

ABSTRACT
A long-standing quest is to define the mechanisms responsible for the mitochondrial dysfunction and accumulation of damaged mitochondria that occur during aging. Indeed, those defects are considered major contributors to the aging process. We have analyzed the effect of aging on the muscle expression of Mfn2 and the impact of Mfn2 ablation on muscle function. Our findings reveal that Mfn2 is repressed in muscle during aging, and that is a determinant for the inhibition of autophagy, and mitochondrial quality control, which lead to the accumulation of damaged mitochondria.
KEYWORDS: aging, mitochondria, mitophagy, muscle, sarcopenia
Aging has major effects on skeletal muscle mass and performance, as well as on energy metabolism. Sarcopenia, or the gradual loss of muscle mass and muscle function during aging, is one of the most relevant alterations that affect the quality of life of the elderly. However, the underlying causes of sarcopenia are mainly unknown, and no treatments are available to reverse this condition. Aging is also characterized by a reduction in autophagy and by mitochondrial dysfunction in several tissues, including skeletal muscle, although the mechanisms are unknown. In this regard, alterations in muscle macroautophagy/autophagy have been reported to cause muscle atrophy, and it has thus been postulated that such alterations are involved in sarcopenia.
To uncover the molecular mechanisms underlying muscle alterations during aging, we addressed the role of the mitochondrial protein MFN2 (mitofusin 2), a protein that is involved in mitochondrial fusion. MFN2 also regulates mitochondrial respiration and oxidative metabolism in vivo, and it is essential for normal glucose homeostasis in mice. In addition, MFN2 has been implicated in autophagy and in mitochondrial quality control.
By monitoring young and old control and muscle-specific Mfn2 knockout (KO) mice, we have determined that Mfn2 repression in muscle during aging is a determinant of autophagy inhibition and the accumulation of damaged mitochondria. These data establish Mfn2 as an underlying factor linking autophagy and mitochondrial quality to the maintenance of muscle fitness during aging.
During aging, healthy mice show a progressive decrease in MFN2 protein expression in skeletal muscle. This decrease is greater in slow-twitch than in fast-twitch fibers, and it is not a consequence of reduced Mfn2 gene expression. Analysis of the impact of Mfn2 ablation on various parameters revealed a worsening of systemic metabolic alterations associated with aging (glucose intolerance, insulin resistance, lower basal metabolic rate and metabolic inflexibility). Furthermore, MFN2 deficiency promoted muscle atrophy in young mice and the development of sarcopenia and frailty in old mice. Muscle atrophy in Mfn2 KO animals was due mainly to a reduction in the size of type IIb fibers, which are those most affected by aging. Muscle function was assessed using both in vivo and ex vivo approaches. Thus, physical capacity (muscle performance) and grip strength were lower in old Mfn2 KO mice compared to controls. Moreover, ex vivo evaluation of muscle mechanical properties indicated that muscles of Mfn2 KO mice show alterations typical of age in rodents and humans (reduction in maximal tetanic force, increase in twitch-to-tetanus ratio, and increase in half-relaxation and contraction times). These data strongly support the notion that MFN2 is involved in aging and that the repression of Mfn2 in young mice leads to loss of muscle quality and unhealthy aging.
In order to unravel the molecular mechanisms linking MFN2 deficiency to muscle atrophy, we focused our attention on autophagy. Autophagy is essential for the maintenance of muscle mass and function, and it decreases during aging. In keeping with this concept, we showed that both aging and MFN2 deficiency are associated with a decrease in autophagy (increased levels of LC3-II, SQSTM1/p62 and BNIP3, together with an increased number of autophagosomes). MFN2 deficiency also reduces autophagic flux both in skeletal muscle and in muscle cells. Interestingly, as observed in old control mice, young Mfn2 KO animals show impaired autophagy, which leads to a dramatic accumulation of autophagosomes in aged mice of this model.
Mitochondrial autophagy is also impaired, as indicated by a lower mitophagic flux and accumulation of mitophagosomes in Mfn2 KO mice and a lower response to CCCP-induced translocation of autophagic proteins into mitochondria in Mfn2 knockdown (KD) muscle cells. Although basal levels of autophagic proteins in mitochondrial fractions are increased in Mfn2 KD muscle cells, mitochondrial mass is slightly increased, suggesting that basal mitophagy is inhibited in MFN2-deficient conditions.
As a result of reduced autophagy and mitophagy, we observed that muscle aging is associated with an accumulation of damaged and dysfunctional mitochondria, thus leading to a decrease in mitochondrial respiration and oxidative capacity and to an increase in oxidative stress. Importantly, all these alterations are present in young Mfn2 KO mice and are further impaired in old Mfn2 KO animals. Therefore, we propose that Mfn2 repression during aging leads to a reduction in autophagy and alterations in mitochondrial quality control, thereby contributing to the accumulation of damaged mitochondria and muscle atrophy (Fig. 1). This model was further confirmed by re-expression of Mfn2 in the skeletal muscle of Mfn2 KO mice, which leads to the restoration of autophagy and reversion of muscle atrophy.
Figure 1.
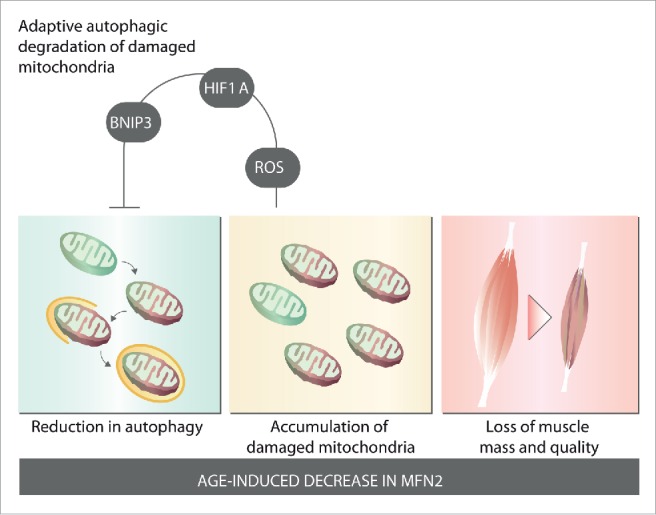
MFN2 controls muscle fitness during aging. Aging is associated with a decrease of MFN2 protein expression in skeletal muscle. Age-induced decrease in MFN2 triggers a decrease in autophagy, which leads to an accumulation of damaged and dysfunctional mitochondria. These alterations lead to a loss of muscle mass and quality, and sarcopenia. Aging and MFN2 deficiency induce a ROS-dependent HIF1A stabilization, which promotes a BNIP3-driven mitophagy pathway; this represents an adaptive mechanism to minimize accumulation of damaged mitochondria.
Interestingly, gene set enrichment analysis of transcriptomic data revealed the activation of a gene pathway both in aging and MFN2 deficiency. Further molecular analysis showed that both conditions trigger an adaptive pathway involving HIF1A/HIF1α activation of BNIP3 expression. The induction of this pathway is dependent on ROS, as treatment with the antioxidant N-acetylcysteine blocks the increase in HIF1A and BNIP3. This adaptive pathway is geared to minimize the accumulation of damaged mitochondria and muscle damage by potentiating BNIP3-driven mitochondrial autophagy. Inhibition of this pathway with a HIF1A inhibitor or with N-acetylcysteine leads to an extensive accumulation of damaged mitochondria, reduced mitochondrial function, and increased muscle atrophy. These observations thus highlight the relevance of this adaptive pathway for ensuring mitochondrial health and muscle fitness.
In summary, our findings reveal that MFN2 regulates the optimal biological properties of skeletal muscle by maintaining mitochondrial quality control and efficient mitochondrial metabolism. The repression of Mfn2 during normal aging causes a reduction in mitochondrial autophagy and an increase in damaged mitochondria, both of which contribute to muscle dysfunction and sarcopenia. Surprisingly, the alterations in mitochondrial function induce mitochondrial retrograde signaling through HIF1A and enhanced BNIP3, thereby contributing to the maintenance of mitochondrial autophagy. Overall, our data strongly support the notion of Mfn2 as a target for the treatment of age-related alterations in skeletal muscle and of sarcopenia.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.