

Abstract
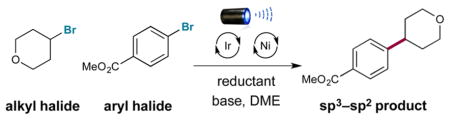
A strategy for cross-electrophile coupling has been developed via the merger of photoredox and transition metal catalysis. In this report, we demonstrate the use of commercially available tris(trimethylsilyl)silane with metallaphotoredox catalysis to efficiently couple alkyl bromides with aryl or heteroaryl bromides in excellent yields. We hypothesize that a photocatalytically generated silyl radical species can perform halogen-atom abstraction to activate alkyl halides as nucleophilic cross-coupling partners. This protocol allows the use of mild yet robust conditions to construct Csp3–Csp2 bonds generically via a unique cross-coupling pathway.
In recent years, the field of synthetic chemistry has witnessed the adoption of nickel as a complementary catalyst to palladium for traditional cross-coupling reactions (i.e., Suzuki–Miyaura, Kumada, Negishi, Stille, and Hiyama couplings).1 In particular, the capacity of nickel to undergo rapid oxidative insertion yet be resistant to β-hydride elimination pathways has been exploited in the development of a variety of Csp3–Csp2 bond-forming technologies.2 Traditionally, the Csp3 fragment employed in these protocols consists of a boronic acid derivative, Grignard reagent, or organozinc reagent. Recently, however, an increasingly popular approach has emerged that involves cross-electrophile coupling, a strategy wherein the union of two bench-stable electrophiles (e.g., a combination of an alkyl and aryl halide) can be accomplished using transition metal catalysis and an external reductant.3 The application of reductive coupling to C–C bond formation first became synthetically viable with the initial reports of Périchon, Gosmini, Jacobi von Wangelin, and Lipshutz.4 Thereafter, seminal papers from Weix, Gong, Molander, and Buchwald demonstrated that use of a metal reductant (i.e., Zn, Mn, or Mg) with either a nickel or palladium catalyst can indeed produce a variety of Csp3–Csp2 bonds from the corresponding halides.5
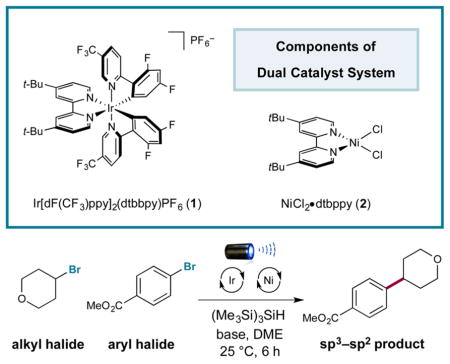
A better understanding of the innate properties of nickel catalysis has enabled the discovery of new fragment-coupling mechanisms.6 One notable example is the merger of photoredox catalysis with nickel catalysis, termed metallaphotoredox catalysis.7 Since its conception, this platform has enabled the development of a variety of new cross-coupling processes on the basis that photoredox catalysts can (i) readily modulate the oxidation states of nickel and (ii) generate a wide variety of reactive radical species under mild conditions that can suitably interface with nickel catalytic cycles.8 On this basis, we recently questioned whether a cross-electrophile coupling could be possible via the use of a metallaphotoredox mechanism and attendant activation of alkyl and aryl halides toward Csp3–Csp2 bond formation. As a critical design element, we focused on the use of silyl radical intermediates—which are classically generated from heat-induced radical initiators (i.e., AIBN, peroxides, etc.)—and their established capacity to abstract halogen atoms from alkyl Csp3–halide bonds at near-diffusion-controlled rates (Figure 1).9 This abstraction step is effectively irreversible given the difference in bond dissociation energies (BDEs) of the Si–Br bond (96 kcal/mol for Me3Si–Br) and the Csp3–Br bond (69 kcal/mol for bromoethane).10 Thus, we hypothesized that a large variety of commercially available alkyl bromides could be converted to open-shell coupling partners for Ni catalysis via the use of transient silyl radicals that are generated at room temperature using photoredox activation (eq 2). Herein we disclose the successful execution of these ideals and present a unique mechanism for cross-electrophile coupling to generate Csp3–Csp2 bonds via the use of silyl halide abstraction in concert with photoredox and nickel catalysis.
Figure 1.

Alkyl bromide–aryl bromide coupling via photoredox.
Design plan
A detailed mechanism for the proposed photoredox-mediated silyl abstraction Ni coupling is shown in Figure 2. It has been established that the commercially available heteroleptic photoredox catalyst [Ir[dF(CF3)ppy]2(dtbbpy)]-PF6 (1)11 readily absorbs photons for excitation to the strongly oxidizing complex *Ir[dF(CF3)ppy]2(dtbbpy)+ (3) ( vs saturated calomel electrode (SCE) in CH3CN).12 On this basis, we hypothesized that single-electron oxidation of bromide ( vs SCE in dimethoxyethane (DME)),13 a dissociable ligand on nickel,14 by photoexcited catalyst 3 to generate bromine radical (5) and the reduced photocomplex (6) should be thermodynamically feasible.
Figure 2.

Mechanistic cycle for silyl-mediated cross-coupling.
It is well-precedented that, once generated, electrophilic bromine radicals can rapidly abstract hydrogen atoms from Si–H bonds.9 Thus, we postulated that hydrogen-atom abstraction from commercially available tris(trimethysilyl)silane (TTMSS) to generate stabilized silyl radical intermediate 7 should occur. Subsequent halogen-atom abstraction from alkyl bromide 8 would then provide the corresponding nucleophilic radical species 9 and the silyl bromide byproduct. At the same time, Ni0 complex 10 can readily undergo oxidative addition into aryl bromide 11 to furnish intermediate 12. Facile oxidative capture of alkyl radical 9 by the nickel catalyst would then furnish the corresponding alkyl–NiIII species 13. Reductive elimination from the latter would afford the requisite Csp3–Csp2 bond and deliver the corresponding NiI catalyst 15. Lastly, single-electron transfer from the available IrII species 6 ( vs SCE in CH3CN) to complex 15 can reduce the metal catalyst back to the starting Ni0 catalyst while simultaneously regenerating the ground-state photocatalyst 1, effectively closing both catalytic cycles. As a key design element, silyl radicals selectively abstract from the weaker alkyl halide bond in the presence of the stronger aryl halide bond. This complements the ability of the nickel catalyst to insert into aryl halides over alkyl halides.15
Initial investigations revealed the feasibility of the bromide oxidation step (4 to 5) via Stern–Volmer experiments.16 With these results in hand, we examined the proposed silyl-mediated photoredox cross-coupling of 4-bromotetrahydropyran and methyl 4-bromobenzoate using 1 mol % photocatalyst 1, 1 equiv of TTMSS, 0.5 mol % Ni catalyst 2, and blue light-emitting diodes (Table 1). We were delighted to find that the desired transformation was possible in a series of solvents (40–67% yield; entries 1–3). Ultimately, the use of sodium carbonate or lithium hydroxide in DME provided superior yields (80% isolated yield; entries 4 and 5). Furthermore, control experiments demonstrated the reaction requires photocatalyst, visible light, nickel catalyst, silane, and base (0–3% yield; entries 6–10). Notably, entry 6 indicates that the silyl radical is likely not generated via a silane-aryl halide energy-transfer complex as previously proposed.17
Table 1.
Optimization of the Silyl-Mediated Couplinga
| |||
|---|---|---|---|
| entry | conditions | base | yieldb |
| 1 | DMA | K2CO3 | 40% |
| 2 | CH3CN | K2CO3 | 54% |
| 3 | as shown | K2CO3 | 67% |
| 4 | as shown | Na2CO3 | 84% (80%) |
| 5 | as shown | LiOH | 85% (80%) |
| 6 | no photocatalyst | Na2CO3 or LiOH | 0% |
| 7 | no light | Na2CO3 or LiOH | 0% |
| 8 | no Ni | Na2CO3 or LiOH | 0% |
| 9 | no silane | Na2CO3 or LiOH | 0% |
| 10 | no base | – | 3% |
Preliminary reactions were run with photocatalyst 1 (1 mol %), NiCl2·dtbbpy (0.5 mol %), aryl halide (1 equiv), alkyl halide (1.5 equiv), TTMSS (1 equiv), and base (2 equiv).
Yields were obtained by 1H NMR analyses of the crude reaction mixtures with mesitylene as an internal standard. Yields in parentheses are isolated yields.
Results
We are glad to report that the optimal catalyst combination developed in Table 1 can be employed for a variety of alkyl and aryl halide substrates (Table 2). Investigations of the aryl bromide scope demonstrated that electron-neutral and electron-rich arenes containing alkyl and methoxy functionality may be employed in excellent yields (77–83% yield; entries 1–6). Moreover, electron-deficient adducts containing nitrile, fluoride, and trifluoromethyl groups were also readily produced (73–78% yield; entries 7–9). Notably, the efficiency of the reaction was not impeded by ortho substituents on the aryl ring (63–94% yield; entries 10–12). Additionally, unprotected aniline substrates can be employed directly (65% yield; entry 10).
Table 2.
Photoredox-Enabled Csp3–Csp2 Coupling from Alkyl and Aryl Halide Precursors: Comprehensive Scope of Productsa

|
All yields are isolated yields using photocatalyst 1 (1 mol %), NiCl2·dtbbpy (0.5 mol %), aryl halide (0.5 mmol), alkyl halide (0.75 mmol), TTMSS
(0.5 mmol), and Na2CO3 (2 equiv).
12 h
LiOH as base.
Aryl chloride.
10 mol % 2.
5 mol % 2.
MOMCl.
MeOTs and LiBr.
Dioxane as solvent.
48 h.
24 h.
The synthesis of substituted heterocyclic arenes—common scaffolds in medicinally relevant targets—also proved successful. Precursors 2-,3-, and 4-bromopyridine are also suitable substrates (50–81% yield; entries 13–16). Multinitrogen containing heterocycles such as pyrazine, pyrimidine, and pyridazine were also tolerated, albeit in slightly diminished yields (50–64% yield, entries 17–19). Extended aromatic systems also readily undergo the photoredox-mediated silyl-abstraction coupling, including quinolone, isoquinoline, and pyrazole-substituted pyrazine (75–86% yield; entries 20–22). Lastly, we were delighted to find that five-membered heteroaryl bromides (N-methylimidazole and N-methylpyrazole), which are notoriously problematic for many cross-coupling technologies, performed well under these conditions (60% and 66% yield, respectively; entries 23 and 24).18
We next examined the generality of the photoredox silyl-abstraction coupling with respect to the alkyl halide fragment. In addition to the model substrate, seven- and five-membered cyclic systems were also tolerated, along with an acyclic secondary alkyl halide (66–80% yield; entries 25–28). Moreover, smaller strained ring systems such as cyclobutane, cyclopropane, azetidine, and oxetane bromides work in good measure (32–92% yield; entries 29–32). Primary alkyl bromide precursors were highly successful reaction partners (82% and 92% yield; entries 33 and 34), and notably, methoxymethyl chloride was viable to deliver an α-oxy adduct in 58% yield (entry 35). Sterically-encumbered neopentyl bromide was also shown to be a competent substrate (77% yield; entry 36). Interestingly, methylation of aryl bromides was successfully achieved with easy-to-handle reagents, methyl tosylate and LiBr (62% yield; entry 37), a result that we believe will have ramifications for isotopic labeling protocols.19 Lastly, tertiary alkyl bromides were found to readily couple with aryl halides in synthetically useful yields (52% and 62% yields; entries 38 and 39, respectively). Given the generality demonstrated in these studies, we expect this light-mediated cross-electrophile protocol to be useful for fragment couplings leading toward a large range of medicinal agents and complex targets.
Preliminary experiments were carried out to gain a deeper understanding of the role of TTMSS. As shown in Table 3, replacement of the silane with Hantzsch ester gave no desired product, and only formation of the biaryl product was observed (entry 1).20 In addition, other commonly used silanes for reduction of Ni intermediates to Ni0 resulted in no observable efficiency (entry 2).21 Moreover, the use of photocatalysts with diminished oxidizing capacity also failed to give the desired coupled adduct (entries 3–5). Together, these observations suggest that our coupling strategy does not operate via a direct reduction of either the photocatalyst or the nickel catalyst. While further studies are currently being carried out to confirm the presence of a silyl radical, we were encouraged to observe a correlation between the reaction efficiency and the Si–H BDE (entries 6 and 7).9 We believe that the dependence on the Si–H BDE is in line with a hydrogen-atom abstraction mechanism. Complete results of our mechanistic studies will be reported in due course.
Table 3.
Examination of Alternative Reductantsa
| |||
|---|---|---|---|
| entry | reductant | photocatalyst | yieldb |
| 1 | Hantzsch ethyl ester | 1 | 0%c |
| 2 | (MeO)3SiH or Et3SiH | 1 | 0% |
| 3 | TTMSS | Ir(ppy)3 | 0% |
| 4 | TTMSS | Ru(bpy)3(PF6)2 | 0% |
| 5 | TTMSS | Ru(phen)3Cl2 | 0% |
| 6 | Ph3SiH | 1 | 50% |
| 7 | Me2(TMS)SiH | 1 | 18% |
See Supporting Information for conditions.
Yields were obtained by 1H NMR analyses with mesitylene as an internal standard.
Gave an 80% yield of the biaryl product.
Supplementary Material
Acknowledgments
Support was provided by NIH NIGMS (R01 GM078201-05) and gifts from Merck and Abbvie.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b04818.
Experimental procedures and spectral data (PDF)
References
- 1.(a) de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2. Wiley-VCH; Weinheim, Germany: 2004. [Google Scholar]; (b) Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For some reviews of the use of nickel in cross-coupling methods, see: Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299. doi: 10.1038/nature13274.Netherton MR, Fu GC. Adv Synth Catal. 2004;346:1525.Rudolph A, Lautens M. Angew Chem, Int Ed. 2009;48:2656. doi: 10.1002/anie.200803611.
- 3.Knappke CEI, Grupe S, Gärtner D, Corpet M, Gosmini C, Jacobi von Wangelin A. Chem - Eur J. 2014;20:6828. doi: 10.1002/chem.201402302. [DOI] [PubMed] [Google Scholar]
- 4.(a) Czaplik WM, Mayer M, Jacobi von Wangelin A. Synlett. 2009;2009:2931. doi: 10.1002/anie.200804434. [DOI] [PubMed] [Google Scholar]; (b) Krasovskiy A, Duplais C, Lipshutz BH. J Am Chem Soc. 2009;131:15592. doi: 10.1021/ja906803t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Amatore M, Gosmini C. Chem - Eur J. 2010;16:5848. doi: 10.1002/chem.201000178. [DOI] [PubMed] [Google Scholar]; (d) Durandetti M, Gosmini C, Périchon J. Tetrahedron. 2007;63:1146. [Google Scholar]
- 5.(a) Everson DA, Jones BA, Weix DJ. J Am Chem Soc. 2012;134:6146. doi: 10.1021/ja301769r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang S, Qian Q, Gong H. Org Lett. 2012;14:3352. doi: 10.1021/ol3013342. [DOI] [PubMed] [Google Scholar]; (c) Molander GA, Traister KM, O’Neill BT. J Org Chem. 2014;79:5771. doi: 10.1021/jo500905m. [DOI] [PubMed] [Google Scholar]; (d) Bhonde VR, O’Neill BT, Buchwald SL. Angew Chem, Int Ed. 2016;55:1849. doi: 10.1002/anie.201509341. [DOI] [PubMed] [Google Scholar]
- 6.(a) Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DWC. Nature. 2015;524:330. doi: 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tellis JC, Primer DN, Molander GA. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu L, Lipshultz JM, MacMillan DWC. Angew Chem, Int Ed. 2015;54:7929. doi: 10.1002/anie.201501908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Noble A, McCarver SJ, MacMillan DWC. J Am Chem Soc. 2015;137:624. doi: 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC. J Am Chem Soc. 2015;137:4896. doi: 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Chatgilialoglu C. Acc Chem Res. 1992;25:188. [Google Scholar]; (b) Chatgilialoglu C, Lalevée J. Molecules. 2012;17:527. doi: 10.3390/molecules17010527. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chatgilialoglu C. Organosilanes in Radical Chemistry. Wiley; Chichester, U.K: 2004. [Google Scholar]; (d) Ballestri M, Chatgilialoglu C, Clark KB, Griller D, Giese B, Kopping B. J Org Chem. 1991;56:678. [Google Scholar]
- 10.For Si–Br BDE, see: Walsh R. Acc Chem Res. 1981;14:246.For C–Br BDE, see: Gordon AJ, Ford RA. The Chemist’s Companion: A Handbook of Practical Data, Techniques, and References. Wiley; New York: 1972.
- 11.dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine; dtbbpy = 4,4′-di-t-Bu-2,2′-bipyridine.
- 12.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712. [Google Scholar]
- 13.See Supporting Information for cyclic voltammogram. For other potential mechanisms to generate the halogen radical, see: Hwang SJ, Powers DC, Maher AG, Anderson BL, Hadt RG, Zheng S-L, Chen Y-S, Nocera DGJ. Am Chem Soc. 2015;137:6472. doi: 10.1021/jacs.5b03192.Galicia M, Gonzalez FJ. J Electrochem Soc. 2002;149:D46.See the Supporting Information for the cyclic voltammogram. For other potential mechanisms to generate the halogen radical, see: Hwang SJ, Powers DC, Maher AG, Anderson BL, Hadt RG, Zheng S-L, Chen Y-S, Nocera DG. J Am Chem Soc. 2015;137:6472. doi: 10.1021/jacs.5b03192.
- 14.For the lability of halides on nickel salts, see: Klein A, Kaiser A, Sarkar B, Wanner M, Fiedler J. Eur J Inorg Chem. 2007;2007:965.
- 15.Biswas S, Weix DJ. J Am Chem Soc. 2013;135:16192. doi: 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For Stern–Volmer data on the bromide, see the Supporting Information.
- 17.Piva da Silva G, Ali A, da Silva RC, Jiang H, Paixão MW. Chem Commun. 2015;51:15110. doi: 10.1039/c5cc06329a. [DOI] [PubMed] [Google Scholar]
- 18.Su M, Buchwald SL. Angew Chem, Int Ed. 2012;51:4710. doi: 10.1002/anie.201201244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller PW, Long NJ, Vilar R, Gee AD. Angew Chem, Int Ed. 2008;47:8998. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 20.Elimination of the silicon component, while maintaining the reducing ability of the system, demonstrates the noninnocent role of the silane.
- 21.Jackson EP, Montgomery J. J Am Chem Soc. 2015;137:958. doi: 10.1021/ja511778a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.