

Abstract
Background
Chiroptera, the bats, are the only order of mammals capable of true self-powered flight. Bats exhibit a number of other exceptional traits such as echolocation, viral tolerance and, perhaps most puzzlingly, extreme longevity given their body size. Little is known about the molecular mechanisms driving their extended longevity particularly at the levels of gene expression and post-transcriptional regulation. To elucidate the molecular mechanisms that may underlie their unusual longevity, we have deep sequenced 246.5 million small RNA reads from whole blood of the long-lived greater mouse-eared bats, Myotis myotis, and conducted a series of genome-wide comparative analyses between bat and non-bat mammals (human, pig and cow) in both blood miRNomes and transcriptomes, for the first time.
Results
We identified 539 miRNA gene candidates from bats, of which 468 unique mature miRNA were obtained. More than half of these miRNA (65.1 %) were regarded as bat-specific, regulating genes involved in the immune, ageing and tumorigenesis pathways. We have also developed a stringent pipeline for genome-wide miRNome comparisons across species, and identified 37 orthologous miRNA groups shared with bat, human, pig and cow, 6 of which were differentially expressed. For bats, 3 out of 4 up-regulated miRNA (miR-101-3p, miR-16-5p, miR-143-3p) likely function as tumor suppressors against various kinds of cancers, while one down-regulated miRNA (miR-221-5p) acts as a tumorigenesis promoter in human breast and pancreatic cancers. Additionally, a genome-wide comparison of mRNA transcriptomes across species also revealed specific gene expression patterns in bats. 127 up-regulated genes were enriched mainly in mitotic cell cycle and DNA repair mechanisms, while 364 down-regulated genes were involved primarily in mitochondrial activity.
Conclusions
Our comprehensive and integrative analyses revealed bat-specific and differentially expressed miRNA and mRNA that function in key longevity pathways, producing a distinct bat gene expression pattern. For the first time, we show that bats may possess unique regulatory mechanisms for resisting tumorigenesis, repairing cellular damage and preventing oxidative stresses, all of which likely contribute to the extraordinary lifespan of Myotis myotis.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3227-8) contains supplementary material, which is available to authorized users.
Keywords: Bats, Illumina miRNA-Seq, Blood miRNA, Comparative miRNome, Myotis myotis
Background
Of all mammals, bats possess some of the most unique and peculiar adaptations that render them as excellent models to investigate the mechanisms of extended longevity and potentially halted senescence. They are considered the ‘Methusalehs’1 among mammals due to their exceptional and surprising longevity given their body size and metabolic rate [1, 2]. Typically mammals that are small have a high metabolic rate (e.g. shrews) and do not live for a long time [1, 3]. However, despite their small size and high metabolic rate bats can live for an exceptionally long time, with the oldest recorded bat (wild caught as an adult) ever recaptured being >41 years old (Myotis brandti, 7 g, Brandt’s bat) [2, 4, 5]. Indeed, to get a positive correlation between longevity and body size in mammals, bats must be removed from the analyses [1]. By comparing the ratio of expected longevity to that predicted from the ‘non-bat placental mammal’ regression line (longevity quotient- LQ) only 19 species of mammals are longer lived than man, one of these species being the naked mole rat and the other 18 are bats [1]. This suggests that bats have some underlying mechanisms that may explain their exceptional longevity.
MicroRNA (miRNA) are a subset of short (~22 bp) endogenous non-coding RNA that play a significant role in post-transcriptional regulation, via repression of translation. Typically, the 5′ end of a mature miRNA (seed region) predominantly determines the specificity and sensitivity in binding the target mRNA through either perfect or partial complementarity to the mRNA 3′-UTR, resulting in mRNA degradation or translational repression [6]. Since the first miRNA, lin-4, was discovered in 1993 [7], a multitude of miRNA have subsequently been identified, and implicated in the regulation of the vast majority of biological pathways including cell cycle regulation, metabolism, tumorigenesis, as well as immune response. However, the role of miRNA regulation in mammalian ageing and the onset of age-related diseases has only recently been established [8].
In mammals, various miRNA have been shown to be differentially expressed during ageing, most of which appear to be generally tissue-specific [8]. It was reported that the expression of miR-93 remarkably increased in liver tissues from extremely old mice and rats [9, 10]. miR-93 targets glutathione-S-transferases (GST) and SIRT1 genes [9, 10], which are vitally important in oxidative stress defense and metabolic homeostasis. Another example is miR-144. It is significantly up-regulated in aged human brains, which targets ataxin-1, the gene associated with spinocerebellar ataxin type 1 (SCA1) [11]. In addition to tissue-specific ageing, it is increasingly evident that many miRNA regulate gene expressions in well-known ageing pathways, most notably in the p53 tumor suppressor pathway (miR-34, miR-29 and miR-217, etc.) and insulin-like growth factor signaling pathway (IIS) (miR-17-92 family and miR-214, etc.) [8]. As altered patterns of miRNA expression can contribute to mammalian ageing, an investigation of the genome-wide miRNA profile (miRNome) may help elucidate the molecular mechanisms behind the extreme longevity of bats.
Despite being the second largest order of mammals (~1200 species) [12], there is a scarcity of genomic and transcriptomic bat resources. To date, only five well-annotated bat genomes (Myotis lucifugus, Myotis brandtii, Myotis davidii, Pteropus alecto and Pteropus vampyrus) are publically available. Phylogenomic studies of bat genomes and other mammalian species reveal that a number of genes are under positive selection in bats [5, 13, 14]. These genic adaptations have been correlated with traits such as echolocation, powered flight, hibernation, immunity and longevity. For example, specific non-synonymous mutations in GHR and IGF1R, key ageing-related genes, were detected in several long-lived vespertilionid bats (M. brandtii, M. lucifugus and Eptesicus fuscus) [5], while a large proportion of genes involved in DNA repair (RAD50, KU80, MDM2 etc.) and the NF-кB pathway (c-REL and ATM2 etc.) were reported to be under positive or divergent selection in M. davidii and P. alecto [14]. These results suggest bats may better detect and repair DNA damage. Intriguingly, positive selection was also detected in mitochondrial-encoded and nuclear-encoded oxidative phosphorylation genes in bats, which may explain their efficient energy metabolism necessary for flight [15]. Apart from comparative genome analysis, only a small number of transcriptomic studies on bats using mRNA-Seq and miRNA-Seq technologies have been carried out, focused primarily on the characteristics of hibernation [16], immunity [17, 18], echolocation [19] and phylogeny [20]. However, the molecular mechanisms of adaptations affecting longevity are still far from understood, especially with respect to gene regulation.
In the present study, we sequenced six small RNA libraries from whole blood sampled from wild-caught greater mouse-eared bats (Myotis myotis) and for the first time made genome-wide comparisons of both miRNomes and mRNA transcriptomes between bat and non-bat mammalian species (human, pig and cow). Whole blood is a nonlethal sample type that can reflect the real time overall physiological status of an individual [21]. Our earlier study showed that only a few drops of whole blood (<200 μl) contains a high percentage of genome-wide and body-wide transcripts [22]. We then compared both the corresponding bat blood miRNome and transcriptome to those of non-bat mammals (human, pig, cow), to elucidate bat-specific gene expression as well as post-transcriptional regulation.
The profiling of the M. myotis blood miRNome showed a large number of bat-specific miRNA involved in regulating important pathways related to immunity, tumorigenesis and ageing. Comparative analyses of both miRNomes and transcriptomes also revealed distinctive longevity mechanisms in bats. Several up-regulated miRNA possibly act as tumor suppressors. Gene Ontology (GO) enrichment analysis of differentially expressed protein-coding genes showed that up-regulated genes in bats compared to other mammals were mainly involved in mitotic cell cycle and DNA damage repair pathways while a high number of down-regulated genes were enriched in mitochondrial metabolism. The results and data presented here show unique regulatory mechanisms for protection against tumorigenesis, reduced oxidative stress, and robust DNA repair systems, likely contribute to the extraordinary longevity of bats.
Results
Bioinformatic analyses of M. myotis blood miRNome
We pooled the raw reads of all six libraries together (two individuals, three technical replicates each) to represent the M. myotis blood miRNome (Fig. 1a). A total of ~246.5 million single-end reads were generated on the Illumina HiSeq 2000 sequencer, with the uniform length of 50 bp. After adaptor trimming, size selection and base-calling filtering, we retained a final set of 202.9 million (82.3 %) high-quality post-processed reads for miRNA identification and further analysis. With strict criteria, the miRDeep2 pipelines predicted 539 pairs of mature miRNA and their corresponding precursors from which 203 were identified as known miRNA, with the remaining 336 predicted to be novel (Additional file 1: Table S1). As the same mature miRNA can be cleaved from different precursors, we acquired 468 unique mature miRNA after removing duplicates (Additional file 1: Table S2).
Fig. 1.
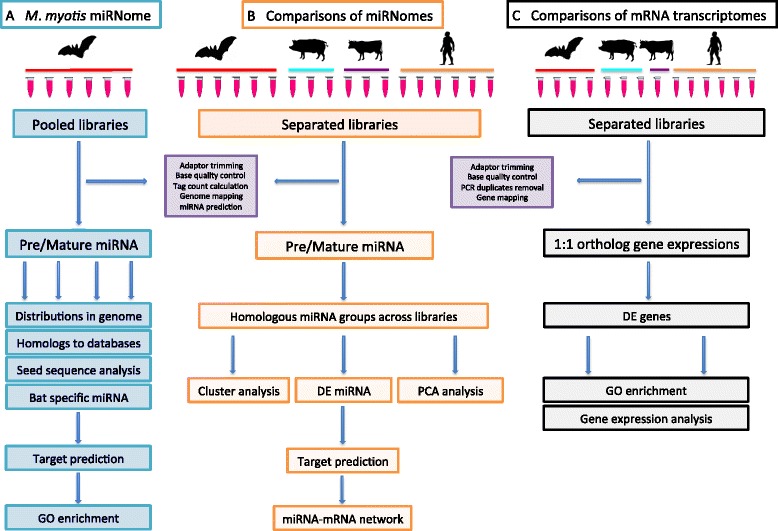
The workflow of analyses and bioinformatic pipelines. a The pipeline for identification and analyses of M. myotis blood miRNA. b The pipeline for comparisons and analyses of blood miRNomes between bat, human, pig and cow. c The pipeline for comparisons and analyses of blood mRNA transcriptomes between bat, human, pig and cow
The bioinformatic analysis indicated that the miRNA (86.1 %) were mainly between 20 bp and 23 bp in length, with the peak at 22 bp (Fig. 2a), and their expression spanned several orders of magnitudes (Fig. 2b). The analysis of the genomic coordinates showed 214 miRNA (39.8 %) were located in the intergenic regions, followed by 196 (36.4 %) in the exonic regions as the second largest category (Fig. 2c). Interestingly, we also detected 18 miRNA traversing the boundaries of exons and introns. In order to annotate and evaluate the M. myotis blood miRNome, the predicted mature miRNA were compared to miRBase (release 21) and a collection of customized bat mature miRNA database (see Methods). Of all 468 unique mature miRNA, only 180 (38.5 %) and 166 (35.5 %) had 100 % identical entries in the miRBase and the customized bat miRNA database respectively, with more than half having no hits in both databases (Fig. 2d, Additional file 2: Figure S1). Typically, silencing of the target mRNA relies mainly on complementarity to bases 2–7 of mature miRNA, the ‘seed region’. We analyzed all seed regions of the blood miRNome, and the result revealed a set of 356 unique seeds, with the most frequent seed ‘gaggua’ and 68 other seeds appearing more than once. Not surprisingly, we found 29 novel seeds that did not exist in miRBase (release 21) (Additional file 1: Table S3).
Fig. 2.
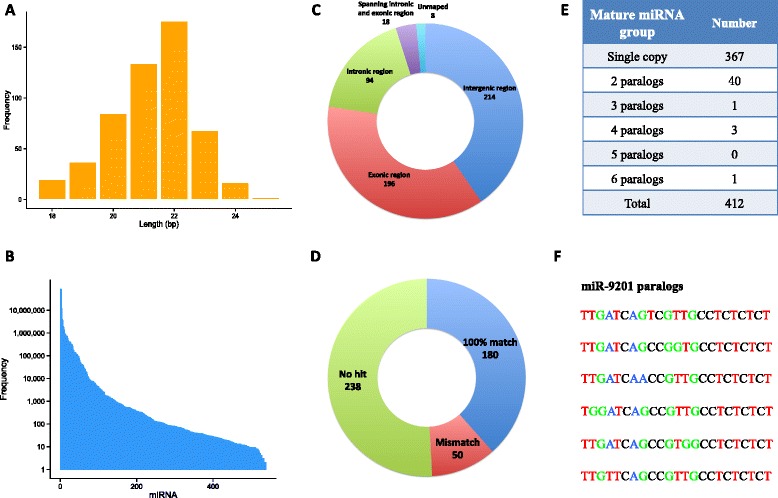
The bioinformatic analyses of the M. myotis blood miRNome. a The length distribution of M. myotis mature miRNA. b The frequency distribution of the M. myotis mature miRNA. c The genome locations of the miRNA genes (precursors). d The homology of the M. myotis mature miRNA to all the entries in miRBase (release 21). e The paralogous groups of M. myotis mature miRNA f) An example of the miRNA paralogous groups. The members in this paralogous group had consistent top BLAST hit, miR-9201
We also ascertained the paralogous groups amongst the identified miRNA, resulting in 412 groups derived from 468 unique mature miRNA using our strict criteria (see Methods). 367 mature miRNA (78.4 %) were identified as single copy while 80 (17.1 %) were grouped into 40 pairs (Fig. 2e). We also found a group (miR-9201) containing 6 miRNA (Fig. 2f). To assess the validity of the procedure of paralog grouping, we validated all the members in 45 miRNA groups that had more than one member by BLAST-ing them against miRBase (release 21). Apart from 9 groups which had no hits, in 34 out of 36 groups (94.4 %) all members consistently had matching top BLAST hits in miRBase, suggesting our paralog grouping procedure was reliable in most cases. Furthermore, 351 miRNA genes (65.1 %) were discovered as bat-specific due to the failure to map to any of the 13 selected non-bat mammalian genomes (Additional file 1: Table S4, S5). However, these novel miRNA had a relatively lower level of expression than that of conserved miRNA, which appears to be a common phenomenon across species (Fig. 3a). The targets of these novel miRNA were subsequently predicted, and Gene Ontology enrichment analysis of the targets was carried out. Ranked by the FDR values, the most significantly enriched GO terms were ‘ATP binding’ (GO: 0005524), followed by ‘zinc ion binding’ (GO: 0008270) and ‘nucleolus’ (GO: 0005730). The top 10 enriched GO terms and their interaction networks were shown in Fig. 3b. However, it is a possibility that different species may use species-specific miRNA but target the same pathway. To ascertain the unique GO terms that bat-specific miRNA were involved in, we also analyzed the GO terms where the target genes of pig-, cow- and human-specific miRNA were enriched, respectively. By comparing the top 20 GO terms, we found that some common biological processes overlapped across species (bat, pig, cow and human), such as ‘ATP binding’ (GO: 0005524) and ‘zinc ion binding’ (GO: 0008270). However, we also noticed that many enriched GO terms were species-specific (Additional file 1: Table S6).
Fig. 3.
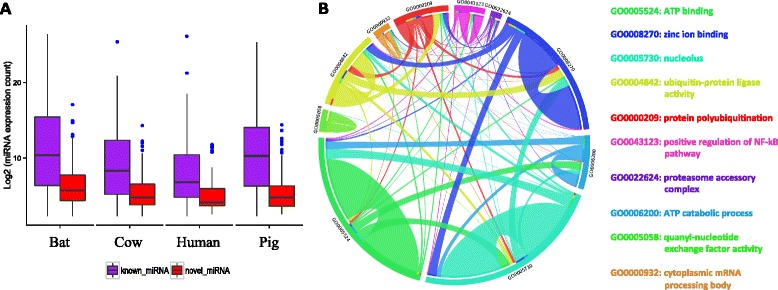
The analyses of bat-specific miRNA. a The distribution of the known miRNA and novel miRNA expression in bat, human, pig and cow, respectively. The expression data were log 2 transformed. b The top 10 enriched GO terms of the targets of the bat-specific miRNA and their interaction networks. The networks show the number of targeted genes which are shared within different GO terms
Several up-regulated miRNA in bats may act as tumor suppressors
Interspecific gene expression variation of conserved miRNA can result from changes in selection pressures or through random genetic drift, which may explain the distinct patterns observed in bats. To identify and compare the conserved homologous miRNA across species, we employed the data from other studies focusing on the blood miRNome of human (Homo sapien), pig (Sus scrofa) and cow (Bos taurus) using miRNA-Seq technology. All the libraries (Nbat = 6, Nhuman = 6, Npig = 3, Ncow = 3) were separately analyzed, and the same quality control steps and miRNA identification pipelines, which were used to identify the bat blood miRNome, were applied. The statistical information on each library is summarized in Additional file 1: Table S7.
According to our stringent pipeline, only 37 conserved miRNA groups were identified across all libraries, 18 of which had more than one paralog in each library. To examine expression differences between species, we conducted a series of analyses on these 37 conserved miRNA groups, including hierarchical clustering analysis, principal component analysis (PCA) and differential expression (DE) analysis. The hierarchical clustering revealed several distinct miRNA expression patterns, suggesting their co-expression and common pathways they may be involved in (Fig. 4a). Not surprisingly, the libraries clustered together based on their respective species of origin (Fig. 4a). PCA also demonstrated that variations in the conserved miRNA expression differed primarily by species, exhibiting specific regions for each species on the plot (Fig. 4b). From the DE miRNA analysis, 12, 15 and 14 candidates were detected as significantly differentially expressed between bat and human, pig, cow respectively, with six DE miRNA common to all (Fig. 4c). The summary of the six DE miRNA common to all species is described in Fig. 5. Briefly, all DE candidates were single copy miRNA across all libraries, and 4 DE miRNA (miR-101-3p, miR-16-5p, miR-143-3p and miR-155-5p) were up-regulated in bats while 2 (miR-125-5p and miR-221-5p) were down-regulated. These DE miRNA are involved in a variety of biological pathways, but are primarily associated with cancer, immunity, apoptosis and ageing pathways. Given that a large abundance of protein-coding genes may be modulated by the same miRNA, we built a miRNA-mRNA interaction network using miRanda with strict filtering parameters (Fig. 6). In this network, 149 pairs of miRNA-mRNA were predicted, corresponding to 145 unique protein-coding genes regulated by 6 DE miRNA.
Fig. 4.

Comparative analyses of blood conserved miRNomes amongst bat, human, pig and cow. a Cluster analysis of conserved miRNA expression levels across all 18 libraries. The raw expression counts were normalized using the quantile method. The rows were clustered using Pearson correlation method (normal distribution, P > 0.05), while the columns were clustered using Spearman correlation method (non-normal distribution, P < 0.05). b Principal component analysis (PCA) of conserved miRNA expression levels across all 18 libraries. The raw expression counts were normalized using the quantile method. The proportion of the variance is indicated for each principal component. c The differentially expressed miRNA between bat and other mammalian species. 6 DE miRNA candidates were obtained after intersecting the results
Fig. 5.
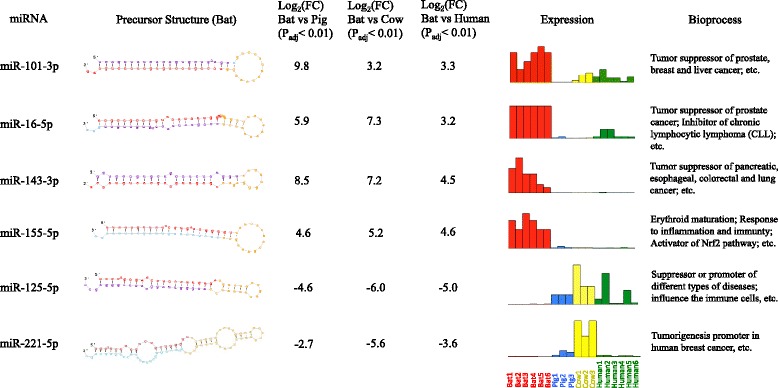
The summary of 6 differentially expressed (DE) miRNA. For the precursor structure, nucleotides highlighted in red indicate observed mature sequences, yellow for predicted stem loop region, blue for predicted star sequence and purple for observed star sequence. The fold changes between bat and other species were calculated by DESeq respectively. The relative miRNA expressions in each library were normalized using quantile normalization from the raw counts. The supporting references for the bioprocesses that miRNA are involved in are listed in the Additional file 2: Text S1
Fig. 6.
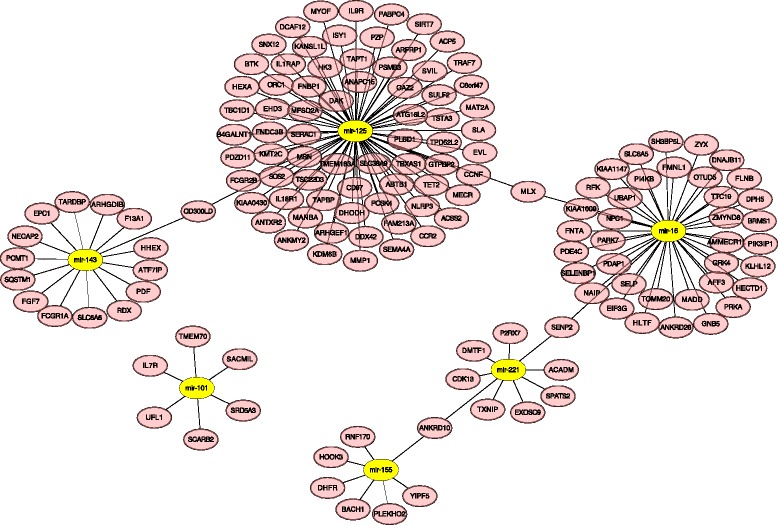
The interaction of 6 differentially expressed (DE) miRNA and their mRNA targets. The ellipses, highlighted in yellow, indicated the 6 DE miRNA, and the ellipses connected with the miRNA represent their mRNA targets
Unique gene expression patterns indicate reduced respiratory activity and robust cellular maintenance systems in bats
To better elucidate the molecular mechanisms underlying the many unique characteristics of bats, we also employed and analyzed the mRNA-Seq transcriptome data for these 4 species (Nbat = 4, Nhuman = 6, Npig = 3, Ncow = 1). For bat, pig and cow, the blood mRNA transcriptomes were generated in the same studies corresponding to their blood miRNomes respectively, while human mRNA transcriptomes and miRNomes were employed from different sources (Table 1, Additional file 1: Table S8). In total, 3150 1:1 single-copy orthologous genes were extracted from their respective genomes, 3065 of which were retained after quality control (see Methods), and subsequently used for DE gene analysis. Analyzed by DESeq, 1366, 864 and 1166 genes were identified as DE genes between bat and human, pig, cow respectively (FDR < 0.05). Among those DE genes, 441, 216 and 364 genes were up-regulated while 802, 925 and 648 were down-regulated in bats compared to human, pig and cow (Fig. 7a and b). Overall, 127 genes were up-regulated and 364 genes were down-regulated in bats compared to other non-bat mammals (Fig. 7a and b). Gene Ontology enrichment analysis showed that up-regulated genes were mainly involved in DNA repair (GO: 0006281), DNA replication initiation (GO: 0006270) and mitotic cell cycle (GO: 0000278) while down-regulated genes were enriched in mitochondrion (GO: 0005739), glycolipid metabolic process (GO: 0006664) and ncRNA processing (GO: 0034470) (Fig. 7c and d).
Table 1.
The publically available data employed for the comparative miRNomes and transcriptomes analyses in this study
| Comparative miRNomes | Comparative transcriptomes | ||||||
|---|---|---|---|---|---|---|---|
| Species | Individual | Library | Data source | Species | Individual | Library | Data source |
| Bat (NInd = 2, NLib = 6) | B1 | Bat1 | this study | Bat (NInd = 2, NLib = 4) | B1 | Huang, et al. | |
| Bat2a | Bat1a | ||||||
| Bat3a | Bat2a | ||||||
| B2 | Bat4 | B2 | |||||
| Bat5a | Bat3a | ||||||
| Bat6a | Bat4a | ||||||
| Pig (NInd = 3, NLib = 3) | P1 | Pig1a | Bao, et al. | Pig (NInd = 3, NLib = 3) | P1 | Pig1a | Bao, et al. |
| P2 | Pig2a | P2 | Pig2a | ||||
| P3 | Pig3a | P3 | Pig3a | ||||
| Cow (NInd = 3, NLib = 3) | C1 | Cow1 | Bao, et al. | Cow (NInd = 3, NLib = 1) | C1 | Cow1b | Bao, et al. |
| C2 | Cow2 | C2 | |||||
| C3 | Cow3 | C3 | |||||
| Human (NInd = 6, NLib = 6) | H1 | Human1 | Leidinger, et al. | Human (NInd = 6, NLib = 6) | H7 | Human1 | Memczak, et al. |
| H2 | Human2 | H8 | Human2 | ||||
| H3 | Human3 | H9 | Human3 | ||||
| H4 | Human4 | H10 | Human4 | ||||
| H5 | Human5 | H11 | Human5 | ||||
| H6 | Human6 | H12 | Human6 | ||||
a in each line indicates miRNome and transcriptome libraries were generated from the same individual
b indicates cow transcriptome was pooled from three individual libraries for sequencing
Fig. 7.
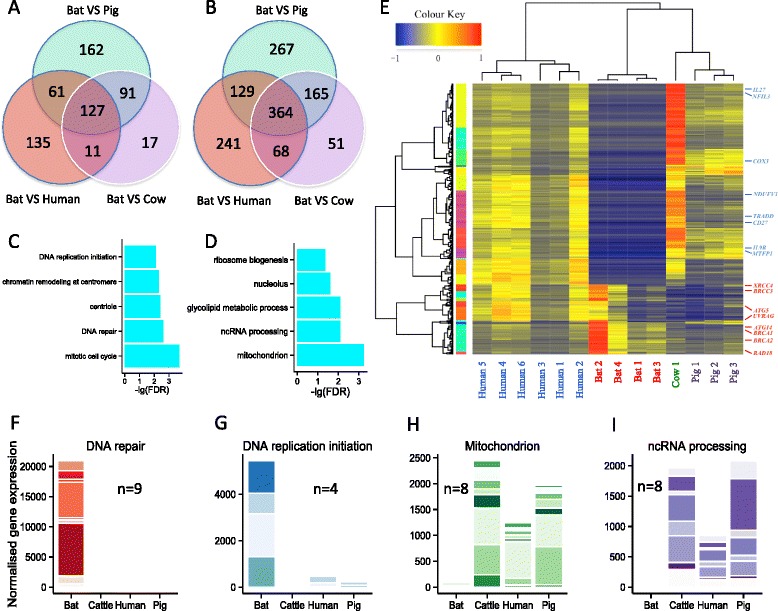
Comparative analyses of mRNA transcriptomes among bat, human, pig and cow. a-b The numbers of up-regulated and down-regulated genes in bat, respectively. The results of the three separate comparisons were intersected (bat vs human, bat vs pig, bat vs cow). c-d The top 5 Gene Ontology enriched terms of up-regulated and down-regulated genes in bat, respectively. The enriched terms with Padj-value < 0.05 were considered statistically significant. e The heatmap of 491 differentially expressed genes between bat and other mammals. The raw expression counts were normalized using the Trimmed of M-value (TMM) method. The rows were clustered using Pearson correlation method (normal distribution, P > 0.05), while the columns were clustered using Spearman correlation method (non-normal distribution, P < 0.05). f-i The relative accumulative gene expressions under certain enriched GO terms across bat, human, pig and cow. The raw expression counts were normalized using Trimmed mean of M-value (TMM) method, and the average gene expressions from the libraries of the same species were calculated and further summed based on the enriched GO terms
Discussion
Mining miRNA from deep sequencing reads is highly dependent on the genome, which enables the prediction of their compact pre-miRNA hairpin structures. In this study, we employed the M. lucifigus genome as a proxy reference since the M. myotis genome has not yet been published. Although suffering some losses in the number, most abundant and conserved miRNA can be identified in a closely related species’ genome [23]. This is also shown by large number of miRNA genes (539) found in this study using the strict pipeline, the number of which falls within the range observed for other bat studies using miRNA-Seq, although different tissues and sequencing depths were used [17, 20]. The distribution of miRNA genes displayed no particular bias for genomic location. However, the largest category of miRNA (39.8 %) was in the intergenic regions (Fig. 2c), which is similar to human (49.9 %) and mouse (44.8 %) [24]. A large number of intragenic miRNA, which were located within introns and exons, were also found, and it is hypothesized that they may co-express with their host genes because of shared promoter sequences [25, 26]. Interestingly, we also detected 18 miRNA genes spanning the boundaries of exons and introns (Fig. 2c). These findings suggest the complexity of transcriptional splicing activity, and that the locations of miRNA can affect their expression and function. In addition, we identified and analyzed the paralogous groups in the M. myotis blood miRNome, observing that a large proportion of mature miRNA (78.4 %) were single-copy miRNA (Fig. 2e). Paralogous miRNA can shed light on gene duplication events, evolutionary history and post-transcriptional RNA editing. However, few studies on mammalian miRNA paralogs are available for comparison and analysis, probably due to the poor annotation of noncoding regions of mammalian genomes.
A significant proportion of miRNA (Fig. 2d) had no homologs in other species, reflected by many novel seed regions and bat-specific miRNA identified in this study. Potentially this could result from a lack of taxon-wide miRNA studies based on well-assembled and annotated genomes. However, a recent study estimated that the proportion of miRNA orthologous pairs between human and mouse was only 16 %, whose genomes are better annotated, leaving a large abundance of species-specific miRNA [24]. miRNA experience a rapid birth and death rate across species, and species-specific miRNA are considered to be evolutionarily recent. Not surprisingly, the expression level of these novel miRNA tended to be significantly lower (P < 0.05) than that of conserved miRNA (Fig. 3a). Abundantly expressed miRNA are highly likely to be functionally important, evolutionarily conserved, and under purifying selection; while novel miRNA with low expression level may have a comparatively weaker effect on their potential target transcripts, yet could convey species-specific adaptations. Occasionally, miRNA with low expression may be selectively favored and will be maintained in the genome. A direct correlation between the numbers of miRNA and biological complexity has been proposed, suggesting that miRNA innovation may play a key role in the emergence of increasingly novel and complex traits [27]. To understand the function of the bat-specific miRNA, we constructed the miRNA-mRNA network and performed function enrichment analysis on their targets. Many of the pathways and functions were associated with metabolism, immunity and protein homeostasis (Fig. 3b). Simultaneously, analyses of species-specific miRNA from other mammals (pig, cow and human) revealed common GO terms shared with bat, suggesting different, convergent species-specific miRNA can target the same pathways. The bat-specific GO terms, however, indicated the acquisition of novel miRNA in bats which regulate ageing and cellular senescence.
For bat, the most representative enriched terms, which are highly related to ageing and cell senescence, were ‘aging’ (GO: 0007568) and ‘guanyl-nucleatide exchange factor activity’ (GO: 0005085). Remarkably, some cell cycle mediators which accelerate cell growth and proliferation were included under the term ‘aging’, such as GRB2, CASP7, SHIP1 and GSN. It is commonly accepted that accelerated cell growth directly coincides with tumorigenesis and cancer progression. Gelsolin (GSN), which functions as a cell growth promotor and apoptosis inhibitor, has been implicated in the oncogenesis of certain cancers [28]. Previous study showed its overexpression promoted the proliferation and mobility of hepatocellular carcinoma (HCC) cells [28]. Another example is GRB2, an activator of Ras/Raf/MEK/mitogen-activated protein kinase pathway (MAPK). It is reported that its downregulation could inhibit breast cancer cell growth [29]. Interestingly, we also noticed SHIP1, which limits blood cell production and immune regulatory cell numbers. The inhibition of SHIP1 was shown to trigger apoptosis of blood cancer cells, suggesting the potential target for the treatment of hematological malignancies [30]. As well as inhibition of cell growth, bats may suppress inflammation and tumorigenesis by targeting guanyl-nucleotide exchange factors (GEFs). GEFs play a fundamental role in many diverse signaling pathways related to immunity, cell growth and tumorigenesis. RasGRP1, 2, 3 and SOS2 are all GEFs which activate Ras small GTPases and are some of the 50 GEFs targeted by bat-specific miRNA predicted in this study. RasGRP1 and 3 are recruited during the signaling cascades instigated by T-cell and B-cell receptor stimulation, respectively [31]. They are essential for the activation of Ras and promoting proliferation in T and B-cells in response to infection. Unsurprisingly, these genes have been shown to be overexpressed in a myriad of cancers, including melanoma, T-cell acute lymphoblastic leukemia (T-ALL) and prostate cancer [32, 33], making inhibitors of these proteins possible therapeutics. These results imply that M. myotis are capable of regulating cell cycle regulators as well as Ras small GTPases through degradation of direct or indirect activators. This may prevent tumor or cancer development and immune-related disorders, contributing to their long lifespan.
Comparisons of conserved miRNA expressions across species can also reveal distinct adaptations. However, there are two major obstacles to compare miRNA across species. First, there were limited miRNA resources that could be used for comparison. We employed data from human, pig and cow because they had both blood miRNomes and transcriptomes sequenced from healthy adults using NGST. This makes the investigation of the correlation between miRNA and mRNA expressions possible. Second, the existence of paralogs made it difficult to accurately quantify the orthologous miRNA across species [34]. Therefore, we calculated the geometric mean to represent the expression of each paralogous group. This is because all these paralogs were under strict inspection that they shared the same seed region, suggesting a high possibility of targeting the same mRNA. Due to a large number of bat-specific miRNA identified, it is not surprising that only 37 conserved orthologous groups were found across all 18 libraries. Cluster analysis and PCA indicated the differences within biological or technical replicates were much smaller than the differences between species (Fig. 4a, b).
Six conserved miRNA were found differentially expressed between bat and non-bat species (human, pig and cow), most of which were highly likely to be biologically functional in oncogenic or immune pathways. Among the 4 up-regulated miRNA, the expression of miR-101 in bat was at least three times than that in other mammals (Fig. 5). It is reported that miR-101, which functions as a tumor suppressor, is down-regulated in several cancers. Its overexpression inhibits cell proliferation and promotes apoptosis in breast cancer cells by inhibiting Jak2 [35]. Similarly, expressions of miR-16 and miR-143 inhibit cell proliferation and suppress tumorigenesis, and miR-143 has been observed to be down-regulated in cervical cancer [36]. Importantly, one down-regulated miRNA, miR-221, was reported to function as a tumorigenesis promoter in human breast cancer. These findings demonstrate M. myotis are likely better at suppressing tumorigenesis than human, pig and cow, achieved by overexpression of miRNA associated with tumor suppression and inhibition of miRNA which promotes tumorigenesis. miR-155 is a multifunctional miRNA regulating inflammation, immunity and oncogenic pathways [37]. Interestingly, miR-155 also targets Bach1, a negative regulator of Nrf2, which is a core activator of the antioxidant response signaling pathway [38]. Activation of this pathway is a major mechanism for cells to defend against oxidative and electrophilic stresses by enhancing cellular antioxidant activity. This indicates that M. myotis efficiently degrade Bach1 mRNA and activate the Nrf2 pathway, thus increasing defenses against cellular damage caused by reactive oxygen species (ROS). These miRNA regulatory mechanisms through which M. myotis can prevent tumorigenesis, inflammatory diseases and cellular oxidative damage, may explain their incredible longevity. Interestingly, transcriptome and proteome analysis of another long-lived mammal, the naked mole rat, also indicated their resistance to tumorigenesis, and insusceptibility to oxidative damage with age [39]. This demonstrates that long-lived mammals may regulate and control common ageing pathways to reach old age.
To address the uniqueness of bats from a different perspective, we also compared the RNA-Seq transcriptomes between bat and non-bat mammals. However, it is also a challenge to compare transcriptomes across species due to the existence of many paralogs. The quantification of paralogs calculated by mapping the clean reads to the genome cannot reflect the true expression, as paralogs are highly similar so that it is impossible to tease apart the reads having multiple mapping coordinates. Therefore, we only obtained all 1:1 single-copy orthologous protein-coding genes across bat, human, pig and cow genomes for analysis. Surprisingly, we identified many more down-regulated genes than up-regulated genes in bat (Fig. 7a, b). It was a concern that the quality of bat transcriptomes might be lower than that of other species, leading to the identification of massive down-regulated genes in bat. However, this concern was obviated by a measure of transcriptome completeness (CEGMA) which showed their qualities were highly comparable (bat: 64.9 %, human: 70 %, pig: 69 % and cow: 65.7 % in completeness).
Based on the enrichment results, a large number of mitochondria-related genes were down-regulated in bat (Fig. 7c, d). Mitochondria are the powerhouse of the cell generating adenosine triphosphate (ATP) from oxygen and the products of glycolysis. However, reactive oxygen species (ROS), such as superoxides and peroxides generated by the premature reduction of oxygen, can damage DNA, RNA and proteins, contributing to the pathophysiology of ageing. It is noteworthy that many of these down-regulated genes encode proteins, such as NDUFV1 and NDUFA1, which are components of electron respiratory chain complex I, the primary site of ROS generation [40]. Reduced expression of mitochondrial-associated transcripts may represent a general mechanism where cells protect themselves against oxidative stress [41]. Our finding is consistent with the study of oxidative stress in another long-lived bat M. lucifugus, concluding that their mitochondria produce half to one-third the amount of hydrogen peroxide per unit of oxygen consumed than that of relatively short-lived mammals of similar size, short-tail shrew and white-footed mouse respectively [42].
On the other hand, Myotis bats seem to have evolved a heightened DNA repair system, showing up-regulation of several DNA repair genes. For example, UVRAG, encodes a protein which interacts with DNA repair enzymes to aid DNA double-strand break repair and plays a fundamental role in centrosomal and chromosomal stability [43]. Interestingly, UVRAG also interacts with Beclin 1 protein, leading to the activation of autophagy and inhibition of tumorigenesis [43]. The tumor suppressor genes BRCA1 and BRCA2, which were both up-regulated in bats, encode proteins that are pivotal in maintaining genomic stability by promoting efficient and accurate repair of DNA double-strand breaks. Mutations in these two genes will lead to the development of various types of cancers [44]. We conclude that decreased levels of oxidative stress and heightened DNA repair activity may protect bats against genomic instability, contributing to halted ageing in the long-lived M. myotis.
Conclusions
In summary, we established the first atlas of the blood miRNome of the long-lived bat, M. myotis, identifying a large number of miRNA involved in key ageing-related pathways. The comparative analyses of miRNomes and transcriptomes between bats and other mammals resulted in the discovery of many bat-specific miRNA, differentially expressed miRNA and mRNA, and their regulatory networks. Our results show that M. myotis exhibit increased protection against tumorigenesis and oxidative damage, heightened cellular damage repair activity and better genomic stability, all of which likely contribute to their exceptional longevity.
Methods
Bat blood sampling, total RNA extraction and miRNA sequencing
Two adult female greater mouse-eared bats (Myotis myotis) were captured in Béganne, western France (47°35′N, 2°14′W). For each bat, 80–200 μL of blood (depending on the weight of the individual) was collected from one of the two uropatagial veins. Total RNA was isolated from the whole blood using RNAzol® BD kit (catalog number RB192, Molecular Biology Center, Inc) according to the manufacturer’s instructions with minor modifications. Bat sampling, blood collection and total RNA extraction were carried out as described in our previous study [22]. The concentration and quality of total RNA were quantified and evaluated for each sample by a Nanodrop-ND-1000 Spectrophotometer (Thermo Scientific) and a 2100 Bioanalyzer (Aligent Technologies), respectively. RNA had a 280/260 ratio >1.8, RNA integrity number (RIN) > 8 and total RNA > 1 μg, and was further processed for Illumina deep sequencing. Six small RNA libraries from two 2-years-old adult female individuals were prepared and constructed from the total RNA using the TruSeq Small RNA Preparation Kit (Illumina) following the manufacturer’s protocols, and were sequenced to generate 50 bp single-end reads on HiSeq 2000 (Illumina, USA) platform by two different companies (Table 2). The raw small RNA reads have been deposited in National Center for Biotechnology Information (NCBI) under the Bioproject: PRJNA267654.
Table 2.
Summary of bat miRNA sequencing and identification
| Library | Bat1 | Bat2 | Bat3 | Bat4 | Bat5 | Bat6 | Pooled |
|---|---|---|---|---|---|---|---|
| Individual | B1 | B1 | B2 | B2 | B1 | B2 | |
| Sequencing Company | 1 | 1 | 1 | 1 | 2 | 2 | |
| No. of Raw Reads | 24662730 | 44380583 | 28680178 | 93925331 | 27924234 | 26889119 | 246462175 |
| No. of miRNA precursors | 257 | 308 | 264 | 390 | 383 | 339 | 539 |
| No. of unique mature miRNA | 216 | 263 | 224 | 336 | 323 | 278 | 468 |
Profiling of M. myotis miRNA
The workflow for miRNA identification and analyses is portrayed in Fig. 1a. The reads from the six libraries were pooled together to represent the blood miRNome of M. myotis. The identification of miRNA sequences was based mainly on the miRDeep2 pipeline. However, the raw reads were initially inspected and filtered as a quality control step. Briefly, the 3′ adaptor sequence (TGGAATTCTCGGGTGCCAAGGAACTCCAA) was cut using cutadapt (v1.3) [45], and only the trimmed reads with the read lengths between 18 and 25 bp were preserved, and further filtered by base quality using NGS-toolkit (v2.3) [46]. The sequences containing unknown bases (N’s) were discarded, and those with at least 80 % of bases, with quality scores above Q30, were retained as clean reads. The identical clean reads were compressed to single entries with the headers designating their frequencies using Mapper.pl, a module of miRDeep2 pipeline [47]. The unique sequence tags were then mapped to the genome and analyzed by miRDeep2.pl to predict mature miRNA and their corresponding precursors. As the genome of M. myotis is not available, the well-annotated M. lucifugus genome was employed in this study. The predicted miRNA candidates were filtered by the tag count and the probability of true positive estimated by miRDeep2. The candidates, with a count less than 5 and true positive probability lower than 60 %, were deemed unreliable and were excluded from the further analysis.
Bioinformatic analyses of M. myotis miRNome
The locations of predicted miRNA genes were determined by mapping the precursors to exonic, intronic and intergenic regions of the M. lucifugus genome using bowtie (v 0.12.7) [48], allowing no mismatches (bowtie -v 0 --best). The exonic, intronic and intergenic regions were extracted from the genome based on the annotation information using Bedtools (v.2.4.3) [49]. In order to better annotate the miRNome, we collected bat miRNA from multiple studies, including P. alecto [17], M. lucifugus [20] and E. fuscus [20]. Mature miRNA were respectively compared to the miRBase (release 21) [50] and this customized bat miRNA dataset using BLASTN, with the parameters optimized for short read alignment (−evalue 0.1 -strand plus -word_size 4 -gapopen −8 -gapextend −6 -penalty −4) [51]. Based on their top BLAST hits, miRNA were categorized into the entire match, mismatch and no-hit groups. The entire match is defined by allowing maximum 2 bp overhanging at both ends for each sequence, while the mismatch group only allows 1 bp overhanging at each end but can tolerate 1 bp mismatch or indel. The sequences with other conditions of alignments and no hits were categorized into the no-hit group. The seed sequences of predicted M. myotis miRNA were analyzed and novel seeds discovered by comparing all seed sequences to those of all mature miRNA in the miRBase database (release 21). To identify groups of paralogous mature miRNA in the M. myotis miRNome, we performed sequence clustering analysis using CD-HIT [52] based on the sequence similarity. The parameters were set as ‘-c 0.9 -aL 0.9’, signifying that the sequences would cluster into groups if they share at least 90 % of both similarities and sequence overlaps in length. These preliminary paralogous groups were manually inspected, and the final set of paralogous groups was determined by excluding the alignments with mismatches > 2 and overhanging > 1 at each end. Each group was validated by comparing its members to the miRBase database (release 21) using miRBase BLAST web service, and their top BLAST hits were inspected.
In addition, bat-specific miRNA genes were determined by mapping all predicted miRNA precursors to 13 non-bat mammalian genomes (Additional file 1: Table S4) using bowtie (v0.12.7), allowing 3 mismatches per alignment. These 13 non-bat mammals were selected to represent the vast ecological and evolutionary diversity within mammals, and their genomes were also sequenced, assembled and annotated to a high standard. miRNA genes that did not map to any of the genomes above were considered to be bat-specific. The targets of these bat-specific miRNA were also predicted and analyzed. Briefly, we obtained the assembled M. myotis blood mRNA-Seq transcriptome from our previous study [22], which also had corresponding miRNome sequenced by Illumina for each individual used in this study (Table 1). The 3′-UTR sequences were extracted from the assembled transcripts which had been annotated against all protein-coding genes in the M. lucifugus genome and further filtered to discard those with lengths shorter than 20 bp. The miRNA-mRNA interactions were then predicted using miRanda [53]. Gene Ontology enrichment analysis of target genes was performed using Blast2GO suite [54], with the M. lucifugus genome as the background reference. The enriched GO terms with false discovery rates (FDR) < 0.05 were considered statistically significant. The interaction networks of GO terms were generated using Circos web service [55]. The human-, pig- and cow-specific miRNA were also respectively identified using the same pipeline described above.
Comparative miRNomes between bat, human, pig and cow
In order to ascertain the potential species-specific adaptations in long-lived bats, genome-wide miRNA expressions in M. myotis blood were analyzed and compared to other mammals. In this study, six human (H. sapien), three cow (B. taurus) and three pig (S. scrofa) blood miRNA-Seq libraries [56, 57] were employed as non-bat mammal comparisons. All individuals used were healthy adults when the blood was collected. The raw small RNA data from human, pig and cow were downloaded from NCBI SRA database (Additional file 1: Table S7), and the pipeline for comparative miRNome analyses is displayed in Fig. 1b. For each library (Nbat = 6, Nhuman = 6, Npig = 3, Ncow = 3), miRNA were processed and predicted through the same quality control and identification pipelines described previously. To obtain the differentially expressed (DE) miRNA, the conserved miRNA were required to be identified amongst bat, human, pig and cow. To do this, the mature miRNA sequences from 18 libraries were merged and subsequently clustered using CD-HIT with the same parameters as above. The homologous miRNA clusters were retrieved and filtered using the same criteria as the identification of paralogous groups but not allowing any mismatches within the seed region. Only homologous miRNA, which were discovered across all 18 libraries, were considered to be conserved miRNA and associated expression data (tag counts) were collected. However, there were some conserved miRNA which had paralogs in each library, leading to inaccurate quantifications of miRNA. Under this circumstance, we calculated the geometric mean of the expression from these paralogs to represent the expression of this conserved group across each library. The formula of geometric mean is described below, where x̄ g represents the geometric mean; x n represents the expression of each member in a certain paralogous group; k is the number of the members in that group.
After obtaining the matrix of conserved miRNA, and their associated expression data in each library, tag counts were normalized using quantile normalization before further analysis. Hierarchical cluster analysis and principal component analysis (PCA) were then implemented using the R packages hclust and prcomp respectively. For cluster analysis, the miRNA (rows) were clustered using Pearson correlation method (normal distribution of the data; P-value > 0.05) while the libraries (columns) were clustered using Spearman correlation method (non-normal distribution of the data; P-value < 0.05). DE miRNA were identified between bat/human, bat/pig and bat/cow using the DESeq’s generalized linear model (GLM) [58]. DE miRNA required an absolute value of log2 (Fold Change) > 2 and a Benjamin-Hochberg adjusted P-value < 0.01, denoting a 99 % probability that each miRNA was differentially expressed to be deemed statistically significant. The overlapping DE miRNA from bat/human, bat/pig and bat/cow were collected, and further investigated. Their mRNA targets were predicted using miRanda.
Comparative mRNA transcriptomes between bat, human, pig and cow
To better understand the expression difference between bats and other mammals, we also employed and analyzed the mRNA-Seq data of the blood transcriptome from these four species (Nbat = 4, Nhuman = 6, Npig = 3, Ncow = 1; libraries from cow were pooled prior to sequencing; Table 1) [22, 56, 59]. The raw mRNA-Seq data of bat, human, pig and cow were obtained from NCBI SRA database (Additional file 1: Table S8), and the workflow for comparative transcriptome analyses is described in Fig. 1c. For the quality control steps, the raw reads were trimmed of adaptor sequences, filtered by base quality and PCR duplicates were removed. Subsequently, the clean reads were pooled by species, assembled, and annotated by their respective genomes. See Additional file 1: Table S4 for genome build details. In this study, the M. lucifugus genome (v2.0) was employed as a reference since the M. myotis genome is not available, and the pipelines applied were as described in our previous study [22]. The quality of assembled transcripts for each species was assessed by the Core Eukaryotic Gene Mapping Approach (CEGMA) system [60].
For the analysis of DE genes, 1:1 single-copy orthologous genes in the genomes of M. lucifugus, H. sapiens, S. scrofa and B. taurus were retrieved using the ensembl web-tool BioMart [61]. For each library (Nbat = 4, Nhuman = 6, Npig = 3, Ncow = 1), these orthologous genes were quantified using Sailfish (v0.7.6) with the default parameters [62]. The raw count of each gene in each library was collected and the genes that were not expressed in any libraries were excluded from DE gene analysis. The raw counts were normalized using Trimmed Mean of M-value (TMM) method [63], and DE genes were detected between bat and other mammals respectively, using the DESeq’s generalized linear model (GLM). The genes, which had an absolute value of log2 (Fold Change) > 2 and a Benjamin-Hochberg adjusted P-value < 0.05, were considered as significant DE genes. The intersections of up-regulated and down-regulated genes between bat and other mammals were analyzed. The Gene Ontology enrichment analysis was conducted using Blast2GO suite. Due to the large number of enriched GO terms obtained, an additional filter of Benjamin-Hochberg adjust P-value < 0.05 was applied.
Acknowledgements
We thank Sébastien Puechmaille, Nicole Foley, Eric Petit, Frédéric Touzalin, Olivier Farcy and Arnaud Le Houédec, and the numerous volunteers and students from BV and University College Dublin for their extensive help in the field, sample collection and the owners/local authorities for allowing access to the sites.
Funding
This study was funded by a European Research Council Research Grant ERC-2012-StG311000 awarded to Emma C. Teeling and a China Scholarship Council studentship (under UCD-CSC funding programme) to Zixia Huang, and supported by the Contrat Nature ‘Etude de la dynamique des populations de grand murin (Myotis myotis) en Bretagne et Pays de Loire’ awarded to Bretagne Vivante (BV).
Availability of data and materials
All the sequencing data generated in this study have been deposited in National Centre for Biotechnology Information (NCBI) under the BioProject: PRJNA267654, and can be accessed in the Sequence Read Archive (SRA) under the accessions SRX1826159, SRX11828114, SRX1828153, SRX1828154, SRX1828397 and SRX1828398. All accession numbers used in this study are in Additional file 1: Table S7 and S8.
Authors’ contributions
ECT, ZH, DJ conceived of the study. Data was generated in ECT laboratory. ZH analyzed the data. ZH, DJ, ECT wrote, edited and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All captures and sample collections were carried out in accordance with the ethical guidelines and permits delivered in ‘Arrêté’ by the Préfet du Morbihan, Bretagne awarded to Eric Petit, Frédéric Touzalin and Sébastien Puechmaille for the time period 15 June-15 September 2013–2017. Full ethics approval and permission (AREC-13-38-Teeling) for capture and field sampling was also awarded by the University College Dublin, ethics committee to Emma Teeling. Access to all field sites was granted by local authorities in collaboration with Bretagne Vivante.
Abbreviations
- CEGMA
Core eukaryotic genes mapping approach
- DE
Differentially expressed
- FDR
False discovery rate
- GEF
Guanyl-nucleotide exchange factor
- GLM
Generalized linear model
- GO
Gene ontology
- GST
Glutathione-S-transferase
- HCC
Hepatocellular carcinoma
- IIS
Insulin-like growth factor signaling pathway
- MAPK
Ras/Raf/MEK/mitogen-activated protein kinase pathway
- miRNA
microRNA
- NCBI
National center for biotechnology information
- NF-кB
Nuclear factor kappaB signaling pathway
- NGST
Next generation sequencing technology
- PCA
Principal component analysis
- ROS
Reactive oxygen species
- SRA
Sequence read archive
- T-ALL
T-cell acute lymphoblastic leukemia
- TMM
Trimmed mean of M-value
- UTR
Untranslated region
Additional files
Contains supplementary Table S1-S11. Table S1. The miRNA precursor sequences and their corresponding mature miRNA sequences predicted from M. myotis blood. Table S2. The unique mature miRNA sequences. Table S3. The novel seed regions identified from the mature miRNA sequences. Table S4. The genomes of selected species used for the identification of bat-specific miRNA, and their mapping results. Table S5. The list of predicted bat-specific miRNA Table S6. The unique enriched GO terms of the targets of the species-specific miRNA (bat, human, pig and cow). Table S7. The summary of the miRNA-Seq data used in this study. Table S8. The summary of the mRNA-Seq data used in this study. Table S9. The list of the predicted species-specific miRNA sequences (human, pig and cow). Table S10. The orthologous miRNA expression data (raw counts) across bat, human, pig and cow. Table S11. The single-copy orthologous gene expression data (raw counts) across bat, human, pig and cow. (XLSX 370 kb)
Contains supplementary Figure S1 and Text S1. Figure S1. The number of homology of M. myotis mature miRNA to the customized bat miRNA dataset. Text S1. The supporting references for the bioprocesses that 6 differentially expressed (DE) miRNA are involved in. (PDF 176 kb)
Footnotes
Methusaleh was a biblical character said to have lived for 969 years.
Contributor Information
Zixia Huang, Email: zixia.huang@ucdconnect.ie.
David Jebb, Email: david.jebb@ucdconnect.ie.
Emma C. Teeling, Email: emma.teeling@ucd.ie
References
- 1.Austad SN. Methusaleh's Zoo: how nature provides us with clues for extending human health span. J Comp Pathol. 2010;142(Suppl 1):S10–21. doi: 10.1016/j.jcpa.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Munshi-South J, Wilkinson GS. Bats and birds: Exceptional longevity despite high metabolic rates. Ageing Res Rev. 2010;9:12–19. doi: 10.1016/j.arr.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Selman C, Blount JD, Nussey DH, Speakman JR. Oxidative damage, ageing, and life-history evolution: where now? Trends Ecol Evol. 2012;27:570–577. doi: 10.1016/j.tree.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Podlutsky AJ, Khritankov AM, Ovodov ND, Austad SN. A new field record for bat longevity. J Gerontol A Biol Sci Med Sci. 2005;60:1366–1368. doi: 10.1093/gerona/60.11.1366. [DOI] [PubMed] [Google Scholar]
- 5.Seim I, Fang X, Xiong Z, Lobanov AV, Huang Z, Ma S, Feng Y, Turanov AA, Zhu Y, Lenz TL, et al. Genome analysis reveals insights into physiology and longevity of the Brandt's bat Myotis brandtii. Nat Commun. 2013;4:2212. doi: 10.1038/ncomms3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/S0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 7.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 8.Smith-Vikos T, Slack FJ. MicroRNAs and their roles in aging. J Cell Sci. 2012;125:7–17. doi: 10.1242/jcs.099200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li N, Muthusamy S, Liang R, Sarojini H, Wang E. Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech Ageing Dev. 2011;132:75–85. doi: 10.1016/j.mad.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Maes OC, An J, Sarojini H, Wang E. Murine microRNAs implicated in liver functions and aging process. Mech Ageing Dev. 2008;129:534–541. doi: 10.1016/j.mad.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Persengiev S, Kondova I, Otting N, Koeppen AH, Bontrop RE. Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis. Neurobiol Aging. 2011;32:2316 e2317–2327. doi: 10.1016/j.neurobiolaging.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Simmons NB. Order chiroptera. 2005. pp. 312–529. [Google Scholar]
- 13.Parker J, Tsagkogeorga G, Cotton JA, Liu Y, Provero P, Stupka E, Rossiter SJ. Genome-wide signatures of convergent evolution in echolocating mammals. Nature. 2013;502:228–231. doi: 10.1038/nature12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA, Fang X, Wynne JW, Xiong Z, Baker ML, Zhao W, et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 2013;339:456–460. doi: 10.1126/science.1230835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen YY, Liang L, Zhu ZH, Zhou WP, Irwin DM, Zhang YP. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc Natl Acad Sci U S A. 2010;107:8666–8671. doi: 10.1073/pnas.0912613107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lei M, Dong D, Mu S, Pan YH, Zhang S. Comparison of Brain Transcriptome of the Greater Horseshoe Bats (Rhinolophus ferrumequinum) in Active and Torpid Episodes. PLoS One. 2014;9:e107746. doi: 10.1371/journal.pone.0107746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cowled C, Stewart CR, Likic VA, Friedlander MR, Tachedjian M, Jenkins KA, Tizard ML, Cottee P, Marsh GA, Zhou P, et al. Characterisation of novel microRNAs in the Black flying fox (Pteropus alecto) by deep sequencing. BMC Genomics. 2014;15:682. doi: 10.1186/1471-2164-15-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glennon NB, Jabado O, Lo MK, Shaw ML. Transcriptome Profiling of the Virus-Induced Innate Immune Response in Pteropus vampyrus and Its Attenuation by Nipah Virus Interferon Antagonist Functions. J Virol. 2015;89:7550–7566. doi: 10.1128/JVI.00302-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong D, Lei M, Liu Y, Zhang S. Comparative inner ear transcriptome analysis between the Rickett's big-footed bats (Myotis ricketti) and the greater short-nosed fruit bats (Cynopterus sphinx) BMC Genomics. 2013;14:916. doi: 10.1186/1471-2164-14-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Platt RN, 2nd, Vandewege MW, Kern C, Schmidt CJ, Hoffmann FG, Ray DA. Large numbers of novel miRNAs originate from DNA transposons and are coincident with a large species radiation in bats. Mol Biol Evol. 2014;31:1536–1545. doi: 10.1093/molbev/msu112. [DOI] [PubMed] [Google Scholar]
- 21.Liew CC, Ma J, Tang HC, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med. 2006;147:126–132. doi: 10.1016/j.lab.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Huang Z, Gallot A, Lao NT, Puechmaille SJ, Foley NM, Jebb D, Bekaert M, Teeling EC. A nonlethal sampling method to obtain, generate and assemble whole blood transcriptomes from small, wild mammals. Mol Ecol Resour. 2016;16:150–162. doi: 10.1111/1755-0998.12447. [DOI] [PubMed] [Google Scholar]
- 23.Etebari K, Asgari S. Accuracy of microRNA discovery pipelines in non-model organisms using closely related species genomes. PLoS One. 2014;9:e84747. doi: 10.1371/journal.pone.0084747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paczynska P, Grzemski A, Szydlowski M. Distribution of miRNA genes in the pig genome. BMC Genet. 2015;16:6. doi: 10.1186/s12863-015-0166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berezikov E. Evolution of microRNA diversity and regulation in animals. Nat Rev Genet. 2011;12:846–860. doi: 10.1038/nrg3079. [DOI] [PubMed] [Google Scholar]
- 28.Deng B, Fang J, Zhang X, Qu L, Cao Z, Wang B. Role of gelsolin in cell proliferation and invasion of human hepatocellular carcinoma cells. Gene. 2015;571:292–297. doi: 10.1016/j.gene.2015.06.083. [DOI] [PubMed] [Google Scholar]
- 29.Tari AM, Hung MC, Li K, Lopez-Berestein G. Growth inhibition of breast cancer cells by Grb2 downregulation is correlated with inactivation of mitogen-activated protein kinase in EGFR, but not in ErbB2, cells. Oncogene. 1999;18:1325–1332. doi: 10.1038/sj.onc.1202422. [DOI] [PubMed] [Google Scholar]
- 30.Brooks R, Fuhler GM, Iyer S, Smith MJ, Park MY, Paraiso KH, Engelman RW, Kerr WG. SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J Immunol. 2010;184:3582–3589. doi: 10.4049/jimmunol.0902844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 32.Ksionda O, Limnander A, Roose JP. RasGRP Ras guanine nucleotide exchange factors in cancer. Front Biol (Beijing) 2013;8:508–532. doi: 10.1007/s11515-013-1276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma A, Fonseca LL, Rajani C, Yanagida JK, Endo Y, Cline JM, Stone JC, Ji J, Ramos JW, Lorenzo PS. Targeted deletion of RasGRP1 impairs skin tumorigenesis. Carcinogenesis. 2014;35:1084–1091. doi: 10.1093/carcin/bgu016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Li L, Guo R, Li X, Lu Y, Guan X, Gitau SC, Wang L, Xu C, Yang B, Shan H. miR-101 promotes breast cancer cell apoptosis by targeting Janus kinase 2. Cell Physiol Biochem. 2014;34:413–422. doi: 10.1159/000363010. [DOI] [PubMed] [Google Scholar]
- 36.Liu L, Yu X, Guo X, Tian Z, Su M, Long Y, Huang C, Zhou F, Liu M, Wu X. miR-143 is downregulated in cervical cancer and promotes apoptosis and inhibits tumor formation by targeting Bcl-2. Mol Med Rep. 2012;5:753–760. doi: 10.3892/mmr.2011.696. [DOI] [PubMed] [Google Scholar]
- 37.Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR-155 gene: a typical multifunctional microRNA. Biochim Biophys Acta. 2009;1792:497–505. doi: 10.1016/j.bbadis.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 38.Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–16900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 39.Kim EB, Fang X, Fushan AA, Huang Z, Lobanov AV, Han L, Marino SM, Sun X, Turanov AA, Yang P, et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature. 2011;479:223–227. doi: 10.1038/nature10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52:159–164. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- 41.Crawford DR, Wang Y, Schools GP, Kochheiser J, Davies KJA. Down-regulation of mammalian mitochondrial RNAs during oxidative stress. Free Radic. Biol. Med. 1997;22:551–559. doi: 10.1016/S0891-5849(96)00380-2. [DOI] [PubMed] [Google Scholar]
- 42.Brunet-Rossinni AK. Reduced free-radical production and extreme longevity in the little brown bat (Myotis lucifugus) versus two non-flying mammals. Mech Ageing Dev. 2004;125:11–20. doi: 10.1016/j.mad.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Z, Oh S, Li D, Ni D, Pirooz SD, Lee J-H, Yang S, Lee J-Y, Ghozalli I, Costanzo V. A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Dev Cell. 2012;22:1001–1016. doi: 10.1016/j.devcel.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–5874. doi: 10.1038/sj.onc.1209874. [DOI] [PubMed] [Google Scholar]
- 45.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 46.Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 48.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 52.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 53.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2004;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conesa A, Gotz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:619832. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bao H, Kommadath A, Sun X, Meng Y, Arantes AS, Plastow GS, Guan LL, Stothard P. Expansion of ruminant-specific microRNAs shapes target gene expression divergence between ruminant and non-ruminant species. BMC Genomics. 2013;14:609. doi: 10.1186/1471-2164-14-609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leidinger P, Backes C, Deutscher S, Schmitt K, Mueller SC, Frese K, Haas J, Ruprecht K, Paul F, Stahler C, et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013;14:R78. doi: 10.1186/gb-2013-14-7-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Memczak S, Papavasileiou P, Peters O, Rajewsky N. Identification and Characterization of Circular RNAs As a New Class of Putative Biomarkers in Human Blood. PLoS One. 2015;10:e0141214. doi: 10.1371/journal.pone.0141214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 61.Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database. 2011;2011:bar030. doi: 10.1093/database/bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patro R, Mount SM, Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat Biotechnol. 2014;32:462–464. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the sequencing data generated in this study have been deposited in National Centre for Biotechnology Information (NCBI) under the BioProject: PRJNA267654, and can be accessed in the Sequence Read Archive (SRA) under the accessions SRX1826159, SRX11828114, SRX1828153, SRX1828154, SRX1828397 and SRX1828398. All accession numbers used in this study are in Additional file 1: Table S7 and S8.