

ABSTRACT
Autophagy plays a critical role in maintaining cell homeostasis in response to various stressors through protein conjugation and activation of lysosome-dependent degradation. MAP1LC3B/LC3B (microtubule- associated protein 1 light chain 3 β) is conjugated with phosphatidylethanolamine (PE) in the membranes and regulates initiation of autophagy through interaction with many autophagy-related proteins possessing an LC3-interacting region (LIR) motif, which is composed of 2 hydrophobic amino acids (tryptophan and leucine) separated by 2 non-conserved amino acids (WXXL). In this study, we identified a new putative LIR motif in PEBP1/RKIP (phosphatidylethanolamine binding protein 1) that was originally isolated as a PE-binding protein and also a cellular inhibitor of MAPK/ERK signaling. PEBP1 was specifically bound to PE-unconjugated LC3 in cells, and mutation (WXXL mutated to AXXA) of this LIR motif disrupted its interaction with LC3 proteins. Interestingly, overexpression of PEBP1 significantly inhibited starvation-induced autophagy by activating the AKT and MTORC1 (mechanistic target of rapamycin [serine/threonine kinase] complex 1) signaling pathway and consequently suppressing the ULK1 (unc-51 like autophagy activating kinase 1) activity. In contrast, ablation of PEBP1 expression dramatically promoted the autophagic process under starvation conditions. Furthermore, PEBP1 lacking the LIR motif highly stimulated starvation-induced autophagy through the AKT-MTORC1-dependent pathway. PEBP1 phosphorylation at Ser153 caused dissociation of LC3 from the PEBP1-LC3 complex for autophagy induction. PEBP1-dependent suppression of autophagy was not associated with the MAPK pathway. These findings suggest that PEBP1 can act as a negative mediator in autophagy through stimulation of the AKT-MTORC1 pathway and direct interaction with LC3.
KEYWORDS: autophagy, ERK pathway, LC3, LIR motif, MTOR, PEBP1/RKIP
Introduction
When deprived of nutrients or growth factors, cells trigger a self-eating process called macroautophagy (hereafter referred to as autophagy) in order to supply the nutrients necessary for cell survival. Upon signaling, cells initiate the formation of phagophores (precursors to autophagosomes), which are double-membraned structures that engulf intracellular components such as cytosolic proteins or organelles, and subsequently fuse with lysosomes, thereby resulting in the formation of autolysosomes.1-4 Eventually, intracellular components are degraded by lysosomal proteases and recycled to support cell growth under starvation conditions. Several signaling pathways regulate autophagy during starvation conditions. For example, deprivation of nutrients in the growth environment deactivates the AKT-MTORC1 pathway and consequently promotes autophagy by stimulating ULK1 and ULK2 activity.5,6 Furthermore, a low level of intracellular ATP activates AMP-dependent protein kinase (AMPK), which in turn inhibits MTORC1 or/and the direct activation of ULK1 and ULK2.7,8
Two ubiquitin-like conjugation processes are required to initiate autophagosome biogenesis. First, ATG12 is activated by the consecutive reactions of 2 autophagy-related proteins, namely ATG7 and ATG10, followed by covalent conjugation with ATG5.9,10 This ATG12–ATG5 conjugate associates with ATG16L1 for a second ubiquitin-like reaction of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β; called Atg8 in yeast) with phosphatidylethanolamine (PE) present in the membranes.11-13 The PE-conjugated LC3 proteins in the membranes (called LC3-II) play a critical role in regulating the formation of autophagosomes and sequestering cellular components into the phagophore lumen.14 As a result, cargo molecules are captured into phagophores either by specific interaction or nonselective engulfment. LC3 lipidation may be a reversible process, because ATG4B protease can catalyze delipidation of LC3-II, and LC3 proteins on the outer surface of the autophagosome are released from the membranes and recycled during autophagy.15-17
Many proteins selectively regulate autophagy by interacting directly with LC3. These proteins contain a common binding motif called the LC3-interacting region (LIR) composed of 2 hydrophobic amino acids, typically tryptophan (W) and leucine (L), separated by 2 nonspecific amino acids (WXXL).18-20 In particular, PE-conjugated LC3 proteins bind SQSTM1/p62 via the LIR-dependent interaction.19 Because the receptor SQSTM1 is commonly found in inclusion bodies containing ubiquitinated protein aggregates, the SQSTM1-LC3 interaction facilitates autophagy-induced protein degradation of cytosolic protein aggregates. In addition, NBR1 (NBR1, autophagy cargo receptor), ATG3, Atg32, and BNIP3L/NIX contain a LIR motif (or the yeast equivalent termed an Atg8-interacting motif) and contribute to the regulation of autophagy by interacting with LC3/Atg8.21,22 Specifically, Atg32 and BNIP3L/NIX play a critical role in regulating mitochondria-targeted autophagy.23
PEBP1/RKIP (phosphatidylethanolamine binding protein 1)24,25 is highly conserved from yeast to humans, and is involved in diverse functions via many cellular signaling pathways. PEBP1 acts as an endogenous inhibitor of the RAF-MEK-MAPK/ERK pathway by directly blocking RAF1 activation.26,27 This protein also functions as a cellular controller for G protein-coupled receptor signaling, as well as NFKB activation by modulating the IKK complex, MAP3K14/NIK (mitogen-activated protein kinase kinase kinase 14), and MAP3K7/TAK1 (mitogen-activated protein kinase kinase kinase 7).28,29 Moreover, PEBP1 is a precursor for the hippocampal neurostimulating peptide (HCNP), which is involved in neuronal differentiation.30,31 Despite the knowledge of its diverse functions in many cellular signaling pathways, there is no direct evidence yet of PEBP1's role in autophagy. However, the binding of PEBP1 to PE, a key component for LC3 lipidation, suggests its possible role in regulating autophagy. Indeed, we previously proposed that PEBP1 may control autophagy because this protein was found localized to autophagosome-like structures when overexpressed under serum starvation conditions.32
In this study, we investigated the mechanism by which PEBP1 regulates autophagy under starvation and found that it binds directly to LC3 via its WXXL motif. In addition, PEBP1 stimulated AKT-MTORC1 activity, which consequently suppressed the initiation of autophagy. Therefore, we conclude that PEBP1 acts as an inhibitor of autophagy.
Results
PEBP1/RKIP interacts directly with LC3 via its LIR motif
Recently, we identified PEBP1 as a cellular inhibitor of the MAPK/ERK pathway with a possible role in the regulation of autophagy.32 In order to examine the actual role of PEBP1 in autophagy during starvation we analyzed the amino acid sequence of PEBP1 and found a putative LIR motif (WDGL; amino acids 55-58 in human PEBP1). Based on sequence comparison between PEBP1 proteins, the LIR motif is very well conserved among species (Fig. 1A). Next, in order to determine whether this putative WXXL motif is involved in interacting directly with LC3 in cells, we transiently overexpressed FLAG-tagged wild-type PEBP1 (FLAG- PEBP1) or FLAG-tagged LIR mutant PEBP1 protein (FLAG-PEBP1-AXXA, where we substituted both the tryptophan [W] and leucine [L] with alanine [A]) into cells infected with recombinant adenoviruses expressing green fluorescent protein (GFP)-tagged LC3B (Ad-GFP-LC3) and then performed co-immunoprecipitation assays using anti-FLAG M2-agarose beads. As shown in Figure 1B, wild-type PEBP1 proteins bound directly to LC3 or GFP-LC3. Specifically, PEBP1 interacted with only LC3-I, a cytosolic unconjugated LC3 form, but not PE-conjugated LC3-II. However, mutation of the conserved LIR motif (WXXL to AXXA) abolished the ability of PEBP1 to bind LC3 (Fig. 1B), suggesting that PEBP1 interacts directly with nonconjugated LC3 protein via the WXXL motif in cells. Besides the direct interaction with LC3, PEBP1 also showed a weak association with ATG16L1 but not other autophagy-related proteins such as ATG3, BECN1/Beclin1, or ULK1 although nonspecific interactions with ATG12–ATG5 have been observed in immunoprecipitation using anti-FLAG M2-agarose beads and immunocytochemistry (Fig. S1). We will discuss later these possible interactions during autophagy.
Figure 1.
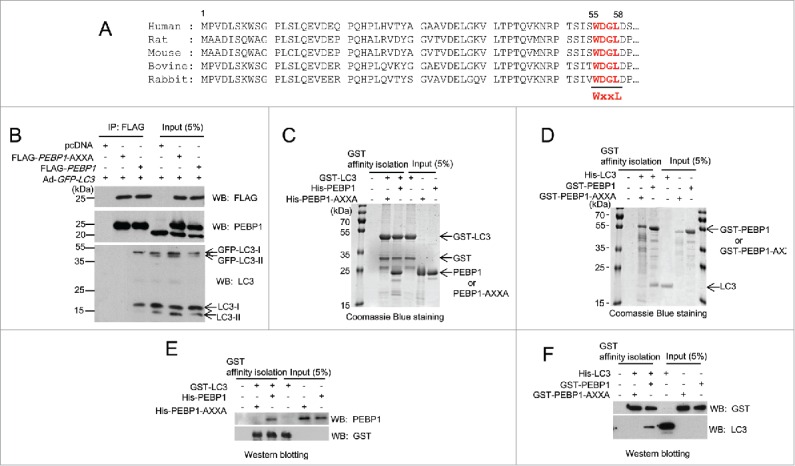
PEBP1 binds directly to LC3 in cells and in vitro. (A) Alignment of PEBP1 amino acid sequences across various species as shown. The N-terminal 60 amino acids of the PEBP1 genes were aligned. The conserved LC3-interacting region (WXXL) of PEBP1 (55th-58th amino acids) is represented in bold. (B) HeLa cells were transiently transfected with plasmids expressing FLAG-PEBP1 or FLAG-PEBP1-AXXA. The pcDNA plasmid was transfected as a negative control. The mutant PEBP1-AXXA protein has alanine residues substituted for Trp55 and Leu58. After 24 h, HeLa cells were infected with recombinant adenoviruses expressing GFP-LC3 (Ad-GFP-LC3). FLAG-tagged PEBP1 proteins (1 mg total protein) were immunoprecipitated with beads conjugated to anti-FLAG M2 antibodies. Bound LC3 proteins were analyzed by western blot using anti-LC3 antibodies. Total PEBP1 proteins (5% input) in whole cell extracts were assessed by western blot using anti- PEBP1 antibody. (C, E) GST-LC3 affinity isolation with His6-tagged PEBP1 in vitro. GST-LC3 protein immobilized to glutathione beads was incubated with either purified His6-PEBP1 or His6-PEBP1-AXXA protein for 1 h at 4°C. Following extensive washing, the bound proteins were subjected to 10% SDS-PAGE and visualized by Coomassie Blue G-250 staining (C) or western blot using anti-PEBP1 or anti-GST antibodies (E). Total protein used for GST affinity isolation is represented as input (5%). (D, F) GST-PEBP1 affinity isolation with His6-tagged LC3 in vitro. GST-PEBP1 or GST-PEBP1-AXXA proteins immobilized to glutathione beads were incubated with purified His6-LC3 proteins for 1 h at 4°C. After washing, bound proteins were analyzed by 10% SDS-PAGE and visualized by Coomassie Blue G-250 staining (D) or western blot using anti-GST or anti-LC3 antibodies (F).
The interaction between PEBP1 and LC3 was also confirmed using in vitro GST affinity isolation assays. We purified different variations of recombinant PEBP1 (GST-PEBP1, GST-PEBP1-AXXA, His6-PEBP1, His6-PEBP1-AXXA) and LC3 (GST-LC3, His6-LC3) proteins from E. coli, immobilized GST-fusion proteins (GST-PEBP1 or GST-PEBP1-AXXA and GST-LC3) to glutathione-sepharose beads, and then examined their interaction by analyzing proteins by SDS-PAGE using Coomassie Blue staining (Fig. 1C and D) and western blotting (Fig. 1E and F). Our data demonstrate the direct interaction between GST-LC3 and His6-PEBP1 (Fig. 1C and E) or between GST-PEBP1 and His6-LC3 (Fig. 1D and F). However, neither His6-PEBP1 LIR mutant proteins (His6-PEBP1-AXXA) nor GST-PEBP1 LIR mutant proteins (GST-PEBP1-AXXA) bound to GST-LC3 or His6-LC3 proteins, respectively, in Coomassie Blue staining or western blotting analyses (Fig. 1C-F). We also found that PEBP1 directly interacts with LC3 through the LIR motif in vitro, consistent with the results obtained from the above immunoprecipitation experiments (Fig. 1B).
Cellular localization of PEBP1 and LC3
LC3, a cytosolic protein, can associate with intracellular membranes through PE-dependent conjugation upon induction of autophagy. Since it ultimately accumulates in autophagosomes, LC3 appears as fluorescent puncta when autophagy is activated in cells expressing fluorescence-tagged LC3 proteins. In this study, we examined the subcellular distribution of PEBP1 and LC3 in HeLa cells transiently expressing GFP-LC3. Endogenous PEBP1 was expressed in the cytoplasm under normal and starved conditions (Fig. 2). However, LC3 proteins were not only randomly distributed throughout the cytoplasm, but accumulated as puncta under starvation conditions. Under normal conditions, LC3 appeared to be diffuse throughout the cytoplasm and nucleus. Therefore, both PEBP1 and LC3 proteins were likely colocalized in cells under normal conditions; however, PEBP1 did not colocalize with GFP-LC3 puncta that formed during starvation. Nevertheless, GFP-LC3 actually merged with PEBP1 in other cytoplasmic areas where GFP-LC3 puncta were not observed during starvation (Fig. 2). These data indicate that PEBP1 could associate with LC3 to regulate autophagy before inclusion of LC3-II proteins within autophagosomes. Indeed, PEBP1 was preferentially bound to LC3-I but not LC3-II as shown in Figure 1B.
Figure 2.
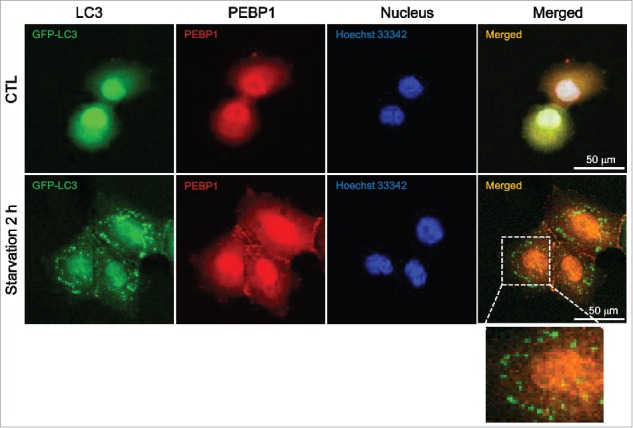
Cellular localization of GFP-LC3 and PEBP1. HeLa cells were transiently transfected with plasmids expressing GFP-tagged LC3 using Lipofectamine 2000. The cellular localization of GFP-LC3 and PEBP1 in control or starved cells was analyzed under a confocal microscope (Olympus FV1000). Endogenous PEBP1 was detected by immunocytochemistry using anti-PEBP1 and TRITC-conjugated secondary anti-rabbit IgG antibodies. Nuclei were stained with Hoechst 33342. Enlargement is a selected area of the merged image under starvation conditions.
PEBP1 inhibits autophagy by reducing autophagosome formation under starvation conditions
Next, we investigated the mechanism of PEBP1-mediated regulation of autophagy. First, we established stable HeLa cell lines overexpressing FLAG-tagged PEBP1 protein (FLAG-PEBP1) and examined the levels of LC3-II and SQSTM1 proteins in these cells during starvation. Control cells (pcDNA) exhibited an increase in LC3-II formation and decrease in SQSTM1 under starvation conditions (Fig. 3A), indicating a regular autophagic response to nutrient deprivation. However, the increased PEBP1 expression (clones #2 and #5) significantly decreased LC3-II production and increased the level of SQSTM1 in starved cells (Fig. 3A). LC3-II formation in PEBP1-overexpressing cells was fairly suppressed even under normal conditions. Furthermore, autophagosome formation during the autophagic process in living cells can be assessed by some additional methods: GFP-LC3 puncta formation, acidic vesicular organelles (AVO) with acridine orange (AO) or monodansylacadaverine (MDC) staining, and electron microscopy analysis. Consistent with the western blotting results (Fig. 3A), overexpression of PEBP1 in HeLa cells (FLAG-PEBP1 #2) caused a significant decrease in GFP-LC3 puncta under starvation conditions compared to control cells (Fig. 3B and C). Moreover, AVO formation (AO- or MDC-positive cells) was largely reduced in PEBP1-overexpressing cells during starvation (Fig. 3D–G). Additionally, LC3 colocalization with LAMP1, a lysosomal protein, and LAMP1 foci, similar to GFP-LC3 puncta, were decreased in PEBP1-overexpressing HeLa cells (Fig. S2). Last, transmission electron micrograph analysis showed a decrease in autophagosome formation in PEBP1-overexpressing cells compared to control cells during starvation (Fig. 3H and I).
Figure 3.
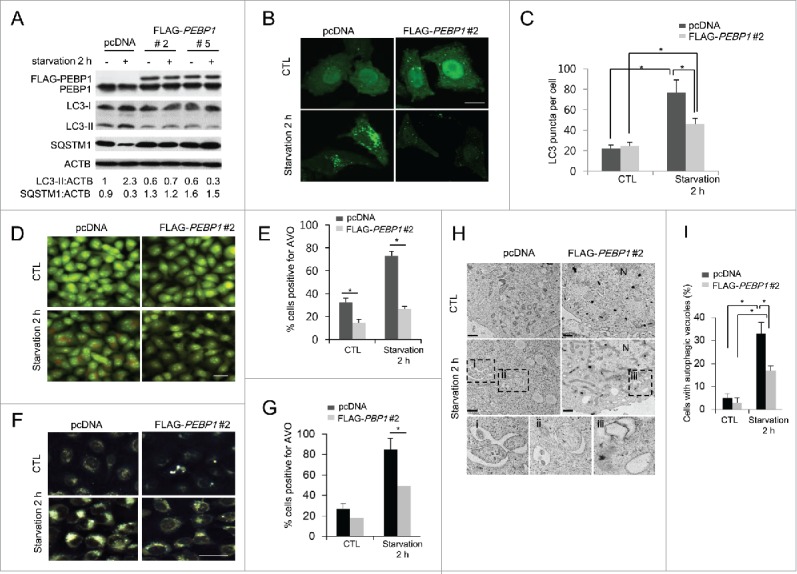
Overexpression of PEBP1 suppresses starvation-induced autophagy. (A) HeLa cells stably selected with control plasmid (pcDNA) or FLAG-tagged PEBP1 plasmid (clones #2 and 5) were starved for 2 h. Total cell extracts (30 μg) were analyzed by 12% SDS-PAGE and western blot using LC3, SQSTM1, and PEBP1 antibodies. ACTB/β-actin was used as a loading control. Proteins were quantified using NIH ImageJ software. Numbers below each lane indicate the relative signal intensity ratio between LC3-II, or SQSTM1 and ACTB, as calculated from 3 independent experiments. (B) Detection of GFP-LC3 puncta in PEBP1-overexpressing cells. HeLa cells stably expressing FLAG- PEBP1 (clone #2) or control vector (pcDNA) were infected with recombinant adenoviral vector expressing GFP-LC3 (Ad-GFP-LC3). After incubation for 24 h, cells were starved for 2 h, and GFP-LC3 puncta were examined under a fluorescence microscope. Quantification of the number of GFP-LC3 puncta per cell is shown in (C). Numbers of GFP-LC3 puncta per cell were determined by counting 20 cells for each sample, which were selected from 6 different areas. Data represent the mean (± SD) of 3 independent experiments (*p < 0.05). Scale bar: 10 μm. (D-G) Detection of acidic vacuoles in PEBP1-overexpressing cells. HeLa cells stably expressing FLAG-PEBP1 (clone #2) or control vector (pcDNA) were starved for 2 h, and acidic vacuoles were detected with AO (D and E) or MDC (F and G) staining. AVO-positive cells were quantified as described in Materials and methods and represented in (E and G). Data represent the mean (± SD) of 3 identical experiments (*p < 0.05). Scale bar: 50 μm. (H) Transmission electron micrograph (TEM) analysis. HeLa cells stably selected with pcDNA or FLAG-PEBP1 were starved for 2 h and subjected to TEM analysis as described in Materials and methods. Sections (90-nm thin) were stained and examined under the 120 kV-transmission electron microscope. The autophagic vacuoles are represented in the enlarged boxes (i, ii and iii). N, nucleus. Scale bar: 1 μm. (I) Quantitative analysis of TEM. The percentage (mean ± SD) of cells containing 5 or more autophagic vacuoles in the cell sections was quantified from at least 50 randomly chosen TEM fields. *p < 0.05.
To further confirm PEBP1-dependent suppression of autophagy, we knocked down the PEBP1 protein in HeLa cells using PEBP1-specific shRNA expressed by a recombinant adenoviral vector (Ad-shPEBP1). An adenoviral vector targeting GFP (Ad-GFP) was used as a control. Our results show that knockdown of PEBP1 expression resulted in a significant increase in LC3-II formation and decrease in total SQSTM1 protein during starvation compared to control cells (Fig. 4A). Consistent with western blotting analysis of LC3-II, GFP-LC3 puncta were remarkably increased in PEBP1-knocked down cells (shPEBP1 RNA) infected with Ad-GFP-LC3 during starvation compared to HeLa cells transfected with control shRNA vectors (shVector) (Fig. 4B and C). These data suggest that PEBP1 negatively regulates the induction of autophagy under starvation conditions.
Figure 4.
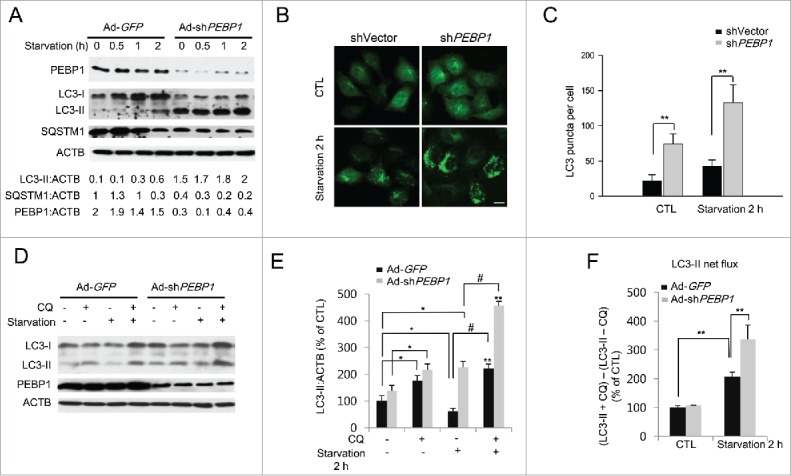
Knockdown of PEBP1 expression stimulates starvation-induced autophagy. (A) HeLa cells were infected with the adenoviral vectors encoding PEBP1 shRNA (Ad-shPEBP1) or control vector (Ad-GFP). After incubation for 72 h, cells were starved for the indicated times, and whole cell extracts were prepared for analysis by western blot. ACTB was used as a loading control. Protein levels were quantified using NIH ImageJ software. Numbers below each lane indicate the relative signal intensity ratio between LC3-II, SQSTM1, or PEBP1 and ACTB, as calculated from 3 independent experiments. (B) Detection of GFP-LC3 puncta in PEBP1-knockdown cells. HeLa cells were transiently transfected with shRNA against PEBP1 (shPEBP1) or control vector DNA (shVector). After 48 h, the cells were infected with Ad-GFP-LC3 (MOI 10) for another 24 h before starvation in HBSS medium for 2 h. The GFP-LC3 puncta were then examined by fluorescence microscopy and quantified as shown in (C). Scale bar: 10 μm. Numbers of GFP-LC3 puncta per cell were determined by counting 20 cells for each sample, which were selected from 6 different areas. Data represent the mean (± SD) of 3 independent experiments (**p < 0.01). (D) Determination of the autophagic flux in PEBP1-knockdown cells. PEBP1 expression in HeLa cells was knocked down by transfection with Ad-shPEBP1 or Ad-GFP vectors for 72 h. Then, cells were starved for 2 h in the absence or presence of 20 μM chloroquine (CQ). Production of LC3-II was analyzed by western blot analysis and quantified as shown in (E). ACTB was used as an internal control. Data represent the relative mean values (± SD) of 3 independent experiments to the control (CTL, the first column; Ad-GFP, no CQ, no starvation). *p < 0.05, **p < 0.01 (compare with control); #p < 0.05 (compared with the presence and absence of CQ under starvation conditions). (F) LC3-II net flux was determined by subtracting the densitometric value of LC3-II in nontreated samples (LC3-II -CQ) from the value corresponding to the chloroquine-treated samples (LC3-II +CQ). **p < 0.01.
The occurrence of an autophagic flux in the presence of lysosomal inhibitors is a critical marker for assessing autophagy.33 Indeed, LC3-II protein turnover (LC3-II net flux) can be determined by comparing the level of LC3-II in the presence and absence of lysosomal inhibitors to account for the amount of LC3-II delivered to the lysosomes. Here we examined the autophagic flux in PEBP1-knocked down cells in the presence of chloroquine (CQ), which inhibits autophagosome-lysosome fusion. Ablation of PEBP1 expression significantly increased LC3-II formation as described above (Fig. 4D and E). Moreover, PEBP1-knockdown greatly stimulated LC3-II net flux in cells treated with a lysosomal inhibitor compared to normal control cells (Fig. 4F), indicating that PEBP1 plays a critical role in regulating autophagy induction.
The WXXL motif of PEBP1 plays a critical role in regulating autophagy under starvation conditions
Next, we investigated whether disrupting the interaction between PEBP1 and LC3 affects the regulation of autophagy during starvation. As shown in Figure 5A, overexpression of the LIR mutant proteins of PEBP1 (PEBP1-AXXA) remarkably increased the LC3-II level and decreased total SQSTM1 protein during starvation compared to the control. Moreover, the formation of GFP-LC3 puncta was significantly elevated in HeLa cells overexpressing FLAG-PEBP1 mutant proteins (PEBP1-AXXA) under starvation conditions compared to control cells (Fig. 5B and C). Besides, we further examined the autophagic flux in HeLa cells expressing LIR mutant proteins in the presence of CQ. Indeed, the autophagic flux (LC3-II turnover) in cells expressing PEBP1-AXXA was significantly elevated compared to control or PEBP1-overexpressing cells (Fig. 5D and E). Additionally, we investigated the effect of PEBP1 in regulating autophagy using another cell line, H1299, which was derived from a human non-small cell lung carcinoma. Similar to the results shown in HeLa cells, PEBP1 overexpression in H1299 cells suppressed the formation of LC3-II puncta and acidic vacuoles during starvation. In contrast, overexpression of LIR mutant PEBP1 proteins (PEBP1-AXXA) induced dramatic autophagosome production in the starved cells (Fig. S3).
Figure 5.
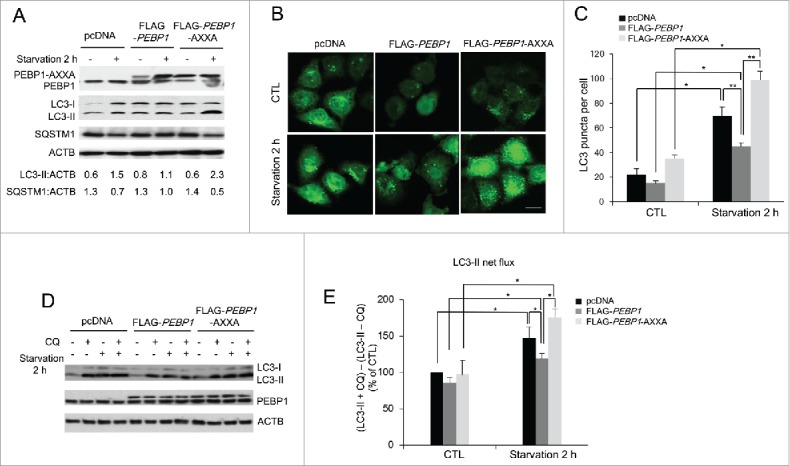
Mutations in the LIR motif (WXXL) of PEBP1 stimulate autophagy. (A) HeLa cells transiently expressing PEBP1 or PEBP1-AXXA mutant proteins were starved for 2 h, and total cell extracts were harvested for 12% SDS-PAGE analysis by western blot using LC3, SQSTM1, and PEBP1 antibodies. ACTB was used as a loading control. Numbers below each lane indicate the relative signal intensity ratio between LC3-II or SQSTM1 and ACTB, as calculated from at least 3 independent experiments. (B) Detection of GFP-LC3 puncta in cells expressing PEBP1-AXXA mutant proteins. HeLa cells transiently overexpressing PEBP1 (FLAG-PEBP1), PEBP1 mutant (FLAG-PEBP1-AXXA) proteins or control (pcDNA) were infected with Ad-GFP-LC3. After 24 h, cells were starved for 2 h, and then GFP-LC3 puncta were examined by fluorescence microscopy and quantified as shown in (C). Scale bar: 10 μm. The data represent the mean (± SD) of 3 independent experiments (*p < 0.05, **p < 0.01). (D) Determination of the autophagic flux in PEBP1-AXXA mutant cells. HeLa cells overexpressing PEBP1 or mutant proteins (FLAG-PEBP1-AXXA) were starved for 2 h in the absence or presence of 20 μM chloroquine (CQ). Production of LC3-II was analyzed by western blot analysis by 4-20% gradient SDS-PAGE and quantified. ACTB was used as an internal control. Data represent the mean (± SD) of 3 independent experiments. (E) LC3-II net flux was determined by subtracting the densitometric value of LC3-II in nontreated samples (LC3-II -CQ) from the value corresponding to the chloroquine-treated samples (LC3-II +CQ). *p < 0.05.
PEBP1 inhibits autophagy by activating the AKT-MTORC1 pathway
When growth factors and nutrients are limited, cells induce autophagy to provide nutrients from intracellular materials by inhibiting the AKT-MTORC1 pathway, a major signaling cascade for cell growth, and subsequently activating many autophagy-related genes.5-8 Therefore, we explored whether PEBP1 can modulate the activation of the AKT-MTORC1 pathway. As expected, overexpression of PEBP1 stimulated the AKT-MTORC1 pathway, which was evidenced by increased phosphorylation of AKT (Ser473) and MTOR (Ser2448). Furthermore, subsequent MTOR-dependent RPS6KB/p70S6K (Thr421/Ser424) phosphorylation was similarly stimulated in both HeLa and H1299 cell lines. Additionally, phosphorylation of ULK1 at Ser757 by MTOR activation, leading to autophagy inhibition, was significantly increased in cells overexpressing FLAG-PEBP1 proteins (Fig. 6A–D). In contrast, phosphorylation of ULK1 at Ser555, stimulating autophagy under conditions of nutrient deprivation, was suppressed in both HeLa and H1299 cells overexpressing FLAG-PEBP1 (Fig. 6A–D). These AKT-MTORC1 and ULK1 activities were specifically regulated in a nutrient-dependent manner. Besides PEBP1 overexpression, knockdown of PEBP1 in HeLa cells using Ad-shPEBP1 significantly inhibited the AKT-MTORC1 activity, consequently stimulating ULK1 phosphorylation at Ser555 and inversely suppressing phosphorylation of ULK1 Ser757 during starvation (Fig. 6E and F).
Figure 6.
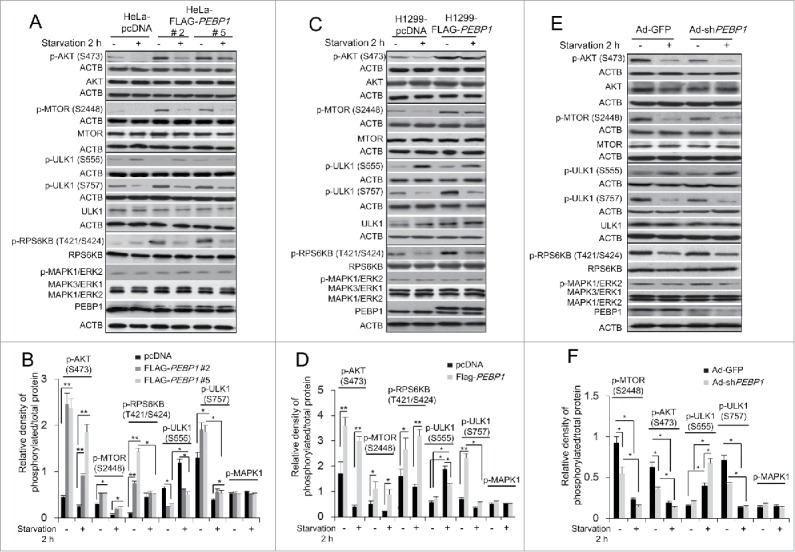
Overexpression of PEBP1 activates the AKT-MTORC1 pathway. (A) HeLa cells stably selected with control pcDNA vector or FLAG-PEBP1-expressing plasmids (clones #2 and #5) were starved for 2 h, and total cell extracts were prepared for analysis by western blot using antibodies against phospho-AKT (S473), phospho-MTOR (S2448), phospho-RPS6KB/p70S6K (T421/S424), phospho-ULK1 (S555 and S757) and phospho-MAPK3/ERK1 and MAPK1/ERK2. ACTB was used as a loading control. Protein and phosphorylation levels were quantified and graphed as shown in (B). (C) H1299 cells stably selected with control pcDNA vector or FLAG-PEBP1-expressing plasmid were analyzed as described above and quantified as shown in (D). (E) HeLa cells were infected with the adenoviral vectors encoding PEBP1 shRNA (Ad-shPEBP1) or control vector (Ad-GFP). After incubation for 72 h, cells were starved for 2 h, and whole cell extracts were analyzed by western blot and quantified as shown in (F). Data represent the mean (± SD) of 3 independent experiments (*p < 0.05, **p < 0.01).
As shown above, we described that PEBP1 could suppress autophagy through activation of AKT-MTORC1 activation under conditions of nutrient deprivation. We further asked whether this PEBP1-dependent suppression of autophagy via AKT-MTORC1 can be alleviated by MTOR inhibitors. Indeed, addition of rapamycin, an MTOR inhibitor, to the PEBP1-overexpressing cells caused a significant decrease of MTOR phosphorylation elevated by PEBP1 overexpression and subsequent activation of ULK1 by increasing phosphorylation at Ser555 and suppressing phosphorylation at Ser757, similar to the starvation response (Fig. S4). Furthermore, we examined how LIR mutant PEBP1 proteins (PEBP1-AXXA) could affect AKT-MTORC1-ULK1 activity. Compared to control (pcDNA) or PEBP1-overexpressing cells (FLAG-PEBP1), overexpression of LIR mutant proteins (FLAG-PEBP1-AXXA) led to a significant decrease of AKT (phosphorylation of S473) and MTOR (phosphorylation of S2448) activity and the inhibitory ULK1 phosphorylation at Ser757, and a large increase of the stimulatory ULK1 phosphorylation at Ser555 under starvation conditions, consequently stimulating LC3-II formation (Fig. S5). Taken together, these data suggest that PEBP1 prevents autophagy by modulating not only LC3-lipidation but also the AKT-MTORC1-ULK1 pathway in HeLa and H1299 cells.
PEBP1 directly inhibits autophagy, independent of its MAPK/ERK inhibitory ability
The MAPK/ERK signaling pathway is also involved in regulating autophagy induced by various cellular conditions.34,35 Studies have demonstrated that PEBP1 is an endogenous inhibitor of MAPK/ERK by direct interaction with RAF1.26,36 Therefore, PEBP1 could control autophagy by modulating MAPK/ERK activity. However, our data indicate that there was no difference in MAPK3/ERK1 and MAPK1/ERK2 activation in starved PEBP1-overexpressing cells or PEBP1-knockdown cells (Fig. 6), and even in LIR mutant PEBP1-overexpressing cells (Fig. S5). Therefore, we examined autophagy in PEBP1-overexpressing HeLa or H1299 cells treated with cisplatin (CDDP), which activates the MAPK/ERK signaling pathway and induces an autophagic response in certain cell types.37,38 Indeed, overexpression of PEBP1 suppressed activation of MAPK/ERK in H1299 cells, especially at 2 and 24 h after cisplatin treatment (Fig. 7B). However, PEBP1 had no effect on cisplatin-mediated MAPK/ERK activation in HeLa cells (Fig. 7A), indicating that they might have different genetic backgrounds responding to cisplatin. Interestingly, regardless of the differential effects of PEBP1 on MAPK/ERK activation in the 2 cell lines, PEBP1 overexpression caused a similar decrease in CDDP-induced LC3-II accumulation and SQSTM1 degradation in both cell lines (Fig. 7A and B). To further confirm these results, we transiently overexpressed a mutant PEBP1 protein (PEBP1-S153A) into the cells. Since PEBP1 phosphorylation at Ser153 triggers dissociation of PEBP1 from the PEBP1-RAF1 complex, mutation at this site leads to potent mitogen-activated protein kinase inhibition.39 We found that overexpression of this mutant decreased LC3-II accumulation and SQSTM1 degradation during starvation (Fig. 7C). We also observed that the autophagic flux (LC3-II turnover) in HeLa cells expressing PEBP1S153A mutant proteins in the presence of CQ was significantly decreased compared to control cells (Fig. S6A and C), suggesting that PEBP1 phosphorylation at Ser153 play a crucial role in the regulation of autophagy. Indeed, according to in vitro GST affinity isolation analyses, a phospho-mimicking PEBP1 mutation (Ser to Asp, S153D) highly stimulated dissociation of PEBP1 from the PEBP1-LC3 complexes compared to wild-type PEBP1 or S153A mutant PEBP1 (Fig. S7). Furthermore, similar to the data shown above, the MAPK/ERK inhibitory effect of PEBP1 had no significant influence on cisplatin-dependent LC3-II formation and the autophagic flux (LC3-II turnover) in cells overexpressing the PEBP1S153A mutant protein (Fig. 7D and E; Fig. S6C and D). These results suggest that PEBP1 inhibits autophagy, independent of its MAPK/ERK inhibitory effect.
Figure 7.
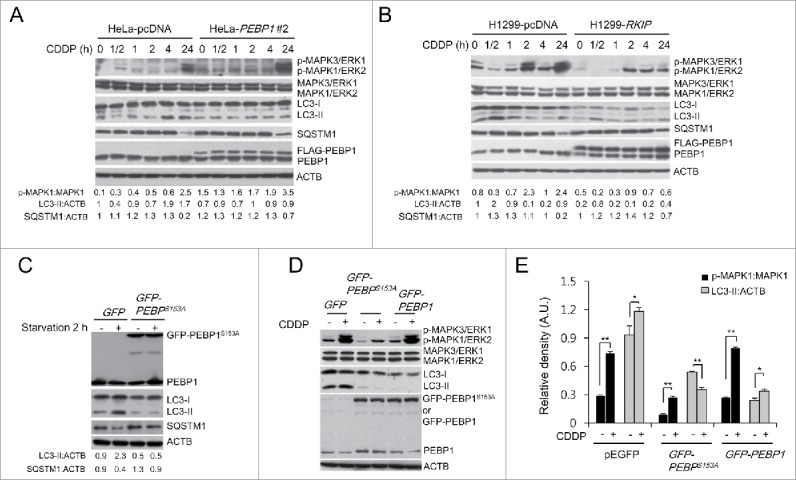
PEBP1 inhibits autophagy in a MAPK/ERK-independent manner. (A, B) HeLa (A) or H1299 cells (B) stably selected with control pcDNA vector or FLAG-PEBP1-expressing plasmid were treated with 20 μM of cisplatin (CDDP) for the indicated times. Protein and phosphorylation levels were examined by western blot using specific antibodies and quantified. ACTB was used as a loading control. Numbers below each lane indicate the relative signal intensity ratio of LC3-II:ACTB, SQSTM1:ACTB, and p-MAPK1/ERK2:MAPK1, as calculated from 3 independent experiments. (C) Effect of PEBP1 mutant proteins (PEBP1S153A) abrogating PEBP1 dissociation from RAF1 on starvation-induced autophagy. HeLa cells were transiently transfected with plasmids encoding mutant PEBP1 (GFP-PEBP1S153A). After 24 h, cells were starved for 2 h, and proteins were then analyzed by western blot and quantified. Numbers below each lane indicate the intensity ratio between LC3-II, or SQSTM1 and ACTB, calculated from 3 independent experiments. (D) Effect of PEBP1 mutant proteins (PEBP1S153A) on CDDP-induced autophagy. HeLa cells transiently expressing GFP-PEBP1S153A, GFP-PEBP1 or GFP were treated with 20 μM CDDP for 24 h, and total cell extracts were analyzed by western blotting using phospho-MAPK/ERK, MAPK/ERK, LC3, and PEBP1 antibodies. (E) The level of protein and phosphorylation was quantified using the NIH ImageJ software (NIH version 1.49). Data represent the mean (± SD) of 3 independent experiments (*p < 0.05, **p < 0.01, compared with untreated control). A.U., arbitrary units.
Discussion
LC3 lipidation with phosphatidylethanolamine, a phospholipid present on the membranes of intracellular organelles, plays a critical role in regulating the initiation of autophagy and subsequent autophagosome formation.9-12 Additionally, the process of sequestering cargo molecules into phagophores apparently determines the 2 characteristics (selective or nonselective) of autophagy through the specific interaction between membrane-bound LC3 and target molecules.40-42 In this report, we demonstrate that PEBP1/RKIP, a member of the PEBP family of proteins and a cellular inhibitor of the MAPK/ERK pathway, contains a novel LIR site (WXXL motif) at the N-terminal region. Our data show that PEBP1 binds specifically to cytosolic LC3. In particular, PEBP1 overexpression significantly inhibits autophagy induced by starvation. In contrast, ablation of PEBP1 stimulates this starvation-induced autophagy, indicating that PEBP1 acts as a negative regulator by modulating the interaction with LC3 via its LIR motif (Fig. 8).
Figure 8.
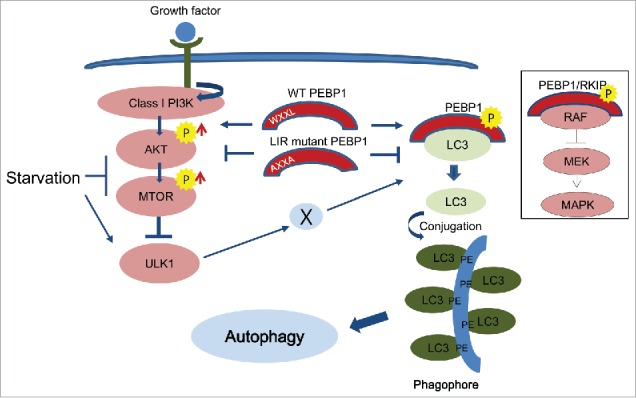
Schematic illustration of PEBP1-mediated autophagy pathways. PEBP1 negatively regulates autophagy during starvation. First, PEBP1 directly interacts with LC3 via its WXXL motif to inhibit LC3 lipidation with phosphatidylethanolamine (PE) and consequently prohibit autophagy. Second, the PEBP1-LC3 complex stimulates the AKT-MTORC1 pathway, which is a major negative regulator for autophagy. Whereas wild-type (WT) PEBP1 tends to form inactive complexes with cytosolic LC3 via a LIR (WXXL) motif, the LIR mutant PEBP1 (AXXA) protein fails to form the PEBP1-LC3 complex, leading to LC3 lipidation for autophagy. In addition, deactivation of the AKT-MTORC1 pathway by the LIR mutant PEBP1 (AXXA) could involve ULK1 activation and consequently promote autophagy through modulation of the PEBP1-LC3 complex and PEBP1 phosphorylation by an unidentified kinase (X). The general role of PEBP1/RKIP and its phosphorylation at Ser153 (indicated by P) as a MAPK/ERK inhibitor is depicted in the rectangle.
The putative WXXL motif of PEBP1 is highly conserved across species. Indeed mutation of this LIR motif results in abolishment of LC3 binding and increased expression of autophagy-associated makers; both phenomena also occur when PEBP1 is knocked down. However, it is not clear how the LIR-dependent interaction of PEBP1 with LC3 controls autophagy. Our data suggest that PEBP1 may be involved in holding LC3 proteins in the PEBP1-LC3 complex, which are not accessible for PE-lipidation. Upon the activation of autophagy, kinases may phosphorylate PEBP1 to liberate LC3 proteins, leading to PE conjugation at the membrane (Fig. 8). In fact, PEBP1 is specifically bound to LC3-I (cytosolic form) but not to LC3-II (membrane-bound form conjugated to PE) as shown in Figure 1B. In addition, PEBP1 mutant proteins (PEBP1-AXXA) lacking the LIR motif fail to interact with LC3 and rather stimulate an autophagic response following nutrient deficiency (Figs. 1 and 5), indicating that the LIR-mutant PEBP1 proteins lose their ability to interact with LC3, subsequently releasing free LC3 for PE lipidation and autophagy activation. Similarly, Ser153 of PEBP1 plays a critical role in maintaining PEBP1-RAF1 complexes through PRKC (protein kinase C)-dependent phosphorylation; that is, phosphorylation at this serine residue triggers the release of RAF1 from inactive complexes and promotes the activation of the MAPK/ERK pathway.39 A similar mechanism has been reported for GRK2 activation.28 According to our results, mutation of Ser153 of PEBP1 (S153A) inhibits autophagy under starvation conditions. In contrast, a phospho-mimicking PEBP1 mutation (PEBP1-S153D) induces LC3 dissociation from PEBP1-LC3 complexes (Fig. S7), indicating that phosphorylation at this site plays an important role in regulation of LC3 lipidation, necessary for autophagy induction.
To date, several steps have been shown to be specifically regulated by interaction between LIR-containing proteins and LC3 (Atg8 in yeast) during the autophagy process. The autophagic receptor SQSTM1 was first discovered as a LIR-binding protein.19,43-45 Since the discovery of SQSTM1, many other autophagy receptors such as BNIP3L/NIX, FUNDC1, NBR1, and CALCOCO2/NDP52 have been identified, and their roles in controlling autophagy have been elucidated.20-22 These autophagy receptors have been suggested to tether cargo molecules to phagophores through the interaction of their LIR motif with LC3, because they possess a ubiquitin-binding domain that can bind ubiquitinated proteins or organelles and deliver them to the degradation sites. In addition, other LIR motif-containing proteins are essential members of the autophagosome biogenesis process. For example, ULK1, which is functionally equivalent to Atg1 in yeast, have canonical LIR motifs and form a large complex with ATG13 and RB1CC1/FIP200, regulating the initial step of autophagosome biogenesis and late events of autophagy.46-48 Other core members of the autophagy machinery, Atg3, Atg32 in yeast or ATG4B, ATG7, and ATG13 in mammals, undergo LIR-dependent interactions with LC3 that potentially contribute to autophagy regulation.17,21,22,49-51 Moreover, some vesicle trafficking proteins, such as the RAB7-interacting protein FYCO1 or the GAP TBC1D5, interact with LC3 and play critical roles in intracellular trafficking of autophagosomes in a LIR-dependent manner.52,53 Based on our data, interaction between PEBP1 and LC3 via the LIR motif might regulate the accessibility of free LC3 proteins for conjugation to PE, which is a critical step necessary for autophagosome biogenesis (depicted in Fig. 8).
PEBP1/RKIP was originally discovered as a member of the PE-binding protein (PEBP) family.24,25 Therefore, its possible role in regulating LC3 lipidation with PE is very conceivable. According to our results, PEBP1 proteins directly interact with PE-unconjugated LC3 protein. Moreover, PEBP1 is distributed evenly throughout the cytoplasm, and colocalizes with LC3 under normal conditions. However, under starvation conditions, although PEBP1 does not colocalize with LC3 puncta in autophagosomes, it likely associates with LC3 in cytoplasmic areas that do not form LC3 puncta (Fig. 2). Furthermore, PEBP1 can sequester LC3 into inactive complexes even under normal conditions, if PEBP1 is capable of binding to LC3. Indeed, the overexpression of PEBP1 causes similar levels of LC3-II production in either normal or starvation conditions. These data suggest that PEBP1 is specifically involved in controlling LC3-lipidation through a LIR-dependent interaction before autophagosome formation. According to a previous work, ATG3-conjugated LC3 intermediates can be recruited to the membrane through direct interaction of ATG3 and ATG12 in the ATG16L1 complexes, which recruits LC3 proteins to the membrane for subsequent PE lipidation.13 If PEBP1 controls the recruitment of LC3 to the membrane, it might be associated with the ATG16L1 complex (ATG12–ATG5-ATG16L1). Indeed, PEBP1 likely interacts with ATG16L1 complexes in cells expressing FLAG-PEBP1 proteins. Therefore, PEBP1 could be a key player in controlling LC3 lipidation through the ATG16L1-PEBP1-LC3 linkage axis.
Generally, nutrient deprivation suppresses the cell growth signals such as AKT-MTORC1 and instead promotes the autophagic process, necessary for cell survival, through the ULK1-dependent pathway.5,6 In this work, besides LIR-dependent regulation of LC3 lipidation, PEBP1 also stimulates AKT-MTORC1 signaling, consequently inhibiting autophagy through both upregulation of ULK1 phosphorylation at serine 757 residue and downregulation of ULK1 phosphorylation at the serine 555 residue. In contrast, knockdown of PEBP1 inhibits the AKT-MTORC1 activity and inversely modulates ULK1 phosphorylation, resulting in facilitation of autophagy under starvation conditions. Indeed, phosphorylation of ULK1 at Ser757 by MTORC1 inhibits autophagy in mammalian cells under conditions of nutrient sufficiency by disrupting the interaction between ULK1 and AMPK. In response to starvation, rapid dephosphorylation of ULK1 at Ser757 promotes AMPK-ULK1 to form an active ULK1 complex together with ATG13, ATG101 and RB1CC1/FIP200, leading to induction of autophagy.8,54 In addition to this ULK1 dephosphorylation at Ser757 following starvation, AMPK-dependent ULK1 phosphorylation and ULK1-dependent phosphorylation of the ULK1 complex proteins are also required for initiation of autophagy. In particular, a critical role of ULK1 phosphorylation at the serine 555 residue by AMPK in autophagy has been suggested in some reports.6,55,56 We also observed that ULK1 phosphorylation at Ser555 was coincidently regulated with occurrence of autophagy. Nevertheless, it is unclear whether PEBP1 can directly activate the AKT-MTORC1 pathway or indirectly through other regulator proteins during autophagy.
Additionally, LIR-mutant PEBP1 (PEBP1-AXXA) proteins, defective in interaction with LC3, increase autophagy not only through liberation of LC3 for lipidation to PE but also suppression of MTORC1 activity (Fig. S5), suggesting that modulation of interaction between PEBP1 and LC3, that is, dissociation of PEBP1 from the PEBP1-LC3 complexes, plays a critical role in regulation of AKT-MTORC1 activity during autophagy. At the moment it is not clear how PEBP1 activates the AKT-MTORC1 pathway. PEBP1-LC3 complexes could directly or indirectly associate with upstream signaling proteins of the AKT-MTORC1 pathway and subsequently activate this signaling pathway. Otherwise, PEBP1 might control other cellular signal pathways linking to AKT-MTORC1. Indeed, PEBP1 is involved in many cellular signal pathways including MAPK/ERK, GRK2, GSK3B and NFKB, which might be associated with control of autophagy.27 In particular, PEBP1, known as a physiological inhibitor of GRK2,28 facilitates G protein signaling that activates the MTORC1 pathway and consequently inhibits autophagy.57,58 Also, PEBP1 directly binds the inhibitory IKK complex to control NFKB signaling. Indeed, IKK has been demonstrated to be an effector of AKT in promoting MTORC1 activity.59,60 This PEBP1-dependent MTORC1-ULK1 regulation in autophagy needs to be further investigated in the future.
MAPK/ERK activity is reciprocally associated with autophagy under some stress conditions. MAPK/ERK stimulates autophagy by direct interaction with autophagy-related proteins.61-63 Also, some autophagy proteins, such as ATG7, lead to MAPK/ERK activation.64 In this context, PEBP1, an endogenous MAPK/ERK inhibitor, could coordinate with the MAPK/ERK pathway to regulate autophagy. However, according to our data, PEBP1 negatively regulated autophagy in a MAPK/ERK-independent manner. Indeed, PEBP1 overexpression in the human cervical cancer cell line HeLa did not influence MAPK/ERK activation, even though autophagy was significantly inhibited under this condition. Furthermore, PEBP1 did not affect MAPK/ERK activation following the treatment with cisplatin, an anticancer drug that generally activates the MAPK/ERK pathway to stimulate cell death in cancer cells and induces autophagy under certain conditions.65-67 However, PEBP1 overexpression in HeLa cells significantly suppressed LC3-II, suggesting that PEBP1-dependent inhibition of autophagy does not require MAPK/ERK activity. These results were also observed in H1299 cells. According to previous studies, phosphorylation of PEBP1 at Ser153 is required for MAPK/ERK activation by releasing RAF1 from inactive PEBP1-RAF1 complexes.28 In fact, the GFP-PEBP1S153A mutant fails to activate MAPK/ERK, but produces a similar PEBP1-mediated suppression of LC3-II production. Moreover, starvation cannot induce LC3-II production in cells overexpressing PEBP1S153A, and a phospho-mimicking PEBP1 mutation facilitates release of LC3 from PEBP1-LC3 complexes. These results suggest that this phosphorylation site of PEBP1 could regulate the interaction between PEBP1 and LC3.
In conclusion, PEBP1/RKIP, a member of the PEBP family, can specifically bind to LC3 via its WXXL motif and inhibit autophagy induced by nutrient deprivation. Overexpression of PEBP1 significantly prevents starvation-induced autophagy. In contrast, knockdown of PEBP1 expression induces high levels of autophagy via stimulation of LC3-lipidation and AKT-MTORC1-associated ULK1 activation. These data all suggest that PEBP1 could play a critical role in regulating autophagy by direct interaction with other autophagy mediators during starvation.
Materials and methods
Reagents
Chloroquine (C6628) and monodansylacadaverine (MDC, 30432) were purchased from Sigma-Aldrich. Acridine orange dye (AO, A1301) was obtained from Life Technologies. Rapamycin (R-5000) was purchased from LC Laboratoreis. Antibodies used in the study were as follows: PEBP1/RKIP (sc-28837), GFP (sc-9996), GST (sc-138), AKT (sc-8312) from Santa Cruz Biotechnology, ATG3 (3415), ATG5 (D5F5U; 12994), ATG12 (D88H11; 4180), ATG16L1 (D6D4; 8089), BECN1 (3495), LAMP1 (D2D11; 9091), MAPK3/ERK1 p44/42 and MAPK1/ERK2 (9102), phospho-p44/42 MAPK3/ERK1 and MAPK1/ERK2 (Thr202/Tyr204; 9101), phospho-AKT (9271), MTOR (2983), phospho-MTOR (Ser2448; 2971), RPS6KB/p70S6 kinase (9202), phospho-RPS6KB/p70S6 Kinase (Thr421/Ser424; 9204), SQSTM1 (5114), ULK1 (D8H5; 8054), phospho-ULK1 (Ser555; 5869), phospho-ULK1 (Ser757; 14202) from Cell Signaling Technology, LC3 (Ab63817) from Abcam, and anti-Flag-M2 (F3165) and ACTB/β-actin (A5441) from Sigma-Aldrich. Secondary antibodies against rabbit (STAR208P), mouse (STAR117P), and goat immunoglobulins (STAR207P) were purchased from Bio-Rad. RPMI-1640 (11875-119), fetal bovine serum (FBS; 16000-044), Dulbecco's modified Eagle's medium (DMEM, 11995-065), Hanks' buffered saline solution (HBSS, 14025-092), Lipofectamine 2000 (11668-500), and G418 (10131-035) were purchased from Gibco and Life Technologies. Anti-FLAG M2 affinity gel (A2220) was from Sigma-Aldrich.
Cell culture and transfection
H1299 (CRL-5803) and HeLa (CCL-2) cell lines were obtained from the American Type Culture Collection. 293A (R705-07) was from Life Technologies. H1299 cells were maintained in RPMI-1640 containing 10% FBS. HeLa cells were grown in DMEM containing 10% FBS. These cells were grown at 37°C in a humidified atmosphere of 5% CO2. Cells were starved in HBSS for the indicated times. H1299 and HeLa cells were transfected using Lipofectamine 2000 as described by the manufacturer's instruction. For generation of stable cell lines, cells transfected with plasmids were selected in medium containing 500 μg/ml G418 for 2 wk.
Construction of plasmids and recombinant adenoviral vectors
The plasmid pcDNA3.1-FLAG-RKIP/PEBP1 was kindly provided by Dr. Walter Kolch (University of Glasgow, Glasgow, United Kingdom). Human MAP1LC3B/LC3 cDNA was amplified by PCR using the human brain cDNA library and then subcloned into pDestpEGFP-C1, pDestmCherry-C1, or pAdCMV/V5–DEST gateway cloning vectors (Invitrogen, V493-20). Gateway LR recombination reactions were carried out as described by the manufacturer's manual (Invitrogen, 11791-019). For construction of bacterial expression vectors, the PCR-amplified PEBP1 or LC3 DNA fragments were cloned into pET41a (GST-fusion vector; Novagen, 70556) and pProEXHTa (His6-tagged vector; BlueGene, G06V0433) vectors. Mutation of PEBP1 to substitute amino acids (S153A or S153D) was performed by the QuikChange site-directed mutagenesis kit (Stratagene, 200518) using pcDNA3.1-FLAG-PEBP1, pET41a-PEBP1, or pProEXHT-PEBP1 plasmids as template. All plasmid constructs were verified by restriction enzyme digestion and DNA sequencing (Cosmo Genetech, Korea).
To generate adenoviral PEBP1 shRNA expression vectors, oligonucleotide sequences (5′-GGATCCCAAATACAGAGAATG-3′) were subcloned using the BLOCK-iT™ Adenoviral RNAi kits (Invitrogen, K4941-00) following the manufacturer's instruction. GFP target sequences were used as a negative control. pAd/BLOCK-iT™–DEST-PEBP1 (Ad-shPEBP1), pAd/BLOCK-iT™–DEST-GFP (Ad-GFP), and pAdCMV/V5–DEST-GFP-LC3B (Adenoviral GFP-LC3 fusion vector, Ad-GFP-LC3) plasmids were transfected into 293A cells using Lipofectamine 2000 to generate viral particles. After 10 d, viral particles were harvested from the cell lysates and culture media. After viral titers were determined, the recombinant viruses were used to infect cells. Cells were plated in 6-well plates at a density of 1 × 105 cells/mL, infected with recombinant adenoviruses at a multiplicity of infection (MOI) of 100 at the following day, and then further incubated for 48 h at 37°C.
Co-immunoprecipitation
pcDNA3.1-FLAG-PEBP1, pcDNA3.1-FLAG-PEBP1-AXXA or pcDNA3.1 plasmids were transiently transfected into H1299 cells using Lipofectamine 2000. After 24 h, cells were infected with recombinant adenoviruses expressing GFP-LC3 (Ad-GFP-LC3). Then, cells were harvested at 48 h after viral infection and lysed in M2 lysis buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 (Thermo Scientific, 85111) and protease inhibitor cocktails (Thermo Scientific, 78441). The FLAG-fused PEBP1 proteins were purified using anti-FLAG M2 affinity gel (Sigma-Aldrich, A2220). After washing with M2 lysis buffer 3 times, bound proteins were eluted by boiling beads in the 2XSDS gel loading buffer and subjected to SDS-PAGE analysis. Proteins were specifically detected by western blot using the indicated primary antibodies.
Bacterial protein purification and in vitro GST affinity isolation
All GST- and His6-tagged proteins were overexpressed in Escherichia coli BL21 (DE3) pLysS by inducing cells with 1 mM IPTG. GST-fused proteins were immobilized to the glutathione-sepharose 4B beads (Amersham Biosciences, 17-0756-01) with incubation of total cell lysates at 4°C for 1 h. Protein-bound beads were washed 5 times with 100 volumes of TEN buffer (20 mM Tris, pH 7.4, 0.1 mM EDTA, 100 mM NaCl), and resuspended in an equal volume of TEN buffer. His6-fused proteins in cell lysates were bound to the Ni2+-nitrotriacetic acid-agarose beads (Qiagen, 30210), then intensively washed with 20 mM imidazole in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4, pH 7.5) and eluted with 200 mM imidazole, 0.3 M NaCl in phosphate buffered saline, pH 7.5. For the GST affinity isolation assay, purified His6-fused proteins were incubated with GST-tagged proteins immobilized to the glutathione-sepharose 4B beads at 4°C for 1 h and then washed 5 times with 100 volumes of TEN buffer. The bound proteins were eluted by boiling beads in the 2X SDS loading buffer and subjected to SDS-PAGE analysis. Gels were stained with Coomassie Brilliant Blue dye or subjected to western blot using the indicated antibodies.
Western blot analysis
Total cellular proteins were prepared by lysing cells in RIPA buffer (Thermo Scientific, 89901) supplemented with protease inhibitor cocktails, and protein concentration of cell lysates was determined using a protein assay kit (Pierce, 23225). Total proteins (30 μg) were separated by 10% SDS-PAGE unless indicated otherwise and transferred to a nitrocellulose membrane using a semi-dry transfer system (Bio-Rad) for 30 min at 15 V. The membrane was blocked for 1 h at room temperature in TBST (10 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween 20) with 5% skim milk. After incubation with primary antibodies overnight at 4°C in TBST with 5% skim milk, the membrane was washed 3 times in TBST for 10 min each and then incubated with secondary antibodies in TBST for 1 h. The membrane was washed with TBST for 10 min 3 times. Specific proteins were visualized using the Enhanced ChemiLuminescence (ECL) detection system (Thermo Scientific, 34080). The densitometric-scanned proteins were quantified using the NIH ImageJ program (version 1.49). For quantification of phospho-proteins, we used separate gels for each analysis of total and phospho-proteins and quantified them on the basis of its own loading control. The graphical data represent the mean (± SD) of at least 3 independent experiments.
GFP-LC3 puncta assay
HeLa cells were seeded on a 42-mm glass cover slip at a density of 1 × 105 cells/mL, infected with recombinant adenoviruses expressing GFP-LC3 proteins (Ad-GFP-LC3) at a MOI of 100, and then incubated at the indicated condition for 24 h. GFP-LC3 puncta were visualized and analyzed under a confocal microscope (FV-1000; Olympus). Numbers of GFP-LC3 puncta were assessed from 6 high-power fields randomly selected. GFP-LC3 puncta were quantified from at least 20 cells per sample, and each experiment was carried out 3 times.
Acridine orange (AO) and monodansylacadaverine (MDC) staining
Cells (1 × 105 cells/mL) were plated in 6-well plates and incubated at 37°C for 24 h. Cells were incubated with AO dye (2.5 μg/mL final concentration) or MDC (50 μM final concentration) at 37°C for 30 min, or 15 min, respectively, and washed with PBS. AO- or MDC-positive acidic vacuoles (AVO) were examined by inverted fluorescence microscopy (ECLIPSE; Nicon). The relative number of acidic vacuoles was quantified by comparing the AO- or MDC-positive cells containing at least 10 AVO aggregates to other cells within a selected area. AO- or MDC-positive acidic vacuoles were determined from at least 3 independent experiments.
Immunocytochemistry
HeLa cells expressing GFP-LC3 or GFP-PEBP1 were cultured on coverslips for 24 h and then starved for 2 h. Cells were fixed with 4% (w/v) paraformaldehyde for 30 min, and permeabilized with PBS containing 0.1% Triton X-100 for 10 min at room temperature. Then, cells were blocked with 2% normal goat serum (Thermo Scientific, PCN5000) in PBS for 1 h and incubated with the corresponding primary antibodies for 2 h. After washing in PBS, cells were further incubated with tetramethylrhodamine-5-isothiocyanate (TRITC)-conjugated secondary anti-rabbit IgG goat antibodies. Fluorescent images were obtained from analyses at the confocal microscope system (FV1000, Olympus) with excitation/emission wavelength of 488/520 nm (EGFP) or 543/570 nm (TRITC).
Electron microscopy analysis
Cells were fixed in 0.1 M PBS with 2.5% glutaraldehyde, and then treated in 1% osmium tetroxide buffer. After dehydration in a series of ethanol, cells were embedded in the EMBed-812 resin (Electron Microscopy Sciences, 14120). Thin sections (90 nm) were cut on a Reichert Ultracut E microtome and stained with a saturated solution of uranyl acetate and lead citrate. Cells were examined under a TECNAI 12 transmission electron microscope (FEI) at 120 kV.
Statistical analysis
Each experiment was independently conducted at least 3 times, and data were expressed as the mean value (± S.D). Difference between 2 groups was assessed by 2-tailed Student t test. Values of P < 0.05 were considered as significant.
Supplementary Material
Abbreviations
- AMPK
AMP-dependent protein kinase
- AO
acridine orange
- ATG
autophagy-related
- CDDP
cisplatin
- DMEM
Dulbecco's modified Eagle's medium
- GFP
green fluorescent protein
- LAMP1
lysosomal-associated membrane protein 1
- LIR
LC3-interacting region
- MAP1LC3B/LC3B
microtubule associated protein 1 light chain 3 β
- MAPK
mitogen-activated protein kinase
- MDC
monodansylacadaverine
- MTORC1
mechanistic target of rapamycin (serine/threonine kinase) complex 1
- PE
phosphatidylethanolamine
- PEBP
phosphatidylethanolamine binding protein
- SQSTM1
sequestosome 1
- ULK
unc-51 like autophagy activating kinase.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Walter Kolch (University of Glasgow, Glasgow, United Kingdom) for kindly providing the pcDNA3.1-FLAG-RKIP/PEBP1 plasmid.
Funding
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education Science and Technology [2012R1A1A2005699 and 2015R1D1A01019753] and by the Ministry of Science, ICT and Future Planning [NRF-2015R1A5A2008833]. It was also partially supported by Gyeongsang National University.
References
- [1].Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct 2002; 27:421-9; PMID:12576635; http://dx.doi.org/ 10.1247/csf.27.421 [DOI] [PubMed] [Google Scholar]
- [2].Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102-9; PMID:17909521; http://dx.doi.org/ 10.1038/ncb1007-1102 [DOI] [PubMed] [Google Scholar]
- [3].Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol 2012; 22:R29-34; PMID:22240478; http://dx.doi.org/ 10.1016/j.cub.2011.11.034 [DOI] [PubMed] [Google Scholar]
- [4].Noda NN, Inagaki F. Mechanisms of Autophagy. Annu Rev Biophys 2015; 44:101-22; PMID:25747593; http://dx.doi.org/ 10.1146/annurev-biophys-060414-034248 [DOI] [PubMed] [Google Scholar]
- [5].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al.. Nutrient-dependent mTORC1 association with the ULK1-ATG13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981-91; PMID:19211835; http://dx.doi.org/ 10.1091/mbc.E08-12-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-ULK1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/ 10.1128/MCB.06159-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Löffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, Campbell DG, Wesselborg S, Alessi DR, Stork B. ULK1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011; 7:696-706; PMID:21460634; http://dx.doi.org/ 10.4161/auto.7.7.15451 [DOI] [PubMed] [Google Scholar]
- [8].Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of ULK1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. An ATG4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 2008; 19:4651-9; PMID:18768752; http://dx.doi.org/ 10.1091/mbc.E08-03-0312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Noda NN, Fujioka Y, Hanada T, Ohsumi Y, Inagaki F. Structure of the ATG12-ATG5 conjugate reveals a platform for stimulating ATG8-PE conjugation. EMBO Rep 2013; 14:206-11; PMID:23238393; http://dx.doi.org/ 10.1038/embor.2012.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12∼ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol 2013; 20:59-66; PMID:23202584; http://dx.doi.org/ 10.1038/nsmb.2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298-302; PMID:17986448; http://dx.doi.org/ 10.1074/jbc.C700195200 [DOI] [PubMed] [Google Scholar]
- [13].Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092-100; PMID:18321988; http://dx.doi.org/ 10.1091/mbc.E07-12-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/ 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805-12; PMID:15169837; http://dx.doi.org/ 10.1242/jcs.01131 [DOI] [PubMed] [Google Scholar]
- [16].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/ 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- [17].Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. The structure of ATG4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J 2009; 28:1341-50; PMID:19322194; http://dx.doi.org/ 10.1038/emboj.2009.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Birgisdottir ÅB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci 2013; 126:3237-47; PMID:23908376 [DOI] [PubMed] [Google Scholar]
- [19].Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 2010; 584:1374-8; PMID:20153326; http://dx.doi.org/ 10.1016/j.febslet.2010.02.017 [DOI] [PubMed] [Google Scholar]
- [20].Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, et al.. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505-16; PMID:19250911; http://dx.doi.org/ 10.1016/j.molcel.2009.01.020 [DOI] [PubMed] [Google Scholar]
- [21].Wild P, McEwan DG, Dikic I. The LC3 interactome at a glance. J Cell Sci 2014; 127:3-9; PMID:24345374; http://dx.doi.org/ 10.1242/jcs.140426 [DOI] [PubMed] [Google Scholar]
- [22].Kalvari I, Tsompanis S, Mulakkal NC, Osgood R, Johansen T, Nezis IP, Promponas VJ. iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy 2014; 10:913-25; PMID:24589857; http://dx.doi.org/ 10.4161/auto.28260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wei H, Liu L, Chen Q. Selective removal of mitochondria via mitophagy:distinct pathways for different mitochondrial stresses. Biochim Biophys Acta 2015; 4889:00114-7. [DOI] [PubMed] [Google Scholar]
- [24].Bernier I, Jollès P. Purification and characterization of a basic 23 kDa cytosolic protein from bovine brain. Biochim Biophys Acta 1984; 790:174-81; PMID:6435678; http://dx.doi.org/ 10.1016/0167-4838(84)90221-8 [DOI] [PubMed] [Google Scholar]
- [25].Banfield MJ, Barker JJ, Perry AC, Brady RL. Function from structure? The crystal structure of human phosphatidylethanolamine-binding protein suggests a role in membrane signal transduction. Structure 1998; 6:1245-54; PMID:9782050; http://dx.doi.org/ 10.1016/S0969-2126(98)00125-7 [DOI] [PubMed] [Google Scholar]
- [26].Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, et al.. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 1999; 401:173-7; PMID:10490027; http://dx.doi.org/ 10.1038/43686 [DOI] [PubMed] [Google Scholar]
- [27].Hagan S, Garcia R, Dhillon A, Kolch W. Raf kinase inhibitor protein regulation of raf and MAPK signaling. Methods Enzymol 2006; 407:248-59; PMID:16757329; http://dx.doi.org/ 10.1016/S0076-6879(05)07021-7 [DOI] [PubMed] [Google Scholar]
- [28].Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003; 426:574-9; PMID:14654844; http://dx.doi.org/ 10.1038/nature02158 [DOI] [PubMed] [Google Scholar]
- [29].Yeung KC, Rose DW, Dhillon AS, Yaros D, Gustafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol 2001; 21:7207-17; PMID:11585904; http://dx.doi.org/ 10.1128/MCB.21.21.7207-7217.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Seddiqi N, Bollengier F, Alliel PM, Périn JP, Bonnet F, Bucquoy S, Jollès P, Schoentgen F. Amino acid sequence of the Homo sapiens brain 21-23-kDa protein (neuropolypeptide h3), comparison with its counterparts from Rattus norvegicus and Bos taurus species, and expression of its mRNA in different tissues. J Mol Evol 1994; 39:655-60; PMID:7807553; http://dx.doi.org/ 10.1007/BF00160411 [DOI] [PubMed] [Google Scholar]
- [31].Mitake S, Ojika K, Katada E, Otsuka Y, Matsukawa N, Fujimori O. Accumulation of hippocampal cholinergic neurostimulating peptide (HCNP)-related components in Hirano bodies. Neuropathol Appl Neurobiol 1995; 21:35-40; PMID:7770119; http://dx.doi.org/ 10.1111/j.1365-2990.1995.tb01026.x [DOI] [PubMed] [Google Scholar]
- [32].Hahm JR, Ahn JS, Noh HS, Baek SM, Ha JH, Jung TS, An YJ, Kim DK, Kim DR. Comparative analysis of fat and muscle proteins in fenofibrate-fed type II diabetic OLETF rats: the fenofibrate-dependent expression of PEBP or C11orf59 protein. BMB Rep 2010; 43:337-43; PMID:20510017; http://dx.doi.org/ 10.5483/BMBRep.2010.43.5.337 [DOI] [PubMed] [Google Scholar]
- [33].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tong Y, Huang H, Pan H. Inhibition of MEK/ERK activation attenuates autophagy and potentiates pemetrexed-induced activity against HepG2 hepatocellular carcinoma cells. Biochem Biophys Res Commun 2015; 456:86-91; PMID:25446102; http://dx.doi.org/ 10.1016/j.bbrc.2014.11.038 [DOI] [PubMed] [Google Scholar]
- [35].Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem 2009; 284:21412-24; PMID:19520853; http://dx.doi.org/ 10.1074/jbc.M109.026013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Trakul N, Rosner MR. Modulation of the MAP kinase signaling cascade by Raf kinase inhibitory protein. Cell Res 2005; 15:19-23; PMID:15686621; http://dx.doi.org/ 10.1038/sj.cr.7290258 [DOI] [PubMed] [Google Scholar]
- [37].Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem 2000; 275:39435-43; PMID:10993883; http://dx.doi.org/ 10.1074/jbc.M004583200 [DOI] [PubMed] [Google Scholar]
- [38].Yeh PY, Chuang SE, Yeh KH, Song YC, Ea CK, Cheng AL. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NFκB activation. Biochem Pharmacol 2002; 63:1423-30; PMID:11996883; http://dx.doi.org/ 10.1016/S0006-2952(02)00908-5 [DOI] [PubMed] [Google Scholar]
- [39].Corbit KC, Trakul N, Eves EM, Diaz B, Marshall M, Rosner MR. Activation of Raf‐1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem 2003; 278:13061-8; PMID:12551925; http://dx.doi.org/ 10.1074/jbc.M210015200 [DOI] [PubMed] [Google Scholar]
- [40].Øverbye A, Fengsrud M, Seglen PO. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy 2007; 3:300-22; http://dx.doi.org/ 10.4161/auto.3910 [DOI] [PubMed] [Google Scholar]
- [41].Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009; 8:1986-90; PMID:19502794; http://dx.doi.org/ 10.4161/cc.8.13.8892 [DOI] [PubMed] [Google Scholar]
- [42].Schwarten M, Mohrlüder J, Ma P, Stoldt M, Thielmann Y, Stangler T, Hersch N, Hoffmann B, Merkel R, Willbold D. Nix directly binds to GABARAP: A possible crosstalk between apoptosis and autophagy. Autophagy 2009; 5:690-98; PMID:19363302; http://dx.doi.org/ 10.4161/auto.5.5.8494 [DOI] [PubMed] [Google Scholar]
- [43].Svenning S, Lamark T, Krause K, Johansen T. Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy 2011; 7:993-1010; PMID:21606687; http://dx.doi.org/ 10.4161/auto.7.9.16389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137:1001-4; PMID:19524504; http://dx.doi.org/ 10.1016/j.cell.2009.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al.. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062-75; PMID:19524509; http://dx.doi.org/ 10.1016/j.cell.2009.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kijanska M, Peter M. Atg1 kinase regulates early and late steps during autophagy. Autophagy 2013; 9:249-51; PMID:23108207; http://dx.doi.org/ 10.4161/auto.22584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Øvervatn A, Kirkin V, Johansen T. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem 2012; 287:39275-90; PMID:23043107; http://dx.doi.org/ 10.1074/jbc.M112.378109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, Stoffel I, Brezovich A, Verma M, Hansmann I, et al.. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J 2012; 31:3691-703; PMID:22885598; http://dx.doi.org/ 10.1038/emboj.2012.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang F, Kumano M, Beraldi E, Fazli L, Du C, Moore S, Sorensen P, Zoubeidi A, Gleave ME. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat Commun 2014; 5:5775; PMID:25503391; http://dx.doi.org/ 10.1038/ncomms6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Suzuki H, Tabata K, Morita E, Kawasaki M, Kato R, Dobson RC, Yoshimori T, Wakatsuki S. Structural basis of the autophagy-related LC3/Atg13 LIR complex:recognition and interaction mechanism. Structure 2014; 22:47-58; PMID:24290141; http://dx.doi.org/ 10.1016/j.str.2013.09.023 [DOI] [PubMed] [Google Scholar]
- [51].Gao W, Chen Z, Wang W, Stang MT. E1-like activating enzyme Atg7 ispreferentially sequestered into p62 aggregates via its interaction with LC3-I. PLoS One 2013; 8:e73229; PMID:24023838; http://dx.doi.org/ 10.1371/journal.pone.0073229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pankiv S, Johansen T. FYCO1: linking autophagosomes to microtubule plus end-directing molecular motors. Autophagy 2010; 6:550-2; PMID:20364109; http://dx.doi.org/ 10.4161/auto.6.4.11670 [DOI] [PubMed] [Google Scholar]
- [53].Popovic D, Akutsu M, Novak I, Harper JW, Behrends C, Dikic I. Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol Cell Biol 2012; 32:1733-44; PMID:22354992; http://dx.doi.org/ 10.1128/MCB.06717-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013; 341:1236566; PMID:23888043; http://dx.doi.org/ 10.1126/science.1236566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J 2011; 440:283-91; PMID:21819378; http://dx.doi.org/ 10.1042/BJ20101894 [DOI] [PubMed] [Google Scholar]
- [56].Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al.. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/ 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wauson EM, Zaganjor E, Cobb MH. Amino acid regulation of autophagy through the GPCR TAS1R1-TAS1R3. Autophagy 2013; 9:418-9; PMID:23222068; http://dx.doi.org/ 10.4161/auto.22911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Michel G, Matthes HW, Hachet-Haas M, El Baghdadi K, de Mey J, Pepperkok R, Simpson JC, Galzi JL, Lecat S. Plasma membrane translocation of REDD1 governed by GPCRs contributes to mTORC1 activation. J Cell Sci 2014; 127:773-87; PMID:24338366; http://dx.doi.org/ 10.1242/jcs.136432 [DOI] [PubMed] [Google Scholar]
- [59].Dan HC, Ebbs A, Pasparakis M, Van Dyke T, Basseres DS, Baldwin AS. Akt-dependent activation of mTORC1 complex involves phosphorylation of mTOR (mammalian target of rapamycin) by IκB kinase α (IKKα). J Biol Chem 2014; 289:25227-40; PMID:24990947; http://dx.doi.org/ 10.1074/jbc.M114.554881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, et al.. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007; 130:440-55; PMID:17693255; http://dx.doi.org/ 10.1016/j.cell.2007.05.058 [DOI] [PubMed] [Google Scholar]
- [61].Corcelle E, Djerbi N, Mari M, Nebout M, Fiorini C, Fénichel P, Hofman P, Poujeol P, Mograbi B. Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environmental carcinogens. Autophagy 2007; 3:57-9; PMID:17102581; http://dx.doi.org/ 10.4161/auto.3424 [DOI] [PubMed] [Google Scholar]
- [62].Corcelle E, Nebout M, Bekri S, Gauthier N, Hofman P, Poujeol P, Fénichel P, Mograbi B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res 2006; 66:6861-70; PMID:16818664; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3557 [DOI] [PubMed] [Google Scholar]
- [63].Kim JH, Hong SK, Wu PK, Richards AL, Jackson WT, Park JI. Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Exp Cell Res 2014; 327:340-52; PMID:25128814; http://dx.doi.org/ 10.1016/j.yexcr.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Martinez-Lopez N, Athonvarangkul D, Mishall P, Sahu S, Singh R. Autophagy proteins regulate ERK phosphorylation. Nat Commun 2013; 4:2799; PMID:24240988; http://dx.doi.org/ 10.1038/ncomms3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yu L, Gu C, Zhong D, Shi L, Kong Y, Zhou Z, Liu S. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett 2014; 355:34-45; PMID:25236911; http://dx.doi.org/ 10.1016/j.canlet.2014.09.020 [DOI] [PubMed] [Google Scholar]
- [66].Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem 2014; 289:17163-73; PMID:24794870; http://dx.doi.org/ 10.1074/jbc.M114.558288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wu ZZ, Sun NK, Chien KY, Chao CC. Silencing of the SNARE protein NAPA sensitizes cancer cells to cisplatin by inducing ERK1/2 signaling, synoviolin ubiquitination and p53 accumulation. Biochem Pharmacol 2011; 82:1630-40; PMID:21903092; http://dx.doi.org/ 10.1016/j.bcp.2011.08.018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.