

ABSTRACT
The N-terminal amino acid of a protein is an essential determinant of ubiquitination and subsequent proteasomal degradation in the N-end rule pathway. Using para-chloroamphetamine (PCA), a specific inhibitor of the arginylation branch of the pathway (Arg/N-end rule pathway), we identified that blocking the Arg/N-end rule pathway significantly impaired the fusion of autophagosomes with lysosomes. Under ER stress, ATE1-encoded Arg-tRNA-protein transferases carry out the N-terminal arginylation of the ER heat shock protein HSPA5 that initially targets cargo proteins, along with SQSTM1, to the autophagosome. At the late stage of autophagy, however, proteasomal degradation of arginylated HSPA5 might function as a critical checkpoint for the proper progression of autophagic flux in the cells. Consistently, the inhibition of the Arg/N-end rule pathway with PCA significantly elevated levels of MAPT and huntingtin aggregates, accompanied by increased numbers of LC3 and SQSTM1 puncta. Cells treated with the Arg/N-end rule inhibitor became more sensitized to proteotoxic stress-induced cytotoxicity. SILAC-based quantitative proteomics also revealed that PCA significantly alters various biological pathways, including cellular responses to stress, nutrient, and DNA damage, which are also closely involved in modulation of autophagic responses. Thus, our results indicate that the Arg/N-end rule pathway may function to actively protect cells from detrimental effects of cellular stresses, including proteotoxic protein accumulation, by positively regulating autophagic flux.
KEYWORDS: ATE1, autophagy, HSPA5, N-end rule pathway, neurodegenerative disease, para-chloroamphetamine, ubiquitin-proteasome system
Introduction
In the ubiquitin (Ub)-proteasome system (UPS), the biochemical nature of substrate selectivity is mainly based on the direct interaction between the degradation signal (degron) of substrates and the cognate recognition component (recognin) of their E3 Ub ligases. The N-end rule pathway uses the destabilizing amino-terminal residues of proteins as essential determinants (N-degrons) of their half-lives in vivo.1,2 The N-degrons are the first defined degradation signals in eukaryotes and have later been identified even in bacteria, which lack Ub.3,4 These findings suggest that N-degron-dependent proteolysis may be an evolutionary precursor of nature's selective degradation machinery through the “chambered proteases” in the cell.5 The N-end rule pathway in mammals has 2 branches, defined by the nature of N-degrons: the acetylation branch (Ac/N-end rule) and the arginylation branch (Arg/N-end rule).6,7 In the Arg/N-end rule pathway, the primary destabilizing residues are positively charged amino acids (type 1), such as Arg, Lys, and His, and bulky, hydrophobic amino acids (type 2), such as Phe, Trp, Leu, Tyr, and Ile. The destabilizing residues of the Ac/N-end rule pathway are usually created through the cotranslational N-terminal acetylation of nascent proteins that bear either conventional N-terminal Met (from the start codon) or small, uncharged residues, such as Ala, Val, Ser, Thr, and Cys.8,9
The N-recognins of the Arg/N-end rule pathway are UBR E3 Ub ligases, which have a UBR box for specific interaction with type 1 primary destabilizing residues and their unacetylated primary amines at the N-termini.10 Among the UBR spalogs (with structural similarities) encoded in the human genome, UBR1 and UBR2 also have an N-domain, a weak ortholog of the bacterial ClpS domain, for interaction with type 2 residues. The N-domain contains a hydrophobic binding pocket, whereas the UBR box uses several negatively charged side chains in binding pockets.11-13 In a previous study, we confirmed that the l-conformation, protonated α-amine group, and hydrophobic side chains are essential N-degron structural components required for direct interaction with N-recognins.14 Based on this conformational information, several small, biocompatible Arg/N-end rule inhibitors have been developed to target both the UBR box and the N-domain simultaneously.7 Among these inhibitors, the sympathomimetic amine derivative, para-chloroamphetamine (PCA) is a potent N-end rule inhibitor, which not only blocks degradation of N-end rule reporter substrates in cultured cells, but also effectively delays degradation of its physiological substrates such as RGS4 (regulator of G-protein signaling 4) in mouse brain tissues.15,16 Because the genetic nullification of key components of the Arg/N-end rule pathway often results in embryonic lethality,7 these inhibitors are expected to provide a more feasible way to study the pathophysiological functions of the pathway.
In the Arg/N-end rule pathway, a main process that generates a primary destabilizing residue is the posttranslational conjugation of Arg to pro-N-degrons such as Asp, Glu, and oxidized Cys. This conjugation is solely mediated by ATE1-encoded Arg-tRNA-protein transferase.17 Constitutional deletion of mouse Ate1 is embryonic-lethal owing to a defective heart development;18 however, postnatal Ate1 deletion produces broad phenotypes, ranging from growth retardation and defective spermatogenesis to neurological deformities and metabolic abnormalities.19 These outcomes indicate that a single component of the N-end rule pathway may be involved in multiple layers of regulatory circuits, probably in temporal cooperation with various downstream regulators of the pathway, such as the UBR proteins. The biological functions of these UBR proteins include the regulation of cardiovascular development,17 chromosomal stability,20 spermatogenesis,21 oxygen sensing,22 muscle wasting,23-26 proteotoxic protein clearance,27 hypertension,28 neuronal tube formation,29 bacterial/viral virulence,30 and apoptosis.31 More recent studies have suggested that the Arg/N-end rule pathway is also implicated in autophagy and mitophagy.32,33 A set of endoplasmic reticulum (ER)-residing chaperone proteins, including HSPA5/BiP/GRP78 (heat shock protein family A [HSP70] member 5), P4HB/PDI (prolyl 4-hydroxylase subunit β), and DNAJC10/ERdj5 (DnaJ heat shock protein family [Hsp40] member C10), were identified to be N-terminally arginylated, retrotranslocated to the cytosol, bound to misfolded proteins, and recognized by the autophagic receptor SQSTM1/p62 (sequestosome 1).34 Although these proteins are likely to be cotargeted to the autophagosome, the biological functions of arginylated ER proteins, their catabolic regulation (for example, via UPS, autophagy, or both), and the implication of these proteins in autophagic flux remain essentially uncharacterized.
Herein we report that the chemical and genetic inhibition of the Arg/N-end rule pathway impairs autophagic maturation at the fusion step of autophagosome and lysosome. Proteasomal degradation of N-terminally arginylated HSPA5 (Arg-HSPA5) through the Arg/N-end rule pathway at the late stage of autophagy may function as a critical checkpoint of autophagic flux. Cells treated with PCA showed elevated levels and aggregation of autophagic markers, such as MAP1LC3/LC3 and SQSTM1, and proteotoxic proteins, such as MAPT (microtubule-associated protein tau) and HTT (huntingtin). Consistently, inhibition of the Arg/N-end rule pathway induced cellular hypersensitivity to proteotoxic stress-induced cytotoxicity. Finally, quantitative mass spectrometry (MS) with stable isotope labeling by amino acids in cell culture (SILAC) revealed that PCA treatment resulted in global proteomic changes that are primarily implicated in cellular stress responses and autophagy regulation. Therefore, the Arg/N-end rule pathway might actively protect cells from the detrimental effects of accumulated toxic proteins through positive regulation of autophagy under various stress conditions.
Results
PCA, a small-molecule Arg/N-end rule inhibitor, potently blocks the fusion of autophagosomes with lysosomes
To investigate whether the Arg/N-end rule pathway is involved in the autophagic pathway, we first treated HeLa cells with PCA in the absence and presence of various autophagic regulators. We found that levels of the autophagic selective substrate LC3-II were significantly elevated in PCA-treated cells, largely in a dose-dependent manner (Fig. 1A). Endogenous LC3-II levels appeared to be saturated at ∼50 μM PCA, without apparent cytotoxicity up to 500 μM (Fig. S1). Levels of SQSTM1, which are subjected to both autophagic and proteasomal degradation, also mildly increased after PCA treatment, which suggested that PCA negatively regulates overall autophagy output in the cell. In a similar time-course experiment using 50 μM PCA, we observed a gradual but robust increase in LC3-II and SQSTM1 levels until 4 h post-PCA treatment (Fig. 1B). The mRNA levels of LC3-II and SQSTM1 were not significantly changed under these conditions (Fig. 1C), indicating that the upregulation of these autophagic markers occurs post-translationally, likely through changes in protein catabolic machineries in the cell. We also found that inhibition of the Arg/N-end rule pathway by PCA did not affect overall cellular proteasome activity, as measured by suc-LLVY-AMC hydrolysis (Fig. 1D). In addition, levels of total Ub conjugates in the cells remained unchanged after PCA treatment (Fig. S2). Thus, these results indicate that the increase in LC3-II and SQSTM1 levels after PCA treatment were not directly linked to cellular UPS activity but to aberrant autophagic regulation in the cell.
Figure 1.

Treatment with para-chloroamphetamine (PCA), an Arg/N-end rule inhibitor, delays the fusion of autophagosomes with lysosomes and led to the accumulation of autophagic markers. (A) Accumulated LC3-II and SQSTM1 in HeLa cells after treatment with 0, 10, 25, 50, 75, or 100 μM PCA for 4 h. Whole cell extracts (WCEs) were collected and subjected to SDS-PAGE and immunoblotting (IB). ACTB (actin, β) was used as the loading control. (B) HeLa cells were treated with PCA (50 µM) for the indicated times. Endogenous LC3-II and SQSTM1 levels were significantly elevated after PCA treatment. (C) mRNA levels of the autophagic markers in HeLa cells were not significantly changed after PCA treatment (50 µM, 4 h), measured by quantitative RT-PCR. PCA was dissolved in PBS, which was used as the control (cont'l). (D) The proteasome activity in the WCEs of HeLa cells was measured with suc-LLVY-AMC before and after PCA treatment (50 µM, 4 h). The proteasome inhibitor MG132 (10 μM) was added to the WCEs to determine the basal fluorescence level. The values plotted are means ± SD of 3 independent experiments. RFU, relative fluorescence unit. (E) Bafilomycin A1 (BafA1, 100 nM) virtually abolished the effect of PCA (0, 25, 50 μM) on endogenous LC3-II and SQSTM1 levels in HeLa cells. (F) Same as in (E), except GFP-LC3 was transiently overexpressed and examined. WCEs were collected 2 d post-transfection and used for SDS-PAGE and IB as indicated. (G and H) Endogenous LC3 (G) and SQSTM1 (H) puncta induced by PCA. HeLa cells were treated with 50 μM PCA for 4 h and immunostained with anti-LC3 or anti-SQSTM1 antibodies. (I) Quantification of percentage of HeLa cells showing YFP (RFP+GFP+)- or RFP (RFP+GFP−)-LC3 puncta after cells were transfected with plasmids expressing mRFP-GFP-LC3 and treated with PCA (50 μM, 4 h). The fractions of YFP- and RFP-LC3 puncta, which represent autophagosomes and autolysosomes respectively, were determined in merged images using ImageJ software. Cells with more than 10 puncta were counted and normalized with total cell numbers (represented as DAPI). Bars are the mean (± SD) of 3 independent determinations from 10 different images (including >1,000 cells). *, P < 0.01. (J) HeLa cells were treated with PCA (50 μM), rapamycin (Rapa, 10 μM), or BafA1 (100 nM), or all of 3 for 4 h. The conversion of LC3-I to LC3-II was examined with SDS-PAGE and IB. (K) The turnover of LC3-II after stimulation by serum starvation was significantly impaired by PCA treatment. Cells were incubated in serum-deficient media for 12 h, followed by incubation in normal (serum-containing) media with 50 μM PCA. Samples were collected at the indicated time points and analyzed with SDS-PAGE/IB.
Autophagy involves the formation of double-membrane vesicles, termed autophagosomes that fuse with lysosomes at a relatively late stage of autophagy, leading to the formation of autolysosomes and the degradation of enclosed biological materials, including LC3-II, SQSTM1, and other transported cargoes.35 To examine whether the elevated LC3-II and SQSTM1 levels we observed were due to the upregulation of overall cellular autophagy or the escape of these substrates from autophagic clearance, we first used bafilomycin A1 (BafA1), which inhibits the fusion of autophagosomes with lysosomes and thus blocks LC3-II degradation.36 We observed that, whereas PCA treatment consistently increased LC3-II and SQSTM1 levels in the absence of BafA1, cotreatment of cells with BafA1 and PCA did not have additive effects or further elevate LC3-II protein levels (Fig. 1E). Estimated from the relative levels of LC3-II, the maximal efficacy of PCA and BafA1 on autophagy were comparable at their working concentrations, suggesting that the inhibition of the Arg/N-end rule pathway may potently block the fusion of autophagosomes with lysosomes and perturb basal autophagic flux in the cell.
Measurements of transiently overexpressed GFP-LC3 showed similar responses after PCA and BafA1 treatments. LC3-II and SQSTM1 levels were significantly elevated after PCA treatment, while, when cells were cotreated with BafA1, the protein levels were unaffected (Fig. 1F), indicating strong autophagic suppression.37 With excess LC3, the efficacy of PCA—assessed by levels of overexpressed GFP-LC3-II and endogenous SQSTM1—appeared to be lower than that of BafA1 (Fig. 1F). These results suggest that the effect of PCA is likely limited by the total LC3 level and probably by the magnitude of other autophagic substrates in the cell as well. To examine PCA-impaired autophagy microscopically, we monitored the formation of LC3 puncta in the presence of PCA. Under normal conditions, LC3 showed broad cytosolic localization and its puncta formation was virtually undetected (Fig. 1G). However, after PCA treatment, significant numbers of GFP-LC3 puncta with dispersed perinuclear clustering were observed. Similar immunostaining analysis showed that PCA treatment also led to increased SQSTM1 puncta formation mostly in the cytosol (Fig. 1H).
To further analyze the effect of PCA on autophagy-lysosome fusion, we transiently overexpressed the pH-sensitive, tandem reporter mRFP-GFP-LC3, in which autophagosomes are labeled in green and red (merged in yellow, RFP+ GFP+) and autolysosomes were labeled in red (RFP+ GFP−) only, because GFP signals are more readily quenched under conditions of low lysosomal pH.38 As shown in Fig. 1I and Fig S3, we observed a markedly increased number of cells with autophagosomes, but a decreased number of cells with autolysosome after PCA treatment. These results collectively indicate that dynamic autophagic regulation in the cell is negatively regulated by PCA, an Arg/N-end rule inhibitor, which appears to be required for the proper maturation of autophagosomes through fusion with lysosomes to form autolysosomes.
To understand how PCA affects basal and induced autophagy in HeLa cells, we used rapamycin, which activates autophagy and mildly increases LC3-II levels.39,40 Cotreatment of cells with rapamycin and either PCA or BafA1 resulted in further accumulation of LC3-II in the cells, but few synthetic effects were observed when the combination of PCA and BafA1 was added to the cells (Fig. 1J). Under normal conditions, metabolic stress (via serum starvation)-induced autophagy is restored to the basal level when cells were supplemented with serum: upregulated LC3-II rapidly disappears and de novo LC3-I and SQSTM1 gradually increase (Fig. 1K).41,42 On the contrary, when cells were pretreated with PCA, additional LC3-II accumulated in the cells even after they recovered from serum-starvation stress. This sharp contrast in LC3-II turnover further indicates that PCA strongly impairs autophagic flux. This effect appeared not to be directly linked to lysosomal function per se because no significant change in lysosomal acidification was observed after PCA treatment (Fig. S4). Moreover, PCA treatment did not alter the lysosomal activity-mediated fluorescene signal generation from self-quenched reporter proteins or the maturation process of lysosomal proteins (Fig. S5 and Fig. S6). Taken together, our results demonstrate that acute treatment of cells with PCA potently increases LC3-II conversion, SQSTM1 accumulation, and LC3 puncta formation by inhibiting the fusion of the autophagosomes with lysosomes.
The inhibitory effect of PCA on autophagic flux is directly dependent on the Arg/N-end rule pathway
The in vivo N-end rule inhibitor PCA directly interacts with UBR1 and UBR2 proteins through their UBR boxes and N-domains.43 To examine whether the inhibitory effect of PCA on autophagosome maturation is specifically mediated by the Arg/N-end rule pathway, we used wild-type (WT) and mutant mouse embryonic fibroblasts (MEFs) lacking its key components, including ate1−/− MEFs and ubr1−/− ubr2−/− MEFs. Constitutional nullification of these genes resulted in embryonic lethality.18,44 WT MEFs showed similar responses to PCA that were observed in HeLa cells—stabilization of LC3-II by PCA but no additive effects with BafA1 (Fig. 2A)—thereby also revealing the inhibitory effect of PCA on autophagosome-lysosome fusion. On the contrary, in ubr1−/− ubr2−/− double-knockout MEFs, in which the major E3 ligase activity of the Arg/N-end rule pathway is deficient,45 LC3-II levels were not significantly altered after PCA treatment (Fig. S7). Compared with WT MEFs, ubr1−/− ubr2−/− MEFs exhibited modestly higher basal levels of LC3-II, likely owing to the long-term compensatory effects of reduced N-end rule activity in the cells. To avoid the possible compensatory effect, we knocked down human ATE1 as well as UBR1 and UBR2 in HeLa cells (Fig. 2B). The resulting knockdown cells showed a significant impairment of autophagic flux with increased LC3-II levels and more LC3 puncta formation, also rendering cells unresponsive to PCA treatment (Fig. 2C and 2D). Overall, these data strongly suggest that Arg/N-end rule E3 Ub ligases, such as UBR 1 and UBR2, are essential to the mediation of PCA inhibitory effects on autophagic flux.
Figure 2.
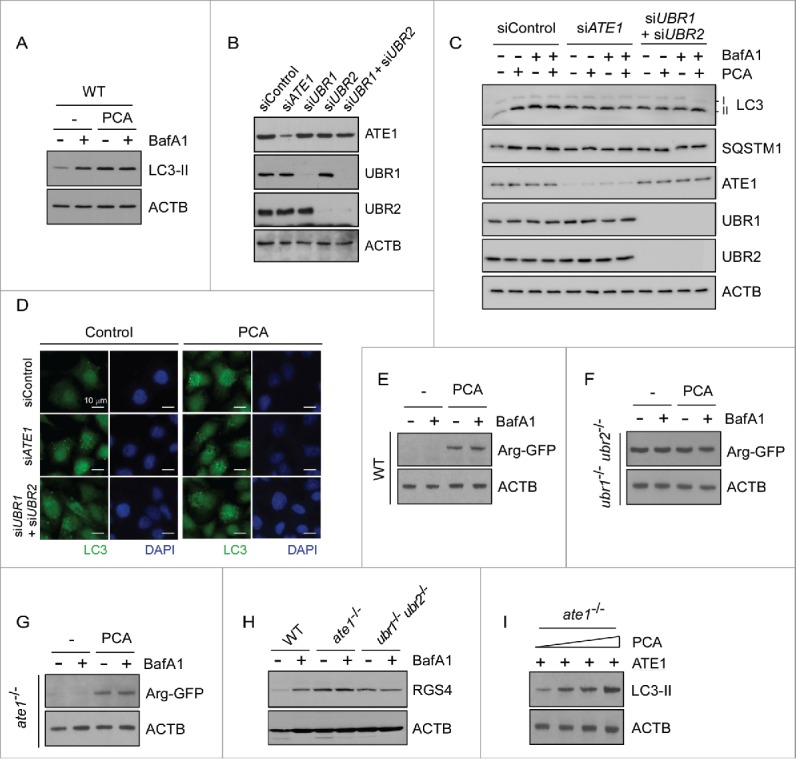
Inhibitory effects of PCA on autophagic flux are mediated by the Arg/N-end rule pathway. (A) PCA treatment results in the accumulation of LC3-II in mouse embryonic fibroblasts (MEFs), but without synthetic effects with BafA1 treatment. Wild-type (WT) MEFs were treated with 50 μM PCA, 100 nM BafA1, or both for 4 h. WCEs were collected and subjected to SDS-PAGE and IB. ACTB was used as a loading control. (B) HeLa cells were transfected with 10 nmol of siRNA for silencing human ATE1 (siATE1), UBR1 (siUBR1), UBR2 (siUBR2), and both UBR1 and UBR2 simultaneously (siUBR1+siUBR2). A scrambled sequence was used as control (siControl). Protein samples were collected 2 d post-transfection and analyzed by SDS-PAGE/IB. (C) Endogenous LC3-II levels were examined from siControl, siATE1, and siUBR1+siUBR2 cells in the presence and absence of PCA (50 μM) and/or BafA1 (100 nM for 4 h). The blots were striped and reprobed with ATE1, UBR1, and UBR2 antibodies to examine their siRNA effects. (D) As in (C), except that endogenous LC3 puncta were examined by immunostaining. (E) Degradation of model substrates of the Arg/N-end rule pathway in WT MEFs. Ub-Arg-GFP was transfected in WT MEFs, which generated Arg-GFP by the action of deubiquitinating enzymes. Note that the N-terminal Arg is not conjugated by ATE1 but exposed by the action of deubiquitinating enzymes. MEFs were treated with 50 μM PCA and/or 100 nM BafA1 for 4 h after 24 h transfection. (F) As in (E), except that the experiment was performed in ubr1−/− ubr2−/− MEFs, in which Arg-GFP was long-lived contrary to the observation in WT MEFs. (G) As in (E), except that the experiment was performed in ate1−/− MEFs, in which Arg-GFP is rapidly degraded through the Arg/N-end rule pathway. (H) Degradation of transiently overexpressed RGS4 (regulator of G-protein signaling 4), a bona fide mammalian N-end rule substrates, in the presence and absence of PCA (50 μM, 4 h) in different MEFs. (I) Add-back experiment with ATE1. ate1−/− MEFs were transfected with plasmids expressing wild-type ATE1. After 24 h, cells were treated with 0, 25, 50, or 100 μM PCA for 4 h. WCEs were then collected and subjected to SDS-PAGE and LC3 IB.
When ate1−/− MEFs were used, we found that PCA failed to increase LC3-II levels, similar to those observed in ubr1−/− ubr2−/− MEFs (Fig. S7). The basal LC3-II levels in ate1−/− MEFs were also higher than in WT MEFs. These findings were consistent with the results of fluorescence microscopy analysis with GFP-LC3 overexpression, which showed significantly elevated levels of LC3-II puncta in both ubr1−/− ubr2−/− and ate1−/− MEFs (Fig. S8). These results occurred possibly because that the direct targets of PCA are UBR1 and UBR2 proteins, not ATE1, an upstream component of the Arg/N-end rule pathway. N-terminal arginylation by ATE1 is usually sufficient for the recognition by UBR proteins and subsequent ubiquitination and degradation in the Arg/N-end rule pathway. Therefore, these results suggested that the Arg/N-end rule-mediated autophagic flux regulator might be a direct substrate of ATE1, rather than UBR1 or UBR2.
The MEFs used and the inhibitory effect of PCA on the Arg/N-end rule pathway was examined using various substrates. For example, Arg-GFP—which is generated from Ub-Arg-GFP fusion proteins46 after translational cleavage of the Ub-Arg junction by deubiquitinating enzymes and therefore not a substrate of ATE1—was short-lived in WT and ate1−/− MEFs but stable in ubr1−/− ubr2−/− MEFs, under normal conditions (Fig. 2E to 2G). PCA treatment in WT and ate1−/− MEFs significantly increased the levels of Arg-GFP. Autophagic inhibitor BafA1 had little effect on the levels of these reporter substrates. Unlike Arg-GFP, RGS4 proteins were stable in both ate1−/− and ubr1−/− ubr2−/− MEFs (Fig. 2H) because the N terminus of RGS4 is modified and recognized by ATE1 and UBR proteins, respectively, for its proteasomal degradation.16 To further examine the role of N-terminal arginylation of ATE1 in autophagy, we added back ATE1 in ate1−/− MEFs through transient transfection and found significant elevation of LC3-II levels in the presence of PCA (Fig. 2I). Therefore, when the biochemical machinery of the Arg/N-end rule pathway in the MEF was normally operative, the effect of PCA on autophagy appeared to specifically depend on the N-terminal arginylation event and the subsequent UBR-dependent ubiquitination of unknown proteasomal substrates.
Arg/N-end rule pathway-dependent degradation of Arg-HSPA5 is a critical regulatory step for autophagosome maturation
To investigate the molecular mechanism of Arg/N-end rule-dependent autophagic inhibition, we examined various potential mediators of PCA action, including BECN1, HDAC6, and UVRAG. However, we observed no significant changes of these proteins by chemical or genetic inhibition of the Arg/N-end rule pathway (data not shown). While investigating the linker protein between PCA and autophagic flux, a recent study suggests that N-terminally arginylated HSPA5 by ATE1 (Arg-HSPA5) may play a key role in SQSTM1 activation, cargo transport, and stress response in autophagy.34 HSPA5 is an ER chaperone protein that regulates calcium storage and release from ER, which are therefore considered sensors of ER stress.47 However, the biochemical implication and consequence of Arg-HSPA5 in the late stage of autophagy remain uncharacterized. We found that HeLa cells treated with geldanamycin, which inhibits HSP90 and induces ER stress, had elevated levels of Arg-HSPA5, which was accompanied with elevated HSPA5 as well, at high concentrations of geldanamycin (Fig. 3A). Under these conditions, LC3-II levels were only modestly or little changed. On the contrary, PCA treatment resulted in significant increases of not only Arg-HSPA5 but also LC3-II levels (Fig. 3B). Again, SQSTM1 levels increased robustly after PCA treatment with Arg-HSPA5 levels.
Figure 3.
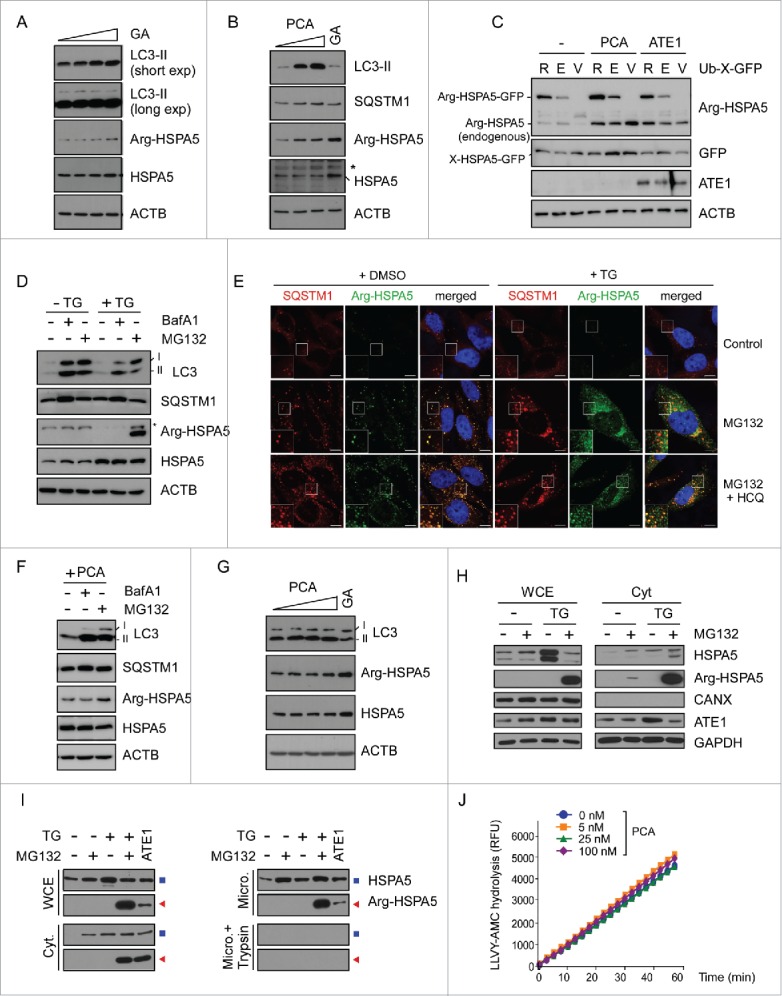
Proteasomal degradation of Arg-HSPA5 by the Arg/N-end rule pathway is a critical checkpoint for autophagic flux. (A) Arg-HSPA5 is induced under ER stress conditions. HeLa cells were treated with 0, 0.5, 1, or 2 µM geldanamycin (GA) for 4 h. WCEs were collected and subjected to SDS-PAGE and IB. (B) HeLa cells treated with PCA (0, 50, 100 μM for 4 h) showed elevated levels of Arg-HSPA5 accompanied by significant upregulation of LC3-II and SQSTM1. Unlike GA treatment (2 μM), PCA treatment did not change total HSPA5 levels. (C) ATE1-dependent N-terminal arginylation of HSPA5 is determined with the Ub-fused constructs, in which Ub is C-terminally conjugated with the N-terminal X residue, such as Arg (R), Glu (E), or Val (V) of HSPA5-GFP. Ub-X-HSPA5-GFP proteins are cleaved by deubiquitinating enzymes at the Ub-HSPA5 junction, producing X-HSPA5-GFP. HeLa cells were cotransfected with either empty vector or ATE1-expressing plasmids, and further treated with 50 μM of PCA for 4 h after 24 h post-transfection. The samples were subject to IB analysis using Arg-HSPA5-specific antibodies, which simultaneously detected overexpressed Arg-HSPA5-GFP and endogenous Arg-HSPA5. (D) Arg-HSPA5 was dramatically stabilized after MG132 treatment, not BafA1, under thapsigargin (TG)-mediated ER stress conditions. HeLa cells were treated with 20 nM TG in the presence and absence of 100 nM BafA1 or 10 μM MG132 for 12 h. (E) N-terminal arginylation of HSPA5 induced by ER-stress-inducing reagent (200 nM of TG for 18 h) is further upregulated by prolonged proteasome inhibition. HeLa cells were treated with the proteasome inhibitor MG132 (3 μM) in the presence and absence of the lysosomal inhibitor hydroxychloroquine (HCQ, 25 μM), Following immunostaining of Arg-HSPA5 and SQSTM1, obtained fluorescence images were merged with Image J for analyzing colocalization of puncta. Counterstained with DAPI. Scale bar: 10 μm. (F) Upregulated Arg-HSPA5 levels after PCA treatment were not affected by autophagic inhibition through BafA1 but mildly increased by subsequent proteasome inhibition. (G) As in (B), except that the experiment was performed with sqstm1−/− MEFs. (H) TG-induced Arg-HSPA5 was significantly stabilized by MG132. (I) Cell fractionation assay indicates that Arg-HSPA5 mainly localizes to cytosolic (Cyt) and microsomal (Micro) fractions. Transient overexpression of ATE1 resulted in significant upregulation of Arg-HSPA5 in these fractions. ▪ and ◂ indicate HSPA5 and Arg-HSPA5, respectively. (J) LLVY-AMC assay using purified human proteasomes and PCA. The hydrolysis of the fluorogenic peptide substrates is for chymotrypsin-like activity in human proteasomes.
Although LC3-II, Arg-HSPA5 and SQSTM1 levels in WT MEFs and HeLa cells were similarly changed after PCA treatment, these proteins were virtually unaffected by PCA in ubr1−/− ubr2−/− MEFs (Fig. S9). Even in ubr1−/− ubr2−/− MEFs, total HSPA5 proteins were significantly upregulated by geldanamycin, as in WT MEFs, indicating that the alteration of LC3-II levels (and possibly autophagic flux as well) is associated with Arg-HSPA5, not HSPA5 (Fig. S9). Consistent with the inhibitory role of PCA on the autophagic flux, ectopically expressed Arg-HSPA5-GFP, generated from Ub-Arg-HSPA5-GFP,46 was also significantly stabilized after PCA treatment, along with endogenous Arg-HSPA5, when examined with anti-Arg-HSPA5-specific antibodies34 (Fig. 3C). A significant portion of Glu-HSPA5-GFP (a precursor of Arg-HSPA5), but not Val-HSPA5-GFP (nonsubstrate of the Arg/N-end rule pathway), was converted to Arg-HSPA5-GFP, which was also stabilized by PCA. Coexpression of ATE1 substantially increased the level of endogenous Arg-HSPA5 (Fig. 3C), possibly mediating more effective arginylation in the cytoplasm. Together these results strongly indicate that the Arg/N-end rule may function critically in the cells by inducing Arg-HSPA5 and transporting target proteins to autophagosomes in cells under various types of autophagic stress.
The Arg/N-end rule pathway appears to degrade Arg-HSPA5 through UPS, particularly at the late stage of autophagy. This was initially supported by our observation that, when HeLa cells were under ER stress conditions through thapsigargin treatment, Arg-HSPA5 was dramatically stabilized in the presence of MG132, whereas BafA1 had little effect (Fig. 3D). Consistently, our immunostaining analysis identified that, under thapsigargin-induced ER stress conditions, the puncta formation of Arg-HSPA5 levels also significantly increased after MG132 treatment (Fig. 3E). The majority of cytosolic Arg-HSPA5 puncta colocalized with SQSTM1 puncta, which significantly increased after MG132 and BafA1 treatments. In the presence of thapsigargin, however, marked accumulation of Arg-HSPA5+ SQSTM1+ cytosolic puncta by MG132 was not affected after autophagic inhibition using hydroxychloroquine. These results collectively suggest that proteasome inhibition stimulates the formation of Arg-HSPA5 and that accumulated Arg-HSPA5 in the cytosol is targeted to autophagy via SQSTM1 bodies. When Arg-HSPA5 was induced by PCA, BafA1 had no effect but MG132 showed a mild additive effect on the level of Arg-HSPA5 (Fig. 3F), indicating that the positive effect of PCA on Arg-HSPA5 stability may be an upstream event in the UPS-mediated proteolysis cascade but independent of autophagic clearance. In sqstm1−/− MEFs, PCA treatment had little effect on LC3-II and Arg-HSPA5 levels, while GA still induced Arg-HSPA5 (Fig. 3G). These results further indicate that SQSTM1 is a critical mediator of the translocation of Arg-HSPA5 and LC3-II to autophagosomes.
Moreover, these data suggest that the effect of PCA on autophagy may require multiple and sequential N-terminal modifications of HSPA5, which eventually leads to its proteasomal degradation at the late stage of autophagy. Under ER stress (and possible other autophagic stress as well), certain signal peptidases may cleave the C terminus of HSPA5 inside the ER, exposing the N-terminal Glu, which is a secondary destabilizing residue in the Arg/N-end rule pathway.48 The cleaved Glu-HSPA5 is translocated to the cytosol, where it is N-terminally arginylated by ATE1 (Fig. 3A to G). Indeed, significant amounts of Arg-HSPA5 were retrieved from the cytosolic and microsomal fractions when cells were under ER-stress conditions and the cellular proteasomes were inhibited by MG132 (Fig. 3H and 3I). Arg-HSPA5 interacts directly with SQSTM1 (Fig. 3E), along with many misfolded cytosolic proteins,34 and is delivered to autophagosomes. Finally, Arg-HSPA5 may be recognized and ubiquitinated by UBR Arg-N-end rule E3 ligases for proteasomal degradation (Fig. 3D to F and Fig. S9), which appears to be an important regulatory step for progression of autophagic flux in the cells. It remains to be determined whether additional ER proteins are involved in relating Arg/N-end rule pathway-dependent proteolysis to cellular autophagy.
Accumulating evidence has shown that the overall activity of the UPS affects autophagic flux in the cells and vice versa. For example, inhibition of the 26S proteasome induces autophagy that is likely mediated by the stabilization of several regulatory proteins responding to proteasomal stress.49,50 The impairment of autophagy ultimately leads to the downregulation of UPS flux, mainly through accumulated SQSTM1, which may function as an overload on the proteasome, clogging its catalytic chamber.51 In addition, we have recently identified that UBR1 and UBR2 proteins are reversibly associated with 26S proteasomes and affect their proteolytic activity (data not shown). For these reasons, we needed to test the possible hypothesis that PCA affects proteasome activity through direct interaction with proteasome-bound UBR proteins, which in turn affects cellular autophagic flux. When the activity of purified human proteasome was measured with suc-LLVY-AMC in the presence of PCA, we observed no significant changes in 26S proteasome activity even when excess amounts of PCA were added to the hydrolysis reaction (Fig. 3J), which is consistent with the previous data that PCA did not alter the overall UPS output in the cells (Fig. 1D and Fig. S2). Therefore, our results collectively indicate that Arg/N-end rule-mediated proteolysis regulates the proteostasis of Arg-HSPA5 in the late stage of autophagy and that the timely degradation of Arg-HSPA5 through the Arg/N-end rule pathway appears to be essential for the fusion of autophagosomes with lysosomes.
Inhibition of the Arg/N-end rule pathway facilitates the formation of MAPT and HTT aggregates, which leads to synergic proteotoxicity
To investigate the biological consequences of Arg/N-end rule pathway inhibition, which accompanies delayed autophagic flux in cells but without affecting UPS function, we first examined the degradation and accumulation of HTT with long polyglutamine (polyQ) repeats, which is the pathological HTT form present in Huntington disease. Under normal conditions, the number of polyQ in HTT is 35 or fewer, whereas an expansion of 50 or more repeats is closely linked to a genetic cause of Huntington disease.52 These mutant HTT proteins are mainly degraded through autophagy instead of via the UPS.52-54 We transiently overexpressed HTT-Q97 in HeLa cells, treated the cells with various concentrations of PCA, and separated the Triton X-100 soluble and insoluble fractions. In the absence of PCA, only low levels of HTT-Q97 proteins were detected in both fractions (Fig. 4A). However, after PCA treatment, these protein levels were dramatically increased, much higher in the Triton X-100 insoluble fraction than those in the soluble fraction. Consistent with previous data, Arg-HSPA5 levels were gradually increased by PCA treatment, indicating abnormal autophagic flux in the cells. Using the HeLa-derived cell lines stably expressing GFP-tagged HTT-Q25 and HTT-Q97 proteins with either nuclear export signals (NES) or nucleus localization signals (NLS), we observed similar effects of PCA on HTT aggregation: whereas HTT-Q25-expressing cells showed no puncta formation regardless of PCA treatment, a significantly increased number of cytoplasmic and nuclear puncta were found in HTT-Q97-expressing cells after treatment with PCA, compared with control cells (Fig. 4B and 4C). These results strongly suggest that impaired autophagic flux, due to inhibition of the Arg/N-end rule pathway and Arg-HSPA5 degradation, is directly responsible for the delayed clearance of HTT-Q97 through autophagy in the cells.
Figure 4.

Inhibition of the Arg/N-end rule pathway facilitates the aggregation of proteotoxic proteins such as MAPT and HTT. (A) HeLa cells were transfected with HTT with 97 polyglutamine repeats (HTT-Q97) and treated with PCA (0, 25, 50, or 100 μM) for 4 h. Protein samples from Triton X100-soluble and -insoluble fractions were prepared separately and used for SDS-PAGE and IB. (B) More inclusions from longQ HTT were observed when the Arg/N-end rule pathway was inhibited by PCA. HeLa cells stably expressing HTT-Q25 and HTT-Q97 with a nuclear export signal (NES) or nuclear localization signal (NLS) were treated with PCA (50 μM, 4 h). (C) Cells with HTT inclusions were quantified. Data represent mean ± SD of 3 independent experiments. (D) Inducible MAPT cell lines were treated with 0, 100, or 200 ng/mL doxycycline (Dox) for 48 h and then protein samples were prepared and subject to SDS-PAGE with (reducing condition) or without reducing reagents β-mercaptoethanol (nonreducing condition). (E) Inducible MAPT cell lines were sequentially treated with Dox (100 ng/mL, 48 h) and PCA (50 μM, 4 h). SDS-PAGE and IB under the reducing and nonreducing conditions. (F) Aggregated MAPT proteins were more prominently detected after PCA treatment (50 μM, 4 h) in HeLa cells transfected with MAPT-GFP. (G) Quantification of cells with MAPT-GFP aggregates. Data represent mean ± SD of 3 independent experiments. ((H)and I). Comparison of MAPT oligomerization after MAPT-BiFC cell lines were treated with PCA. Representative images (H) and quantification (I) of cells exhibiting Venus fluorescence, which represent oligomeric MAPT.57 Values represent the means ± SD of 3 independent cultures including a total of ∼10,000 cells. (J) Cytotoxicity from overexpressed MAPT was measured in the presence and absence of PCA. MAPT proteins were induced by Dox (100 nM, 24 h), followed by treatment with PCA (50 μM, 4 h). Values represent means ± SD (n = 3).
We then examined the degradation and aggregation of another aggregation-prone protein, MAPT, which is implicated in Alzheimer disease. MAPT proteins, similar to many other proteotoxic proteins, are regulated through the UPS when they are in soluble states. However, higher-order oligomers and aggregates are not likely to be processed by the 26S proteasome, and are expected to be degraded via autophagy.55 We used a HEK293-derived cell line expressing the longest isoform of human MAPT upon doxycycline (Dox) induction (cell line expressing an inducible MAPT).56 These cells produced SDS-resistant MAPT aggregates, a pathological hallmark of Alzheimer disease, under nonreducing gel conditions, ∼2 d after treatment with 100 nM Dox (Fig. 4D). Interestingly, induction of MAPT proteins resulted in increased amounts of LC3-II and SQSTM1 in the cells, which probably reflects cellular adaptive responses to the toxic, aggregation-prone proteins. After PCA treatment and under the MAPT induction condition, the inducible MAPT cells showed not only further increased levels of MAPT oligomers but also more SQSTM1 oligomers (Fig. 4E) in a nonreducing gel. LC3-II levels were consistently accumulated by PCA in a dose-dependent manner. SQSTM1 directly interacts with the N-terminal Arg of HSPA5 through its ZZ domain.34 Therefore, the induction of SQSTM1 and MAPT aggregates by PCA is expected to originate from the excess amounts of Arg-HSPA5, which not only interferes with the delivery of cargo proteins such as MAPT or HTT to autophagosomes but also directly inhibits autophagic flux.
Consistent with these results, treatment of HeLa cells with PCA after transient overexpression of GFP-MAPT significantly increased MAPT aggregates (Fig. 4F and 4G). For quantitative analysis of MAPT oligomerization in living cells, we utilized a HEK293-based MAPT-expressing cell line with bimolecular fluorescence complementation (BiFC).57 MAPT-BiFC cells treated with PCA had significantly elevated BiFC signals of MAPT, indicating more MAPT aggregates formation (17.8% after 25 μM PCA treatment vs. 10.1% after control treatment) (Fig. 4H and 4I). In addition, we examined the effect of PCA on proteotoxic protein-mediated cytotoxicity. The working concentration of PCA (50 μM) or overexpression of MAPT with excess Dox (1 μM) resulted in only negligible cytotoxicity (Fig. 4J). However, the combination of proteotoxic stress by MAPT and Arg-N-end rule inhibition by PCA led to a significant loss of inducible MAPT cells (∼60.2% cell viability compared with controls) (Fig. 4J). Thus, our results suggest that the autophagic system may be the major catabolic pathway for proteotoxic proteins such as MAPT and HTT aggregates, and that the Arg-N-end rule pathway has a protective role in preventing aggregation through positive regulation of autophagic flux and the clearance of these aggregates from the cell.
SILAC analysis for global proteomic changes in PCA-treated cells
The global effects of PCA-mediated Arg/N-end rule pathway inhibition in HeLa cells were characterized with quantitative proteomics based on SILAC. For SILAC-MS analysis, control cells were supplemented with stable heavy isotopes substitutes for Lys and Arg, and PCA-treated cells were labeled with light isotopes (Fig. 5A). Protein samples were independently obtained from duplicate experiments, and analyzed with multiple rounds of liquid chromatography/tandem MS (LC-MS/MS), which showed relatively good reproducibility when evaluated with intra- and inter-group component analysis (Fig. 5B). A total of 6,647 proteins were identified from 2 SILAC experiments and 3,867 proteins were found in both (74.4% overlapped with the 1st SILAC result and 72.8% from the 2nd) (Fig. 5B and Table S1). At a threshold cutoff of 1.5-fold change in expression with a P value less than 0.05, total 324 proteins showed significant changes, which is ∼5% of all identified proteins and indicates that the consequence of Arg/N-end rule inhibition is specific on certain target proteins (Fig. 5B and Table S2). Of these differentially expressed proteins (DEPs), 145 proteins were upregulated after PCA treatment, and 179 were downregulated in the same condition (Fig. 5B, Table S3 and Table S4), likely mediated in a nonproteolytic manner or through secondary effects.
Figure 5.

Quantitative proteomic analysis using PCA. (A) Overall work flow of the SILAC approach used to identify the targets of PCA. PCA-treated HeLa cell lysate (light) and stable-isotope-labeled amino acid-treated lysate (heavy) were mixed with equal amounts of total proteins. The trypsin-digested sample was analyzed with high-resolution LC-MS/MS. The analyzed sample was subjected to the search program “MaxQuant” and differentially expressed proteins (DEPs) were selected according to a P value standard of< 0.05, and 1.5-fold up- or downregulation. These processes are duplicated in an independent manner. (B) Venn diagram summary of total proteins and DEPs identified by SILAC. The intersections of 2 sets indicate commonly identified proteins from 2 SILAC analysis. (C) Immunoblotting analysis of DEPs. (D) and (E) GO analysis was carried out with the “DAVID” web-based program with parameters such as minimal count of 5 and P value of < 0.05. Important biological pathways (D) and cellular localizations (E) were enriched from DEPs. Analyzed DEPs were displayed by count number.
To validate the legitimacy of the target proteins that are sensitive to Arg/N-end rule inhibition, we used immunoblotting analysis to examine several proteins with significant changes in HeLa cells with PCA from SILAC analysis (Fig. 5C). The levels of many DEPs were confirmed to behave as conjectured from the quantitative mass spectrometry. For example, CAV1/caveolin-1, whose function involves the modulation of mechanical stress and oxidative stress in cells,58,59 were significantly upregulated after PCA treatment, along with the endogenous Arg/N-end rule substrate RGS4 (Fig. 5C). MDC1 and CDK7, both sensing DNA damage and replication stress,60,61 were also upregulated after PCA treatment, while those of proteasome subunits and stable proteins, such as PSMA4, ADRM1, and ACTB were unchanged (Fig. 5C). These data provide strong evidence that many protein targets from our SILAC analysis are directly related with the Arg/N-end rule pathway-mediated stress regulation.
The obtained global proteomic profiles were further analyzed through gene ontology (GO) enrichments. We found that a significant portion of DEP function is involved in cellular responses to stress, organic substance, DNA damage, drug, and nutrients (Fig. 5D and Table S5). In addition, DEPs are primarily localized in the ER, caveolae, chromosomes, and membrane-enclosed lumen (Fig. 5E and Table S5). These MS data strongly suggest that many of the DEPs obtained from our SILAC analysis are involved in the modulation of autophagy, which is often prosurvival process responding to cell stressors. PCA did not affect UPS flux or related processes in the cell, based on the result of the SILAC analysis. Taken together, these data indicate that our proteomic approach provided a broad view of Arg/N-end rule function in autophagy, which also reflects its critical role as a common denominator of both the UPS and autophagy possibly to protect the cells from various internal and external stresses. The underlying molecular mechanism of this global effect remains to be elucidated.
Discussion
Herein, we reported that inhibition of the Arg/N-end rule pathway by PCA resulted in impaired autophagic flux, possibly through delayed degradation of Arg-HSPA5 at the autophagosome. Because autophagy is a major catabolic process that reduces pathological accumulation of toxic, aggregation-prone proteins in the cells, the Arg/N-end rule pathway appears to have critical neuroprotective roles against MAPT and HTT aggregation (see Fig. 4). Our global proteomic studies also indicate that the Arg/N-end rule is implicated in a number of cellular response mechanisms toward various types of stress (see Fig. 5). PCA is a unique small-molecule inhibitor of the Arg/N-end rule pathway, targeting both the UBR box and the N-domain of UBR proteins, and it has a capacity to efficiently pass the blood-brain barrier (BBB).15 We observed that PCA potently blocked the degradation of Arg-HSPA5 and other known substrates such as Arg-GFP and RGS4 (see Fig. 3). However, the inhibitory function of PCA on autophagy appeared to be specific to the ATE1-mediated arginylation and proteasomal degradation of HSPA5 (see Fig. 2 and Fig. 3). Considering that most of the components of Arg/N-end rule pathway are essential for normal development, PCA can be used to identify other unknown in vivo substrates and their related cellular regulatory mechanisms governed by the Arg/N-end rule pathway.
As illustrated in Figure 6, in this autophagy regulatory mechanism, the ER chaperone protein HSPA5 is cleaved under stress conditions (and other autophagy-induction conditions) to generate Glu-HSPA5, which is converted to Arg-HSPA5 by ATE1 in the cytoplasm. The cytosolic Arg-HSPA5 protein, alone or associated with SQSTM1, may escort autophagic cargo proteins to the autophagosome. Therefore, the Arg/N-end rule pathway has the first regulatory function in autophagy for substrate selection.34 However, at the late stage of autophagy, Arg-HSPA5 appears to be degraded through UPS-mediated proteolysis to facilitate the fusion of autophagosomes with lysosomes. The exact inhibitory mechanism of Arg-HSPA5 remains to be determined. Taken all together, these data suggest that the Arg/N-end rule pathway positively regulates autophagic flux. Thus the Arg/N-end rule pathway may have the essential cellular function of suppressing the cytotoxicity and related pathophysiology of various aggregation-prone proteins. Considering that the core autophagy machinery, which requires membranes and lysosomes, is expected to have originated evolutionarily later than the Arg/N-end rule pathway, autophagy must not be a simple survival mechanism of eukaryotic cells under starvation conditions. As is extensively identified, autophagy provides more delicate and complex regulatory schemes for intracellular protein degradation, for example, in neurodegeneration. From a pragmatic point of view, it will be interesting to develop pharmacological methods that enhance the overall capacity of the Arg/N-end rule pathway and apply them to various models of neurodegenerative diseases.
Figure 6.
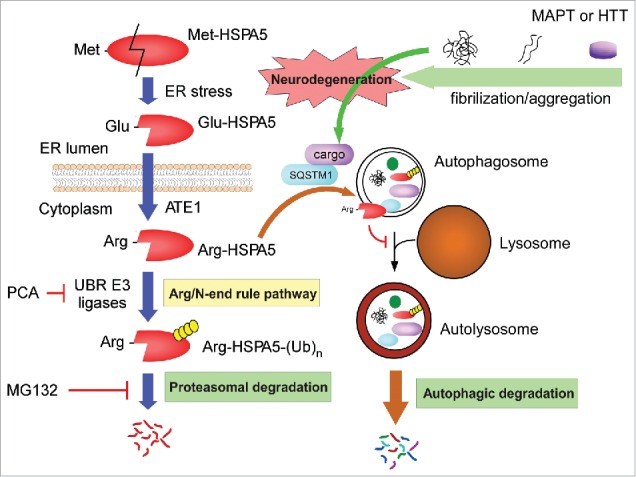
Regulatory roles of the Arg/N-end rule pathway in autophagic flux and protein aggregate clearance. Model illustrating the pro-autophagic and possible neuroprotective roles of the Arg/N-end rule pathway. The ER-residing chaperone protein HSPA5 is cleaved to Glu-HSPA5 under ER stress and translocates to the cytoplasm. ATE1 transfers an Arg residue to the N terminus of Glu-HSPA5, generating Arg-HSPA5. Arg-HSPA5 directly interacts with SQSTM1 and ultimately other cargo proteins, which include various aggregation-prone proteins such as MAPT and HTT, and guides them into the autophagosome for autophagic clearance. At the relatively late stage of autophagy, Arg-HSPA5 is recognized by UBR N-end rule E3 ligases and degraded by the 26S proteasome, which allows autophagosome-lysosome fusion. Therefore, the Arg/N-end rule pathway is a positive regulator of cellular autophagic flux and may have a protective role against proteotoxic protein-mediated neurodegeneration.
Materials and methods
Measurement of proteasome activity with fluorogenic peptide substrates
Hydrolysis of the fluorogenic substrate N-succinyl-Leu-Leu-Val-Tyr-7-amidomethylcroumarin (suc-LLVY-AMC; Enzo Life Sciences, BML-P802) was measured to determine the proteolytic activity of the chymotrypsin-like sites of proteasomes, which typically represents total proteasomal activity. Briefly, 10 μg whole cell extracts mixed with 12.5 μM suc-LLVY-AMC in a buffer containing 50 nM Tris-HCl, pH 7.5, 1 mM ethylenediaminetetraacetic acid (Daejung Chemicals, 4000-4405), 1 mg/mL bovine serum albumin (Bovogen Biologicals, BSA025), 1 mM ATP (Biobasic, AB0020), and 1 mM dithiothreitol (Geogiachem, CDT10). Proteasomal activity was monitored by measuring free AMC fluorescence in a black 96-well plate with a TECAN infinite m200 fluorometer (TECAN, Männedorf, Switzerland).
Cell cultures and transient overexpression
The mammalian cells used in this study were cultured in Dulbecco's modified Eagle's medium (Welgene, LM001-05) supplemented with 10% fetal bovine serum (Tissue Culture Biologics, 101), 2 mM glutamine (Mediatech, 25-005-CI), and 100 units/mL penicillin/streptomycin (Mediatech, 30-002-CI). Stable HEK293 cell lines expressing X-GFP were generated as previously reported,46 with minor modifications. When the cells were >95% confluent or reached a density of 106 cells/well, they were transfected with 2 to 4 μg total plasmid DNA in a 6-well culture plate for 4 h with Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific, 11668-019). Cell lysates were prepared 36 to 48 h post-transfection in RIPA buffer and used for immunoblotting. To obtain cytosolic and microsomal proteins, cells were harvested and fractionated using the Subcellular Protein Fractionation Kit (Thermo Fisher Scientific, 78840) according to the manufacturer's instructions. For fluorescence studies, HeLa cells were treated with 50 μM PCA (Sigma-Aldrich, C9635) for 4 h and fixed in 4% paraformaldehyde in phosphate-buffered saline (137 mM NaCl, 50 mM NaH2PO4, pH 7.4) for 10 min at 36 h post-transfection of GFP-LC3, GFP-MAPT, or RFP-GFP-LC3. Lysosome activity was monitored by the accumulation of LysoTracker Red (Thermo Fisher Scientific, L7528) or DQ-BSA Green (Thermo Fisher Scientific, D12050). All of the fluorescence micrographic images are representative of the total cell population.
RNA interference
The small interfering RNAs (siRNA) including the control scrambled siRNA were ordered from Genolution (Seoul, Korea). siRNA transfection was performed using G-fectin (Genolution) following the manufacturer's instructions. HeLa cells were plated at 20% to 30% confluency in 6-well plates and the siRNA were added to the final concentration of 10 nM in transfection. After 48 h, whole cell extracts (WCEs) were prepared for immunoblotting analysis. siRNA sequences used are as follows: for control (5′-CCUCGUGCCGUUCCAUCAGGUAGUU-3′ for sense, 5′-CUACCUGAUGGAACGGCACGAGGUU-3′ for antisense), for ATE1 (5′-GUGACUUUGCAUUGAUAAAUU-3′ for sense, 5′-UUUAUCAAUGCAAAGUCACUU-3′ for antisense), for UBR1 (5′-CUUACUUGUGGCUUAUAAAUU-3′ for sense, 5′-UUUAUAAGCCACAAGUAAGUU-3′ for antisense), and for UBR2 (5′-CAAGAAACCUGGAUUAACAUU-3′ for sense, 5′-UGUUAAUCCAGGUUUCUUGUU-3′ for antisense).
Assay of MAPT aggregation in cultured cells
HEK293-trex-MAPT cells and HeLa-based NES/NLS-HTT-GFP cells were generated and cultured as described previously.56,62 At ∼80% confluence, cells were treated with 50 μM PCA for 4 h, lysed in buffer A (20 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100 [Daejung Chemicals, 8566-4405], and protease inhibitor cocktail [Sigma-Aldrich, P2714]), and centrifuged at 200 × g for 15 min at 4°C to generate Triton X-100-soluble fractions. The pellets were suspended in buffer B (20 mm Tris, pH 6.8, 2% sodium dodecyl sulfate [Sigma-Aldrich, L4509], and protease inhibitor cocktail) to generate Triton X-100-insoluble fractions. Both fractions were resuspended in sample buffer for immunoblotting with anti-MAPT (Thermo Fisher Scientific, AHB0042) and anti-HTT antibodies (EMD Millipore, MAB5374).
Quantitative RT-PCR analysis
Total RNA from cultured cells was prepared using the TRIzol reagent (Thermo Fisher Scientific, 15596018), followed by further purification through RNeasy mini-column (Qiagen, 74124) with on-column DNase I (Qiagen, 79254) treatment. The cDNA samples were prepared by reverse transcription using Accupower® RT-pre mix (Bioneer, K-2055). Real-time PCR reaction was then performed using StepOne Real-Time PCR machine (Applied Biosystems, Foster City, CA, USA) with a diluted cDNA, SYBR Green qPCR master mixture (Enzynomics, RT501M), and 10 pmole of gene-specific primers. Thermal cycling conditions were comprised of 95°C for 10 min for enzyme activation, 40 cycles at 95°C for 15 sec, 60°C for 1 min. After 40 cycles, perform the other 95°C for 15 sec, 60°C for 1 min and at last keep at 95°C for 15 sec. Primer sequences were as follows: for SQSTM1, forward (5′-CCGGCTGATTGAGTCCCTC-3′) and reverse (5′-CCCCGATGTCGTAATTCTTG-3′); for MAP1LC3/LC3, forward (5′-AGCAGCATCCAACCAAAATC-3′) and reverse (5′-CTGTGTCCGTTCACCAACAG-3′); for GAPDH, forward (5′-AGGTCGGTGTGAACGGATTTG-3′) and reverse (5′-GGGGTCGTTGATGGCAACA-3′). Each mRNA level was normalized to that of GAPDH and the values were plotted as means ± SD of 3 independent experiments.
MAPT-BiFC cell analysis
A HEK293-derived stable cell line (MAPT-BiFC) that constitutively expressed both the C terminus and the N terminus of Venus protein independently fused with MAPT was used.63 MAPT-BiFC cells were seeded in a 96-well plate at a density of 105 cells/well and treated with 30 nM okadaic acid (LC Laboratories, O-5857) for 24 h to accelerate MAPT oligomerization. The cells were then treated with PCA (50 μM) for 4 h. Fluorescence images were quantified with ImageJ software (ver. 1.48k, National Institutes of Health).
Assessment of cell viability
Cell viability was assessed with a modified thiazolyl blue tetrazolium bromide (MTT) assay, as previously reported.64 Briefly, HEK293-based MAPT inducible cells were treated with 100 nM Dox (Biosesang, D1121) for 24 h to induce excess amounts of MAPT, followed by treatment with 50 μM PCA for additional 4 h. MTT solution was added to the media and incubated for 3 h at 37°C to form blue MTT-formazan products, which were assessed by measuring absorbance measurement (570 nm for test and 630 nm for reference wavelength).
Sample preparation for LC-MS/MS analysis
For LC-MS/MS analysis, the filter-aided sample preparation method for tryptic digestion was conducted as previously described.65 Briefly, 150 μg of protein samples (labeled heavy or light isotopes; Thermo Fisher Scientific, 89988 for Lys labeling, 88210 for Arg labeling) were mixed with 0.1 M dithiothreitol (Roche Diagnostics, 10708984001), 0.1 M Tris-HCl (pH 7.6, Duchefa Biochemie, T1501), and 4% sodium dodecyl sulfate for protein denaturation. The denatured proteins were incubated for 45 min at 37°C, boiled for 10 min, sonicated for 10 min, and then centrifuged at 14,000 × g for 90 min at 20°C, with a 30 k centrifugal filter (EMD Millipore, UFC800324). Urea buffer (8 M urea [Sigma-Aldrich, U5128] in 0.1 M Tris-HCl, pH 7.6) was added to the centrifugal filter and further centrifuged at 14,000 × g for 90 min. This step was repeated twice. After centrifugation, samples were treated with 0.05 M iodoacetamide (Sigma-Aldrich, I16125) at 37°C for 25 min to block disulfide bond formation. The samples were washed twice with 0.05 M ammonium bicarbonate buffer (Sigma-Aldrich, A6141), and then treated with MS-grade modified trypsin (Thermo Fisher Scientific, 90057) at 37°C for 12 h. Samples were collected via centrifugation at 14,000 × g for 40 min and vacuum-dried. Dried samples were rehydrated with 5% acetonitrile (J.T. Baker Chemicals, 9017) in water with 0.5% formic acid (Thermo Fisher Scientific, 28905) equilibration buffer, desalted with a C18 spin column (Thermo Fisher Scientific, 89870), and fractionated with an Agilent 3100 OFFGEL fractionator (Agilent Technologies, Santa Clara, CA, USA). Eluted samples were dried with a vacuum until LC-MS/MS analysis was performed.
LC-MS/MS analysis
Prepared samples were injected into a Q-Exactive (Thermo Fisher Scientific, Waltham, MA, USA) mass spectrometer coupled with an easy-nano LC 1000 high-performance liquid chromatography (HPLC) (Thermo Fisher Scientific, Waltham, MA, USA) in a 10-μl volume after dissolving in solvent A (0.1% formic acid in HPLC-grade water [J.T. Baker Chemicals, 4218). A 50-cm column (75-μm internal diameter) packed with 2 μm, 100-Å pore size C18 resin (Thermo Fisher Scientific, ES803) was used. Peptides were eluted in gradient solvent B (0.1% formic acid in HPLC acetonitrile) from 5% to 40% with a 250-nm flow rate. Separated peptides were ionized with 2.0-kV high voltage and scanned with a top-10 data-dependent mode. Tandem MS/MS was conducted in the high-energy collision dissociation cell with 27% normalized collision energy.
Database search and go analysis
The obtained raw data acquired by LC-MS/MS were subjected to a analysis process with the “MaxQuant” (version 1.5.2.8), a “Andromeda” algorithm packaged program66 with the Homo sapiens sequence database acquired from Uniprot. The parameters used in the process were as follows: maximum missed cleavages 2, variable oxidation on Met (+16 Da), and fixed carbamidomethylation in Cys (+57 Da). Arg +6 Da and Lys +6 Da labels were designated as heavy labels for quantification. Initial search peptide tolerance, main search tolerance, and isotope match tolerance were 20 ppm, 4.5 ppm, and 2 ppm, respectively. The searched, queried data were subjected to a statistical analysis process with “Perseus” (version 1.5.1.6). Data were normalized and contaminants and razor peptides were removed, then enriched DEPs with the standards of fold change below < 1.5 or above > 1.5 and a P value of < 0.05 were screened. Enriched DEPs were processed for GO biological processes and cellular compartments, using the DAVID website.67 All GO biological processes and cellular compartments were displayed by official gene names in HUGO Gene Nomenclature Committee (http://genenames.org) with P values of < 0.05 and a minimal count 5. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE68 partner repository with the data set identifier PXD003813.
Statistical analysis
The statistical significance of differences between various groups was determined with one-way analysis of variance followed by the Bonferroni post hoc test, in most data, by using GraphPad Prism 5 (GraphPad Software). Differences were considered significant when P was < 0.05.
Supplementary Material
Abbreviations
- ACTB
actin, β
- ADRM1
adhesion-regulating molecule 1
- Arg-HSPA5
N-terminally arginylated heat shock protein family A (Hsp70) member 5
- ATE1
arginyltransferase 1
- BafA1
bafilomycin A1
- BiFC
biomolecular fluorescence complementation
- CANX
calnexin; CAV1, caveolin 1
- CDK7
cyclin-dependent kinase 7
- DEP
differentially expressed protein
- Dox
doxycycline
- ER
endoplasmic reticulum
- FDR
false discovery rate
- GA
geldanamycin
- GO
gene ontology
- HCQ
hydroxychloroquine
- HSPA5
heat shock protein family A (Hsp70) member 5
- HTT
huntingtin
- LAMP1
lysosomal associated membrane protein 1
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MAPT
microtubule-associated protein tau
- MDC1
mediator of DNA damage checkpoint 1
- MEF
mouse embryonic fibroblast
- MS
mass spectrometry
- P4HB
prolyl 4-hydroxylase subunit β
- PCA
para-chloroamphetamine; PSMA4, proteasome subunit α 4
- RGS4
regulator of G protein signaling 4
- SILAC
stable isotope labeling by amino acids in cell culture
- siRNA
small interfering RNA
- suc-LLVY-AMC
N-succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin
- TG
thapsigargin
- Ub
ubiquitin
- UBR
ubiquitin protein ligase E3 component n-recognin
- UPS
ubiquitin-proteasome system
- WCE
whole cell extract
Disclosure of potential conflict of interest
The authors declare that there is no potential conflict of interest.
Accession code
Proteomic raw data are available via ProteomeXchange with identifier PXD003813.
Funding
This work was supported by a Disease-Oriented Translational Research grant (HI14C0202 to M.J.L.) and Korea-UK R&D Collaboration grant (HI14C2036 to M.J.L.) of the Korea Health Industry Development Institute. This work also supported by grants of the National Research Foundation of Korea (2013R1A1A2059793, 2016R1C1B2011367 to J.H.L., 2014M3A9B5073938 to B.Y.K, 2013R1A2A2A01014170 to Y.T.K., 2012M3A9B6055305 to K.P.K, 2016R1A2B2006507 to M.J.L.), WCI program (to B.Y.K.), KRIBB Research Initiative program (to B.Y.K), SNU Nobel Laureates Invitation program (to Y.T.K.), the Creative-Pioneering Researchers Program of SNU (to. M.J.L.), the SNUH Research Fund (04-2015-0380 to M.J.L.), and the Brain Research Program (2016M3C7A1913895 to M.J.L.). J.L., J.H.K., and W.H.C. received a scholarship from the BK21-plus program from NRF of Korea.
References
- [1].Sriram SM, Kim BY, Kwon YT. The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Biol 2011; 12:735-47; PMID:22016057; http://dx.doi.org/ 10.1038/nrm3217 [DOI] [PubMed] [Google Scholar]
- [2].Varshavsky A. The N-end rule pathway and regulation by proteolysis. Protein Sci 2011; 20(8):1298-345; PMID:21633985; http://dx.doi.org/ 10.1002/pro.666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cerda-Maira FA, Pearce MJ, Fuortes M, Bishai WR, Hubbard SR, Darwin KH. Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol Microbiol 2010; 77:1123-35; PMID:20636328; http://dx.doi.org/ 10.1111/j.1365-2958.2010.07276.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Humbard MA, Miranda HV, Lim JM, Krause DJ, Pritz JR, Zhou G, Chen S, Wells L, Maupin-Furlow JA. Ubiquitin-like small archaeal modifier proteins (SAMPs) in Haloferax volcanii. Nature 2010; 463:54-60; PMID:20054389; http://dx.doi.org/ 10.1038/nature08659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol 2004; 5:177-87; PMID:14990998; http://dx.doi.org/ 10.1038/nrm1336 [DOI] [PubMed] [Google Scholar]
- [6].Varshavsky A. The ubiquitin system, an immense realm. Annu Rev Biochem 2012; 81:167-76; PMID:22663079; http://dx.doi.org/ 10.1146/annurev-biochem-051910-094049 [DOI] [PubMed] [Google Scholar]
- [7].Lee JH, Jiang Y, Kwon YT, Lee MJ. Pharmacological modulation of the N-End Rule Pathway and its Therapeutic Implications. Trends Pharmacol Sci 2015; 36(11):782-97; PMID:26434644; http://dx.doi.org/ 10.1016/j.tips.2015.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hwang CS, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science 2010; 327:973-7; PMID:20110468; http://dx.doi.org/ 10.1126/science.1183147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shemorry A, Hwang CS, Varshavsky A. Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol Cell 2013; 50:540-51; PMID:23603116; http://dx.doi.org/ 10.1016/j.molcel.2013.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tasaki T, Mulder LC, Iwamatsu A, Lee MJ, Davydov IV, Varshavsky A, Muesing M, Kwon YT. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol Cell Biol 2005; 25:7120-36; PMID:16055722; http://dx.doi.org/ 10.1128/MCB.25.16.7120-7136.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Choi WS, Jeong BC, Joo YJ, Lee MR, Kim J, Eck MJ, Song HK. Structural basis for the recognition of N-end rule substrates by the UBR box of ubiquitin ligases. Nat Struct Mol Biol 2010; 17:1175-81; PMID:20835240; http://dx.doi.org/ 10.1038/nsmb.1907 [DOI] [PubMed] [Google Scholar]
- [12].Matta-Camacho E, Kozlov G, Li FF, Gehring K. Structural basis of substrate recognition and specificity in the N-end rule pathway. Nat Struct Mol Biol 2010; 17:1182-7; PMID:20835242; http://dx.doi.org/ 10.1038/nsmb.1894 [DOI] [PubMed] [Google Scholar]
- [13].Roman-Hernandez G, Grant RA, Sauer RT, Baker TA. Molecular basis of substrate selection by the N-end rule adaptor protein ClpS. Proc Natl Acad Sci U S A 2009; 106:8888-93; PMID:19451643; http://dx.doi.org/ 10.1073/pnas.0903614106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jiang Y, Pore SK, Lee JH, Sriram S, Mai BK, Han DH, Agarwalla P, Zakrzewska A, Kim Y, Banerjee R, et al.. Characterization of mammalian N-degrons and development of heterovalent inhibitors of the N-end rule pathway. Chem Sci 2013; 4:3339-46; http://dx.doi.org/ 10.1039/c3sc51059j [DOI] [Google Scholar]
- [15].Jiang Y, Choi WH, Lee JH, Han DH, Kim JH, Chung YS, Kim SH, Lee MJ. A neurostimulant para-chloroamphetamine inhibits the arginylation branch of the N-end rule pathway. Sci Rep 2014; 4:6344; PMID:25212999; http://dx.doi.org/ 10.1038/srep06344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee MJ, Tasaki T, Moroi K, An JY, Kimura S, Davydov IV, Kwon YT. RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc Natl Acad Sci U S A 2005; 102:15030-5; PMID:16217033; http://dx.doi.org/ 10.1073/pnas.0507533102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee MJ, Kim DE, Zakrzewska A, Yoo YD, Kim SH, Kim ST, Seo JW, Lee YS, Dorn GW 2nd, Oh U, et al.. Characterization of arginylation branch of N-end rule pathway in G-protein-mediated proliferation and signaling of cardiomyocytes. J Biol Chem 2012; 287:24043-52; PMID:22577142; http://dx.doi.org/ 10.1074/jbc.M112.364117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kwon YT, Kashina AS, Davydov IV, Hu RG, An JY, Seo JW, Du F, Varshavsky A. An essential role of N-terminal arginylation in cardiovascular development. Science 2002; 297:96-9; PMID:12098698; http://dx.doi.org/ 10.1126/science.1069531 [DOI] [PubMed] [Google Scholar]
- [19].Brower CS, Varshavsky A. Ablation of arginylation in the mouse N-end rule pathway: loss of fat, higher metabolic rate, damaged spermatogenesis, and neurological perturbations. PLoS One 2009; 4:e7757; PMID:19915679; http://dx.doi.org/ 10.1371/journal.pone.0007757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].An JY, Kim E, Zakrzewska A, Yoo YD, Jang JM, Han DH, Lee MJ, Seo JW, Lee YJ, Kim TY, et al.. UBR2 of the N-end rule pathway is required for chromosome stability via histone ubiquitylation in spermatocytes and somatic cells. PLoS One 2012; 7:e37414; PMID:22616001; http://dx.doi.org/ 10.1371/journal.pone.0037414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].An JY, Kim EA, Jiang Y, Zakrzewska A, Kim DE, Lee MJ, Mook-Jung I, Zhang Y, Kwon YT. UBR2 mediates transcriptional silencing during spermatogenesis via histone ubiquitination. Proc Natl Acad Sci U S A 2010; 107:1912-7; PMID:20080676; http://dx.doi.org/ 10.1073/pnas.0910267107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hu RG, Sheng J, Qi X, Xu Z, Takahashi TT, Varshavsky A. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature 2005; 437:981-6; PMID:16222293; http://dx.doi.org/ 10.1038/nature04027 [DOI] [PubMed] [Google Scholar]
- [23].Solomon V, Baracos V, Sarraf P, Goldberg AL. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proc Natl Acad Sci U S A 1998; 95:12602-7; PMID:9770532; http://dx.doi.org/ 10.1073/pnas.95.21.12602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Solomon V, Lecker SH, Goldberg AL. The N-end rule pathway catalyzes a major fraction of the protein degradation in skeletal muscle. J Biol Chem 1998; 273:25216-22; PMID:9737984; http://dx.doi.org/ 10.1074/jbc.273.39.25216 [DOI] [PubMed] [Google Scholar]
- [25].Lecker SH, Solomon V, Price SR, Kwon YT, Mitch WE, Goldberg AL. Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J Clin Invest 1999; 104:1411-20; PMID:10562303; http://dx.doi.org/ 10.1172/JCI7300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang G, Lin RK, Kwon YT, Li YP. Signaling mechanism of tumor cell-induced up-regulation of E3 ubiquitin ligase UBR2. FASEB J 2013; 27:2893-901; PMID:23568773; http://dx.doi.org/ 10.1096/fj.12-222711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Brower CS, Piatkov KI, Varshavsky A. Neurodegeneration-associated protein fragments as short-lived substrates of the N-end rule pathway. Mol Cell 2013; 50:161-71; PMID:23499006; http://dx.doi.org/ 10.1016/j.molcel.2013.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Park SE, Kim JM, Seok OH, Cho H, Wadas B, Kim SY, Varshavsky A, Hwang CS. Control of mammalian G protein signaling by N-terminal acetylation and the N-end rule pathway. Science 2015; 347:1249-52; PMID:25766235; http://dx.doi.org/ 10.1126/science.aaa3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xie Y, Varshavsky A. The E2-E3 interaction in the N-end rule pathway: the RING-H2 finger of E3 is required for the synthesis of multiubiquitin chain. Embo J 1999; 18:6832-44; PMID:10581257; http://dx.doi.org/ 10.1093/emboj/18.23.6832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schnupf P, Zhou J, Varshavsky A, Portnoy DA. Listeriolysin O secreted by Listeria monocytogenes into the host cell cytosol is degraded by the N-end rule pathway. Infect Immun 2007; 75:5135-47; PMID:17682039; http://dx.doi.org/ 10.1128/IAI.00164-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Piatkov KI, Brower CS, Varshavsky A. The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc Natl Acad Sci U S A 2012; 109:E1839-47; PMID:22670058; http://dx.doi.org/ 10.1073/pnas.1207786109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy 2013; 9:1758-69; PMID:24121706; http://dx.doi.org/ 10.4161/auto.24633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tasaki T, Kim ST, Zakrzewska A, Lee BE, Kang MJ, Yoo YD, Cha-Molstad HJ, Hwang J, Soung NK, Sung KS, et al.. UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc Natl Acad Sci U S A 2013; 110:3800-5; PMID:23431188; http://dx.doi.org/ 10.1073/pnas.1217358110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cha-Molstad H, Sung KS, Hwang J, Kim KA, Yu JE, Yoo YD, Jang JM, Han DH, Molstad M, Kim JG, et al.. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol 2015; 17:917-29; PMID:26075355; http://dx.doi.org/ 10.1038/ncb3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol 2013; 105:49-59; PMID:23528736; http://dx.doi.org/ 10.1016/j.pneurobio.2013.03.001 [DOI] [PubMed] [Google Scholar]
- [36].Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008; 4:849-50; PMID:18758232; http://dx.doi.org/ 10.4161/auto.6845 [DOI] [PubMed] [Google Scholar]
- [37].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/ 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/ 10.4161/auto.4451 [DOI] [PubMed] [Google Scholar]
- [39].Sarkar S, Krishna G, Imarisio S, Saiki S, O'Kane CJ, Rubinsztein DC. A rational mechanism for combination treatment of Huntington's disease using lithium and rapamycin. Hum Mol Genet 2008; 17:170-8; PMID:17921520; http://dx.doi.org/ 10.1093/hmg/ddm294 [DOI] [PubMed] [Google Scholar]
- [40].Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, et al.. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36:585-95; PMID:15146184; http://dx.doi.org/ 10.1038/ng1362 [DOI] [PubMed] [Google Scholar]
- [41].Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al.. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/ 10.1083/jcb.200412022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/ 10.1091/mbc.E03-09-0704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sriram S, Lee JH, Mai BK, Jiang Y, Kim Y, Yoo YD, Banerjee R, Lee SH, Lee MJ. Development and characterization of monomeric N-end rule inhibitors through in vitro model substrates. J Med Chem 2013; 56:2540-6; PMID:23432203; http://dx.doi.org/ 10.1021/jm400046q [DOI] [PubMed] [Google Scholar]
- [44].An JY, Seo JW, Tasaki T, Lee MJ, Varshavsky A, Kwon YT. Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc Natl Acad Sci U S A 2006; 103:6212-7; PMID:16606826; http://dx.doi.org/ 10.1073/pnas.0601700103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tasaki T, Sohr R, Xia Z, Hellweg R, Hortnagl H, Varshavsky A, Kwon YT. Biochemical and genetic studies of UBR3, a ubiquitin ligase with a function in olfactory and other sensory systems. J Biol Chem 2007; 282:18510-20; PMID:17462990; http://dx.doi.org/ 10.1074/jbc.M701894200 [DOI] [PubMed] [Google Scholar]
- [46].Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol 2000; 18:538-43; PMID:10802622; http://dx.doi.org/ 10.1038/75406 [DOI] [PubMed] [Google Scholar]
- [47].Baumeister P, Luo S, Skarnes WC, Sui G, Seto E, Shi Y, Lee AS. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol Cell Biol 2005; 25:4529-40; PMID:15899857; http://dx.doi.org/ 10.1128/MCB.25.11.4529-4540.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tasaki T, Sriram SM, Park KS, Kwon YT. The N-end rule pathway. Annu Rev Biochem 2012; 81:261-89; PMID:22524314; http://dx.doi.org/ 10.1146/annurev-biochem-051710-093308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171:513-24; PMID:17620365; http://dx.doi.org/ 10.2353/ajpath.2007.070188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhu K, Dunner K Jr, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010; 29:451-62; PMID:19881538; http://dx.doi.org/ 10.1038/onc.2009.343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517-27; PMID:19250912; http://dx.doi.org/ 10.1016/j.molcel.2009.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, Krishna G, Davies JE, Ttofi E, Underwood BR, et al.. Huntington's disease: from pathology and genetics to potential therapies. Biochem J 2008; 412:191-209; PMID:18466116; http://dx.doi.org/ 10.1042/BJ20071619 [DOI] [PubMed] [Google Scholar]
- [53].Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/ 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bhutani N, Venkatraman P, Goldberg AL. Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. EMBO J 2007; 26:1385-96; PMID:17318184; http://dx.doi.org/ 10.1038/sj.emboj.7601592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2012; 8:108-17; http://dx.doi.org/ 10.1038/nrneurol.2011.200 [DOI] [PubMed] [Google Scholar]
- [56].Bandyopadhyay B, Li G, Yin H, Kuret J. Tau aggregation and toxicity in a cell culture model of tauopathy. J Biol Chem 2007; 282:16454-64; PMID:17428800; http://dx.doi.org/ 10.1074/jbc.M700192200 [DOI] [PubMed] [Google Scholar]
- [57].Tak H, Haque MM, Kim MJ, Lee JH, Baik JH, Kim Y, Kim DJ, Grailhe R, Kim YK. Bimolecular fluorescence complementation; lighting-up tau-tau interaction in living cells. PLoS One 2013; 8:e81682; PMID:24312574; http://dx.doi.org/ 10.1371/journal.pone.0081682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, Stan RV, Butler-Browne G, Vedie B, Johannes L, et al.. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 2011; 144:402-13; PMID:21295700; http://dx.doi.org/ 10.1016/j.cell.2010.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mougeolle A, Poussard S, Decossas M, Lamaze C, Lambert O, Dargelos E. Oxidative stress induces caveolin 1 degradation and impairs caveolae functions in skeletal muscle cells. PLoS One 2015; 10:e0122654; PMID:25799323; http://dx.doi.org/ 10.1371/journal.pone.0122654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang J, Gong Z, Chen J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J Cell Biol 2011; 193:267-73; PMID:21482717; http://dx.doi.org/ 10.1083/jcb.201010026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ganuza M, Saiz-Ladera C, Canamero M, Gomez G, Schneider R, Blasco MA, Pisano D, Paramio JM, Santamaria D, Barbacid M. Genetic inactivation of Cdk7 leads to cell cycle arrest and induces premature aging due to adult stem cell exhaustion. EMBO J 2012; 31:2498-510; PMID:22505032; http://dx.doi.org/ 10.1038/emboj.2012.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci U S A 2005; 102:13135-40; PMID:16141322; http://dx.doi.org/ 10.1073/pnas.0505801102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Han DH, Na HK, Choi WH, Lee JH, Kim YK, Won C, Lee SH, Kim KP, Kuret J, Min DH, et al.. Direct cellular delivery of human proteasomes to delay tau aggregation. Nat Commun 2014; 5:5633; PMID:25476420; http://dx.doi.org/ 10.1038/ncomms6633 [DOI] [PubMed] [Google Scholar]
- [64].Choi WH, de Poot S, Lee JH, Kim JH, Han DH, Kim YK, Finley D, Lee MJ. Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nat Commun 2016; 7:10963; PMID:26957043; http://dx.doi.org/ 10.1038/ncomms10963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature Methods 2009; 6:359-62; PMID:19377485; http://dx.doi.org/ 10.1038/nmeth.1322 [DOI] [PubMed] [Google Scholar]
- [66].Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 2011; 10:1794-805; PMID:21254760; http://dx.doi.org/ 10.1021/pr101065j [DOI] [PubMed] [Google Scholar]
- [67].Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/ 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- [68].Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al.. 2016 update of the PRIDE database and related tools. Nucleic Acids Res 2016; 44:D447-56; http://dx.doi.org/ 10.1093/nar/gkv1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.