

Abstract
Patients with atopic dermatitis (AD) have an abnormal skin barrier and are frequently colonized by S. aureus. In this study we investigated if S. aureus penetrates the epidermal barrier of subjects with AD and sought to understand the mechanism and functional significance of this entry. S. aureus was observed to be more abundant in the dermis of lesional skin from AD patients. Bacterial entry past the epidermis was observed in cultured human skin equivalents and in mice, but found to be increased in the skin of cathelicidin knockout (Camp−/−) and ovalbumin-sensitized filaggrin mutant (FLGft/ft) mice. S. aureus penetration through the epidermis was dependent on bacterial viability and protease activity as killed bacteria or a protease-null mutant strain of S. aureus was unable to penetrate. Entry of S. aureus directly correlated with increased expression of IL4, IL13, IL22, TSLP and other cytokines associated with AD, and with decreased expression of cathelicidin. These data illustrate how abnormalities of the epidermal barrier in AD can alter the balance of S. aureus entry into the dermis and provides an explanation for how such dermal dysbiosis results in increased inflammatory cytokines and exacerbation of disease.
Keywords: Atopic dermatitis, Cathelicidin, Microbiome, Staphylococcus aureus
INTRODUCTION
The microbial community “microbiome” can have both beneficial and detrimental functions (Gallo and Nakatsuji, 2011). For example, Staphylococcus epidermidis, a predominant resident on healthy human skin, can suppress inflammation after skin injury, maintain immune tolerance to commensals, modify cutaneous T-cell development, and enhance innate immune defense by inducing expression of antimicrobial peptides (AMPs) (Cogen et al., 2010; Lai et al., 2010; Li et al., 2013; Naik et al., 2015; Naik et al., 2012; Scharschmidt et al., 2015; Wang et al., 2012; Wanke et al., 2011). Conversely, imbalance of the microbiome (dysbiosis), appears to contribute to the pathogenesis of some skin diseases. Strong associations have been shown between dysbiosis and the clinical phenotype of atopic dermatitis (AD) (Leung and Guttman-Yassky, 2014). For example, AD subjects are well known to have increased colonization by Staphylococcus aureus (S. aureus) (Leyden et al., 1974) and a loss in bacterial diversity on the skin (Kong et al., 2012). Furthermore, recent mechanistic studies have demonstrated that S. aureus can drive development of AD-like lesions in mice (Kobayashi et al., 2015). These findings suggest that a better understanding of how bacteria influence skin immunity may provide important clues to improve management of AD.
S. aureus can cause inflammation by inducing T cell-independent B cell expansion, initiating the production of proinflammatory cytokines such as thymic stromal lymphopoietin (TSLP) from keratinocytes, and stimulating mast cell degranulation, resulting in TH2 skewing (Bekeredjian-Ding et al., 2007; Nakamura et al., 2013; Vu et al., 2010). S. aureus also disrupts proteolytic balance in the skin by inducing multiple metalloproteases in dermal fibroblasts (Kanangat et al., 2006). However, because of the complex structures and cell networks that comprise mammalian skin, the mechanism by which S. aureus disrupts cutaneous inflammatory homeostasis is incompletely understood. It appears that most beneficial and detrimental actions of skin bacteria are dependent on their capacity to interact with host cells that reside under the surface stratum corneum. Until recently it was unclear how skin surface microbes could influence immunological responses through a stratum corneum structure.
We recently observed that bacteria residing on the epidermis can be observed within the dermis of healthy normal human skin (Nakatsuji et al., 2013). This surprising observation that bacteria can penetrate the epidermis illustrated how bacteria position themselves to directly influence immune responses. The epidermis apparently acts as a regulator of microbiome entry rather than as an absolute barrier to microbes. This suggests that epidermal barrier defects such as the loss-of-function mutations found within the filaggrin gene (FLG) (Bisgaard et al., 2008; Palmer et al., 2006; Sandilands et al., 2007; Smith et al., 2006) could promote disease. In addition, the epidermis of AD subjects can have a decreased capacity to produce AMPs such as cathelicidin and β-defensins (Hata et al., 2010; Howell et al., 2006a; Howell et al., 2006b; Mallbris et al., 2010; Ong et al., 2002). Such antimicrobial or physical barrier defects may facilitate physical penetration of the epidermis by bacteria that otherwise would not trigger inflammation on normal skin.
In this study we sought to determine the mechanism by which S. aureus penetrates the epidermis. We hypothesized that the altered physical and antimicrobial barrier of the skin in AD will result in enhanced penetration of S. aureus across the epidermal surface, and that this can contribute to the loss of immune homeostasis. Such an interaction between bacteria on the skin surface and cells in the dermis provides a unifying hypothesis to explain why genetic or environmental defects in the skin barrier drive immunologic abnormalities of AD.
RESULTS
Dysbiosis of the bacterial community in the dermis of patients with atopic dermatitis
To examine the microbial community in the dermis of skin from subjects with AD, skin biopsies from non-AD control subjects and lesional and nonlesional sites of AD patients were obtained. Biopsies of lesional AD skin did not include skin sites that were excoriated. Skin samples were separated into epidermal and dermal tissues by laser-capture microdissection (LCM) (Figure S1a) and bacterial DNA contained in each tissue was analyzed by qPCR and pyrosequencing. The absolute abundance of 16S rDNA in the LCM-dissected epidermis and dermis was higher from lesional skin than that of non-AD and nonlesional AD skin (Figure 1a). S. aureus was detected by qPCR in both epidermis and dermis of lesional skin, but not in non-AD and nonlesional AD skin (Figure 1b). S. epidermidis DNA was also detected in epidermis and dermis of non-AD and AD skin, and was higher in dermis of lesional skin than that of non-AD and nonlesional AD skin (Figure 1c). Pyrosequencing for 16S rDNA detected relatively higher abundance of Firmicutes (phylum of Staphylococcus species) in epidermal and dermal compartments of lesional AD skin than those of nonlesional AD skin (Figure 1d). To detect potential contamination, non-tissue controls (NTC) prepared from the same tissue blocks and assay reagents, and DNA was not detected in these samples (Figure 1a–c).
Figure 1. Dysbiosis of the subepidermal compartments from skin of AD patients.
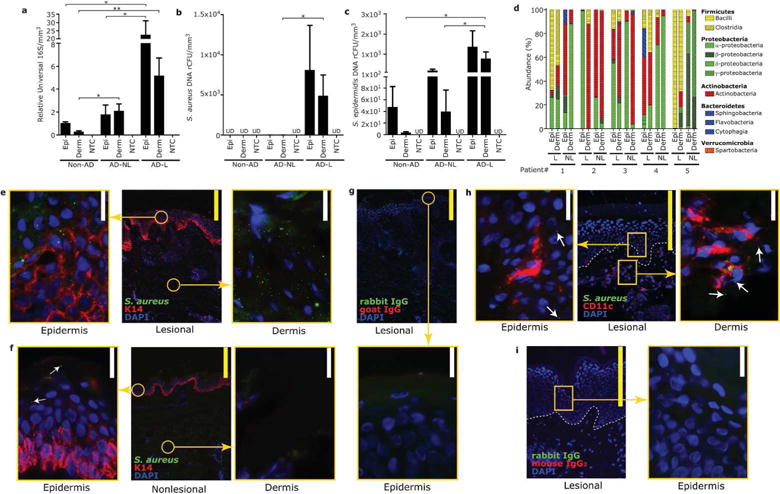
(a) qPCR results for relative abundance of DNA for 16S rRNA, (b) S. aureus (c) and S. epidermidis detected in epidermal or dermal compartments, isolated by laser-capture microdissection (LCM), of skin from normal skin of non-AD subjects, nonlesional and lesional skin of patients with AD. Non-tissue controls (NTC) were simultaneously processed. Data represent mean ± SEM of 11 subjects. *P<0.05, **P<0.01. Epi=epidermis, Derm=dermis, UD=undetectable. (d) 16S rRNA pyrosequencing results from samples isolated by LCM of the epidermis and dermis of nonlesional (NL) and lesional (L) skin of AD subjects. Each bacterial phylum is shown in different color. (e–g) Immunofluorescence for S. aureus and keratin-14 in lesional (e) or nonlesional (f) skin of AD subject. Staining with isotype control (g). (h,i) Immunofluorescense for S. aureus and CD11c in lesional skin of AD subject. Staining with isotype control (i). Arrows indicate S. aureus staining detected outside of CD11c+ immune cells. Immunostaining shown is a representative of 3 biopsies from different donors. Scale bar= 20μm (white) or 200μm (yellow).
To further confirm these observations, lesional and nonlesional skin was stained for S. aureus, and this directly demonstrated that S. aureus was more abundant within the epidermis and dermis of lesional skin compared to nonlesional samples (Figure 1e–f). Notably, S. aureus was independently detected in the epidermis and dermis of lesional AD skin and not associated with CD11c+ immune cells (Figure 1h), thus suggesting a penetration mechanism independent from classical phagocytic cells. No immunoreactivity was seen with control IgG (Figure 1g,i).
S. aureus penetrates cultured human skin equivalents
To begin to understand how S. aureus may enter the dermis, we next compared the capacity of live and dead S. aureus to penetrate the epidermis of an organotypic human skin equivalent. Immunohistochemical analysis was done on samples processed at 13, 24 and 48 hours after S. aureus application. Immunostaining of the skin equivalent demonstrated that S. aureus were present at progressively deeper layers in the epidermis in a time-dependent manner, and detectable below the basement membrane by 48hrs (Figure 2a–c). In contrast, despite application of a larger bacterial load, bacteria that were killed by UV radiation remained on the surface at 48 hrs (Figure 2d). These data demonstrated that S. aureus must be viable to penetrate the epidermis.
Figure 2. S. aureus actively penetrates human skin organotypic equivalents.
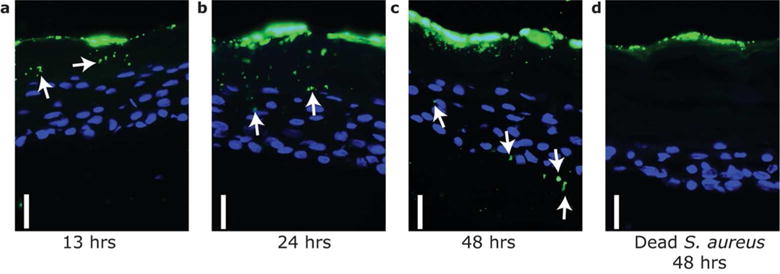
Time-dependent entry of S. aureus across the epidermis of a human skin organotypic construct. Viable S. aureus (1×106 CFU) (a–c) or UV-killed S. aureus (1×107 CFU) (d) were applied on the stratum corneum surface and individual constructs fixed at the indicated time after bacterial application. Skin constructs were then sectioned and stained with anti-S. aureus (green) to visualize bacteria. Keratinocyte nuclei were counter stained with DAPI (4′,6-diamidino-2-phenylindole) (blue). Scale bar=20 μm. Arrows indicate immunoreactivity for S. aureus under the epidermal surface when live bacteria were applied to the surface.
Cathelicidin inhibits bacterial entry into dermis
Since we observed that penetration of skin required viable bacteria, we hypothesized that AMP activity may act to limit bacterial entry into the dermis. To test this, S. aureus was applied to dorsal skin of cathelicidin (Camp) knock out or wild type (WT) mice. Mouse skin was then separated by LCM into control, epidermal, dermal and dermal adipose compartments (Figure S1b). After application to the skin surface, S. aureus abundance was not statistically different on the epidermis of WT and Camp−/− mice (Figure 3a). However, S. aureus was significantly more abundant in the dermis and adipose tissue of Camp−/− mice than WT mice (Figure 3b–d). These data suggest that the antimicrobial barrier provided by cathelicidin limits entry of S. aureus into the skin and is one variable controlling bacterial penetration into the dermis.
Figure 3. Cathelicidin inhibits S. aureus entry into the mouse dermis.
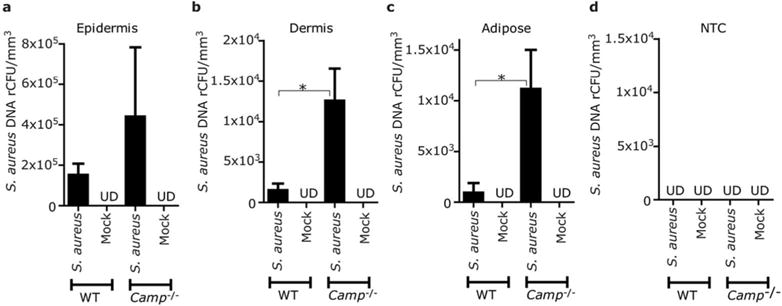
(a–c) S. aureus (ATCC35556) or vehicle (mock) were loaded in agar discs and applied on dorsal skin of Camp−/− or WT mice for 20 hrs. Skin was then excised and DNA extracted from epidermis (a), dermis (b) or adipose tissue (c) isolated by LCM. Relative colony forming units (rCFU) of S. aureus DNA was determined by real-time qPCR by comparison to a standard of known CFUs of S. aureus (ATCC35556). (d) As negative control a non-tissue control (NTC) was simultaneously processed with the same reagents from embedding material adjacent to each tissue section. The data were normalized against tissue volume excised by LCM. Data represent mean±SEM of results from 4 independent experiments.*P<0.05. UD=undetectable
S. aureus penetration is enhanced in a mouse loss-of-function of filaggrin model of atopic dermatitis
We next evaluated entry of S. aureus in loss-of-function of filaggrin (FLGft/ft) mice (Fallon et al., 2009). Similar to previous reports, FLGft/ft mice developed AD-like skin inflammation after mechanical barrier disruption by tape-stripping and repeated application of ovalbumin (OVA), whereas WT mice or FLGft/ft without OVA sensitization had much lesser inflammation (Figure S2). Consistent with this observation, the skin of FLGft/ft mice after tape-stripping and OVA sensitization showed enhanced transepidermal water loss (TEWL) compared to WT mice (Figure 4a). Twenty hrs after application of S. aureus to the skin of these mice there was only a minimal difference in S. aureus detected in the surface epidermal compartment of the FLGft/ft mice (Figure 4b), whereas a significantly increased amount of S. aureus was detected in the dermis and adipose tissue of FLGft/ft than WT mice with similar tape-strip and OVA sensitization or PBS-treated FLGft/ft mice (Figure 4c–d). S. aureus was undetectable in NTC (Figure 4e). These data demonstrate that in the setting of increased inflammation and enhanced TWEL, a loss-of-function mutation in FLG facilitates bacterial entry through the epidermal barrier.
Figure 4. A loss-of-function mutation in filaggrin increases S. aureus entry into the mouse dermis after ovalbumin sensitization.
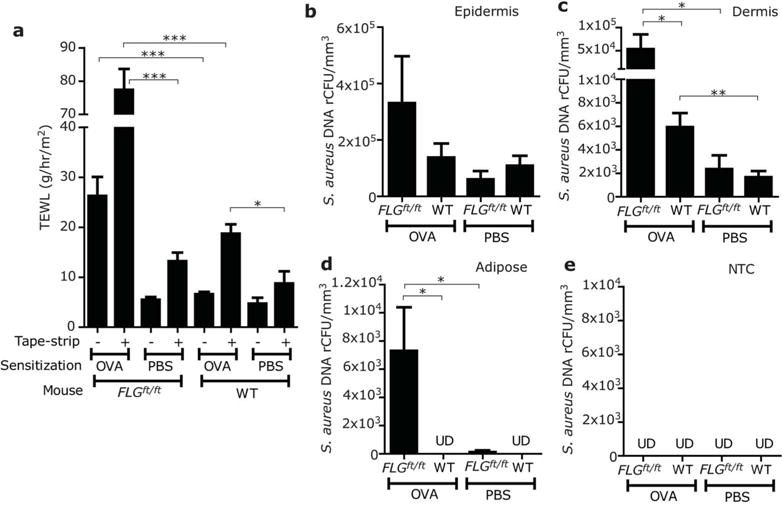
(a) Transepidermal water loss (TEWL) determined before (−) or after tape-stripping (+) on the dorsal skin of FLGft/ft Balb/c or WT mice which were treated by repeated applications of OVA or PBS. (b–e) The backs of FLGft/ft Balb/c mice, or wild type (WT) Balb/c mice were treated with tape-stripping and OVA as described in panel (a). Abundance of S. aureus (ATCC3555) in epidermis (b), dermis (c) and adipose tissue (d) was measured by qPCR and LCM as described in Figure 3. (e) NTC was simultaneously processed as negative control. Data represent mean ± SEM of results from 6 independent experiments. *P<0.05, **P<0.01, ***P<0.001. UD=undetectable
S. aureus proteolytic activity enhances penetration of the epidermal barrier
As Staphylococcus extracellular proteases have been known to interact with the epidermal barrier components, we hypothesized that bacteria enter the dermis through the action of their proteases. To address this, we compared entry of WT methicillin-resistant S. aureus (MRSA) USA300 to an extracellular protease-mutant of this strain which lacks 10 major proteolytic enzymes, including aureolysin metalloprotease, V8 and SspA serine proteases, ScpA and SspB cysteine proteases, and 6 other serine-like protease homologs (Kolar et al., 2013). We confirmed that the culture supernatant of the mutant strain contained less proteolytic activity than that of WT of the same background strain (Figure S3). First, as in Figure 2, an equal number of bacteria were applied on the epidermis of cultured human skin equivalents to compare the capacity of each strain to penetrate the epidermis. Both strains grew at the same rate in this model since after 48 hrs, comparable colony-forming units (CFUs) of the live WT and mutant strains were detected in full-thickness biopsies of the skin construct (Figure 5a). More WT S. aureus was detected in the dermis than the protease deficient mutant strain. Next, when these bacteria were applied to mice in the AD skin model of OVA-sensitized and tape-stripped FLGft/ft mice shown in Figure 4, a similar behavior of S. aureus was observed. Both bacterial strains were detected in similar quantities in the mouse epidermis (Figure 5b), whereas the only the WT strain showed a significant capacity to enter the dermis and adipose tissue (Figure 5c–e). These data suggest that skin entry by bacteria is, at least in part, facilitated by microbial proteases.
Figure 5. S. aureus protease activity is required for penetration of the epidermis and induction of inflammatory cytokines.
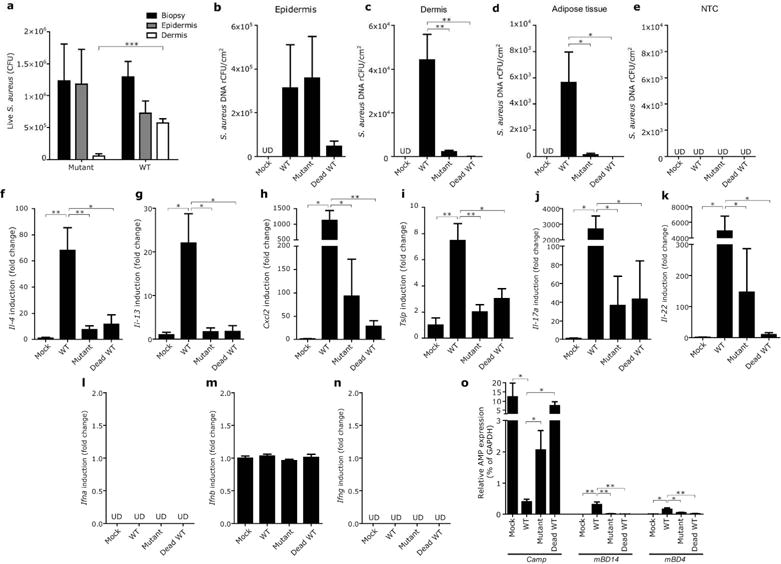
(a) Entry of WT or an extracellular protease-null mutant strain of MRSA into organotypic human skin constructs . (b–e) Entry of WT, extracellular protease-null mutant strain and UV-killed WT strain of MRSA into epidermis (b), dermis (c) and adipose tissue (d) of FLGft/ft Balb/c mice sensitized by OVA was tracked as described in Figure 3. NTC was processed as negative control (e). (f–o) To correlate entry of MRSA strains with cutaneous immune response, gene expression of indicated cytokines (f–n) and indicated AMPs (o) was measured in the same whole skin biopsies from panel (b–d). To compare relative expression level of each AMP, data was shown as relative to GAPDH expression. Data represent mean ± SEM of results from 5–6 independent experiments. *P<0.05, **P<0.01, ***P<0.001.
Entry of S. aureus into the dermis triggers immune abnormalities seen in AD skin
With the observation that WT and protease deficient strains of S. aureus penetrated differently below the epidermis, it became possible to examine if a correlation exists between entry of bacteria into the dermis and an immunological response. The expression of mRNA for inflammatory cytokines (IL-4, IL-13, CXCL2, TSLP, IL-17a, IL-22 and IFNs), and cathelicidin (Camp) was therefore measured in each condition. In accordance with the capacity to enter the skin as seen in Figure 5b–c, live WT S. aureus induced more expression of IL-4, IL-13, CXCL2, TSLP, IL-17 and IL-22, but not IFNs (Figure 5f–n). Furthermore, similar to observations in human subjects with AD, S. aureus entry resulted in suppression of Camp expression (Figure 5o). In contrast, S. aureus entry was correlated with a slight increase in β-defensins-14 and -4 expression, but the relative expression level of these β-defensins was much lower than that of Camp.
Application of a barrier repair cream decreases bacterial penetration of skin
An essential element in the therapy of AD is the topical application of moisturizers or barrier repair products. This can result in significant decrease in inflammation, but the immunological mechanism responsible for this improvement is unclear. To examine if restoration of skin barrier function could benefit the immune response by limiting bacterial entry into the dermis, AD-like skin lesions in OVA-sensitized FLGft/ft mice were treated with a barrier repair formulation consisting of optimized ceramide-triple lipid mixture (Man et al., 1996; Mao-Qiang et al., 1995) or vehicle. Subsequently, S. aureus was applied to the surface and entry was tracked with LCM and qPCR as described previously. Treatment with the ceramide-triple lipid mixture restored barrier function as measured by TEWL (Figure 6a). Following barrier repair, S. aureus penetration decreased into the subepidermal tissues in comparison to the control skin treated with vehicle (Figure 6b–e). Application of the ceramide-triple lipid mixture did not exert antimicrobial effect on the S. aureus colonized on the epidermis (Figure 6b). In addition, barrier repair partially limited cytokine induction and failure of Camp induction caused by S. aureus application (Figure 6f–i).
Figure 6. Skin barrier repair decreases S. aureus entry into the dermis and suppresses subsequent immune response in skin with FLG mutation.
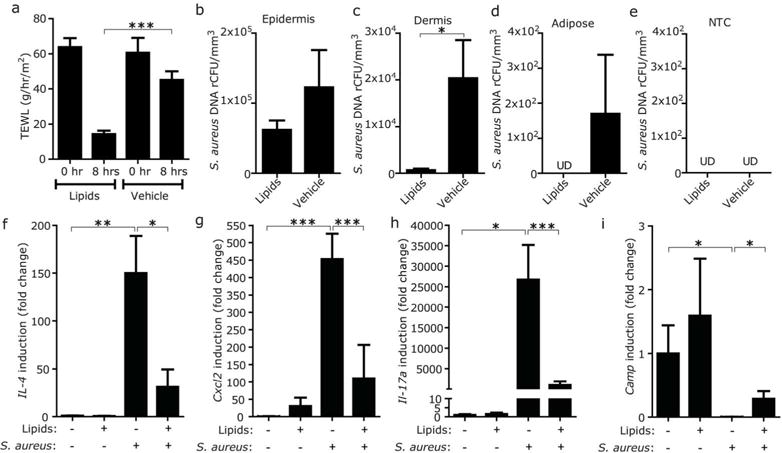
(a) Effect of the application of barrier repair formula of ceramide-triple lipid mixture on TEWL from FLGft/ft Balb/c mice sensitized with OVA. TEWL was measured 4 hrs after application of barrier repair lipids or vehicle. (b–e) Effect of the application of barrier repair lipid mixture on entry of S. aureus into the skin of FLGft/ft Balb/c mice with AD-like inflammation. Entry of S. aureus (ATCC3555) into epidermis (b), dermis (c) and adipose tissue (d) was tracked 4 hrs after application of barrier repair lipids or vehicle. NTC was simultaneously processed as negative control (e). Data represent mean ± SEM of results from 5–6 independent experiments. (f–i) Effect of the application of barrier repair lipid mixture on cytokine and Camp inductions after epicutaneous application of S. aureus on the OVA-sensitized skin of FLGft/ft Balb/c mice. Data represent mean ± SEM of results from 6–7 independent experiments. *P<0.05, **<0.01, ***P<0.001. UD=undetectable
DISCUSSION
Defects in the skin barrier have been associated with the pathogenesis of AD and are frequently associated with colonization by S. aureus, a factor that exacerbates disease (De Benedetto et al., 2011; Leung and Guttman-Yassky, 2014). It was unclear how dysbiosis at the skin surface could trigger inflammation that arises below the stratum corneum. We report herein that S. aureus penetrates the epidermis by a proteolytic mechanism and that failure of the antimicrobial or physical skin barrier of the epidermis enhances entry of S. aureus into the dermis. Entry of bacteria enables them to come into direct contact with viable immunocytes and stimulates production of pro-inflammatory cytokines. These data directly demonstrate how the skin barrier controls the interaction between the microbiome and the cutaneous immune system, and illustrate how abnormal penetration of surface microbes can mediate immune dysregulation associated with AD. Importantly, since this immune dysregulation further disrupts the barrier function of the skin, this relationship provides an explanation for the chronic nature of inflammation observed in this disorder.
An important variable that influenced microbial entry into the dermis was AMP expression. Cathelicidin exhibits direct antimicrobial action against a wide range of pathogens (Dorschner et al., 2001; Gallo et al., 1994; Nizet et al., 2001). In healthy skin, cathelicidin expression is increased upon infection, inflammation and injury (Gallo and Hooper, 2012; Lai and Gallo, 2009). However, the skin of patients with AD has been shown in some studies to have a decreased capacity to produce an adequate amount of this AMP and β-defensins-2 and -3 (Hata et al., 2010; Howell et al., 2006a; Howell et al., 2006b; Mallbris et al., 2010; Ong et al., 2002). This AMP deficiency may result in an inability of the skin to resist infection by several pathogens including S. aureus. However, it was not known if the constitutive presence of cathelicidin expressed in healthy skin contributed to antibacterial defense or how this influenced colonization of skin microbes. In this study, cathelicidin was shown to regulate entry of S. aureus. This finding may be clinically relevant when considered with observations that cathelicidin expression is enhanced by vitamin D and improves clinical outcome in AD (Liu et al., 2006; Schauber et al., 2007; Schauber et al., 2006; Camargo et al., 2014; Hata et al., 2008). Therefore, although many variables may confound this response, the capacity of vitamin D to enhance the antimicrobial barrier may partially explain reported benefits of vitamin D3 in AD and other allergic disorders (Goetz, 2011; Malley et al., 2009).
Another variable found here to influence penetration of S. aureus was the expression of filaggrin. Filaggrin is a structural protein that is fundamental in the development and maintenance of the physical skin barrier (Sandilands et al., 2009). Loss-of-function mutations in FLG represent a significant genetic factor predisposing the development of AD in some populations (Bisgaard et al., 2008; Palmer et al., 2006; Sandilands et al., 2007; Smith et al., 2006). Two of the most studied mutations (R501X and 2282del4) are common in European populations and result in loss-of-function (Weidinger et al., 2006). The flaky-tail mouse used here has a naturally-occurring single-base-pair deletion (FLGft/ft) that induces a premature stop codon and also results in loss-of-function (Fallon et al., 2009; Moniaga and Kabashima, 2011). This mouse differs from the original flaky-tail mouse that has an additional mutation in Tmem79/Matt, a mutation that is also associated with AD in humans (Saunders et al., 2013). In contrast to the double mutation original flaky-tail mouse, this FLGft/ft mouse line does not develop spontaneous inflammation (Hoff et al., 2015). However, out data illustrate how mutations in only FLG increase the risk of inflammation and enhance S. aureus entry. Other epidermal barrier proteins, such as envoplakin, periplakin, and involucrin, may also control microbiome penetration into the skin (Natsuga, et al., 2016). Barrier defects permitted increased S. aureus entry and subsequent enhanced expression of TH2 cytokines, IL-17 and TSLP, and decreased expression of cathelicidin. These results are consistent with previous reports demonstrating that TH2 cytokines directly downregulate the induction of cathelicidin in the skin (Howell et al., 2006b). Such changes are characteristic of AD and may illustrate how mutations of FLG in the human population may confer risk of AD by enabling the abnormal entry of microbes into the dermis.
We previously reported that most of the microbes in the dermis of human skin are not present within classical CD11c+ phagocytic immune cells (Nakatsuji et al., 2013). Similarly S. aureus was detected outside of CD11c+ cells in lesional AD skin. These data suggest that these microbes entered across the epidermal barrier rather than carried in by phagocytosis. Here, we directly demonstrated in cultured skin equivalents that antigen presenting cells are not required for S. aureus to enter the dermis. In contrast penetration of the stratum corneum by S. aureus was dependent on protease activity. Future work will determine if a specific protease mediates entry although there are several proteases produced by Staphylococcal species that are of interest. For example, S. aureus produces an extracellular zinc-calcium-dependent metalloproteinase called aureolysin which can proteolytically degrade cathelicidin and neutralize its antimicrobial activity (Sieprawska-Lupa et al., 2004). S. aureus also produces a serine protease, commonly referred to as V8 protease, that is known to impair epidermal barrier function in mice (Hirasawa et al., 2010) and has been known as the “epidermolytic toxin” (Dancer et al., 1990; Redpath et al., 1991). S. aureus strains isolated from AD patients have found to produce high extracellular proteolytic activity, and aureolysin and V8 protease are predominantly contribute to their total proteolytic activity (Miedzobrodzki et al., 2002). In the current study we showed that the extracellular protease-null mutant of MRSA USA300, which lacks both proteases, had less capacity to break through the epidermal barrier of the murine skin and organotypic human skin constructs. Thus, aureolysin and the V8 serine protease are two prime specific candidates to examine for their role to permit bacteria to penetrate the skin, and may explain the higher amount of S. aureus seen in the dermis of AD subjects. Blocking specific bacterial proteases could therefore be a useful therapeutic approach for AD though this therapeutic use of protease inhibitors for AD has been controversial (Foelster Holst et al., 2010; Wachter and Lezdey, 1992).
This study demonstrates that S. aureus can directly penetrate the stratum corneum and epidermis, a behavior that explains how this microbe can disrupt skin immune homeostasis. Defects in the skin barrier enable enhanced entry from the surface, and although this entry does not show characteristics of infection, this penetration alters cytokine and AMP responses. Such inflammatory triggers may then further alter the surface microbiome and perpetuate disease by adding to the process of barrier disruption. Notably, we demonstrated that barrier repair reduces S. aureus penetration into skin and normalizes immune abnormality triggered by the S. aureus penetration. Thus, these observations provide clinical insight into how physical and innate immune barrier defects influence the pathogenesis of AD and provide guidance for optimizing therapeutic approaches to this disorder.
MATERIALS AND METHODS
Analysis of skin from human subjects with AD
All sample acquisition, including biopsies of lesional and nonlesional skin from patients with AD, and normal skin from non-AD subjects, was approved and performed in accordance with the Human Research Protections Program at the University of California, San Diego (reference number: 071032). Written informed consent was obtained from all subjects prior to performing the skin biopsies. Demographic data of AD patients recruited are shown in Table S1. Subepidermal compartments of lesional and nonlesional skin from patients with AD were excised from the skin biopsy by using LCM without inclusion of appendageal structures. Total genomic DNA was extracted from LCM sections and subjected to real-time qPCR or pyrosequencing for 16S rRNA using universal 16S rRNA or species-specific primers/probe (Table S2). For details, see Supplementary Material.
Tracking bacteria entry into mouse skin
All animal protocols were reviewed and approved by the UC San Diego (approval number: S09074). After shaving and disinfecting the dorsal skin of OVA-sensitized FLGft/ft or Camp−/− mice, an agar disk (6 mm) containing S. aureus ATCC35556 or MRSA USA300 LAC strains (1×106 CFU) was applied on the skin, and the entire dorsal skin was then covered with wound dressing film for 20 hrs. An agar disc without bacteria or with UV-killed S. aureus (1×107 CFU equivalent) was used as controls. Following disk application, skin was carefully cleaned with alcohol swabs and frozen in tissue embedding compound. Epidermis, dermis and adipose tissue sections were excised by LCM without inclusion of appendageal structures. Total genomic DNA was extracted from each section for qPCR. For details, see Supplementary Material.
Skin barrier disruption and restoration
A sterile patch with OVA solution or PBS was placed on tape-stripped dorsal skin of FLGft/ft Balb/c mice for 8 days (the patch was replaced every 2 days) as described previously (Jin et al., 2009). Each mouse received three 8-day exposures in 2 week intervals. Twenty four hrs after the third sensitization, dorsal skin was tape-stripped. For skin barrier repair experiments an optimized formula of cholesterol, ceramide, linoleate and palmitate (3:1:1:1), or propylene glycol-ethanol (7:3) (vehicle) was applied twice every 4 hrs as previously described (Man et al., 1996; Mao-Qiang et al., 1995). S. aureus was applied 4 hrs after the second application of barrier repair mixture or vehicle. For details, see Supplementary Material.
Data access
The 16S pyrosequencing data for this study has been submitted to DDBJ Sequence Read Achieve (http://trace.ddbj.nig.ac.jp/dra/index_e.html) through the BioProject ID PRJDB4882 and published under the Accession Code DRA004759.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 software (GraphPad, La Jolla, CA). Independent t-test was used for significance of differences.
Supplementary Material
Acknowledgments
We thank Dr. Peter M. Elias at the UC San Francisco for providing technical advice. This study was funded by Atopic Dermatitis Research Network (HHSN272201000017C), and National Institute of Health R01 grants, (R01AR064781, R01AI116576, and R01AI052453 to RLG).
Abbreviations used
- AD
Atopic dermatitis
- AMP
Antimicrobial peptide
- CFU
Colony-forming unit
- DAPI
4′,6-diamidino-2-phenylindole
- IFN
Interferon
- IL
Interleukin
- LCM
Laser-capture microdissection
- MRSA
Methicillin-resistant Staphylococcus aureus
- NTC
Non-tissue control
- OVA
Ovalbumin
- TEWL
Transepidermal water loss
- TSLP
Thymic stromal lymphopoietin
- WT
Wild type: WT
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Bekeredjian-Ding I, Inamura S, Giese T, Moll H, Endres S, Sing A, et al. Staphylococcus aureus protein A triggers T cell-independent B cell proliferation by sensitizing B cells for TLR2 ligands. J Immunol. 2007;178:2803–12. doi: 10.4049/jimmunol.178.5.2803. [DOI] [PubMed] [Google Scholar]
- Bisgaard H, Simpson A, Palmer CN, Bonnelykke K, McLean I, Mukhopadhyay S, et al. Gene-environment interaction in the onset of eczema in infancy: filaggrin loss-of-function mutations enhanced by neonatal cat exposure. PLoS Med. 2008;5:e131. doi: 10.1371/journal.pmed.0050131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo CA, Jr, Ganmaa D, Sidbury R, Erdenedelger K, Radnaakhand N, Khandsuren B. Randomized trial of vitamin D supplementation for winter-related atopic dermatitis in children. J Allergy Clin Immunol. 2014;134:831–5 e1. doi: 10.1016/j.jaci.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Cogen AL, Yamasaki K, Sanchez KM, Dorschner RA, Lai Y, MacLeod DT, et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J Invest Dermatol. 2010;130:192–200. doi: 10.1038/jid.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancer SJ, Garratt R, Saldanha J, Jhoti H, Evans R. The epidermolytic toxins are serine proteases. FEBS Lett. 1990;268:129–32. doi: 10.1016/0014-5793(90)80990-z. [DOI] [PubMed] [Google Scholar]
- De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:773–86. e1–7. doi: 10.1016/j.jaci.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nardo A, Yamasaki K, Dorschner RA, Lai Y, Gallo RL. Mast cell cathelicidin antimicrobial peptide prevents invasive group A Streptococcus infection of the skin. J Immunol. 2008;180:7565–73. doi: 10.4049/jimmunol.180.11.7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, et al. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol. 2001;117:91–7. doi: 10.1046/j.1523-1747.2001.01340.x. [DOI] [PubMed] [Google Scholar]
- Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009;41:602–8. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foelster Holst R, Reitamo S, Yankova R, Worm M, Kadurina M, Thaci D, et al. The novel protease inhibitor SRD441 ointment is not effective in the treatment of adult subjects with atopic dermatitis: results of a randomized, vehicle-controlled study. Allergy. 2010;65:1594–9. doi: 10.1111/j.1398-9995.2010.02417.x. [DOI] [PubMed] [Google Scholar]
- Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. 2012;12:503–16. doi: 10.1038/nri3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo RL, Nakatsuji T. Microbial symbiosis with the innate immune defense system of the skin. J Invest Dermatol. 2011;131:1974–80. doi: 10.1038/jid.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo RL, Ono M, Povsic T, Page C, Eriksson E, Klagsbrun M, et al. Syndecans, cell surface heparan sulfate proteoglycans, are induced by a proline-rich antimicrobial peptide from wounds. Proc Natl Acad Sci U S A. 1994;91:11035–9. doi: 10.1073/pnas.91.23.11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz DW. Idiopathic itch, rash, and urticaria/angioedema merit serum vitamin D evaluation: a descriptive case series. W V Med J. 2011;107:14–20. [PubMed] [Google Scholar]
- Hata TR, Kotol P, Boguniewicz M, Taylor P, Paik A, Jackson M, et al. History of eczema herpeticum is associated with the inability to induce human beta-defensin (HBD)-2, HBD-3 and cathelicidin in the skin of patients with atopic dermatitis. Br J Dermatol. 2010;163:659–61. doi: 10.1111/j.1365-2133.2010.09892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata TR, Kotol P, Jackson M, Nguyen M, Paik A, Udall D, et al. Administration of oral vitamin D induces cathelicidin production in atopic individuals. J Allergy Clin Immunol. 2008;122:829–31. doi: 10.1016/j.jaci.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa Y, Takai T, Nakamura T, Mitsuishi K, Gunawan H, Suto H, et al. Staphylococcus aureus extracellular protease causes epidermal barrier dysfunction. J Invest Dermatol. 2010;130:614–7. doi: 10.1038/jid.2009.257. [DOI] [PubMed] [Google Scholar]
- Hoff S, Oyoshi MK, Macpherson A, Geha RS. The microbiota is important for IL-17A expression and neutrophil infiltration in lesional skin of Flg(ft/ft) mice. Clin Immunol. 2015;156:128–30. doi: 10.1016/j.clim.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell MD, Boguniewicz M, Pastore S, Novak N, Bieber T, Girolomoni G, et al. Mechanism of HBD-3 deficiency in atopic dermatitis. Clin Immunol. 2006a;121:332–8. doi: 10.1016/j.clim.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Jin H, He R, Oyoshi M, Geha RS. Animal models of atopic dermatitis. J Invest Dermatol. 2009;129:31–40. doi: 10.1038/jid.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanangat S, Postlethwaite A, Hasty K, Kang A, Smeltzer M, Appling W, et al. Induction of multiple matrix metalloproteinases in human dermal and synovial fibroblasts by Staphylococcus aureus: implications in the pathogenesis of septic arthritis and other soft tissue infections. Arthritis Res Ther. 2006;8:R176. doi: 10.1186/ar2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, et al. Dysbiosis and Staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity. 2015;42:756–66. doi: 10.1016/j.immuni.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolar SL, Ibarra JA, Rivera FE, Mootz JM, Davenport JE, Stevens SM, et al. Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. Microbiologyopen. 2013;2:18–34. doi: 10.1002/mbo3.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–9. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, et al. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol. 2010;130:2211–21. doi: 10.1038/jid.2010.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Gallo RL. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009;30:131–41. doi: 10.1016/j.it.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DY, Guttman-Yassky E. Deciphering the complexities of atopic dermatitis: shifting paradigms in treatment approaches. J Allergy Clin Immunol. 2014;134:769–79. doi: 10.1016/j.jaci.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyden JJ, Marples RR, Kligman AM. Staphylococcus aureus in the lesions of atopic dermatitis. Br J Dermatol. 1974;90:525–30. doi: 10.1111/j.1365-2133.1974.tb06447.x. [DOI] [PubMed] [Google Scholar]
- Li D, Lei H, Li Z, Li H, Wang Y, Lai Y. A novel lipopeptide from skin commensal activates TLR2/CD36-p38 MAPK signaling to increase antibacterial defense against bacterial infection. PLoS One. 2013;8:e58288. doi: 10.1371/journal.pone.0058288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–3. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- Mallbris L, Carlen L, Wei T, Heilborn J, Nilsson MF, Granath F, et al. Injury downregulates the expression of the human cathelicidin protein hCAP18/LL-37 in atopic dermatitis. Exp Dermatol. 2010;19:442–9. doi: 10.1111/j.1600-0625.2009.00918.x. [DOI] [PubMed] [Google Scholar]
- Malley RC, Muller HK, Norval M, Woods GM. Vitamin D3 deficiency enhances contact hypersensitivity in male but not in female mice. Cell Immunol. 2009;255:33–40. doi: 10.1016/j.cellimm.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Man MM, Feingold KR, Thornfeldt CR, Elias PM. Optimization of physiological lipid mixtures for barrier repair. J Invest Dermatol. 1996;106:1096–101. doi: 10.1111/1523-1747.ep12340135. [DOI] [PubMed] [Google Scholar]
- Mao-Qiang M, Brown BE, Wu-Pong S, Feingold KR, Elias PM. Exogenous nonphysiologic vs physiologic lipids. Divergent mechanisms for correction of permeability barrier dysfunction. Arch Dermatol. 1995;131:809–16. doi: 10.1001/archderm.131.7.809. [DOI] [PubMed] [Google Scholar]
- Miedzobrodzki J, Kaszycki P, Bialecka A, Kasprowicz A. Proteolytic activity of Staphylococcus aureus strains isolated from the colonized skin of patients with acute-phase atopic dermatitis. Eur J Clin Microbiol Infect Dis. 2002;21:269–76. doi: 10.1007/s10096-002-0706-4. [DOI] [PubMed] [Google Scholar]
- Moniaga CS, Kabashima K. Filaggrin in atopic dermatitis: flaky tail mice as a novel model for developing drug targets in atopic dermatitis. Inflamm Allergy Drug Targets. 2011;10:477–85. doi: 10.2174/187152811798104881. [DOI] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. 2015;520:104–8. doi: 10.1038/nature14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, et al. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337:1115–9. doi: 10.1126/science.1225152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, et al. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503:397–401. doi: 10.1038/nature12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun. 2013;4:1431. doi: 10.1038/ncomms2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Gallo RL. Antimicrobial peptides: old molecules with new ideas. J Invest Dermatol. 2012;132:887–95. doi: 10.1038/jid.2011.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuga K, Cipolat S, Watt FM. Increased Bacterial Load and Expression of Antimicrobial Peptides in Skin of Barrier-Deficient Mice with Reduced Cancer Susceptibility. J Invest Dermatol. 2016;136:99–106. doi: 10.1038/jid.2015.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, et al. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414:454–7. doi: 10.1038/35106587. [DOI] [PubMed] [Google Scholar]
- Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–60. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–6. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- Redpath MB, Foster TJ, Bailey CJ. The role of the serine protease active site in the mode of action of epidermolytic toxin of Staphylococcus aureus. FEMS Microbiol Lett. 1991;65:151–5. doi: 10.1016/0378-1097(91)90295-l. [DOI] [PubMed] [Google Scholar]
- Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122:1285–94. doi: 10.1242/jcs.033969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilands A, Terron-Kwiatkowski A, Hull PR, O’Regan GM, Clayton TH, Watson RM, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39:650–4. doi: 10.1038/ng2020. [DOI] [PubMed] [Google Scholar]
- Saunders SP, Goh CS, Brown SJ, Palmer CN, Porter RM, Cole C, et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J Allergy Clin Immunol. 2013;132:1121–9. doi: 10.1016/j.jaci.2013.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A Wave of Regulatory T Cells into Neonatal Skin Mediates Tolerance to Commensal Microbes. Immunity. 2015;43:1011–21. doi: 10.1016/j.immuni.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauber J, Dorschner RA, Coda AB, Buchau AS, Liu PT, Kiken D, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest. 2007;117:803–11. doi: 10.1172/JCI30142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauber J, Dorschner RA, Yamasaki K, Brouha B, Gallo RL. Control of the innate epithelial antimicrobial response is cell-type specific and dependent on relevant microenvironmental stimuli. Immunology. 2006;118:509–19. doi: 10.1111/j.1365-2567.2006.02399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother. 2004;48:4673–9. doi: 10.1128/AAC.48.12.4673-4679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson CL, Kojima S, Getsios S. RNA interference in keratinocytes and an organotypic model of human epidermis. Methods Mol Biol. 2010;585:127–46. doi: 10.1007/978-1-60761-380-0_10. [DOI] [PubMed] [Google Scholar]
- Smith FJ, Irvine AD, Terron-Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38:337–42. doi: 10.1038/ng1743. [DOI] [PubMed] [Google Scholar]
- Vu AT, Baba T, Chen X, Le TA, Kinoshita H, Xie Y, et al. Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the Toll-like receptor 2-Toll-like receptor 6 pathway. J Allergy Clin Immunol. 2010;126:985–93. e1–3. doi: 10.1016/j.jaci.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Wachter AM, Lezdey J. Treatment of atopic dermatitis with alpha 1-proteinase inhibitor. Ann Allergy. 1992;69:407–14. [PubMed] [Google Scholar]
- Wang Z, MacLeod DT, Di Nardo A Commensal bacteria lipoteichoic acid increases skin mast cell antimicrobial activity against vaccinia viruses. J Immunol. 2012;189:1551–8. doi: 10.4049/jimmunol.1200471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanke I, Steffen H, Christ C, Krismer B, Gotz F, Peschel A, et al. Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J Invest Dermatol. 2011;131:382–90. doi: 10.1038/jid.2010.328. [DOI] [PubMed] [Google Scholar]
- Weidinger S, Illig T, Baurecht H, Irvine AD, Rodriguez E, Diaz-Lacava A, et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J Allergy Clin Immunol. 2006;118:214–9. doi: 10.1016/j.jaci.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Kubo A, Fujita H, Yokouchi M, Ishii K, Kawasaki H, et al. Distinct behavior of human Langerhans cells and inflammatory dendritic epidermal cells at tight junctions in patients with atopic dermatitis. J Allergy Clin Immunol. 2014;134:856–64. doi: 10.1016/j.jaci.2014.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.