

Abstract
Currently, over 10% of the US population is taking antidepressants. Numerous antidepressants such as amitriptyline are known to inhibit acid sphingomyelinase (Asm), an enzyme that is known to mediate leukocyte function and homeostasis. Severe burn injury can lead to an immunosuppressive state that is characterized by decreased leukocyte function and numbers as well as increased susceptibility to infection. Based upon the intersection of these facts, we hypothesized that amitriptyline-treated, scald-injured mice would have an altered immune response to injury as compared with untreated scald mice. Prior to burn, mice were pretreated with amitriptyline. Drug- or saline-treated mice were subjected full thickness dorsal scald- or sham-injury. Immune cells from spleen, thymus, and bone marrow were subsequently harvested and characterized. We first observed that amitriptyline prior to burn injury increased body mass loss and spleen contraction. Both amitriptylinetreatment and burn injury resulted in a 40% decrease of leukocyte Asm activity. Following scald injury, we demonstrate increased reduction of lymphocyte precursors in the bone marrow and thymus, as well as mature leukocytes in the spleen in mice that were treated with amitriptyline. We also demonstrate that amitriptyline treatment prior to injury reduced neutrophil accumulation following peptidoglycan stimulus in scald-injured mice. These data show that Asm alterations can play a significant role in mediating alterations to the immune system after injury. The data further suggest that those taking antidepressants may be at a higher risk for complications following burn injury.
Keywords: Acid sphingomyelinase, burn injury, neutrophil, T cell
INTRODUCTION
Trauma is the leading cause of death in the United States for individuals between the ages of one and 46 (1). Burn injury is the fourth most common type of trauma worldwide. In the United States, burn injury results in more than 486,000 hospital admissions and emergency department visits annually. Severe burn injury results in a catabolic state, resulting in weight loss and worse clinical outcome (2, 3) as well as a suppressed immune system. A significant cause of the morbidity and mortality associated with burn injury is the development of subsequent infections and sepsis.
One key aspect of immune suppression in critically ill burn patients is the persistent reduced number of T lymphocytes. In healthy subjects, T-cell development begins in the bone marrow with the production of common lymphoid progenitor (CLP) cells. These cells migrate from the bone marrow to the thymus. Following differentiation in the thymus, mature lymphoid cells migrate to peripheral lymphoid tissues, such as the spleen. The spleen is a critical organ of the immune system as it has a large number and mixture of leukocytes. Previously, we demonstrated that scald injury can reduce spleen mass as well as splenic T-cell numbers (4). In addition, it has been reported that prevention of T-cell apoptosis during sepsis decreases bacterial load and mortality in sepsis (5, 6). Finally, it has been shown that although the adaptive immune system is not essential for survival after burn injury, the adaptive immune arm is required for normal defense against bacterial infections (7). In addition to the immune adaptive arm, neutrophils also play a critical role in combating infection. Neutrophils and their precursors develop in the bone marrow and are recruited to the site of infection by interleukin-17 (IL-17) and CXCL1 (8). Integrin molecules are necessary for the proper trafficking and retention of neutrophils at the site of infection.
Autophagy is a conserved catabolic process that allows cells under stress to break down and recycle cellular components and damaged organelles. It is a crucial pathway that allows leukocytes to avoid apoptosis, maintain cell homeostasis, and play an immunologic role in sepsis and burn injury (9). However, excessive up-regulation of autophagy results in damage of essential proteins and organelles that can lead to cell death. During autophagy, cellular components are stored in a double membrane phospholipid bilayer called an autophagosome, which combines with lysosomes to form an autolysosome. Sequestosome 1, otherwise known as and will be referred to henceforth as p62, is an autophagy-related protein that serves as a scaffold for autophagosomes during the maturation process. Microtubule-associated proteins 1A/1B light chain 3B (LC3) is a protein that is also involved in autophagosome formation. Accumulation of p62 in neutrophils serves as a marker of inefficient autophagic flux, and as a result, these cells transition to apoptosis instead of playing a vital role in the inflammatory response to burn injury and sepsis (10–12).
Amitriptyline is a tricyclic antidepressant that was initially introduced by Merck in 1961 for the treatment of major depressive disorder and additionally used for migraine prophylaxis, neuropathic pain disorders, fibromyalgia, nocturnal enuresis, and irritable bowel syndrome (13). A recent study reported that 22% of trauma patients older than the age of 45 were taking one or more antidepressants at the time of injury (14). Additionally, the use of psychotropic medications, including antidepressants, has increased in adolescents (15). Amitripyline as well as most of the antidepressants currently on the market functionally inhibits acid sphingomyelinase (Asm), a lipid signaling enzyme, by competitively displacing the enzyme from lysosomal membranes resulting in degradation of the released enzyme. Due to this mechanism of action, antidepressants can induce a 40% to 70% inhibition of Asm (16). Immunologically, Asm is a potential mediator of leukocyte apoptosis (17–19) and neutrophil chemotaxis (20). Altogether, changes in Asm activity and its potential impact upon the immune status have not been characterized after trauma or burn injury. Altogether, we hypothesize that ongoing amitriptyline treatment prior to burn injury reduces Asm activity resulting in changes to the immune system.
MATERIALS AND METHODS
Ethical statement
All murine experiments were performed using protocols approved by the Institutional Animal Care and Use Committee of the University of Cincinnati (protocol number: 08-09-19-01).
Scald burn injury
Male CF-1 mice (Charles River Laboratories, Wilmington, MA) weighing 32 g to 36 g (5–7 weeks of age) were used for scald and sham experiments. After purchase, mice were allowed to acclimate for 7 to 10 days prior to experimentation. Mice were housed in standard environmental conditions in groups of four with corn-cob bedding and fed with a standard pellet diet and water ad libitum. Mice underwent full-thickness scald injury as previously described (21). Briefly, the sham- and scald procedures were always conducted between 8 and 11 am. Mice were weighed and then anesthetized with 4.5% isoflurane in oxygen. Hair was clipped from their dorsal surface and mice were then placed in a template that exposed 28% of their total body surface area. The mice were immersed in 90°C water for 9 s resulting in a well-demarcated full-thickness scald injury. Following injury, mice received 1.5 mL of 0.9% saline, intraperitoneally. The mice were then placed on a 42.0°C heating pad and allowed to recover for 3 h. Sham-treated mice received the same treatment, except they were immersed in room temperature water. We did not use analgesia after inducing a full-thickness scald burn injury in these mice, consistent with our previous work (4, 21). Our justification comes from extensive literature demonstrating changes in immune function to include altered neutrophil chemotaxis, oxidative burst, phagocytic function, leukocyte homeostasis, and outcome in sepsis following opioid treatment in mice (22).
Amitriptyline treatment
For pretreatment with amitriptyline before scald, mice were injected intraperitoneally with 10 mg/kg amitriptyline (Sigma-Aldrich, St. Louis, MO) twice daily for 2 days as previously described (23). The last dose was given 1 h before injury. Mice were weighed 24 h following scald or sham treatment. Elavil (amitriptyline HCI) dosage for outpatients ranges from 75 mg to 150 mg a day while hospitalized patients may require 100 mg to 300 mg a day.
Flow cytometry for surface and intracellular staining
Analyses of cell surface antigen expression were performed as previously described (24) on bone marrow, thymus, spleen, and peritoneal lavage samples. Flow cytometry data acquisition and analysis were performed on an Attune Acoustic Focusing Cytometer using Attune Cytometric Software v2.1. The following antibodies were used: CD4 (Clone: RM4-5, BD Biosciences, San Jose, CA), CD8 (Clone: 53–6.7, BD Biosciences), CD69 (Clone: H1.2F3, BD Biosciences), CD44 (Clone: IM7, BD Biosciences), Ly-6G (Clone: 1A8, BD Biosciences), CD11b (Clone: M1/70, BD Biosciences), CD29 (Clone HM β1-1, BD Biosciences), CD18 (Clone: C71/16, BD Biosciences), CD61 (Clone: 2C9.G2, BD Biosciences), CD117 (Clone: 2B8, BD Biosciences), Ly-6A/E (CloneD7, BD Biosciences), and FITC Lineage cocktail: CD3/Gr-1/Cd11b/CD45R(B220)/Ter-119 (BioLegend, San Diego, CA). Common Lymphoid Progenitor cells were identified as lineage negative, CD117+, Ly6A/E +. T-cell subsets were defined as follows: CD4+ naive (CD62LHi/CD44Lo), CD4+ central memory (CD62LHi/CD44Hi), CD4+ effector memory (CD62LLo/CD44Hi), CD8+ naive (CD62LHi/ CD44Lo), CD8+ central memory (CD62LHi/CD44Hi), CD8+ effector memory (CD62LLo/ CD44Hi).
Asm activity
Four hours following scald injury, the spleens were removed, tissue was homogenized, and samples frozen. Asm activity was assessed as previously described (13). Briefly, the samples were diluted to 250 mM sodium acetate (pH 5.0) 0.1% NP40 and incubated with 50 nCi per sample C14-sphingomyelin at 37°C for 30 min. The sample was dried and resuspended in 250 mM sodium acetate and 0.1% NP40 and sonicated for 10 min to obtain micelles. The addition of 800 μL of chloroform/methanol (2:1) was added to terminate the enzyme reaction. Phases were separated by centrifugation. Liquid scintillation counting was used to measure radioactivity of the aqueous phase to determine the release of C14-phosphorylcholine from C14-sphingomyelin as a measure of Asm activity.
Ceramide measurements
Spleens were harvested 4 h following scald injury. Spleens were homogenized and splenocytes frozen for ceramide analysis as described previously (25). Briefly, ceramide concentrations were measured in the spleen using the diacylglycerol kinase method. Lipids were separated on Thin Layer Chromatography plates and then analyzed by autoradiography. Ceramide amounts were determined by comparison with a standard curve using C16-ceramide as substrate.
Neutrophil recruitment
Twenty-four hours following the scald injury, mice received intraperitoneal injection of 250 μg of peptidoglycan Staphylococcus aureus cell wall component (Sigma-Aldrich, St. Louis, MO). Four hours later, mice were sacrificed and peritoneal lavage fluid was collected. The cells were enumerated by an automated cell counter. Flow cytometry was performed on peritoneal lavage fluid as described above using anti-Ly6G and anti-CD11b to indentify the neutrophils.
Cytokine and chemokine measurement by ELISA
Twenty-four hours following scald injury, cell-free peritoneal lavages were obtained as described above. CXCL1/KC (R&D, Minneapolis, MN) and IL-17 (eBioscience, San Diego, CA) levels were analyzed by ELISA according to manufacturers' protocol.
Autophagy measurement by FACS
Bone marrow from WT and Asm heterozygous mice were harvested between 8 and 11 am. Bone marrow neutrophil isolation was performed using a Histopaque gradient of 1077/1119 (Sigma Life Science, St. Louis, MO), and samples were incubated for 48 h. LysoTracker Green (Life Technologies, Eugene, OR) percentage and MFI were analyzed by FACS according to manufacturer's protocol.
p62, LC3, and Bcl-2 measurement by western blot
Isolated bone marrow neutrophils (5 × 106 cells) were incubated for 48 h, pelleted and resuspended in lysis buffer containing sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer with 5% beta-mercaptoethanol, as previously described (6). Samples were sonicated and heated at 60°C for 20 min. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was carried out using 12% NuPAGE Novex Bis-Tris gels according to the manufacturer's protocols (Invitrogen, Carlsbad, Calif). Separated proteins were transferred to PVDF membranes and blocked with 5% nonfat milk in phosphate-buffered saline. The membranes were incubated with 1:1,000 of anti-p62, anti-LC3, or anti-Bcl-2 rabbit primary antibody at 4°C overnight. After washing, the membranes were incubated for 2 h at room temperature with 1:200 anti-FITC rabbit secondary antibody for p62 and LC3, and 1:200 anti-FITC mouse secondary antibody for Bcl-2. After washing, the membranes were incubated for 1 h at room temperature with 1:5,000 anti-FITC antibody. After washing, the membrane was developed with ECF substrate (Thermo Scientific, Rockford, IL) and visualized on an Alpha Innotech FluorChem 8,900 imager (San Leandro, CA).
Statistical analyses
Statistical comparisons were performed using Student t test (two groups), or one way ANOVA with Tukey post-hoc analysis (more than two groups) using GraphPad Prism 6.0 (Graphpad Software, La Jolla, CA). The mean and standard error of the mean were calculated in experiments containing multiple data points. A value of P ≤0.05 was considered statistically significant.
RESULTS
Severity of scald injury following amitriptyline treatment
At the time of injury, approximately 20% of trauma patients are frequently taking one or more antidepressants. Many antidepressants, including amitriptyline, inhibit the Asm-mediated conversion of sphingomyelin into ceramide (26–28). We first ascertained whether amitriptyline, by itself, was sufficient to alter physiological or immunological parameters. We found that this was not the case (data not shown). However, the potential impact of amitriptyline treatment on the host response to burn injury is unknown. Upon testing, we observed that on post-burn day 1 (PBD1), amitriptyline pretreated scald mice had a two-fold increase in weight loss compared with untreated scald mice (Fig. 1A). Spleen mass after burn injury was reduced approximately 30% as compared with sham-injured mice. Spleens from mice with ongoing amitriptyline treatment decreased an additional 15% compared with injured mice on PBD1 (Fig. 1B). Finally, splenic white blood cell counts in amitriptyline-treated mice were decreased an additional 30% as compared with untreated burn-injured controls (Fig. 1C). Of note, sham-injured mice pretreated with amitriptyline did not demonstrate significant differences in either body or spleen masses (data not shown). Altogether, amitriptyline pretreated mice sustained significantly more weight loss and splenic contraction 1 day after burn injury.
Fig. 1. Amitriptyline treatment results in greater weight loss, smaller spleen mass, and fewer spleen WBCs following scald injury.

Mice were weighed and then subjected to scald injury. Mice were pretreated with 10 mg/kg amitriptyline or saline vehicle. Twenty-four hours after burn injury, (A) weight change, (B) wet spleen mass, and (C) spleen WBC were recorded. Spleen mass was normalized to sham levels and percent of sham level is depicted. The sample size was 16 to 17 per group. Data are expressed as the mean ± SEM. Statistical comparisons were made using Student t test. *, P <0.05 as compared with scald group. Dotted line indicates sham levels. AMIT indicates amitriptyline; WBCs, white blood cells.
Acid sphingomyelinase and ceramide in scald injury
We have previously observed a significant increase in splenic T-cell caspase activity as early as 6 h after scald injury (4). We therefore chose to determine Asm activity and ceramide levels prior to that 6-h period. In sham, amitriptyline pretreated mice, we observed Asm activity is reduced by 35.2% in amitriptyline treated mice compared with sham, untreated mice. Four hours following scald injury, there is a 32.8% reduction in Asm activity compared with sham mice. When scald injured mice were pretreated with amitriptyline, there was no further reduction in Asm activity compared with untreated scald mice (Fig. 2A). Similarly, there is a 35.3% reduction in splenocyte ceramide levels in amitriptyline treated mice compared with sham. Scald injury alone also resulted in a 35.3% reduction in ceramide levels. Again, when scald injured mice are pretreated with amitriptyline, there is no significant difference in ceramide levels compared with untreated scald mice (Fig. 2B). Amitriptyline treatment and scald injury both decrease Asm activity and ceramide levels prior to 6 h, albeit there is not an additive effect.
Fig. 2. Acid sphingomyelinase activity and ceramide levels decreased by prior amitriptyline treatment and scald injury.

Mice were pretreated with amitriptyline or saline vehicle and underwent sham or scald injury as described in the Materials and Methods. (A) Acid sphingomyelinase activity and (B) ceramide levels were recorded 6 h after injury. Sample size was six to eight mice per group. Data are expressed as the mean ± SEM. Statistical comparisons were made using ANOVA with Tukey post-hoc comparison. *, P <0.05 as compared with sham. AMIT indicates amitriptyline; Asm, acid sphingomyelinase.
Amitriptyline exacerbates burn-induced lymphocyte loss in multiple compartments
Due to increased splenic mass loss observed with amitripty-line-treated scald-injured mice, we anticipated that amitripty-line treatment would magnify leukocyte depletion following injury. To evaluate the effect of amitriptyline on the immune response in scald injury, we enumerated lymphoid cell populations in various compartments 24 h following burn injury. We first observed a 49.4% decrease in CLP cell populations in amitriptyline treated scald mice compared with untreated scald mice, 24 h following injury. Scald injury alone did not reduce this population when compared with sham levels (Fig. 3). Of note, sham-injured mice pretreated with amitriptyline did not demonstrate significant differences in bone marrow CLP cells compared with untreated mice (data not shown). We next examined thymus CD4+ and CD8+ single positive T cells, and observed that amitriptyline treated scald mice had a 25% and 23.5% reduction in the respective lymphocyte populations compared with scald injured mice. Scald with and without amitriptyline treatment were both reduced compared with sham mice (Figs. 4A and 5A, respectively). We next examined splenic CD4+ and CD8+ T cells, specifically examining the naive (CD62LHi/ CD44Lo), central memory (CD62LHi/ CD44Hi), and effector memory (CD62LLo/ CD44H) T lymphocyte subsets. Again, scald with and without amitriptyline treatment were reduced compared with sham mice. Compared with untreated scald-injured mice, amitriptyline treated scald mice had a significant reduction in each of the CD4+ lymphocyte subsets (Fig. 4, B–D) as well as the CD8+ splenic lymphocyte subsets (Fig. 5, B–D). Of note, sham-injured mice pretreated with amitriptyline did not demonstrate significant differences in thymic or splenic lymphocytes compared with untreated mice (data not shown). Altogether, amitriptyline treatment resulted in the increased loss of both immature and mature T cells following burn injury.
Fig. 3. Common lymphoid progenitor cells in bone marrow are reduced in amitriptyline treated mice after scald injury.
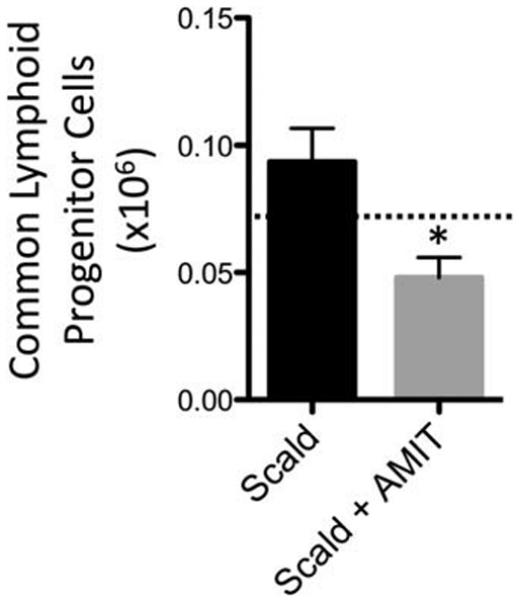
Mice were pretreated with amitriptyline or saline vehicle and underwent sham or scald injury as described in the Materials and Methods. Common lymphoid progenitor cells from bone marrow were enumerated and characterized 24 h after injury. Sample size was 7 to 12 mice per group. Data are expressed as the mean ± SEM. Statistical comparisons were made using Student t test. *, P <0.05 as compared with scald group. Dotted line indicates sham levels. AMIT indicates amitriptyline.
Fig. 4. Prior amitriptyline treatment exacerbates scald-induced CD4+ T-cell depletion in thymus and spleen.

Mice were pretreated with amitriptyline or saline vehicle and underwent sham or scald injury as described in the Materials and Methods. Thymocyte or splenocytes were harvested and (A) CD4+ single positive thymocytes, (B) naive CD4+ splenocyte, (C) central memory (CM) CD4+ splencoyte, and (D) effector memory CD4+ splenocyte subsets were characterized and enumerated as describe in the Materials and Methods. Sample size was 8 to 14 mice per group. Data are expressed as the mean ± SEM. Statistical comparisons were made using Student t test. *, P <0.05 as compared with scald group. Dotted line indicates sham levels. AMIT indicates amitriptyline.
Fig. 5. Prior amitriptyline treatment exacerbates scald-induced CD8+ T-cell depletion in thymus and spleen.

Mice were pretreated with amitriptyline or saline vehicle and underwent sham or scald injury as described in the Materials and Methods. Thymocyte or splenocytes were harvested and (A) CD8+ single positive thymocytes, (B) naive CD8+ splenocyte, (C) central memory (CM) CD8+ splencoyte, and (D) effector memory CD8+ splenocyte subsets were characterized and enumerated as describe in the Materials and Methods. Sample size was 8 to 14 mice per group. Data are expressed as the mean ± SEM. Statistical comparisons were made using Student t test. *, P <0.05 as compared with scald group. Dotted line indicates sham levels. AMIT indicates amitriptyline.
Amitriptyline treatment decreases neutrophil recruitment associated with reduced bone marrow neutrophil numbers
We next sought to determine if amitriptyline can alter the neutrophil response in injured mice with prior amitriptyline treatment. To test this, we induced neutrophil recruitment by peptidoglycan (PepG) injection and enumerated neutrophils in the peritoneum. Both amitriptyline treated and untreated scald injured mice that did not receive PepG demonstrated no significant peritoneal neutrophil accumulation. However, upon PepG administration, amitriptyline-treated scald mice had a 30.8% reduction in the number of elicited peritoneal neutrophils compared with untreated scald injured mice (Fig. 6A). We next sought to determine whether neutrophil chemotractant levels were altered by amitriptyline pretreatment. However, we determined that prior amitriptyline treatment did not alter IL-17 or CXCL1/KC levels (Fig, 6, B and C). Enumeration of bone marrow neutrophils revealed a 41.3% reduction in prior amitriptyline-treated injured mice compared with untreated scald injured mice (Fig. 6D). Altogether, PepG-induced peritoneal neutrophil accumulation was blunted in amitriptyline pretreated injured mice potentially due to reduced neutrophils available in the bone marrow.
Fig. 6. Prior amitriptyline treatment reduces peptidylglycan-mediated peritoneal neutrophil recruitment and bone marrow numbers following scald injury.

Mice were pretreated with amitriptyline or saline vehicle and underwent sham or scald injury as described in the Materials and Methods. Twenty-four hours after injury, mice were injected with 250 μmg of peptidoglycan (PepG). After another 24-h period, peritoneal cells and fluid for (A) neutrophil numbers, (B) IL-17 levels, and (C) KC levels. The dashed line indicates scald mice injected with vehicle. Bone marrow cells were isolated and characterized for (D) neutrophil numbers 24 h after injury. Sample size was five to nine mice per group. Data are expressed as the mean ± SEM. Dotted line indicates sham levels. Statistical comparisons were made either using Student t test. *, P <0.05 as compared with scald group. AMIT indicates amitriptyline; IL, interleukin.
Reduced Asm activity results in inefficient autophagic flux of neutrophils
As prior amitriptyline treatment exacerbated neutrophil loss in scald injured mice, we postulated that reduced Asm activity may alter neutrophil autophagy. To determine the role of Asm in autophagy, we utilized Asm heterozygous mice that have an approximate 50% reduction in Asm activity. Upon autophagy inducement, bone marrow neutrophils from Asm heterozygous mice had a significant increase in the number of cells with augmented acidification as well as the intensity as determined by lysotracker green analysis (Fig. 7, A and B). To compare autophagic flux between WT and Asm heterozygous neutrophils, we determined p62 levels (Fig. 7, C and D) and observed that Asm heterozygous neutrophils had a 1.7-fold increase in p62 expression, suggesting blunted autophagic flux. We next determined that Asm heterozygous neutrophils had a 3.5-fold decrease in the prosurvival Bcl-2 protein. (Fig. 7, C and E). Of interest, we did not observe a change in LC3 2 expression suggesting both cell types had a similar number of autophagosomes. Thus, Asm reduction in neutrophils is associated with blunted autophagic flux and decreased Bcl-2 expression.
Fig. 7. Asm heterozygous mice have blunted autophagy and decreased Bcl-2 expression.

Bone marrow neutrophils from WT and Asm mice were cultured in complete media for 48 h. Samples underwent lysotracker G (LTG) analysis and (A) percentage LTG expressing neutrophils, and (B) LTG mean fluorescence intensity (MFI) determined. Western Blot was utilized to determine protein concentrations of (C) p62, LC3, and Bcl-2. Quantification of (D) p62 and (E) Bcl-2 of the indicated neutrophils. Sample size was four mice per group and representative of two experiments. Data are expressed as the mean ± SEM. Statistical comparisons were made using Student t test. *, P <0.05 as compared with WT group.
DISCUSSION
Here, we investigated the impact of prior amitriptyline usage upon the immune response following scald injury. Our initial observation was that prior treatment with amitriptyline may result in increasing the catabolic response to burn as well as evidenced by increased weight loss and immune suppression as reflected by reduced spleen white blood cell counts. Upon Asm activity and ceramide level analysis, we determined that scald injury alone reduces Asm activity and ceramide levels. Amitriptyline treatment does not result in a further decrease in activity suggesting that Asm activity is maximally decreased in amitriptyline treated mice at the time of scald injury. Further analysis using mice with ongoing amitriptyline treatment revealed a decrease in immature and mature T-cell numbers along the entire axis of differentiation/education. Finally, we observed that prior amitriptyline treatment resulted in blunted PepG-mediated recruitment, likely due to decreased neutrophils that have blunted autophagic flux. Altogether, these data demonstrate prior amitriptyline treatment will exacerbate immune suppression following burn injury.
It is known that burn injury induces a catabolic state that results in increased weight loss following injury (2). Further, a greater weight loss is associated with more severe injury and worse clinical outcome (2, 3). Here, we show that prior amitriptyline treatment exacerbated the weight loss following scald, suggesting that these animals responded with increased catabolic response. The spleen plays an important role in the immune response and spleen mass has previously been demonstrated to be reduced with scald injury (4). We demonstrate on post burn day one, the spleens of scalded mice treated with amitriptyline had decreased mass and cell counts as compared with spleens from untreated scald mice. Thus, we conclude that with a similar injury model, ongoing amitriptyline treatment magnified body and spleen mass loss.
Asm is an enzyme that drives the conversion of sphingomyelin to ceramide (29, 30). Amitriptyline treatment results in the reduction Asm activity, thereby reducing ceramide levels. Ceramide is a key signaling molecule that has been shown to induce apoptosis (31, 32). Our data demonstrate that scald injury and amitriptyline alone reduce Asm activity and ceramide production by 30% to 40%. However, there is not an additive effect when scald mice are pretreated with amitriptyline. This suggests that Asm activity and ceramide levels are maximally decreased following amitriptyline treatment and this reduction is not further exacerbated by scald injury. However, we postulate that the amitriptyline-dependent decreased Asm activity prior to burn injury may drive additional leukocyte depletion. Previously, it has been demonstrated that scald injury alone reduces immature and mature T lymphocyte populations (4, 33, 34). In the current study, following scald injury, there is not a reduction in CLP cells when compared with sham treated mice. However, when scald mice are pretreated with amitriptyline, we observed a significant reduction in CLP cells as well as thymic and splenic T lymphocytes. We postulate that amitriptyline-induced reduction of CLPs does not allow for rapid repopulation of lymphoid compartments as similarly seen (35). Future studies are necessary to fully evaluate the effect of amitriptyline/Asm activity on leukocyte homeostasis following burn injury and the mechanisms underlying such potential changes.
Previously, it has been demonstrated that alterations in neutrophil production alter the ability to clear bacterial pathogens following burn injury, resulting in increased susceptibility to infections (21, 36–38). Here, we demonstrate that prior amitriptyline treatment results in a blunted PepG-mediated neutrophil recruitment following burn injury. We demonstrated that the reduced neutrophil recruitment was likely due to reduction of neutrophils in the bone marrow rather than chemoattractant levels or changes in adhesion molecules. As was true for lymphocytes, amitriptyline treatment results in reduced reserves leading to greater immunosuppression at the time of injury. Additionally, we demonstrate blunted neutrophil autophagy in Asm heterozygous mice. We speculate that Asm is necessary for the fusion of the autophagosome and the lysosome for efficient autophagy. This is similar to a recent study where control of lysosome trafficking and fusion by ASM is essential to a normal autophagic flux in coronary arterial smooth muscle cells. Here, the authors postulate that the deficiency of ASM-mediated regulation of autophagy results in the imbalance of arterial smooth muscle cell homeostasis (39). Future studies are currently ongoing to further elucidate the functionality of neutrophils with reduced Asm and their ability to clear bacterial pathogens.
A limitation to this study is a lack of mechanism linking the reduction of Asm activity and burn injury. Currently, such a mechanism has not been reported in any model system. The elucidation of such of a mechanism may allow for Asm manipulation that could be of benefit in either increasing or decreasing inflammation to combat infection or suppress autoimmunity, respectively.
In summary, our data demonstrate that reduction in Asm activity and ceramide levels caused by prior amitriptyline treatment is associated with increased injury severity following burn. Although Asm is similarly reduced by burn injury and amitriptyline treatment, the notable difference is that with amitriptyline treatment, Asm is reduced prior to burn injury. In contrast, in burn injury, Asm is in the process of being reduced at the time of injury. This prior reduction of Asm is associated with reduced lymphocyte numbers and neutrophil recruitment that correlates with reduced reserves of these cell populations. Altogether, these data suggest that amitriptyline treatment can result in greater immunosuppression following injury and therefore increased susceptibility to secondary infections.
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Rhee P, Joseph B, Pandit V, Aziz H, Vercruysse G, Kulvatunyou N, Friese RS. Increasing trauma deaths in the United States. Ann Surg. 2014;260(1):13–21. doi: 10.1097/SLA.0000000000000600. [DOI] [PubMed] [Google Scholar]
- 2.Hart DW, Wolf SE, Mlcak R, Chinkes DL, Ramzy PI, Obeng MK, Ferrando AA, Wolfe RR, Herndon DN. Persistence of muscle catabolism after severe burn. Surgery. 2000;128(2):312–319. doi: 10.1067/msy.2000.108059. [DOI] [PubMed] [Google Scholar]
- 3.Mlcak RP, Jeschke MG, Barrow RE, Herndon DN. The influence of age and gender on resting energy expenditure in severely burned children. Ann Surg. 2006;244(1):121–130. doi: 10.1097/01.sla.0000217678.78472.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tschop J, Martignoni A, Reid MD, Adediran SG, Gardner J, Noel GJ, Ogle CK, Neely AN, Caldwell CC. Differential immunological phenotypes are exhibited after scald and flame burns. Shock. 2009;31(2):157–163. doi: 10.1097/SHK.0b013e31817fbf4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang KC, Unsinger J, Davis CG, Schwulst SJ, Muenzer JT, Strasser A, Hotchkiss RS. Multiple triggers of cell death in sepsis: death receptor and mitochondrial-mediated apoptosis. FASEB J. 2007;21(3):708–719. doi: 10.1096/fj.06-6805com. [DOI] [PubMed] [Google Scholar]
- 6.Kasten KR, Prakash PS, Unsinger J, Goetzman HS, England LG, Cave CM, Seitz AP, Mazuski CN, Zhou TT, Morre M, et al. Interleukin-7 (IL-7) treatment accelerates neutrophil recruitment through gamma delta T-cell IL-17 production in a murine model of sepsis. Infect Immun. 2010;78(11):4714–4722. doi: 10.1128/IAI.00456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shelley O, Murphy T, Paterson H, Mannick JA, Lederer JA. Interaction between the innate and adaptive immune systems is required to survive sepsis and control inflammation after injury. Shock. 2003;20(2):123–129. doi: 10.1097/01.shk.0000079426.52617.00. [DOI] [PubMed] [Google Scholar]
- 8.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9(8):556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsieh YC, Athar M, Chaudry IH. When apoptosis meets autophagy: deciding cell fate after trauma and sepsis. Trends Mol Med. 2009;15(3):129–138. doi: 10.1016/j.molmed.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin CW, Lo S, Hsu C, Hsieh CH, Chang YF, Hou BS, Kao YH, Lin CC, Yu ML, Yuan SS, et al. T-cell autophagy deficiency increases mortality and suppresses immune responses after sepsis. PLos One. 2014;9(7):e102066. doi: 10.1371/journal.pone.0102066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komatsu M, Waguri S, Koike M, Sou Y, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 13.Beckmann N, Sharma D, Gulbins E, Becker KA, Edelmann B. Inhibition of acid sphingomyelinase by tricyclic antidepressants and analogons. Front Physiol. 2014;5:331. doi: 10.3389/fphys.2014.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wisler JR, Springer AN, Hateley K, Mo XM, Evans DC, Cook CH, Gerlach AT, Murphy CV, Eiferman DS, Steinberg SM, et al. Pre-injury neuro-psychiatric medication use, alone or in combination with cardiac medications, may affect outcomes in trauma patients. J Postgrad Med. 2014;60(4):366–371. doi: 10.4103/0022-3859.143957. [DOI] [PubMed] [Google Scholar]
- 15.Olfson M, Druss BG, Marcus SC. Trends in mental health care among children and adolescents. N Engl J Med. 2015;372(21):2029–2038. doi: 10.1056/NEJMsa1413512. [DOI] [PubMed] [Google Scholar]
- 16.Gulbins E, Palmada M, Reichel M, Luth A, Bohmer C, Amato D, Muller CP, Tischbirek CH, Groemer TW, Tabatabai G, et al. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nat Med. 2013;19(7):934–938. doi: 10.1038/nm.3214. [DOI] [PubMed] [Google Scholar]
- 17.Cifone MG, Migliorati G, Parroni R, Marchetti C, Millimaggi D, Santoni A, Riccardi C. Dexamethasone-induced thymocyte apoptosis: apoptotic signal involves the sequential activation of phosphoinositide-specific phospholipase C, acidic sphingomyelinase, and caspases. Blood. 1999;93(7):2282–2296. [PubMed] [Google Scholar]
- 18.Lepine S, Lakatos B, Courageot MP, Le Stunff H, Sulpice JC, Giraud F. Sphingosine contributes to glucocorticoid-induced apoptosis of thymocytes independently of the mitochondrial pathway. J Immunol. 2004;173(6):3783–3790. doi: 10.4049/jimmunol.173.6.3783. [DOI] [PubMed] [Google Scholar]
- 19.Tischner D, Theiss J, Karabinskaya A, van den Brandt J, Reichardt SD, Karow U, Herold MJ, Luhder F, Utermohlen O, Reichardt HM. Acid sphingomyelinase is required for protection of effector memory T cells against glucocorticoid-induced cell death. J Immunol. 2011;187(9):4509–4516. doi: 10.4049/jimmunol.1100911. [DOI] [PubMed] [Google Scholar]
- 20.Suchard SJ, Hinkovska-Galcheva V, Mansfield PJ, Boxer LA, Shayman JA. Ceramide inhibits IgG-dependent phagocytosis in human polymorphonuclear leukocytes. Blood. 1997;89(6):2139–2147. [PubMed] [Google Scholar]
- 21.Adediran SG, Dauplaise DJ, Kasten KR, Tschop J, Dattilo J, Goetzman HS, England LG, Cave CM, Robinson CT, Caldwell CC. Early infection during burn-induced inflammatory response results in increased mortality and p38-mediated neutrophil dysfunction. Am J Physiol Regul Integr Comp Physiol. 2010;299(3):R918–R925. doi: 10.1152/ajpregu.00132.2010. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Barke RA, Ma J, Charboneau R, Roy S. Opiate abuse, innate immunity, and bacterial infectious diseases. Arch Immunol Ther Exp (Warsz) 2008;56(5):299–309. doi: 10.1007/s00005-008-0035-0. [DOI] [PubMed] [Google Scholar]
- 23.Peng H, Li C, Kadow S, Henry BD, Steinmann J, Becker KA, Riehle A, Beckmann N, Wilker B, Li PL, et al. Acid sphingomyelinase inhibition protects mice from lung edema and lethal Staphylococcus aureus sepsis. J Mol Med. 2015;93(6):675–689. doi: 10.1007/s00109-014-1246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, Sitkovsky MV. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol. 2001;167(11):6140–6149. doi: 10.4049/jimmunol.167.11.6140. [DOI] [PubMed] [Google Scholar]
- 25.Ziobro R, Henry B, Edwards MJ, Lentsch AB, Gulbins E. Ceramide mediates lung fibrosis in cystic fibrosis. Biochem Biophys Res Commun. 2013;434(4):705–709. doi: 10.1016/j.bbrc.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kornhuber J, Muehlbacher M, Trapp S, Pechmann S, Friedl A, Reichel M, Muhle C, Terfloth L, Groemer TW, Spitzer GM, et al. Identification of novel functional inhibitors of acid sphingomyelinase. PLos One. 2011;6(8):e23852. doi: 10.1371/journal.pone.0023852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kornhuber J, Tripal P, Reichel M, Muhle C, Rhein C, Muehlbacher M, Groemer TW, Gulbins E. Functional inhibitors of acid sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem. 2010;26(1):9–20. doi: 10.1159/000315101. [DOI] [PubMed] [Google Scholar]
- 28.Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J, Gulbins E. Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model. J Med Chem. 2008;51(2):219–237. doi: 10.1021/jm070524a. [DOI] [PubMed] [Google Scholar]
- 29.Gulbins E, Kolesnick R. Raft ceramide in molecular medicine. Oncogene. 2003;22(45):7070–7077. doi: 10.1038/sj.onc.1207146. [DOI] [PubMed] [Google Scholar]
- 30.Quintern LE, Schuchman EH, Levran O, Suchi M, Ferlinz K, Reinke H, Sandhoff K, Desnick RJ. Isolation of cDNA clones encoding human acid sphingomyelinase: occurrence of alternatively processed transcripts. EMBO J. 1989;8(9):2469–2473. doi: 10.1002/j.1460-2075.1989.tb08382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez-Munoz A, Gangoiti P, Arana L, Ouro A, Rivera IG, Ordonez M, Trueba M. New insights on the role of ceramide 1-phosphate in inflammation. Biochim Biophys Acta. 2013;1831(6):1060–1066. doi: 10.1016/j.bbalip.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Saba JD, Obeid LM, Hannun YA. Ceramide: an intracellular mediator of apoptosis and growth suppression. Philos Trans R Soc Lond B Biol Sci. 1996;351(1336):233–240. doi: 10.1098/rstb.1996.0021. [DOI] [PubMed] [Google Scholar]
- 33.Patenaude J, D'Elia M, Hamelin C, Garrel D, Bernier J. Burn injury induces a change in T cell homeostasis affecting preferentially CD4+ T cells. J Leukoc Biol. 2005;77(2):141–150. doi: 10.1189/jlb.0703314. [DOI] [PubMed] [Google Scholar]
- 34.Burleson DG, Mason AD, Jr, Pruitt BA., Jr Lymphoid subpopulation changes after thermal injury and thermal injury with infection in an experimental model. Ann Surg. 1988;207(2):208–212. doi: 10.1097/00000658-198802000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prockop SE, Petrie HT. Regulation of thymus size by competition for stromal niches among early T cell progenitors. J Immunol. 2004;173(3):1604–1611. doi: 10.4049/jimmunol.173.3.1604. [DOI] [PubMed] [Google Scholar]
- 36.Engelich G, Wright DG, Hartshorn KL. Acquired disorders of phagocyte function complicating medical and surgical illnesses. Clin Infect Dis. 2001;33(12):2040–2048. doi: 10.1086/324502. [DOI] [PubMed] [Google Scholar]
- 37.Chen LW, Huang HL, Lee IT, Hsu CM, Lu PJ. Thermal injury-induced priming effect of neutrophil is TNF-alpha and P38 dependent. Shock. 2006;26(1):69–76. doi: 10.1097/01.shk0000209531.38188.18. [DOI] [PubMed] [Google Scholar]
- 38.Parihar A, Parihar MS, Milner S, Bhat S. Oxidative stress and anti-oxidative mobilization in burn injury. Burns. 2008;34(1):6–17. doi: 10.1016/j.burns.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Xu M, Pitzer AL, Xia M, Boini KM, Li PL, Zhang Y. Control of autophagy maturation by acid sphingomyelinase in mouse coronary arterial smooth muscle cells: protective role in atherosclerosis. J Mol Med. 2014;92(5):473–485. doi: 10.1007/s00109-014-1120-y. [DOI] [PMC free article] [PubMed] [Google Scholar]