetric diphoshine bridging ligand. Studies of their interactions with DNA and BSA. Eur J Med Chem 73:8–17. [DOI] [PubMed] [Google Scholar]
- 19.Pan Y, Breidt F Jr. 2007. Enumeration of viable Listeria monocytogenes cells by real-time PCR with propidium monoazide and ethidium monoazide in the presence of dead cells. Appl Environ Microbiol 73:8028–8031. doi: 10.1128/AEM.01198-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taskin B, Gozen AG, Duran M. 2011. Selective quantification of viable Escherichia coli bacteria in biosolids by quantitative PCR with propidium monoazide modification. Appl Environ Microbiol 77:4329–4335. doi: 10.1128/AEM.02895-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed, vol 3, p E3–E4. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 22.Soejima T, Minami J-I, Yaeshima T, Iwatsuki K. 2012. An advanced PCR method for the specific detection of viable total coliform bacteria in pasteurized milk. Appl Microbiol Biotechnol 95:485–497. doi: 10.1007/s00253-012-4086-0. [DOI] [PubMed] [Google Scholar]
- 23.Soejima T, Schlitt-Dittrich F. November 2011. Method and kit for detection of microorganism. Japanese patent JP4825313 (B2)-2011-11-30.
- 24.Soejima T, Xiao J-Z, Abe F. 2016. A novel mechanism for direct real-time polymerase chain reaction that does not require DNA isolation from prokaryotic cells. Sci Rep 6:28000. doi: 10.1038/srep28000 http://www.nature.com/articles/srep28000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakano S, Kobayashi T, Funabiki K, Matsumura A, Nagao Y, Yamada T. 2003. Development of a PCR assay for detection of Enterobacteriaceae in foods. J Food Prot 66:1798–1804. [DOI] [PubMed] [Google Scholar]
- 26.Bickley J, Short JK, McDowell DG, Parkes HC. 1996. Polymerase chain reaction (PCR) detection of Listeria monocytogenes in diluted milk and reversal of PCR inhibition caused by calcium ions. Lett Appl Microbiol 22:153–158. doi: 10.1111/j.1472-765X.1996.tb01131.x. [DOI] [PubMed] [Google Scholar]
- 27.Hillerton JE, Berry EA. 2004. Quality of the milk supply: European regulations versus practice, p 207–214. Proceedings of the National Mastitis Council 43rd Annual Meeting, 1–4 February 2004, Charlotte, North Carolina National Mastitis Council, New Prague, MN. [Google Scholar]
- 28.Chu G. 1994. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J Biol Chem 269:787–790. [PubMed] [Google Scholar]
- 29.Kawai K, Kamatani N, Georges E, Ling V. 1990. Identification of a membrane glycoprotein overexpressed in murine lymphoma sublines resistant to cis-diamminedichloroplatinum(II). J Biol Chem 265:13137–13142. [PubMed] [Google Scholar]
- 30.Pettijohn DE. 1988. Histone-like proteins and bacterial chromosome structure. J Biol Chem 263:12793–12796. [PubMed] [Google Scholar]
- 31.Caires AC. 2007. Recent advances involving palladium (II) complexes for the cancer therapy. Anticancer Agents Med Chem 7:484–491. doi: 10.2174/187152007781668661. [DOI] [PubMed] [Google Scholar]
- 32.Jószai V, Nagy Z, Osz K, Sanna D, Di Natale G, La Mendola D, Pappalardo G, Rizzarelli E, Sóvágó I. 2006. Transition metal complexes of terminary protected peptides containing histidyl residues. J Inorg Biochem 100:1399–1409. doi: 10.1016/j.jinorgbio.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Shultz MD, Lassig JP, Gooch MG, Evans BR, Woodward J. 1995. Palladium—a new inhibitor of cellulose activities. Biochem Biophys Res Commun 209:1046–1052. doi: 10.1006/bbrc.1995.1603. [DOI] [PubMed] [Google Scholar]
- 34.Milović NM, Dutca LM, Kostić NM. 2003. Combined use of platinum(II) complexes and palladium(II) complexes for selective cleavage of peptide and proteins. Inorg Chem 42:4036–4045. doi: 10.1021/ic026280w. [DOI] [PubMed] [Google Scholar]
- 35.Hoang HN, Bryant GK, Kelso MJ, Beyer RL, Appleton TG, Fairlie DP. 2008. Linkage isomerism in the binding of pentapeptide Ac-His(Ala)3His-NH2 to (ethylenediamine)palladium(II): effect of the binding mode on peptide conformation. Inorg Chem 47:9439–9449. doi: 10.1021/ic800970p. [DOI] [PubMed] [Google Scholar]
- 36.Craig JP, Garrett AG Jr, Williams HB. 1954. The ovalbumin-chloroauric acid reaction. J Am Chem Soc 76:1570–1575. doi: 10.1021/ja01635a028. [DOI] [Google Scholar]
- 37.Orlando V, Strutt H, Paro R. 1997. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods 11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]