

Abstract
Background
Sphingosine-1-phosphate (S1P) is a bioactive phospholipid that acts as a signal transducer by binding to S1P receptors (S1PR) 1 to 5. The S1P/S1PRs pathway has been associated with remodeling and allergic inflammation in asthma, but the expression pattern of S1PR and its effects on non-immune cells have not been completely clarified. The aim of this study was to examine the contribution of the signaling of S1P and S1PRs expressed in airway epithelial cells (ECs) to asthma responses in mice.
Methods
Bronchial asthma was experimentally induced in BALB/c mice by ovalbumin (OVA) sensitization followed by an OVA inhalation challenge. The effects of S1PR antagonists on the development of asthma were analyzed 24 h after the OVA challenge.
Results
Immunohistological analysis revealed S1PR1-3 expression on mouse airway ECs. Quantitative real-time polymerase chain reaction demonstrated that S1P greatly stimulated the induction of CCL3 and TIMP2 mRNA in human airway ECs, i.e., BEAS-2B cells, in a dose-dependent manner. Pretreatment with the S1PR2 antagonist JTE013 inhibited the CCL3 gene expression in BEAS-2B cells. Immunohistological analysis also showed that the expression level of CCL3 was attenuated by JTE013 in asthmatic mice. Furthermore, JTE013 as well as anti-CCL3 antibody attenuated allergic responses. Intratracheal administration of JTE013 also attenuated eosinophilic reactions in bronchoalveolar lavage fluids. S1P induced transcription factor NFκB activation, while JTE013 greatly reduced the NFκB activation.
Conclusions
JTE013 attenuated allergic airway reactions by regulating CCL3 production from bronchial ECs. The intratracheal administration of JTE013 may be a promising therapeutic strategy for bronchial asthma.
Electronic supplementary material
The online version of this article (doi:10.1186/s12931-016-0465-x) contains supplementary material, which is available to authorized users.
Keywords: Sphingosine-1-phosphate, epithelial cells, BEAS-2B, JTE013, CCL3
Background
Sphingosine-1-phosphate (S1P) is a bioactive phospholipid metabolite [1]. It is produced by most kinds of cells through phosphorylation by sphingosine kinases (SPHK1 and SPHK2), and it works as a signal transducer of intracellular and extracellular cell homeostasis and functions, such as cell differentiation, inflammation, and apoptosis [2]. Outside of the cell, S1P binds to five G-protein-coupled receptors, i.e., S1P receptors (S1PR) 1 to 5, as a ligand. These receptors are expressed mainly by inflammatory cells, and they play an important role in immunity. The individual expression pattern of S1PRs on each cell creates diverse immunological responses [3]. However, the expression pattern of S1PR and its effects on non-immune cells remain unclear. Therefore, we performed a functional analysis of sphingolipids focusing on the roles of S1P and S1PRs on non-immune cells. First, we reported that TGF-β increases SPHK1, and consequently S1P, which transactivates S1PR2 and S1PR3 on fibroblasts, and promotes their transformation to myofibroblasts through Rho kinase [4, 5]. We further reported that SPHK1 mediates the transformation of airway epithelial cells (ECs) to goblet cells [6]. We also clarified that the intratracheal administration of N,N-dimethylsphingosine (DMS), a S1P synthesis inhibitor, restrained allergic inflammation and remodeling [7]. Thus, S1P and S1PRs play crucial roles in remodeling and allergic inflammation in asthma.
Bronchial asthma is a cytokine-based inflammatory dysfunction that involves airway ECs, endothelial cells, and inflammatory cells, such as Th2-type lymphocytes, dendritic cells, and mast cells [8]. Healthy airway ECs, which provide a physical barrier, can keep mast cells in a quiescent state and inhibit mast cell degranulation [3]. However, damage to airway ECs due to inflammation initiates innate immune responses, and stimulates Th2 responses by directly attracting and activating immune cells, such as Th2 cells, eosinophils, and mast cells [9–13]. Clarification of the functions of airway ECs and efforts to minimize airway EC damage are important for asthma treatment.
In this study, we investigated the expression of S1PRs on airway ECs and the function of airway ECs in the signaling pathway of the S1P/S1PRs axis by using an experimental bronchial asthma mouse model. We used human bronchial ECs, i.e., BEAS-2B cells, stimulated with S1P for identifying related cytokines to investigate the signaling pathway, and to evaluate the potential of S1PR inhibitors as a remedy for bronchial asthma.
Methods
Reagents
S1P (PeproTech, Rocky Hill, NJ) was used. A S1PR2 antagonist (also known as JTE013; Cayman, Ann Arbor, MI), and a S1PR3 antagonist (also known as VPC23019; Calbiochem, Darmstadt, Germany) were purchased. The antibodies used were anti-S1PR1-5 (Cayman) and anti-CC chemokine ligand (CCL) 3 (R&D Systems, Minneapolis, MN).
Animals
Female BALB/c mice were purchased from SLC Japan (Shizuoka, Japan). All mice were raised in sterile cages, and were used at 7 to 10 weeks of age. Our studies were approved by the Institutional Animal Care and Use Committee (Permit Numbers: P130610-R1, and P150804), and carried out in accordance with the Kobe University Animal Experimentation Regulations.
Experimental mouse models
The experimental bronchial asthma model mice were prepared by sensitization with intraperitoneal (i.p.) injections of 10 μg of ovalbumin (OVA; Sigma-Aldrich, St. Louis, MO) and 2 mg of aluminum hydroxide (Sigma-Aldrich) in 0.5 ml of sterile phosphate-buffered saline (PBS) on days 0 and 7. Then, on days 21 and 22, the mice were exposed to aerosolized sterile PBS with or without 1.0 % OVA for 30 min with ultrasonic nebulizer, Omron NE-U07 (OMRON, Kyoto, Japan) (the aerosolized particle size ranges 1–8 μm). Mice were sacrificed on day 23 for all mice experimentations. To prepare JTE013-treated or anti-CCL3 antibody-treated mice, mice were given JTE013 (4 mg/kg) or anti-CCL3 antibody (0.15 mg/kg) via i.p. injection before OVA sensitization on days 21 and 22. In the JTE013 intratracheal administration experiment, mice were intratracheally instilled with 10 μg of JTE013 or PBS.
Analysis of bronchoalveolar lavage fluids
Bronchoalveolar lavage (BAL) fluids were collected by lavaging lungs three times with 800 μl of PBS, and samples were stored at −80 °C after centrifugation. The concentrations of interleukin (IL)-4, IL-5, IL-13, and IFN-γ in the BAL fluids were measured using the Mouse ELISA Kit (R&D Systems) according to the manufacturer’s instructions.
Histology
Lungs were harvested to prepare paraffin-embedded sections for histological analysis. PBS was perfused through the lungs of mice under deep anesthesia, and the lungs were then fixed by intratracheal instillation of phosphate-buffered paraformaldehyde (Wako, Osaka, Japan). Airway EC areas were evaluated in five randomly selected sections, and each section was subjected to hematoxylin and eosin staining and immunostaining, as previously described [14, 15].
Cell culture and stimulation
BEAS-2B cells (ATCC, Manassas, VA) were incubated with the BEGM Bullet Kit (Lonza, Walkersville, MD) under a humidified atmosphere containing 5 % CO2 at 37 °C. To analyze the secretion of cytokines, the cells were stimulated with or without S1P (100 nM or 1 μM) for 3 h. In other experiments, cells were treated with 1 μM S1P and 10 μM JTE013, 10 μM VPC23019, 50 μM S3I-201 (STAT3 inhibitor, Cosmo Bio Co, Tokyo, Japan), 2.9 μM IKK inhibitor III (Calbiochem), or dimethyl sulfoxide (DMSO).
Quantitative real-time polymerase chain reaction analysis
Total cellular RNA was prepared and quantitative real-time polymerase chain reaction (qRT-PCR) was performed as previously described [16]. Relative human mRNA levels were calculated with the ΔΔCt method using glyceraldehyde-3-phosphate dehydrogenase mRNA as an internal control. The sequences of the sense and antisense primers are listed in Additional file 1: Table S1.
Western blotting analysis
The preparation of cell lysates, sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and immunoblotting were performed as previously described [17]. Primary antibodies against NFκB (Santa Cruz Biotechnology, Dallas, TX) and β-actin (Cell Signaling, Danvers, MA), and secondary antibodies (Cell Signaling) were used.
Statistical analysis
Data are shown as the mean ± standard error of the mean (SEM). Statistical analyses were performed using the Student’s unpaired t-test to compare two groups. The significances of all statistical tests are provided in the figures. A p-value of <0.05 was considered to be statistically significant.
Results
S1PR1-3 is expressed on airway epithelial cells in vivo
Previous studies suggested the existence of S1PRs on airway ECs, because the administration of DMS to ECs mitigated allergic reactions [6, 7]. Thus, we investigated the localization of S1PRs on ECs by immunostaining with S1PRs antibodies. As shown in Fig. 1, S1PR1, S1PR2, and S1PR3 were expressed in airway ECs, including bronchial ECs and alveolar ECs. These results suggest that the S1P/S1PR1-3 axis in airway ECs has a possible role in airway inflammation.
Fig. 1.
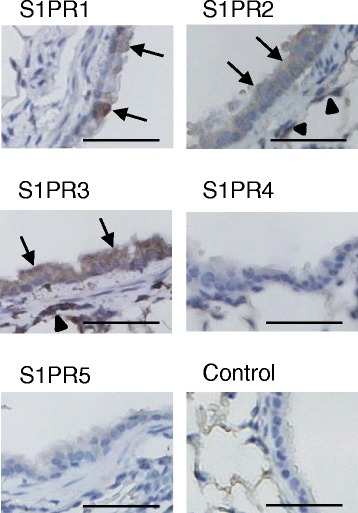
S1PR1-3 is expressed on airway epithelial cells in vivo. Lung sections from BALB/c mice immunostained with antibodies against S1PR1-5 and without these antibodies (control IgG). Bronchial ECs are stained with S1PR1-3 antibodies (arrows), and alveolar ECs are stained with S1PR2 and 3 antibodies (arrowheaads)
S1P stimulation of airway ECs evidently induces CCL3 and TIMP2 gene expression in vitro
Next, we analyzed the function of the S1P/S1PR1-3 axis in cytokine secretion by using BEAS-2B human airway ECs. Comparison by qRT-PCR of the cytokine mRNA levels of S1P- or DMSO-treated BEAS-2B cells indicated that stimulation with S1P promoted the expression of CCL3 and TIMP2 (Fig. 2a).
Fig. 2.
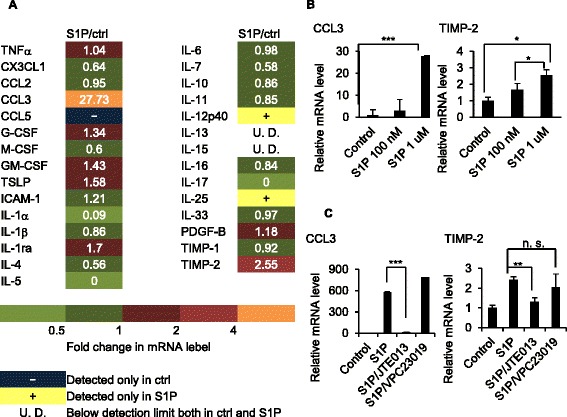
S1P stimulation of airway ECs induces CCL3 and TIMP2 gene expression, and CCL3 and TIMP2 are S1P-dependent in vitro. BEAS-2B cells were cultured with or without S1P (100 nM). The mRNA expression of 29 cytokines was analyzed by quantitative real-time RT-PCR. Data represent the ratio between the relative mRNA level of S1P-treated cells and that of S1P-untreated cells (a). BEAS-2B cells were treated with S1P (100 nM or 1 μM) (b), S1P (1 μM) and JTE013 (10 μM), or S1P (1 μM) and VPC23019 (10 μM) (c) for 3 h, and CCL3 and TIMP2 gene expression was analyzed by quantitative real-time RT-PCR
CCL3 and TIMP2 are S1P-dependent in vitro and in vivo
We further analyzed the dose-dependent CCL3 and TIMP2 gene expression in BEAS-2B cells after stimulation with S1P. As shown in Fig. 2b, CCL3 and TIMP2 gene expression in BEAS-2B cells increased in proportion to the S1P concentration, and they were attenuated by JTE013, a S1PR2 antagonist (Fig. 2c). In contrast, neither was attenuated by VPC23019, a S1PR1 and S1PR3 antagonist (Fig. 2c). Immunohistological analysis also showed that CCL3 and S1PR2 were co-expressed on the airway ECs in the experimental asthma mouse model, and the expression level of CCL3 was attenuated by JTE013, although the expression level of S1PR2 was not attenuated by JTE013 because JTE013 only inhibits S1P binding to S1PR2 (Fig. 3a). further analyzed the effect of CCL3 on airway allergic response using the experimental asthma mouse model. As shown in Fig. 3b, airway eosinophilia and the levels of IL-4, IL-5, and IL-13 were attenuated by the anti-CCL3 antibody. These results suggest that S1P induced the secretion of CCL3, which has a crucial role in bronchial asthma through the S1P/S1PR2 axis in airway ECs.
Fig. 3.
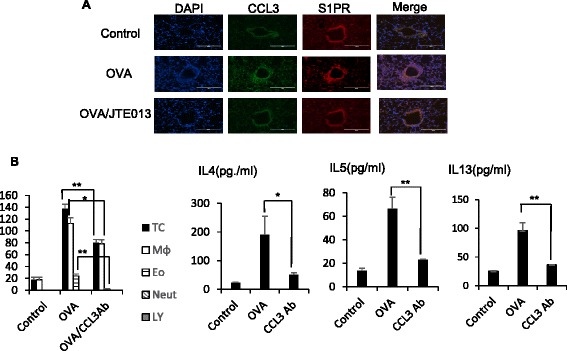
CCL3 and TIMP2 are S1P-dependent in vivo. Immunofluorescent microscopic images show OVA-treated lung sections stained with FITC-conjugated anti-CCL3 (green), and Alexa 594-conjugated anti-S1PR2 (red) antibodies. Co-localization of CCL3 and S1PR2 is seen in yellow (a). BAL Fluids were obtained from BALB/c mice treated with vehicle, OVA, or OVA/mCCL3 antibody (3 μg/cavity), and examined by Diff-Quick staining. The total cell counts and cell differentials in the BAL fluids are shown, and the concentrations of IL-4, IL-5, and IL-13 in the BAL fluids were measured by using ELISA kits (b). Data are expressed as the mean ± standard error (SE) of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
JTE013 decreases allergic responses
To investigate the effects of the S1P/S1PR2 axis on bronchial asthma, we employed an experimental asthma mouse model and evaluated the therapeutic effects of JTE013. As expected, OVA-treated mice showed a marked increase in the number of eosinophils in the BAL fluids (Fig. 4a). In addition, the levels of IL-4, IL-5, and IL-13 were increased in the BAL fluids from OVA-treated mice (Fig. 4b). In contrast, JTE013 administration resulted in a significant decrease in Th2 inflammatory reactions, although there was no statistical difference in the levels of IL-4 (Fig. 4a and b). Consistent with the BAL fluids analysis, histological analysis demonstrated a marked decrease in inflammatory cell infiltration around the bronchiole in OVA/JTE013-treated mice when compared to OVA-treated mice (Fig. 4c).
Fig. 4.
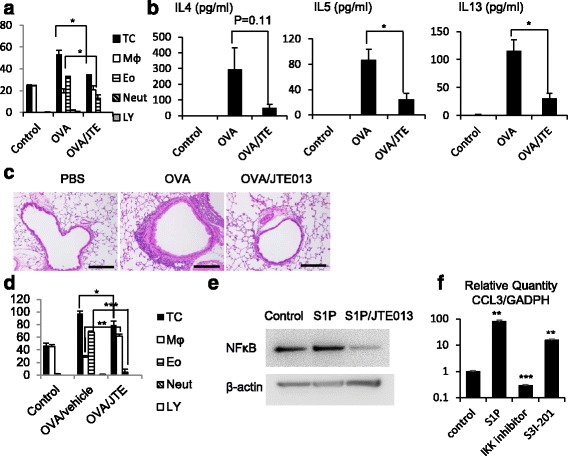
JTE013 decreases allergic responses. BAL fluids were obtained after OVA or vehicle exposure, and examined by Diff-Quick staining. Total cell counts and cell differentials in the BAL fluids are shown (a), and the concentrations of IL-4, IL-5, IL-13, and IFN-γ in the BAL fluids were measured by using ELISA kits (n = 5) (b). Lung sections from BALB/c mice treated with vehicle, OVA, or OVA/JTE013 (4 mg/kg) were subjected to hematoxylin and eosin staining (c). BAL fluids were obtained after OVA or vehicle exposure with intratracheal administration of vehicle or JTE013 (10 μg/trachea), and the total cell counts and cell differentials in the BAL fluids were examined by Diff-Quick staining (n = 3 to 5) (d). BEAS-2B cells were treated with vehicle, S1P (1 μM), or S1P/JTE013 (1 μM) for 4 h. Phosphorylation of p65 was evaluated by western blot analysis. Representative images from three independent experiments are shown (e). BEAS-2B cells were treated with vehicle, S1P (1 μM), IKK inhibitor, or S3I-201. Each CCL3 gene expression was analyzed by quantitative real-time RT-PCR, representing the relative mRNA level of CCL3 (f). Data are expressed as the mean ± SE of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
Moreover, intratracheal administration of JTE013 also resulted in a marked decrease in eosinophilic reactions in the BAL fluids (Fig. 4d). This suggests that intratracheal treatment can inhibit allergic bronchial inflammation as well as i.p. treatment.
JTE013 inhibits CCL3 gene expression by attenuating NFκB and STAT3 activation in vitro
Next, we analyzed the signaling pathways downstream of S1PR2, and investigated the activation of transcription factors, NFκB and STAT3. Previous study reported that S1PR2 can activate the transcription factor, STAT3 in mice lung [18], which regulates CCL3 expression in macrophage [19], and the transcription factor NFκB also induces CCL3 synthesis in nucleus pulposus cells [20]. In this study, using BEAS-2B cells, we first assessed the activity of NFκB downstream of the S1P/S1PR2 signaling pathway, and next analyzed the CCL3 expression downstream of NFκB and STAT3 activation. The results are shown in Fig. 4e and f. The expression of NFκB increased after stimulation with S1P, while it decreased with JTE013 (Fig. 4e), and CCL3 gene expression in BEAS-2B cells increased with the S1P concentration, while it was attenuated by IKK inhibitor, and a STAT3 inhibitor, S3I-201. Taken together, these results suggest that JTE013 inhibits CCL3 expression through NFκB and STAT3 transcription.
Discussion
The aim of this study was to elucidate the role of S1P in bronchial asthma by focusing on airway ECs. Pathological analysis showed that S1PR1-3 are expressed on airway ECs, and airway ECs that expressed S1PR2 also expressed CCL3 at a high level in response to the OVA inhalation challenge. Our previous research showed the inflammation induced by OVA exposure decreases in time-dependent manner [17]. Therefore, we adopted our time schedule as appropriate duration time.
In vitro analysis of cytokine expression in human bronchial ECs also showed that S1P led to the transcriptional activation of the CCL3 gene through NFκB and STAT3 activation. Previous studies reported that S1PR2 is coupled to GαI, Gα12/13, and GαQ, which activates phospholipase C (PLC) [21, 22]. PLC plays a role in the regulation of intracellular signaling pathways through calcium release and protein kinase C activation [22]. PLC also plays a role in proinflammatory gene expression through the activation of NFκB [23]. These results suggest that the S1P/S1PR2 axis has a possible role in cytokine expression via the activation of NFκB downstream of PLC. Furthermore, STAT3 is directly activated by S1PR2 in a Rho-dependent manner [18, 24], which is dependent on the S1PR2-coupled G protein Gα12/13 [21].
We used human bronchial ECs instead of mouse bronchial ECs as previous literatures [25–27], because previous reports showed that approximately 99 % of mouse genes have a homologue in the human genome [28], and bioinformatic analysis and human curation of 60,770 full-length mouse cDNA clones showed similarity (70–85 % identity) to known human-disease genes [29].
CCL3 expression is up-regulated by NFκB and STAT3. Consistent with our results, Jianru W et al. also demonstrated that NFκB introduces CCL3 synthesis, by binding to the proximal promotor of CCL3 major nuclear proteins in nucleus pulposus cells [20]. Furthermore, Zhang C et al. also reported that STAT3 is a key regulator of CCL3 expression in macrophages [19]. Taken together, we infer that the S1P/S1PR2 axis activates STAT3 and NFκB to form a complex with CCL3 nuclear protein binding sites, which in turn produces CCL3 through Gα12/13 and GαQ (Fig. 5).
Fig. 5.
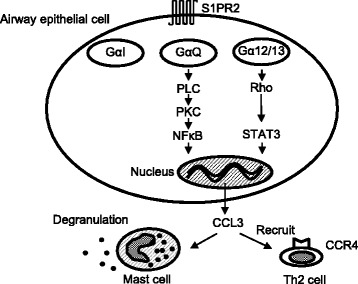
Simplified scheme of the S1PR2 signaling events in airway epithelial cells. On airway epithelial cells, S1P binds to S1PR2, which is coupled to GaI, Ga12/13, and GaQ. GaQ in turn activates PLC, activating PKC. PKC phosphorylates NFκB, which binds to the proximal promotor of CCL3 major nuclear proteins. In addition, another S1PR2-coupled G protein, Ga12/13, also up-regulates CCL3 expression through STAT3 activation in a Rho-dependent manner
CCL3 is a member of the C-C chemokine family, and is a chemoattractant for eosinophils, lymphocytes, leukocytes, and monocytes. Previous studies reported that an increased level of CCL3 was observed in BAL fluids from patients with asthma [30], and that CCL3 evoked local T cell recruitment and induced skin reactions in patients with atopic dermatitis [31]. Furthermore, Elena et al. demonstrated significantly higher levels of CCL3 in BAL fluids from patients with corticosteroid-resistant asthma [32].
CCL3 is also a ligand for CCR1 and CCR4. CCR1 is expressed in mast cells and basophils, and the CCL3/CCR1 axis is important for the activation of mast cells [33]. A previous study reported that CCL3 acted as a co-stimulator for FcεRI-mediated degranulation in conjunctival mast cells in CCL3-deficient mice [34]. Moreover, CCL3 synergistically enhanced FcεRI-mediated degranulation and the production of CCL2, which is a chemoattractant for T cells and eosinophils in bone marrow-derived murine mast cells and the rat basophilic leukemia 2H3 cell line [33, 35–39]. Taken together, there is crosstalk between CCR1-mediated and FcεRI-mediated signaling cascades in mast cells, which plays an important role in allergic disorders as FcεRI cross-linking is a key event of activation. On the other hand, CCR4 mediates allergy-promoting Th2 cell recruitment [23]. A recent publication by Oskeritzian et al. demonstrated that S1P neutralization or a S1PR2 antagonist strongly attenuated early T-cell recruitment to lung perivascular lesions and T-cell chemokine production, including CCL3, in murine mast cell-dependent acute airway allergic responses [23], suggesting that the S1P/S1PR2 axis regulates early T-cell recruitment via chemokine production. In our study, immunohistochemical analysis showed that CCL3 was abundantly produced by airway ECs. Therefore, CCL3 on airway ECs may be a promising target for the treatment of airway allergic inflammation, which is mediated by T-cell recruitment. In fact, our study showed that CCL3 neutralization actually reduced Th2-type allergic responses.
The importance of S1P in an experimental asthma mouse model is well established [4–7]. Past reports have suggested that the S1P/S1PR2 axis also plays an important role in antigen-induced mast cell degranulation [40], myofibroblast contraction [41], and vasoconstriction [42]. Our study first verified the contribution of the signaling of the S1P/S1PR2 axis in airway ECs. However, the intraperitoneal administration of S1PR2 antagonist to investigate the in vivo reaction of airway ECs can also induce a systemic reaction, as described above. Although we examined the intratracheal administration of a S1PR2 antagonist, the possibility cannot be excluded that the method could also induce systemic reaction through resident mast cells around airway ECs. In order to determine the effects of the S1PR2 antagonist on airway ECs in vivo, an appropriate experimental mouse model, such as S1PR2-deficient mice [43], should be employed.
Our in vitro study using primary cultures demonstrated that the S1P/S1PR2 axis promotes airway inflammation. Intriguingly, Yapeng et al. showed that S1PR2 induced the extrusion of apoptotic kidney ECs to maintain an intact barrier [44]. In addition, Kono et al. showed that S1PR2-knockout mice had defective epithelial barrier function in the cochlea [45]. These results suggest that inhibition of the S1P/S1PR2 pathway disturbs the barrier function of ECs, and may aggravate asthmatic reactions. We have two hypotheses to account for these contradictory findings. One hypothesis is that the nature of airway ECs is different from those of other ECs. The other hypothesis is that the quantity of JTE013 used was enough to restrain inflammatory responses, but not enough to restrain the extrusion of airway ECs. Further studies investigating the effects of the S1PR2 antagonist at different intratracheal dosage levels on asthma would be of great interest.
In addition, we investigated the allergic reaction by intratrachial administration of aerosolized OVA and JTE013 to show the potential of intratrachial administration of JTE013 as the remedy of bronchial asthma. However, Kannan et al. have revealed that the deposition of particle substance to airway and lung depends on many factors including the property of the particle, anatomical geometry of the airway, and respiratory pattern [46, 47]. And it may be possible to obtain more analysis from the computational Euler-Langrangian particle delivery approach of Kannan et al. [47]. In this point, there are some limitations to interpret our in vivo data. Though extrapolating from our studies with intratrachial administration in mouse model to the allergic reaction in humans requires cautious interpretation, our data can provide significant promise to JTE013 as the remedy because previous study showed that asthma mouse model can be used to investigate the main point of our pathogenesis, allergic reaction through the cytokine production [48] and previous report showed the intratrachial administration of drugs such as budesonide in mice has similar efficacy in humans [49]. Therefore, I think our results can lead to the development of worthwhile therapeutic interventions.
Conclusion
This study demonstrated that the S1PR2 antagonist JTE013 inhibits asthmatic allergic reactions by suppressing the S1PR2-mediated NFκB activation and CCL3 production in the bronchial epthelium, and that the intratracheal administration of JTE013 may be a promising remedy for bronchial asthma.
Acknowledgements
We thank Mr. Kei Kunimasa for his technical support.
Funding
This work was supported by JSPS KAKENHI 24591131 and Research Grants from MSD and AstraZeneca K.K. to Yoshihiro Nishimura, and JSPS KAKENHI 23591119 to Kazuyuki Kobayashi.
Availability of data and materials
All data generated or analysed during this study are included in this article and its supplementary information files.
Authors’ contributions
TT, KK, and TN analyzed the data and TT drafted the manuscript. KK conceived of the study, and participated in its design. TN participated in its design and helped to draft the manuscript. YK assisted with the histological studies. DT and KN participated in the statistical analysis. MY, TM, and YN participated in the design and assited with the technical advice. ALL the authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
All animal experiments were approved by the Institutional Animal Care and Use Committee (Permit Numbers: P130610-R1, and P150804), and carried out in accordance with the Kobe University Animal Experimentation Regulations.
Abbreviations
- BAL
Bronchoalveolar lavage
- CCL
CC chemokine ligand
- DMS
N,N-dimethylsphingosine
- DMSO
Dimethyl sulfoxide
- ECs
Epithelial cells
- i.p.
Intraperitoneal
- IL
Interleukin
- OVA
Ovalbumin
- PBS
Phosphate buffered saline
- PLC
Phospholipase C
- qRT-PCR
Quantitative real-time polymerase chain reaction
- S1P
Sphingosine-1-phosphate
- S1PR
Sphingosine-1-phosphate receptor
- SEM
Standard error of the mean
- SPHK
Sphingosine kinase
Additional file
The primers used for qRT-PCR. (DOCX 30 kb)
References
- 1.Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8:753–63. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rivera J, Olivera A. Src family kinases and lipid mediators in control of allergic inflammation. Immunol Rev. 2007;217:255–68. doi: 10.1111/j.1600-065X.2007.00505.x. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez T, Hla T. Structual and functional characteristics of S1P receptors. J Cell Biochem. 2004;92:913–22. doi: 10.1002/jcb.20127. [DOI] [PubMed] [Google Scholar]
- 4.Urata Y, Nishimura Y, Hirase T, Yokoyama M. Sphingosin 1-phosphate induces alpha-smooth muscle actin expression in lung fibroblasts via Rho-kinase. Kobe J Med Sci. 2005;51(1–2):17–21. [PubMed] [Google Scholar]
- 5.Kono Y, Nishiuma T, Nishimura Y, Kotani Y, Okada T, Nakamura S, et al. Sphingosine kinase 1 regulates differentiation of human and mouse lung fibroblasts mediated by TGF-β1. Am J Respir Cell Mol Biol. 2007;27:395–404. doi: 10.1165/rcmb.2007-0065OC. [DOI] [PubMed] [Google Scholar]
- 6.Kono Y, Nishiuma T, Okada T, Kobayashi K, Funada Y, Kotani Y, et al. Sphingosine kinase 1 regulates mucin production via ERK phosphorylation. Pulm Pharmacol Ther. 2010;23:36–42. doi: 10.1016/j.pupt.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Nishiuma T, Nishimura Y, Okada T, Kuramoto E, Kotani Y, Jahangeer S, et al. Inhalation of sphingosine kinase inhibitor attenuates airway inflammation in asthmatic mouse model. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1085–93. doi: 10.1152/ajplung.00445.2007. [DOI] [PubMed] [Google Scholar]
- 8.Peter JB. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8:183–92. doi: 10.1038/nri2254. [DOI] [PubMed] [Google Scholar]
- 9.Holgate ST. The epithelium is central to the pathogenesis of asthma. Allergol Int. 2008;57:1–10. doi: 10.2332/allergolint.R-07-154. [DOI] [PubMed] [Google Scholar]
- 10.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–90. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J AllergyClin Immunol. 2007;120:1279–84. doi: 10.1016/j.jaci.2007.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sansonetti PJ. War and peace at mucosal surfaces. Nat Rev Immunol. 2004;4:953–64. doi: 10.1038/nri1499. [DOI] [PubMed] [Google Scholar]
- 13.Bulek K, Swaidani S, Aronica M, Li X. Epithelium: the interplay between innate and Th2 immunity. Immunol Cell Biol. 2010;88:257–68. doi: 10.1038/icb.2009.113. [DOI] [PubMed] [Google Scholar]
- 14.Nagano T, Yasunaga M, Goto K, Kenmotsu H, Koga Y, Kuroda J, et al. Antitumor activity of NK012 combined with cisplatin against small cell lung cancer and intestinal mucosal changes in tumor-bearing mouse after treatment. Clin Cancer Res. 2009;15(13):4348–55. doi: 10.1158/1078-0432.CCR-08-3334. [DOI] [PubMed] [Google Scholar]
- 15.Kenmotsu H, Yasunaga M, Goto K, Nagano T, Kuroda J, Koga Y, et al. The antitumor activity of NK012, an SN-38-incorporating micelle, in combination with bevacizumab against lung cancer xenografts. Cancer. 2010;116(19):4597–604. doi: 10.1002/cncr.25233. [DOI] [PubMed] [Google Scholar]
- 16.Nagano T, Yasunaga M, Goto K, Kenmotsu H, Koga Y, Kuroda J, et al. Synergistic antitumor activity of the SN-38-incorporating polymeric micelles NK012 with S-1 in a mouse model of non-small cell lung cancer. Int J Cancer. 2010;127(11):2699–706. doi: 10.1002/ijc.25282. [DOI] [PubMed] [Google Scholar]
- 17.Kuramoto E, Nishiuma T, Kobayashi K, Yamamoto M, Kono Y, Funada Y, et al. Inhalation of urokinase-type plasminogen activator reduces airway remodeling in a murine asthma model. Am J Physiol Lung Cell Mol Physiol. 2009;296(3):L337–46. doi: 10.1152/ajplung.90434.2008. [DOI] [PubMed] [Google Scholar]
- 18.Oskeritzian CA, Hait NC, Wedman P, Chumanevich A, Kolawole EM, Price MM, et al. The sphingosine-1-phosphate/sphingosine-1-phosphate receptor 2 axis regulates early airway T-cell infiltration in murine mast cell-dependent acute allergic responses. J Allergy Clin Immunol. 2015;135(4):1008–18. doi: 10.1016/j.jaci.2014.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J Biol Chem. 2013;288:1489–99. doi: 10.1074/jbc.M112.419788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jianru W, Ye T, Kate LEP, Neil C, Gail H, Rowena AB, et al. Tumor necrosis factor α– and interleukin-1β–dependent induction of CCL3 expression by nucleus pulposus cells promotes macrophage migration through CCR1. Arthritis & Rheumatism. 2013;65(3):832–42. doi: 10.1002/art.37819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohamad A, Daniel C, Yusuf AH, Lina MO. Sphingosine-1-phosphate receptor 2. FEBS J. 2013;280:6354–66. doi: 10.1111/febs.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aarthi JJ, Darendelier MA, Pushparaj PN. Dissecting the role of the S1P/S1PR axis in health and disease. J Dental Res. 2011;90:841–54. doi: 10.1177/0022034510389178. [DOI] [PubMed] [Google Scholar]
- 23.Nagano T, Edamatsu H, Kobayashi K, Takenaka N, Yamamoto M, Sasaki N, et al. Phospholipase cε, an effector of ras and rap small GTPases, is required for airway inflammatory response in a mouse model of bronchial asthma. PLoS One. 2014;9(9):e108373. doi: 10.1371/journal.pone.0108373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loh KC, Leong WI, Carlson ME, Oskouian B, Kumar A, Fyrst H, et al. Sphingosine-1-phosphate enhances satellite cell activation in dystrophic muscles through a S1PR2/STAT3 signaling pathway. PLoS One. 2012;7:e37218. doi: 10.1371/journal.pone.0037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liou CJ, Cheng PY, Huang WC, Chan CC, Chen MC, et al. Oral lovastatin attenuates airway inflammation and mucus secretion in ovalbumin-induced murine model of asthma. Allergy, Asthma Immunol Res. 2014;6(6):548–57. doi: 10.4168/aair.2014.6.6.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan GH, Choi YH. Salidroside attenuates allergic airway inflammation through negative regulation of nuclear factor-kappa B and p38 mitogen-activated protein kinase. J Pharmacol Sci. 2014;126(2):126–35. doi: 10.1254/jphs.14037FP. [DOI] [PubMed] [Google Scholar]
- 27.Shin IS, Park JW, Shin NR, Jeon CM, Kwon OK, Kim JS, et al. Melatonin reduces airway inflammation in ovalbumin-induced asthma. Immunobiology. 2014;219(12):901–8. doi: 10.1016/j.imbio.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Mouse Genome Sequencing Consortium. Waterston RH, Lindblad-Toh K, Birney E, Rogers J, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 29.Silva DG, Schonbach C, Brusic V, Socha LA, Nagashima T, Petrovsky N. Identification of “pathologs” (disease-related genes) from the RIKEN mouse cDNA dataset using human curation plus FACTS, a new biological information extraction system. BMC Genomics. 2004;5(1):28. doi: 10.1186/1471-2164-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karolien B, Sandra V, Rosette VDH, Hilda W, Inge N, Greet S. The allergic cascade: Review of the most important molecules in the asthmatic lung. Immunol Lett. 2007;113:6–18. doi: 10.1016/j.imlet.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Gaga M, Ong YE, Benyahia F, Aizen M, Barkans J, Kay AB. Skin reactivity and local cell recruitment in human atopic and nonatopic subjects by CCL2/MCP-1 and CCL3/MIP-1α. Allergy. 2008;63:703–11. doi: 10.1111/j.1398-9995.2007.01578.x. [DOI] [PubMed] [Google Scholar]
- 32.Elena G, Pia J, Clifton FH, Andrew HL, David WHR, Richard JM, et al. Corticosteroid resistant asthma is associated with classical anti-microbial activation of airway macrophages. J Allergy Clin Immunol. 2008;122(3):550–9. doi: 10.1016/j.jaci.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toda M, Kuo CH, Borman SK, Richardson RM, Inoko A, Inagaki M, et al. Evidence that formation of vimentin-mitogen-activated protein kinase (MAPK) complex mediates mast cell activation following FCeRI/CC chemokine receptor 1 cross-talk. J Biol Chem. 2012;287(29):24516–24. doi: 10.1074/jbc.M111.319624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyazaki D, Nakamura T, Toda M, Cheung-Chau KW, Richardson RM, Ono SJ, et al. Macrophage inflammatory protein-1α as a costimulatory signal for mast cell-mediated hypersensitivity reactions. J Clin Invest. 2005;115:434–42. doi: 10.1172/JCI18452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toda M, Dawson M, Nakamura T, Munro PM, Richardson RM, Bailly M, et al. Impact of engagement of FcεRI and CC chemokine receptor 1 on mast cell activation and motility. J Biol Chem. 2004;279:48443–8. doi: 10.1074/jbc.M408725200. [DOI] [PubMed] [Google Scholar]
- 36.Fifadara NH, Aye CC, Raghuwanshi SK, Richardson RM, Ono SJ. CCR1 expression and signal transduction by murine BMMC results in secretion of TNF-α, TGFβ-1, and IL-6. Int Immunol. 2009;21:991–1001. doi: 10.1093/intimm/dxp066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laffargue M, Calvez R, Finan P, Trifilieff A, Barbier M, Altruda F, et al. Phosphoinositide 3-kinaseγ is an essential amplifier of mast cell function. Immunity. 2012;16:441–51. doi: 10.1016/S1074-7613(02)00282-0. [DOI] [PubMed] [Google Scholar]
- 38.Aye CC, Toda M, Morohoshi K, Ono SJ. Identification of genes and proteins specifically regulated by costimulation of mast cell FcεRI receotirI and chemokine receptor1. Exp Mol Pathol. 2012;92:267–74. doi: 10.1016/j.yexmp.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Wang J, Tian Y, Phillips KL, Chiverton N, Haddock G, Bunning RA, et al. Tumor necrosis factor α- and interleukin-1β-dependent induction of CCL3 expression by nucleus pulposus cells promotes macrophage migration through CCR1. Arthritis Rheum. 2013;65(3):832–42. doi: 10.1002/art.37819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puneet SJ, Meryem B, Ana O, Claudia GE, Richard LP, Juan R, et al. Transactivation of sphingosine-1-phosphate receptors by FcεRI triggering is required for normal mast cell degranulation and chemotaxis. J Exp Med. 2004;199(7):959–70. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang XQ, Mao LJ, Fang QH, Kobayashi T, Kim HJ, Sugiura H, et al. Sphingosylphosphorylcholine induces α-smooth muscle actin expression in human lung fibroblasts and fibroblast-mediated gel contraction via S1P2 receptor and Rho/Rho-kinase pathway. Prostaglandins Other Lipid Mediat. 2014;108:23–30. doi: 10.1016/j.prostaglandins.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 42.William SS, Bruce RP. Bryan JM.S1P2 receptor-dependent Rho-kinase activation mediates vasoconstriction in the murine pulmonary circulation induced by sphingosine 1-phosphate. Am J Physiol Lung Cell Mol Physiol. 2010;299(1):L137–45. doi: 10.1152/ajplung.00233.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Du W, Takuwa N, Yoshioka K, Okamoto Y, Gonda K, Sugihara K, et al. S1P2, the G protein–coupled receptor for Sphingosine-1-Phosphate, negatively regulates tumor angiogenesis and tumor growth in vivo in mice. Cancer Res. 2010;70(2):772–81. doi: 10.1158/0008-5472.CAN-09-2722. [DOI] [PubMed] [Google Scholar]
- 44.Yapeng G, Tetyana F, Roger S, Jody R. Epithelial cell extrusion requires the sphingosine-1-phosphate receptor 2 pathway. J Cell Biol. 2011;193(4):667–76. doi: 10.1083/jcb.201010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kono M, Belyanseva IA, Skoura A, Frolenkov GI, Starost MF, Dreier JL, et al. Deafness and stria vascularis defects in S1P2 receptor-null mice. J Biol Chem. 2007;282:10690–6. doi: 10.1074/jbc.M700370200. [DOI] [PubMed] [Google Scholar]
- 46.Kannan RR, Chen ZJ, Singh N, Przekwas A, Delvadia R, Tian G, et al. A Quasi-3D wire approach to model pulmonary airflow in human airways. Int J Numer Method Biomed Eng. 2016 doi: 10.1002/cnm.2838. [DOI] [PubMed] [Google Scholar]
- 47.Kannan RR, Guo P, Przekwas A. Particle transport in the human respiratory tract: formulation of a nodal inverse distance weighted Euler-Lagrangian transport and implementation of the Wind-Kessel algorithm for an oral delivery. Int J Numer Methods in Biomed Eng. 2016 doi: 10.1002/cnm.2746. [DOI] [PubMed] [Google Scholar]
- 48.Kumar RK, Foster PS. Are mouse models of asthma appropriate for investigating the pathogenesis of airway hyper-responsiveness? Front Physiol. 2012;3(312):1–7. doi: 10.3389/fphys.2012.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Y, Wang J, Li H, Sun L, Wang Y, Han X. The effects of budesonide on angiogenesis in a murine asthma model. Arch Med Sci. 2013;9(2):361–7. doi: 10.5114/aoms.2013.33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this article and its supplementary information files.