

Abstract
Increasingly, children and adolescents with dyslipidemia qualify for pharmacologic intervention. As they are for adults, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitors (statins) are the mainstay of pediatric dyslipidemia treatment when lifestyle modifications have failed. Despite the overall success of these drugs, the magnitude of variability in dose-exposure-response profiles contributes to adverse events and treatment failure. In children, the cause of treatment failures remains unclear. This review describes the updated guidelines for screening and management of pediatric dyslipidemia and statin disposition pathway to assist the provider in recognizing scenarios where alterations in dosage may be warranted to meet patients' specific needs.
INDEX TERMS: atorvastatin, dyslipidemia, fluvastatin, lovastatin, pediatrics, pharmacogenomics, pharmacokinetics, pravastatin, rosuvastatin, simvastatin, statin
INTRODUCTION
Cardiovascular disease remains the number one cause of mortality in the United States.1 Despite significant advances in medical and surgical management for heart disease and stroke, the burden of cardiovascular disease remains alarming. Coronary artery disease (CAD) alone accounts for 1 of every 7 deaths in the United States.1 Although CAD has historically been perceived as a disease of middle to late adulthood, data now support onset at a much younger age. Clinically silent precursors to CAD, fatty streaks, have been observed in children as young as 3 years of age with coronary involvement identified at adolescence.2 By the time individuals reach their 20s studies suggest that the incidence of coronary atherosclerosis can range from 45% to 75%.3,4 Importantly, several studies confirm that the risk factors observed in adults (e.g., elevated low-density lipoprotein [LDL], obesity, hypertension, tobacco exposure, and diabetes) also contribute to atherosclerosis in children.5,6 Collectively, these studies have illuminated the need for preventive cardiovascular services in children and young adults.
Trends in circulating lipid profiles support a role for screening in children as part of preventative care. The prevalence of total plasma cholesterol (TC) concentrations in excess of 200 mg/dL has risen to 10% in adolescents,7,8 a far cry from the estimated 0.2% of the population that can attribute this laboratory finding to familial hypercholesterolemia.9,10 This may be explained, in part, by the rate of overweight/obesity in children, which as in adults, can be associated with elevated cholesterol levels.11 Importantly, most adolescents with elevated TC will continue to have elevated TC into adulthood, and those who are overweight have a 2-fold higher relative risk of CAD mortality, independent of adult weight.12,13 When pediatric weight and lipid profiles are considered together, the prevalence of symptomatic CAD in young to middle-aged adults is expected to increase by 5% to 16% over the next 2 decades.14 This will likely contribute to an additional 100,000 cases of early coronary heart disease that are specifically due to childhood obesity.
SCREENING AND MANAGEMENT GUIDELINES
Since the last comprehensive review of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) by Eiland et al,15 the pediatric screening and management guidelines have changed, prompting this update for pediatric providers who make recommendations related to prescribing statins.
In 1992, the National Cholesterol Education Program (NCEP) began recommending targeted lipid screening in pediatric patients with risk factors for premature atherosclerotic cardiovascular disease.16 This strategy exposed numerous cases of asymptomatic dyslipidemia that previously would have been neglected for decades. However, additional evidence suggests that simply relying on family history alone will miss at least 30% of pediatric patients with moderate dyslipidemia.17 These previous NCEP guidelines also focused on LDL screening, essentially ignoring the combined dyslipidemic patterns that are observed in obese pediatric patients (i.e., increased triglycerides, increased LDL, decreased high-density lipoprotein [HDL]).
Realizing that a large proportion of at-risk children would remain unidentified, the National Heart, Lung, and Blood Institute convened an expert panel on Cardiovascular Health and Risk Reduction in Children and Adolescents to update the pediatric preventive cardiovascular guidelines, including modifications to lipid screening and management in childhood and adolescence.18 The most striking modification in the updated NCEP guidelines resides in the domain on lipid screening where the panel now recommends universal lipid screening for all children between the ages 9 and 11 years and again between 17 and 21 years of age.18 These age groups were targeted specifically to screen patients prior to and after puberty, when it is observed that TC and LDL can fluctuate with growth and sexual maturity.19,20 The updated guidelines also suggest that lipid profiles can be obtained in either the fasting or non-fasting state given the reliability with either method.21 This offers the benefit of facilitating screening in busy clinic settings where non-fasting lipid profiles may be easier to obtain.
Treatment guidelines were also clarified in the new guidelines with the goal of minimizing the burden of CAD in young adults. As expected, diet and exercise are the first steps in which a provider managing children with lipid abnormalities should implement a change. When lifestyle modifications fail to improve lipid profiles over a 6-month period, pharmacologic therapy may be warranted to reverse lipid abnormalities. In children older than 10 years of age, use of pharmacologic management should be based on the average results of 2 lipid profiles obtained at least 2 weeks apart but no more than 3 months apart. The thresholds used to determine when drug therapy should be initiated are mirrored from the 1992 NCEP guidelines outlined in Table 1. The treatment algorithm is based on a combination of LDL level, family history, and/or associated risk factors and/or risk conditions (Table 2).
Table 1.
Treatment Cutpoints for Statin Therapy *
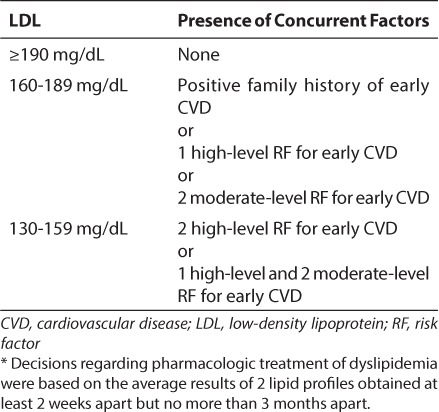
Table 2.
Risk Factor Definitions for Dyslipidemia Guidelines18

Contrasted with the adult guidelines which establish a threshold for treatment at ≥190 mg/dL, the implications of expanded drug use in children below this threshold are self-evident. A recent publication quantified this impact in just the adolescent population (17–21 years of age), which effectively spans both pediatric and adult criteria. Applying the pediatric recommendations to this population would result in 6-fold more patients qualifying for statin therapy than would be eligible based on the adult guidelines (2.5% vs. 0.4%, respectively). This equates to approximately 400,000 adolescents.22 This discrepancy illustrates the challenge faced by providers who care for adolescents who are transitioning into adulthood; specifically, whether the risk of exposing significantly more children to chronic lipid-lowering medications is offset by the anticipated reduction in morbidity and mortality from CAD. The risk of this chronic extrahepatic exposure of statins in the developing child is described briefly in Distribution below. Undoubtedly, additional investigations will be needed to clarify future guideline updates and risk of statin exposure in a developing child.
OVERVIEW OF THE STATINS
As shown by the guidelines, HMG-CoA reductase inhibitors are now the mainstay of pharmacologic treatment for dyslipidemia in both adults and children, due to their demonstrated efficacy in the primary and secondary prevention of CAD coupled with a relatively mild side effect profile.15,23–28 The first 3 statins approved in the United States (lovastatin in 1987, simvastatin in 1991, and pravastatin in 1991) are fungus-derived (Tables 3–5) semisynthetic agents, whereas the remaining U.S. Food and Drug Administration-approved compounds (fluvastatin in 1993, atorvastatin in 1996, rosuvastatin in 2003, and pitavastatin in 2009) are synthetic agents (Table 6).29
Table 3.
Lovastatin: Summary of Safety and LDL reduction in Pediatric Trials
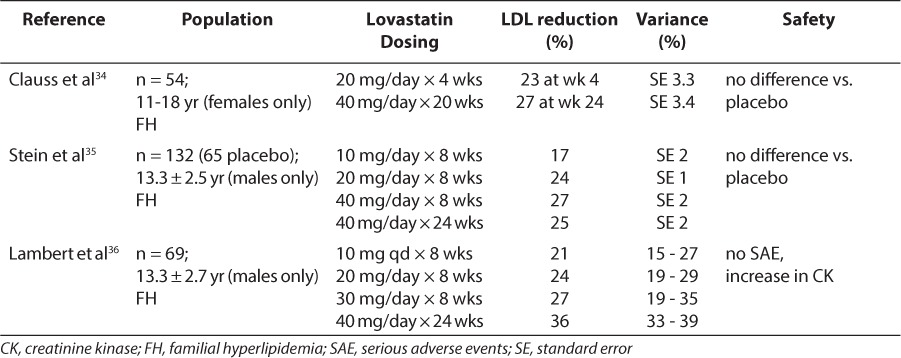
Table 6.
Synthetic Statins: Summary of Safety and LDL Reduction in Pediatric Trials
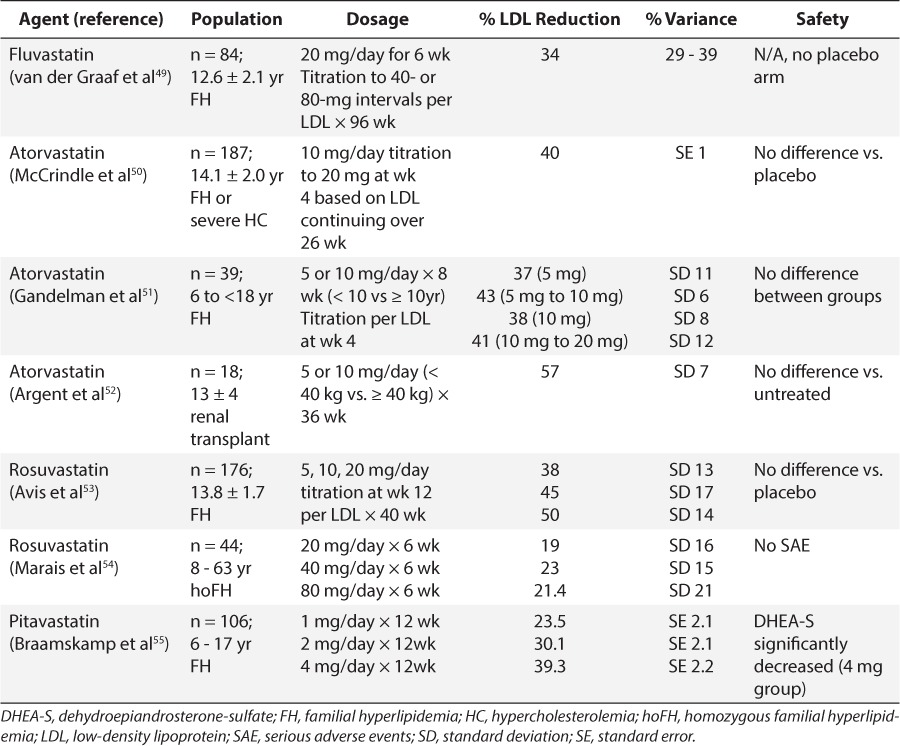
Table 4.
Pravastatin: Summary of Safety and LDL Reduction in Pediatric Trials
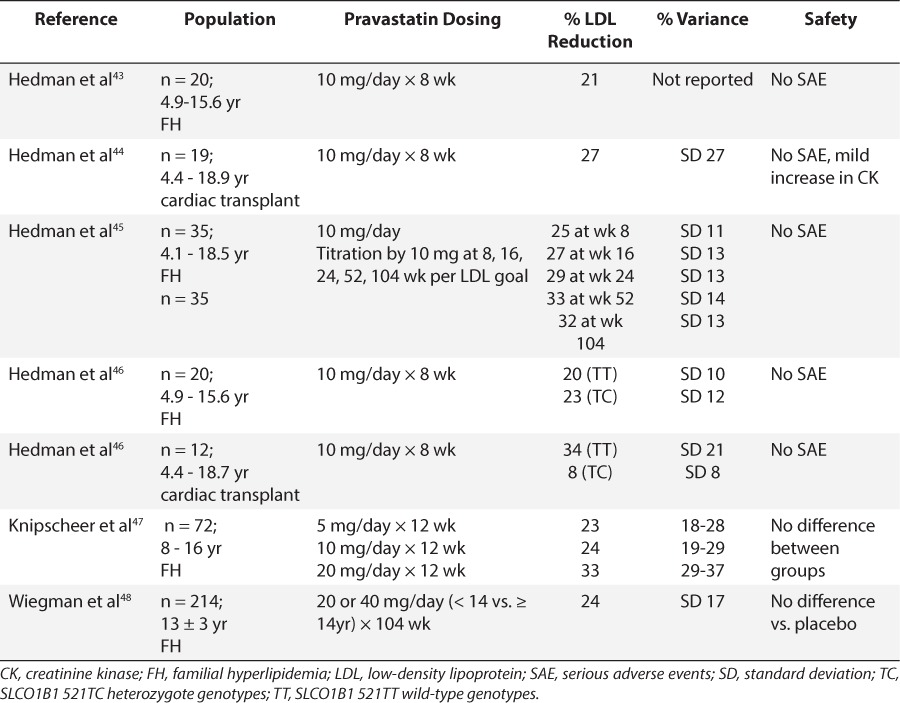
Table 5.
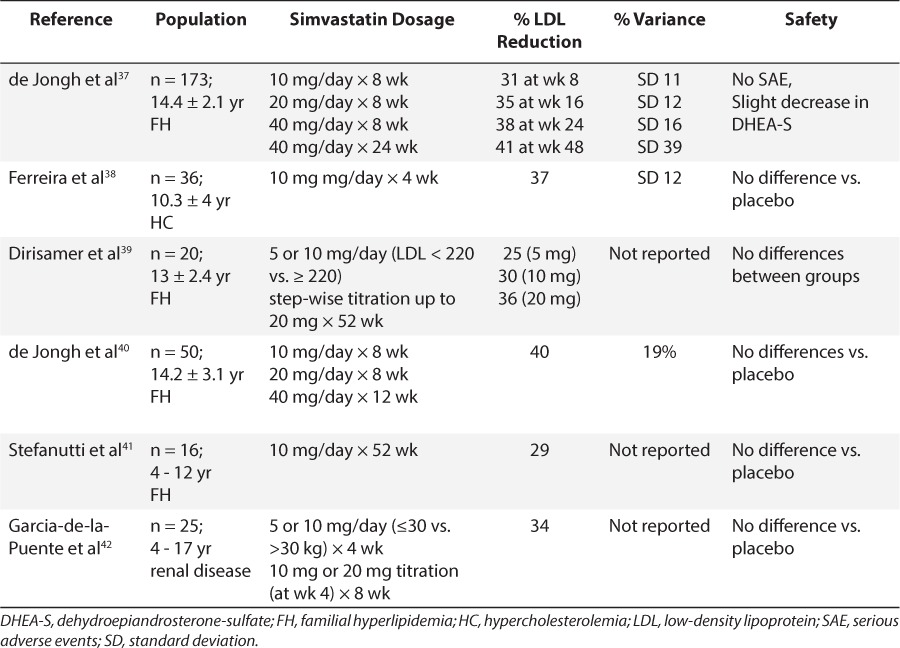
Simvastatin Summary of Safety and LDL Reduction in Pediatric Trials
Pharmacology
Statins decrease the hepatic synthesis of cholesterol by blocking the conversion of HMG-CoA to mevalonate, the rate-limiting step in cholesterol synthesis. In response to a subsequent decrease in intracellular sterols, expression of the genes encoding the cell-surface LDL receptor is up-regulated. This, in turn, enhances the hepatic uptake of LDL and reduces the circulating levels of LDL in the serum.30 However, statins appear to possess other effects including a decrease in inflammatory mediators downstream from HMG-CoA reductase (see Future Considerations below).31–33 Thus, it remains debated whether the reduction in CAD and plaque formation occurs as a result of the statins' lipid-lowering effects or other anti-inflammatory effects.
Efficacy in Children
Clinical trials of statins in children have included lovastatin, simvastatin, pravastatin, fluvastatin, rosuvastatin, atorvastatin, and pitavastatin and most of the studies focused on lipid-lowering and safety (Tables 3–6). With few exceptions, exposure to the statins conferred no added safety risk compared with placebo. However, the trials described in Tables 3 to 6 ranged from 1 month to 2 years and thus, a paucity of data describing the safety of chronic exposure to statins initiated during childhood exists. Moreover, for nearly all agents, reductions in LDL exceeded 20%, and some agents achieved reductions in excess of 40% to 50%. There also appeared to be some degree of dose dependence in LDL response within this class of drugs. However, in many studies, the variability associated with mean response profiles was exceedingly large, almost equivalent in magnitude to the response itself (Tables 3–6). At present, the cause of this variability remains unknown.
With such a high degree of variability in LDL reduction at a given statin dose and the unknown long-term developmental consequences of regular pediatric statin use, identifying the dose that maximizes efficacy and minimizes the risk of toxicity (i.e., dose optimization) is of great clinical importance for a developing child. Notably, all studies presented used a “one-size-fits-all” dosage scheme, effectively ignoring the contributions of ontogeny and genetic variation in statin disposition that are assuredly present in pediatric patients.
Given that the use of statins will inevitably increase as a result of mandatory lipid screening programs and the observed difficulties with adherence to dietary/behavioral modifications, the pediatric community should proactively pursue a more comprehensive understanding of these agents in children and adolescents before their widespread use. The following section discusses developmental, physicochemical, and pharmacogenetic factors that influence the dose-exposure profile for the statins. Notably, the paucity of data for pediatric statin disposition requires extrapolation from in vitro and adult data.
DISPOSITION
Physiochemical Considerations
Despite sharing a common mechanism of action, the statins differ in their physicochemical properties (e.g., octanol-water partition coefficient, pH stability/solubility). These properties are incredibly important to the overall disposition of each agent and explain why the statins should be considered independently when tailoring dosage to individual patient populations. Two statin agents (lovastatin and simvastatin) are formulated as lactone prodrugs which require hydrolysis to become activated inhibitors of HMG-CoA reductase.56,57 The remaining statins are administered in their active hydroxy acid forms.31,58,59 Consequently, lovastatin and simvastatin are the most lipophilic as delivered (simvastatin > lovastatin), readily translocating across membranes,60,61 whereas pravastatin and rosuvastatin are the most hydrophilic agents, requiring transporter-mediated disposition.58,60,61
Another unique element of the statins lies with the pH-dependent chemical interconversion that can occur at any step in the disposition pathway and heavily influences the amount of active drug available at the target. For instance, formation of the inactive 3-alpha-hydroxy-pravastatin acid and lactone isomers in the acidic environment of the stomach prior to absorption can disrupt the amount of pravastatin acid delivered to the drug target (i.e., the liver).62,63 Not surprisingly, isomer formation influences the pharmacodynamic effects of these drugs and is highly variable among healthy human subjects.63,64 Although these data require replication in a larger cohort before changes can be made to the drug label, the extent of chemical interconversion should be taken into consideration in populations where statin response is highly variable.
Absorption
All statins are administered orally, thus, the extent of their systemic availability is determined by the aforementioned physicochemical properties of the drug, the physicochemical milieu of the patient's gastrointestinal environment, and the functional status of their intestinal transporters, which can be influenced by ontogeny and genetics.
Pravastatin preferentially undergoes transporter-mediated absorption, conferring a relatively robust absorption rate despite its hydrophilic properties.65 In vitro, pravastatin appears to be a substrate for the influx transporters OATP1A2 and OATP2B1. Notably, OATP2B1 uptake appears to be pH-sensitive, diminishing greatly as the pH increases from 5.0 to 7.4.66–68 This observation suggests that the primary impact of OATP2B1 translocation occurs at the level of the enterocyte, where it is exposed to lower pH values, as opposed to the hepatocyte, where the systemic pH is higher and less uptake is expected. Pravastatin is not a substrate for the efflux transporters MDR1 and BCRP but, in vitro, appears to be a substrate for the efflux transporter MRP2, which is located on the apical surface of the enterocyte and liver.68–71 In vivo, increased expression of MRP2 conferred by a ‘gain of function’ sequence variation (ABCC2 c.1446C>G), increases presystemic clearance and reduces the bioavailability of pravastatin at the level of the enterocyte.72
Rosuvastatin similarly undergoes transporter-mediated absorption and, although not fully characterized, also appears be a substrate for OATP2B1 and BCRP.73,74 However, rosuvastatin does not display the same pH sensitivity suggesting that OATP2B1 may be relevant to rosuvastatin disposition at the level of both the intestine and liver.73 In vivo, a genetic variation in the gene encoding BCRP (ABCG2 c.421C>A) contributes to an increase in rosuvastatin exposure by way of diminished export back into the intestinal lumen and into the biliary canaliculus.75,76
Fluvastatin, moderately more lipophilic than pravastatin or rosuvastatin, undergoes passive diffusion but in vitro is a substrate of OATP2B1.73 Similarly, in vitro data reveal that atorvastatin is an OATP2B1 substrate at acidic and neutral pH; however, the high passive diffusion rates that are observed and lack of disruption in absorption by known inhibitors of OATP2B1 suggest that this transporter plays a very minor role in the absorption of atorvastatin.73 Pitavastatin, also moderately lipophilic, undergoes passive diffusion and there is no evidence that transporter-mediated influx significantly influences the disposition of this drug. However, pitavastatin absorption can be attenuated presystemically by P-glycoprotein (P-gp).77,78 Finally, there is no reported transporter-mediated absorption influencing the simvastatin or lovastatin lactones.
Another factor for consideration with respect to the absorption of statins is the impact of coad-ministered meals. Regardless of whether the drug is delivered by solution or capsule, concurrent administration of fluvastatin with food markedly reduces exposure and delays absorption (area under the curve [AUC], −17% to −24%; Cmax, −60% to −73%; Tmax, +56%).79 This was also observed with pravastatin (AUC, −30%; Cmax, −49%; Tmax, +50%)80 and rosuvastatin (AUC, −93%; Cmax, −93%; Tmax, +10%).81 However, meals markedly slow the rate of absorption for atorvastatin (Tmax, +124%) and pitavastatin (Tmax, +143%). In fed states, atorvastatin Cmax (−48%) and pitavastatin Cmax (−55%) are reduced, although the impact on the extent of exposure for atorvastatin (AUC, −13%) and pitavastatin (AUC, −15%) is less pronounced.82,83 In contrast, lovastatin concentrations drop when administered under fasting conditions (~33%),84 whereas simvastatin can be taken without regard to meals.85
A final observation is the differential effect of morning versus evening dosage for the statin agents. When pravastatin is given in the evening, the Cmax and AUC are reduced by approximately 60% compared with those for morning dosage.86 Similarly, the Cmax and AUC of atorvastatin are reduced by roughly 30% when administered in the evening.87 Fluvastatin concentrations are reported to be higher following evening dosage,88 while no significant differences were observed for rosuvastatin.89 These differences in drug exposure relative to the timing of dosage could be secondary to physiologic patterns of gastric emptying. Circadian changes in drug absorption have been observed in response to increased gastric emptying times in the evening.90 Additionally, the diurnal pattern of cholesterol biosynthesis (peak, 12:00 midnight to 4:00 am) in relation to an evening dose could increase amount of statin used by the hepatocyte and thereby affect the plasma exposure of a statin.91,92 Whether these differences definitely arise as a result of changes in absorption, distribution or elimination or intrinsic cholesterol production patterns remains unclear; however the observation that these patterns do not appreciably alter lipid-lowering properties of the affected statins limits the clinical relevance of these findings.
Concurrent with and subsequent to oral absorption, the statins (with the exception of pitavastatin) are subject to extensive first-pass extraction, effectively reducing their bioavailability.29,59,93–96 Because drug-metabolizing enzymes mediate statin metabolism, these reactions are reviewed in Metabolism below. However, we point out here that when first-pass occurs at the level of the intestinal enterocyte, the absolute bioavailability of these agents is reduced, influencing both efficacy and toxicity. In contrast, when hepatocytes are the principal mediators of first pass, a more favorable scenario is set where concentrations at the target organ (i.e., the liver) increase while peripheral exposure decreases, thereby leading to enhanced efficacy and fewer side effects (e.g., myalgias).97
Distribution
Hepatic uptake for the highly lipophilic statin lactones occurs by passive diffusion,60 but for most of the statins, it is facilitated by transporter-mediated processes. OATP1B1, encoded by the solute-carrier organic anion transporter gene SLCO1B1, is the principle transporting protein into the hepatocyte for most statins and has been reviewed extensively.60,98–100 Among the transporters with a minor role in statin disposition, pitavastatin, rosuvastatin, and fluvastatin are substrates for OATP1B3 (SLCO1B3),60,101,102 rosuvastatin and fluvastatin appear to be substrates for OATP2B1 (SLCO2B1),73,103,104 and rosuvastatin also appears to be a substrate for the sodium-dependent cotransporting polypeptide (NTCP) which may account for as much as 35% of its hepatic uptake.103 Fluvastatin also appears to enter the hepatocyte by passive diffusion.105–107 As above, cellular uptake of the simvastatin and lovastatin lactones relies primarily on passive diffusion; however, simvastatin and lovastatin acid are substrates of OATP1B1 in vitro and in vivo.108,109 Notably, the simvastatin and lovastatin lactones appear to inhibit OATP1B1-mediated transport.110,111
The clinical relevance of OATP-mediated statin disposition has been demonstrated in a number of drug-drug interaction studies. A 7-fold increase in the AUCs of atorvastatin acid and 2-hydroxy atorvastatin acid and a 3-fold increase in AUC of 4-hydroxy atorvastatin acid were observed when this statin was coadministered with rifampin (a known inhibitor of OATP1B1 and OATP1B3).112–114 In the presence of cyclosporine (a potent inhibitor of OATP1B1 and CYP3A4) atorvastatin AUCs were 6- to 15-fold increased,115–117 fluvastatin AUC was 3-fold increased,118 lovastatin AUC was 20-fold increased,119 pitavastatin AUC was 5-fold increased,99 pravastatin AUC was 5- to 10-fold increased,44,119,120 rosuvastatin AUC was 7-fold increased,121 and simvastatin AUC was 3- to 8-fold increased.122,123 Certainly, CYP3A4 inhibition from cyclosporine can contribute to the overall increases observed in statin exposure; however, this can be concluded to play a minor role given that rosuvastatin, pravastatin, and pitavastatin are not significantly metabolized by CYP3A4.124–128 In fact, pravastatin, the most hydrophilic compound, had a 10-fold increase in AUC when administered to children and adolescents who were taking triple immunosuppressive therapy containing cyclosporine and no other CYP substrates.44 Gemfibrozil, also an inhibitor of OATP1B1 and CYP2C8, produced a 2-fold increase in AUCs of atorvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin, and simvastatin.99,129–133
Cumulatively, the data from the above-described studies provide compelling evidence that OATP1B1 is a critically important determinant of drug disposition for most of the statins. Consequently, functional polymorphisms in the SLCO gene families are also expected to influence statin disposition and, thus, have been the subject of considerable interest.99,134 Much of this work stems from a study of pravastatin pharmacokinetics, where extreme outliers were attributed to 2 single-nucleotide variants in SLCO1B1.135 These mutations were observed in the promoter region (−11187G>A) and in exon 5 (c.521T>C) and were associated with a 50% reduction in non-renal clearance.136 This finding was independently confirmed in heterozygous carriers of SLCO1B1*15 (containing the 388A>G and 521T>C variants) who demonstrated mean pravastatin exposures (AUC0–12) that were 93% higher and heterozygous carriers of the *17 haplotype (containing the −11187G>A, 388A>G, and 521T>C variants) who had exposures that were 130% higher than non-carriers.137
Ultimately, the functional consequence of SLCO1B1 sequence variations on statin exposure are reflected by the dependence of the individual statin on OATP1B1 for cellular uptake. Heterozygosity for SLCO1B1*5 and *15 haplotypes is associated with a 3-fold, 2.5-fold, and 2-fold increase in exposure for simvastatin acid, atorvastatin and pravastatin, respectively, with very little effect on fluvastatin.99 SLCO1B1 genotype also influences the effect of rifampin on atorvastatin exposure wherein a 9-fold increase in AUC is observed in patients with a fully functional 521TT genotype versus a 5-fold increase in AUC observed for the 521CC genotype.138 We would be remiss not to allude to the in vitro data which suggest that the C800T variant of NTCP may confer enhanced uptake of rosuvastatin, but there are no clinical data to support a role for this mutation in vivo.103
These studies underscore the critical role of OATP1B1 in statin disposition. By extension, this has important implications for drug safety, where an increase in systemic exposure mediated by reduced OATP1B1 activity can increase the risk of myopathy in statin-treated patients. The Statin Response Examined by Genetic Haplotype Markers (STRENGTH) trial demonstrated this with the observation that patients who were heterozygous for a non-coding sequence variation in linkage disequilibrium with c.521T>C experienced a 4.5-fold increase in risk of myopathy. Patients who were homozygous for this mutation experienced a 16.9 increase in risk of myopathy.139
All statins, except for pravastatin, are extensively protein bound. 56,57,65,95,96,125,140,141 Therefore, the circulating concentration of free drug is relatively low for most agents in this class. However, the extent of distribution into peripheral tissues in humans is not well characterized. In theory, the statins with reduced lipid solubility (e.g., pravastatin) should demonstrate less extensive tissue distribution, which would ostensibly provide a safer alternative in children, where brain and gonadal tissues are still maturing. In vitro and in vivo data support this supposition, demonstrating that lower exposures are observed for pravastatin than for lovastatin and simvastatin in the central nervous system,142,143 and pravastatin also manifests a lower risk of myopathy than simvastatin and atorvastatin do.144–149 However, there are contradictory data which suggest that pravastatin can influence gene expression in the central nervous system to the same extent as some of the other statins.143 Until the active transporters responsible for tissue distribution of the statins and their ontogeny in children are more fully elucidated, practitioners will need to rely on the adverse event profiles reported from clinical studies.
Metabolism
Although in vitro reaction phenotyping studies suggest that cytochromes P450 (CYP) 2C8, 2C9, 2C19, 2D6, 3A4, and 3A5 are all capable of metabolizing the statins, current data suggest that CYP3A4 is a major contributor to simvastatin, lovastatin, and atorvastatin metabolism.95,150–152 In the presence of the CYP3A4/5 inhibitor troleandomycin, simvastatin acid metabolism is decreased by 90%.150 When administered concurrently with the CYP3A4 inhibitor itraconazole, 15- to 19-fold increases are observed in simvastatin and lovastatin AUCs.98,126–128 The impact of itraconazole on the AUC of atorvastatin is more modest (+47%),151 and the coadministration of CYP3A4 inhibitors has no significant effect on clearance of pravastatin, fluvastatin, rosuvastatin, or pitavastatin, consistent with the limited role of CYP3A4 in the metabolism of these compounds.153 In vitro and in vivo data suggest that fluvastatin is a substrate for CYP2C9,154–156 whereas pravastatin, pitavastatin, and rosuvastatin do not undergo appreciable CYP-mediated metabolism.
Despite the fact that CYP3A4 activity is highly variable, mutations driving this variability have not been fully elucidated.157 However, a sequence variation in intron 6 of this gene associated with reduced CYP3A4 expression and activity (rs35599367 C>T, designated CYP3A4*22) has also been associated with the need for 0.2- to 0.6-fold lower doses of atorvastatin, lovastatin, and simvastatin to adequately manage lipid profiles.158,159 With fluvastatin, patients homozygous for the *3 allele of CYP2C9 (which confers reduced activity in this enzyme) demonstrate 3-fold lower clearance values of the active fluvastatin enantiomer. Notably, the resultant lipid profiles were not correlated with CYP2C9 genotype.160 Collectively, these studies support a role for allelic variations in drug-metabolizing enzymes influencing the pharmacokinetics and, in some cases, pharmacodynamics of the statins that rely on these pathways for clearance.
UDP-glucuronosyl transferase (UGT)-catalyzed conjugation is the primary route by which statins and their metabolites are further biotransformed in hepatocytes.161,162 The open acids are conjugated by UGT to form an acyl glucuronide that subsequently cyclizes to form a lactone ring (i.e., lactonization). This process results in a loss of pharmacologic activity and is common to all statins present in the open acid form. Notably, carboxyl esterase can reverse the lactonization process thereby regenerating the open acids. Alternatively, the lactones can be directly metabolized by the CYPs in a process that appears to occur more rapidly than is observed for open acids.60,163 Although important in the disposition of statins, the overall contribution of UGTs is quantitatively less substantial than that of the CYPs.163 As above, pravastatin, rosuvastatin, and pitavastatin do not undergo extensive UGT-mediated conjugation.
Although conjugation plays a more limited role in statin disposition, recent data suggest that allelic variants of UGT may have a modest effect of statin activity. The UGT1A3*2 allele has been associated with increased lactonization activity for atorvastatin.164 Homozygosity of the UGT1A3*2 allele was accompanied by a 1.7- and 2.7-fold increase in AUC of the parent and 2-hydroxyatorvastatin lactones, respectively, compared to that in patients who are homozygous for UGT1A3*1. Furthermore, this increase in lactone formation correlated with a reduction in the maximal effect of atorvastatin on total and LDL cholesterol-lowering from baseline.165
Excretion
Biliary excretion of the UGT-conjugated statins occurs through several transporters, including multidrug resistance 1 (MDR1; ABCB1), multidrug resistance-associated protein 2 (MRP2; ABCC2), breast cancer resistance protein (BCRP; ABCG2), and bile salt exporting pump (BSEP; ABCB11). However, the quantitative importance of these efflux transporters in the overall disposition profile of the statins has yet to be fully elucidated. Nonetheless, the consequences of genetic variations in the efflux transporters relevant to the statins have also recently been examined. In vitro, there is no consensus regarding MDR1 expression or activity in the common allelic variants of ABCB1 (c.1236C>T, c.2677G>T/A, c.3435C>T).166 In vivo, these allelic variants do not appear to significantly influence the inter-individual variability in fluvastatin, pravastatin, lovastatin, and rosuvastatin pharmacokinetics,167 but significantly increase the exposure of simvastatin and atorvastatin acid by 60% and 55%, respectively.168
Conversely, the ABCG2 c.421C>A variant, which has been associated with transport activity in vitro,169 appears to increase the exposure of atorvastatin, fluvastatin, simvastatin lactone, and rosuvastatin by 72%, 72%, 111%, and 144%, respectively, in subjects with the AA genotype compared to those in patients with the wild-type CC genotype.75,170 Note that this genotype does not appreciably impact the pharmacokinetics of simvastatin acid or pravastatin.170 As discussed in Absorption above, pravastatin is subject to MRP2-mediated transport in vitro at the level of the enterocyte and hepatocyte.69–71In vivo , the ABCC2 c.1446C>G variant decreases the exposure of pravastatin (AUC, −68%) compared to wild-type controls secondary to a “gain of function” mutation.72 It remains unknown whether this decrease in exposure is due to enhanced pre-systemic and/or hepatic clearance. Conversely, Mrp2-deficient rats have significantly diminished biliary clearance of pravastatin,71 and in vitro data suggest that BSEP may be an alternative mechanism by which pravastatin is cleared from the hepatocyte.171
Renal clearance is far less pronounced than biliary excretion. Most of the statin agents have minimal renal clearance (< 10%) after an orally administered dose,56,57,59,79,124,172 except for pravastatin in which 20% is renally cleared.62 The exposure of pravastatin acid is not impacted by diminished renal function; however, exposure of the 3-alpha-hydroxy-pravastatin metabolite was significantly increased (AUC, +48%) compared to that in subjects with normal renal function.173 Halstenson et al173 suggest that this increased interconversion occurs secondary to decreased gastric pH, which is a direct result of kidney-related metabolic changes. Hepatic conversion to 3-alpha-5-beta, 6-beta-trihydroxy isomeric metabolite occurs more frequently in severe renal impairment, suggesting that more pravastatin acid is cleared hepatically. Although renal impairment does not appear to alter the exposure of pravastatin acid, the impact of both metabolites on statin disposition and response require further investigation. In vitro, pravastatin is a substrate of organic anion transporter 3 (OAT3), a transporter located on basolateral membrane of the proximal tubule, and it is responsible for its uptake in the kidney.174,175 In vitro, gemfibrozil inhibits pravastatin uptake in OAT3-expressing cells.176 In vivo, coadministration of pravastatin and gemfibrozil lead to an increase in pravastatin exposure (AUC, +202%) and decreased renal clearance (−40%).131 This 40% reduction in renal clearance does not solely explain the increase in pravastatin exposure, but it could serve as a contribution to pravastatin disposition. Further investigation by Nishizato et al136 found that several OAT3 single-nucleotide polymorphisms did not affect pravastatin pharmacokinetics.136 However, the single-nucleotide polymorphisms included in this analysis have not been associated with decreased transporter function. Overall, patients with renal impairment do not require dose adjustments, but the impact of renal clearance with pravastatin administration requires further elucidation.
Given the current state of our knowledge of the disposition pathways for the available statins (most of which mature prior to adolescence) and the relative absence of data on the ontogeny of transporter expression which could influence recommendations for statin dosage in children, considerations for the selection of statin agents in the pediatric population will largely reflect the same considerations used with adult patients. To maximize the dose-exposure profile, considerations include whether the patient is receiving gastric acid-modifying therapy and whether greater adherence is anticipated to a regimen that requires medication administration with or without meals. To influence the exposure-response profile, one should consider the genetic constitution of the patient, the concurrent administration of drugs that compete as substrates for transport pathways, and comorbidities that may alter circulating protein stores of the presence of protein-binding displacers in the circulation. Future studies on the pharmacokinetics of statins in the pediatric population, and an expansion of our knowledge on the developmental patterns of transport expression, will permit clinicians to further individualize the selection and dosage of statins in this population.
FUTURE CONSIDERATIONS
Owing to their pleiotropic effects, the statins have been extensively evaluated for non-hyperlipidemic conditions, a few of which are detailed in Table 7, and many of which can impact children. For example, patients with sickle cell disease can develop oxidative stress and chronic inflammation to their distal vasculature as a result of transient vaso-occlusion and subsequent reperfusion injury.177 Hoppe et al178 found that biomarkers of vascular dysfunction, including C-reactive protein and interleukin 6, were decreased in adolescents with sickle cell disease from 50% to 70% after a 3-week trial of low (20 mg) or moderate (40 mg) doses of simvastatin.
Table 7.
Statin Studies Under Non-Hyperlipidemic Conditions
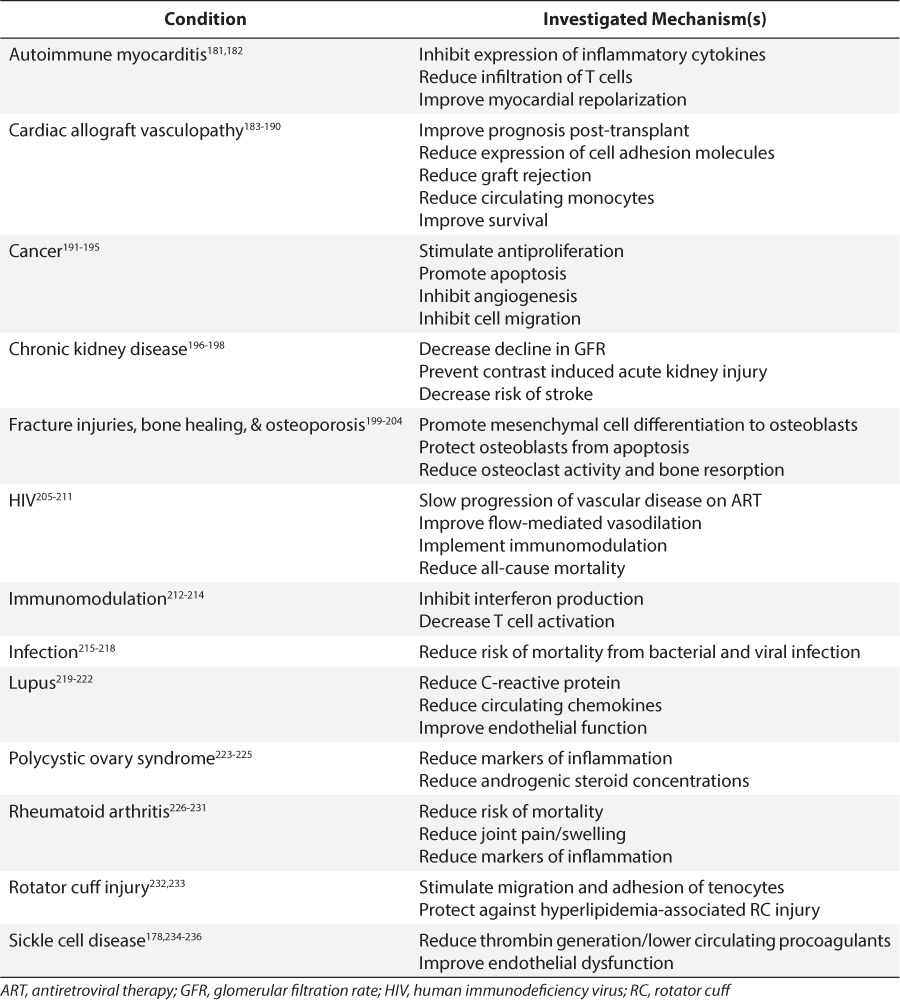
Additionally, statins have been used after cardiac transplantation to prevent coronary allograft vasculopathy (CAV). In pediatric cardiac transplantation, the prevalence of CAV has been reported to be as high as 17% in one retrospective analysis.179 Greater-than-optimal LDL concentrations (>100 mg/dL) post transplantation have been reported in 39% of pediatric patients 1 year after transplantation,180 which can be secondary to post-transplantation steroid and immunosuppressive therapy. The addition of pravastatin therapy in pediatric cardiac transplant recipients yielded a lower incidence of CAV.179
Most of the remaining conditions for which statins have been explored exploit the anti-inflammatory and antiproliferative effects of these drugs (Table 7). Thus, it would not be unexpected to see statin coadministration in the presence of infections, fractures, and malignancies in children. However, it should be appreciated that there are an equally large number of publications that refute a role for statins in these same conditions (Table 7). In the absence of sufficient prospective clinical trials to inform the role of these agents for indications other than hyperlipidemia, the practitioner must carefully weigh the risk-benefit ratio of these agents and thoughtfully examine the in vitro concentration-effect profiles to inform whether and at what dose these agents should be used in pediatric patients.
CONCLUSIONS
With precursors of CAD appearing in childhood, the establishment of pediatric preventive cardiology services is rapidly emerging. However, the most appropriate management of those children and adolescents, where lifestyle changes fail, remains challenging. Despite the overall success of statins, variability in drug response in the pediatric cohort remains concerning. Although not discussed above, genes involved with drug response may contribute to some of the variability in LDL reduction among children and adults receiving statin therapy.237–241 However, it remains unknown whether a consistent statin concentration (i.e., exposure) at the drug target was achieved in these studies. Therefore, future investigations must be designed to characterize these dose-exposure relationships in the developing child so that exposure can be controlled when attempting to determine response in this population. Once the covariates that influence statin disposition in children are validated, future clinical trials will be better informed to fully characterize the entire dose-exposure-response relationship. With these data, dosage will be optimized to maximize efficacy while minimizing toxicity in the individual pediatric patient. In the interim, understanding the statin disposition pathway will assist pediatric providers who make recommendations related to statin prescribing where alteration of drug delivery and dosage may be appropriately tailored to meet their specific patient needs.
Glossary
Abbreviations
- AUC
area under the curve
- BCRP
breast cancer resistance protein
- BSEP
bile salt exporting pump
- CAD
coronary artery disease
- CAV
coronary allograft vasculopathy
- Cmax
maximal concentration
- CVD
cardiovascular disease
- CYP
cytochrome p450
- HDL
high-density lipoprotein
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- LDL
low-density lipoprotein
- MDR1
multi-drug resistance gene
- MRP2
multi-drug resistance-associated protein 2
- NCEP
National Cholesterol Education Program
- NTCP
sodium-dependent co-transporting polypeptide
- OAT
organic anion transporter
- OATP
organic anion transporting polypeptide
- TC
total cholesterol
- Tmax
time of maximal concentration
- UGT
UDP-glucuronosyl transferase
Footnotes
Disclosure The authors declare no conflicts or financial interest in any product or service mentioned in the manuscript, including grants, equipment, medications, employment, gifts, and honoraria.
REFERENCES
- 1. Mozaffarian D, Benjamin EJ, Go AS. et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015; 131( 4): e29– 322. [DOI] [PubMed] [Google Scholar]
- 2. Holman RL, Mc GH, Jr, Strong JP, Geer JC. The natural history of atherosclerosis: the early aortic lesions as seen in New Orleans in the middle of the of the 20th century. Am J Pathol. 1958; 34( 2): 209– 235. [PMC free article] [PubMed] [Google Scholar]
- 3. Enos WF, Holmes RH, Beyer J. Coronary disease among United States soldiers killed in action in Korea: preliminary report. J Am Med Assoc. 1953; 152( 12): 1090– 1093. [DOI] [PubMed] [Google Scholar]
- 4. McNamara JJ, Molot MA, Stremple JF, Cutting RT. Coronary artery disease in combat casualties in Vietnam. JAMA. 1971; 216( 7): 1185– 1187. [PubMed] [Google Scholar]
- 5. Newman WP, 3rd, Freedman DS, Voors AW. et al. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N Engl J Med. 1986; 314( 3): 138– 144. [DOI] [PubMed] [Google Scholar]
- 6. McGill HC, Jr, McMahan CA. Determinants of atherosclerosis in the young. Pathobiological Determinants of Atherosclerosis in Youth (PDAY) research group. Am J Cardiol. 1998; 82( 10B): 30T– 36T. [DOI] [PubMed] [Google Scholar]
- 7. Hickman TB, Briefel RR, Carroll MD. et al. Distributions and trends of serum lipid levels among United States children and adolescents ages 4–19 years: data from the Third National Health and Nutrition Examination Survey. Prev Med. 1998; 27( 6): 879– 890. [DOI] [PubMed] [Google Scholar]
- 8. Kit BK, Carroll MD, Lacher DA, Sorlie PD, DeJesus JM, Ogden C. Trends in serum lipids among US youths aged 6 to 19 years, 1988–2010. JAMA. 2012; 308( 6): 591– 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab. 2012; 97( 11): 3956– 3964. [DOI] [PubMed] [Google Scholar]
- 10. Goldberg AC, Hopkins PN, Toth PP. et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011; 5( 3): 133– 140. [DOI] [PubMed] [Google Scholar]
- 11. Ogden CL, Carroll MD, Flegal KM. High body mass index for age among US children and adolescents, 2003–2006. JAMA. 2008; 299( 20): 2401– 2405. [DOI] [PubMed] [Google Scholar]
- 12. Lauer RM, Clarke WR. Use of cholesterol measurements in childhood for the prediction of adult hypercholesterolemia. The Muscatine study. JAMA. 1990; 264( 23): 3034– 3038. [PubMed] [Google Scholar]
- 13. Must A, Jacques PF, Dallal GE, Bajema CJ, Dietz WH. Long-term morbidity and mortality of overweight adolescents. A follow-up of the Harvard growth study of 1922 to 1935. N Engl J Med. 1992; 327( 19): 1350– 1355. [DOI] [PubMed] [Google Scholar]
- 14. Bibbins-Domingo K, Coxson P, Pletcher MJ, Lightwood J, Goldman L. Adolescent overweight and future adult coronary heart disease. N Engl J Med. 2007; 357( 23): 2371– 2379. [DOI] [PubMed] [Google Scholar]
- 15. Eiland LS, Luttrell PK. Use of statins for dyslipidemia in the pediatric population. J Pediatr Pharmacol Ther. 2010; 15( 3): 160– 172. [PMC free article] [PubMed] [Google Scholar]
- 16. National Cholesterol Education Program (NCEP): highlights of the report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. Pediatrics. 1992; 89( 3): 495– 501. [PubMed] [Google Scholar]
- 17. Ritchie SK, Murphy EC, Ice C. et al. Universal versus targeted blood cholesterol screening among youth: The CARDIAC project. Pediatrics. 2010; 126( 2): 260– 265. [DOI] [PubMed] [Google Scholar]
- 18. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011; 128( suppl 5): S213– 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bertrais S, Balkau B, Charles MA. et al. Puberty-associated differences in total cholesterol and triglyceride levels according to sex in French children aged 10–13 years. Ann Epidemiol. 2000; 10( 5): 316– 323. [DOI] [PubMed] [Google Scholar]
- 20. Kouda K, Nakamura H, Fan W, Takeuchi H. Negative relationships between growth in height and levels of cholesterol in puberty: a 3-year follow-up study. Int J Epidemiol. 2003; 32( 6): 1105– 1110. [DOI] [PubMed] [Google Scholar]
- 21. Doran B, Guo Y, Xu J. et al. Prognostic value of fasting versus nonfasting low-density lipoprotein cholesterol levels on long-term mortality: insight from the National Health and Nutrition Examination Survey III (NHANES-III). Circulation. 2014; 130( 7): 546– 553. [DOI] [PubMed] [Google Scholar]
- 22. Gooding HC, Rodday AM, Wong JB. et al. Application of pediatric and adult guidelines for treatment of lipid levels among US adolescents transitioning to young adulthood. JAMA Pediatr. 2015; 169( 6): 569– 574. [DOI] [PubMed] [Google Scholar]
- 23. Baigent C, Keech A, Kearney PM. et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005; 366( 9493): 1267– 1278. [DOI] [PubMed] [Google Scholar]
- 24. Baigent C, Blackwell L. et al; Cholesterol Treatment Trialists C Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010; 376( 9753): 1670– 1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mills EJ, Wu P, Chong G. et al. Efficacy and safety of statin treatment for cardiovascular disease: a network meta-analysis of 170,255 patients from 76 randomized trials. QJM. 2011; 104( 2): 109– 124. [DOI] [PubMed] [Google Scholar]
- 26. Tonelli M, Lloyd A, Clement F. et al. Efficacy of statins for primary prevention in people at low cardiovascular risk: a meta-analysis. CMAJ. 2011; 183( 16): E1189– 1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Gorman CS, Higgins MF, O'Neill MB. Systematic review and metaanalysis of statins for heterozygous familial hypercholesterolemia in children: evaluation of cholesterol changes and side effects. Pediatr Cardiol. 2009; 30( 4): 482– 489. [DOI] [PubMed] [Google Scholar]
- 28. Vuorio A, Kuoppala J, Kovanen PT. et al. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev. 2014; 7: CD006401. [DOI] [PubMed] [Google Scholar]
- 29. Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005; 19( 1): 117– 125. [DOI] [PubMed] [Google Scholar]
- 30. Bilheimer DW, Grundy SM, Brown MS, Goldstein JL. Mevinolin and colestipol stimulate receptor-mediated clearance of low density lipoprotein from plasma in familial hypercholesterolemia heterozygotes. Proc Natl Acad Sci U S A. 1983; 80( 13): 4124– 4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Corsini A, Bellosta S, Baetta R, Fumagalli R, Paoletti R, Bernini F. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol Ther. 1999; 84( 3): 413– 428. [DOI] [PubMed] [Google Scholar]
- 32. Corsini A, Mazzotti M, Raiteri M. et al. Relationship between mevalonate pathway and arterial myocyte proliferation: in vitro studies with inhibitors of HMG-CoA reductase. Atherosclerosis. 1993; 101( 1): 117– 125. [DOI] [PubMed] [Google Scholar]
- 33. Bernini F, Didoni G, Bonfadini G, Bellosta S, Fumagalli R. Requirement for mevalonate in acetylated LDL induction of cholesterol esterification in macrophages. Atherosclerosis. 1993; 104( 1–2): 19– 26. [DOI] [PubMed] [Google Scholar]
- 34. Clauss SB, Holmes KW, Hopkins P. et al. Efficacy and safety of lovastatin therapy in adolescent girls with heterozygous familial hypercholesterolemia. Pediatrics. 2005; 116( 3): 682– 688. [DOI] [PubMed] [Google Scholar]
- 35. Stein EA, Illingworth DR, Kwiterovich PO., Jr et al. Efficacy and safety of lovastatin in adolescent males with heterozygous familial hypercholesterolemia: a randomized controlled trial. JAMA. 1999; 281( 2): 137– 144. [DOI] [PubMed] [Google Scholar]
- 36. Lambert M, Lupien PJ, Gagne C. et al. Treatment of familial hypercholesterolemia in children and adolescents: effect of lovastatin. Canadian Lovastatin in Children Study Group. Pediatrics. 1996; 97( 5): 619– 628. [PubMed] [Google Scholar]
- 37. de Jongh S, Ose L, Szamosi T. et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized, double-blind, placebo-controlled trial with simvastatin. Circulation. 2002; 106( 17): 2231– 2237. [DOI] [PubMed] [Google Scholar]
- 38. Ferreira WP, Bertolami MC, Santos SN. et al. One-month therapy with simvastatin restores endothelial function in hypercholesterolemic children and adolescents. Pediatr Cardiol. 2007; 28( 1): 8– 13. [DOI] [PubMed] [Google Scholar]
- 39. Dirisamer A, Hachemian N, Bucek RA. et al. The effect of low-dose simvastatin in children with familial hypercholesterolaemia: a 1-year observation. Eur J Pediatr. 2003; 162( 6): 421– 425. [DOI] [PubMed] [Google Scholar]
- 40. de Jongh S, Lilien MR, op't Roodt J. et al. Early statin therapy restores endothelial function in children with familial hypercholesterolemia. J Am Coll Cardiol. 2002; 40( 12): 2117– 2121. [DOI] [PubMed] [Google Scholar]
- 41. Stefanutti C, Lucani G, Vivenzio A, Di Giacomo S. Diet only and diet plus simvastatin in the treatment of heterozygous familial hypercholesterolemia in childhood. Drugs Exp Clin Res. 1999; 25( 1): 23– 28. [PubMed] [Google Scholar]
- 42. Garcia-de-la-Puente S, Arredondo-Garcia JL, Gutierrez-Castrellon P. et al. Efficacy of simvastatin in children with hyperlipidemia secondary to kidney disorders. Pediatr Nephrol. 2009; 24( 6): 1205– 1210. [DOI] [PubMed] [Google Scholar]
- 43. Hedman M, Neuvonen PJ, Neuvonen M, Antikainen M. Pharmacokinetics and pharmacodynamics of pravastatin in children with familial hypercholesterolemia. Clin Pharmacol Ther. 2003; 74( 2): 178– 185. [DOI] [PubMed] [Google Scholar]
- 44. Hedman M, Neuvonen PJ, Neuvonen M. et al. Pharmacokinetics and pharmacodynamics of pravastatin in pediatric and adolescent cardiac transplant recipients on a regimen of triple immunosuppression. Clin Pharmacol Ther. 2004; 75( 1): 101– 109. [DOI] [PubMed] [Google Scholar]
- 45. Hedman M, Matikainen T, Fohr A. et al. Efficacy and safety of pravastatin in children and adolescents with heterozygous familial hypercholesterolemia: a prospective clinical follow-up study. J Clin Endocrinol Metab. 2005; 90( 4): 1942– 1952. [DOI] [PubMed] [Google Scholar]
- 46. Hedman M, Antikainen M, Holmberg C. et al. Pharmacokinetics and response to pravastatin in paediatric patients with familial hypercholesterolaemia and in paediatric cardiac transplant recipients in relation to polymorphisms of the SLCO1B1 and ABCB1 genes. Br J Clin Pharmacol. 2006; 61( 6): 706– 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Knipscheer HC, Boelen CC, Kastelein JJ. et al. Short-term efficacy and safety of pravastatin in 72 children with familial hypercholesterolemia. Pediatr Res. 1996; 39( 5): 867– 871. [DOI] [PubMed] [Google Scholar]
- 48. Wiegman A, Hutten BA, de Groot E. et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial. JAMA. 2004; 292( 3): 331– 337. [DOI] [PubMed] [Google Scholar]
- 49. van der Graaf A, Nierman MC, Firth JC. et al. Efficacy and safety of fluvastatin in children and adolescents with heterozygous familial hypercholesterolaemia. Acta Paediatr. 2006; 95( 11): 1461– 1466. [DOI] [PubMed] [Google Scholar]
- 50. McCrindle BW, Ose L, Marais AD. Efficacy and safety of atorvastatin in children and adolescents with familial hypercholesterolemia or severe hyperlipidemia: a multicenter, randomized, placebo-controlled trial. J Pediatr. 2003; 143( 1): 74– 80. [DOI] [PubMed] [Google Scholar]
- 51. Gandelman K, Glue P, Laskey R. et al. An eight-week trial investigating the efficacy and tolerability of atorvastatin for children and adolescents with heterozygous familial hypercholesterolemia. Pediatr Cardiol. 2011; 32( 4): 433– 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Argent E, Kainer G, Aitken M. et al. Atorvastatin treatment for hyperlipidemia in pediatric renal transplant recipients. Pediatr Transplant. 2003; 7( 1): 38– 42. [DOI] [PubMed] [Google Scholar]
- 53. Avis HJ, Hutten BA, Gagne C. et al. Efficacy and safety of rosuvastatin therapy for children with familial hypercholesterolemia. J Am Coll Cardiol. 2010; 55( 11): 1121– 1126. [DOI] [PubMed] [Google Scholar]
- 54. Marais AD, Raal FJ, Stein EA. et al. A dose-titration and comparative study of rosuvastatin and atorvastatin in patients with homozygous familial hypercholesterolaemia. Atherosclerosis. 2008; 197( 1): 400– 406. [DOI] [PubMed] [Google Scholar]
- 55. Braamskamp MJ, Stefanutti C, Langslet G. et al. Efficacy and safety of pitavastatin in children and adolescents at high future cardiovascular risk. J Pediatr. 2015; 167( 2): 338– 343. [DOI] [PubMed] [Google Scholar]
- 56. Duggan DE, Chen IW, Bayne WF. et al. The physiological disposition of lovastatin. Drug Metab Dispos. 1989; 17( 2): 166– 173. [PubMed] [Google Scholar]
- 57. Vickers S, Duncan CA, Chen IW, Rosegay A, Duggan DE. Metabolic disposition studies on simvastatin, a cholesterol-lowering prodrug. Drug Metab Dispos. 1990; 18( 2): 138– 145. [PubMed] [Google Scholar]
- 58. McTaggart F, Buckett L, Davidson R. et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitor. Am J Cardiol. 2001; 87( 5A): 28B– 32B. [DOI] [PubMed] [Google Scholar]
- 59. Kajinami K, Mabuchi H, Saito Y. NK-104: a novel synthetic HMG-CoA reductase inhibitor. Expert Opin Investig Drugs. 2000; 9( 11): 2653– 2661. [DOI] [PubMed] [Google Scholar]
- 60. Shitara Y, Sugiyama Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther. 2006; 112( 1): 71– 105. [DOI] [PubMed] [Google Scholar]
- 61. Serajuddin AT, Ranadive SA, Mahoney EM. Relative lipophilicities, solubilities, and structure-pharmacological considerations of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors pravastatin, lovastatin, mevastatin, and simvastatin. J Pharm Sci. 1991; 80( 9): 830– 834. [DOI] [PubMed] [Google Scholar]
- 62. Everett DW, Chando TJ, Didonato GC. et al. Biotransformation of pravastatin sodium in humans. Drug Metab Dispos. 1991; 19( 4): 740– 748. [PubMed] [Google Scholar]
- 63. Triscari J, O'Donnell D, Zinny M, Pan HY. Gastrointestinal absorption of pravastatin in healthy subjects. J Clin Pharmacol. 1995; 35( 2): 142– 144. [DOI] [PubMed] [Google Scholar]
- 64. Ito MK. Effects of extensive and poor gastrointestinal metabolism on the pharmaco-dynamics of pravastatin. J Clin Pharmacol. 1998; 38( 4): 331– 336. [DOI] [PubMed] [Google Scholar]
- 65. Singhvi SM, Pan HY, Morrison RA, Willard DA. Disposition of pravastatin sodium, a tissue-selective HMG-CoA reductase inhibitor, in healthy subjects. Br J Clin Pharmacol. 1990; 29( 2): 239– 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nozawa T, Imai K, Nezu J. et al. Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J Pharmacol Exp Ther. 2004; 308( 2): 438– 445. [DOI] [PubMed] [Google Scholar]
- 67. Kobayashi D, Nozawa T, Imai K. et al. Involvement of human organic anion transporting polypeptide OATP-B (SLC21A9) in pH-dependent transport across intestinal apical membrane. J Pharmacol Exp Ther. 2003; 306( 2): 703– 708. [DOI] [PubMed] [Google Scholar]
- 68. Shirasaka Y, Suzuki K, Nakanishi T, Tamai I. Intestinal absorption of HMG-CoA reductase inhibitor pravastatin mediated by organic anion transporting polypeptide. Pharm Res. 2010; 27( 10): 2141– 2149. [DOI] [PubMed] [Google Scholar]
- 69. Kivisto KT, Grisk O, Hofmann U. et al. Disposition of oral and intravenous pravastatin in MRP2-deficient TR- rats. Drug Metab Dispos. 2005; 33( 11): 1593– 1596. [DOI] [PubMed] [Google Scholar]
- 70. Kivisto KT, Niemi M. Influence of drug transporter polymorphisms on pravastatin pharmacokinetics in humans. Pharm Res. 2007; 24( 2): 239– 247. [DOI] [PubMed] [Google Scholar]
- 71. Yamazaki M, Akiyama S, Ni'inuma K. et al. Biliary excretion of pravastatin in rats: contribution of the excretion pathway mediated by canalicular multispecific organic anion transporter. Drug Metab Dispos. 1997; 25( 10): 1123– 1129. [PubMed] [Google Scholar]
- 72. Niemi M, Arnold KA, Backman JT. et al. Association of genetic polymorphism in ABCC2 with hepatic multidrug resistance-associated protein 2 expression and pravastatin pharmacokinetics. Pharmacogenet Genomics. 2006; 16( 11): 801– 808. [DOI] [PubMed] [Google Scholar]
- 73. Varma MV, Rotter CJ, Chupka J. et al. pH-sensitive interaction of HMG-CoA reductase inhibitors (statins) with organic anion transporting polypeptide 2B1. Mol Pharm. 2011; 8( 4): 1303– 1313. [DOI] [PubMed] [Google Scholar]
- 74. Huang L, Wang Y, Grimm S. ATP-dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab Dispos. 2006; 34( 5): 738– 742. [DOI] [PubMed] [Google Scholar]
- 75. Keskitalo JE, Zolk O, Fromm MF. et al. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2009; 86( 2): 197– 203. [DOI] [PubMed] [Google Scholar]
- 76. Zhang W, Yu BN, He YJ. et al. Role of BCRP 421C>A polymorphism on rosuvastatin pharmacokinetics in healthy Chinese males. Clin Chim Acta. 2006; 373( 1–2): 99– 103. [DOI] [PubMed] [Google Scholar]
- 77. Shirasaka Y, Suzuki K, Nakanishi T, Tamai I. Differential effect of grapefruit juice on intestinal absorption of statins due to inhibition of organic anion transporting polypeptide and/or P-glycoprotein. J Pharm Sci. 2011; 100( 9): 3843– 3853. [DOI] [PubMed] [Google Scholar]
- 78. Shirasaka Y, Suzuki K, Shichiri M. et al. Intestinal absorption of HMG-CoA reductase inhibitor pitavastatin mediated by organic anion transporting polypeptide and P-glycoprotein/multidrug resistance 1. Drug Metab Pharmacokinet. 2011; 26( 2): 171– 179. [DOI] [PubMed] [Google Scholar]
- 79. Smith HT, Jokubaitis LA, Troendle AJ. et al. Pharmacokinetics of fluvastatin and specific drug interactions. Am J Hypertens. 1993; 6( 11 Pt 2): 375S– 382S. [DOI] [PubMed] [Google Scholar]
- 80. Pan HY, DeVault AR, Brescia D. et al. Effect of food on pravastatin pharmacokinetics and pharmacodynamics. Int J Clin Pharmacol Ther Toxicol. 1993; 31( 6): 291– 294. [PubMed] [Google Scholar]
- 81. Li Y, Jiang X, Lan K. et al. Pharmacokinetic properties of rosuvastatin after single-dose, oral administration in Chinese volunteers: a randomized, open-label, three-way crossover study. Clin Ther. 2007; 29( 10): 2194– 2203. [DOI] [PubMed] [Google Scholar]
- 82. Radulovic LL, Cilla DD, Posvar EL. et al. Effect of food on the bioavailability of atorvastatin, an HMG-CoA reductase inhibitor. J Clin Pharmacol. 1995; 35( 10): 990– 994. [DOI] [PubMed] [Google Scholar]
- 83. Shang D, Deng S, Yao Z. et al. The effect of food on the pharmacokinetic properties and bioequivalence of two formulations of pitavastatin calcium in healthy Chinese male subjects. Xenobiotica. 2015: 1– 6. [DOI] [PubMed] [Google Scholar]
- 84. Lovastatin Mevacor(R) [package insert]. Merck & Co., Inc., Kenilworth, New Jersey: 1987. http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/019643s088l-bl.pdf. November 10, 2015. [Google Scholar]
- 85. Simvastatin. Zocor(R) [package insert]. Merck & Co., Inc. Kenilworth, New Jersey; 1991. 2015. [Google Scholar]
- 86. Triscari J, Rossi L, Pan HY. Chronokinetics of pravastatin administered in the PM compared with AM dosing. Am J Ther. 1995; 2( 4): 265– 268. [DOI] [PubMed] [Google Scholar]
- 87. Cilla DD, Jr, Gibson DM, Whitfield LR, Sedman AJ. Pharmacodynamic effects and pharmacokinetics of atorvastatin after administration to normocholesterolemic subjects in the morning and evening. J Clin Pharmacol. 1996; 36( 7): 604– 609. [DOI] [PubMed] [Google Scholar]
- 88. Fauler G, Abletshauser C, Erwa W. et al. Time-of-intake (morning versus evening) of extended-release fluvastatin in hyperlipemic patients is without influence on the pharmacodynamics (mevalonic acid excretion) and pharmacokinetics. Int J Clin Pharmacol Ther. 2007; 45( 6): 328– 334. [DOI] [PubMed] [Google Scholar]
- 89. Martin PD, Mitchell PD, Schneck DW. Pharmacodynamic effects and pharmacokinetics of a new HMG-CoA reductase inhibitor, rosuvastatin, after morning or evening administration in healthy volunteers. Br J Clin Pharmacol. 2002; 54( 5): 472– 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Goo RH, Moore JG, Greenberg E, Alazraki NP. Circadian variation in gastric emptying of meals in humans. Gastroenterology. 1987; 93( 3): 515– 518. [DOI] [PubMed] [Google Scholar]
- 91. Pappu AS, Illingworth DR. Diurnal variations in the plasma concentrations of mevalonic acid in patients with abetalipoproteinaemia. Eur J Clin Invest. 1994; 24( 10): 698– 702. [DOI] [PubMed] [Google Scholar]
- 92. Jones PJ, Schoeller DA. Evidence for diurnal periodicity in human cholesterol synthesis. J Lipid Res. 1990; 31( 4): 667– 673. [PubMed] [Google Scholar]
- 93. Tse FL, Jaffe JM, Troendle A. Pharmacokinetics of fluvastatin after single and multiple doses in normal volunteers. J Clin Pharmacol. 1992; 32( 7): 630– 638. [DOI] [PubMed] [Google Scholar]
- 94. Pan HY, DeVault AR, Wang-Iverson D. et al. Comparative pharmacokinetics and pharmacodynamics of pravastatin and lovastatin. J Clin Pharmacol. 1990; 30( 12): 1128– 1135. [DOI] [PubMed] [Google Scholar]
- 95. Lennernas H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003; 42( 13): 1141– 1160. [DOI] [PubMed] [Google Scholar]
- 96. Martin PD, Warwick MJ, Dane AL. et al. Absolute oral bioavailability of rosuvastatin in healthy white adult male volunteers. Clin Ther. 2003; 25( 10): 2553– 2563. [DOI] [PubMed] [Google Scholar]
- 97. Voora D, Shah SH, Spasojevic I. et al. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol. 2009; 54( 17): 1609– 1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Neuvonen PJ, Backman JT, Niemi M. Pharmacokinetic comparison of the potential over-the-counter statins simvastatin, lovastatin, fluvastatin and pravastatin. Clin Pharmacokinet. 2008; 47( 7): 463– 474. [DOI] [PubMed] [Google Scholar]
- 99. Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011; 63( 1): 157– 181. [DOI] [PubMed] [Google Scholar]
- 100. Wagner J, Leeder JS. Pediatric pharmacogenomics: a systematic assessment of ontogeny and genetic variation to guide the design of statin studies in children. Pediatr Clin North Am. 2012; 59( 5): 1017– 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fujino H, Saito T, Ogawa S, Kojima J. Transporter-mediated influx and efflux mechanisms of pitavastatin, a new inhibitor of HMG-CoA reductase. J Pharm Pharmacol. 2005; 57( 10): 1305– 1311. [DOI] [PubMed] [Google Scholar]
- 102. Hirano M, Maeda K, Shitara Y, Sugiyama Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther. 2004; 311( 1): 139– 146. [DOI] [PubMed] [Google Scholar]
- 103. Ho RH, Tirona RG, Leake BF. et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006; 130( 6): 1793– 1806. [DOI] [PubMed] [Google Scholar]
- 104. Kitamura S, Maeda K, Wang Y, Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos. 2008; 36( 10): 2014– 2023. [DOI] [PubMed] [Google Scholar]
- 105. Kopplow K, Letschert K, Konig J. et al. Human hepatobiliary transport of organic anions analyzed by quadruple-transfected cells. Mol Pharmacol. 2005; 68( 4): 1031– 1038. [DOI] [PubMed] [Google Scholar]
- 106. Niemi M, Pasanen MK, Neuvonen PJ. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin Pharmacol Ther. 2006; 80( 4): 356– 366. [DOI] [PubMed] [Google Scholar]
- 107. Deng JW, Song IS, Shin HJ. et al. The effect of SLCO1B1*15 on the disposition of pravastatin and pitavastatin is substrate dependent: the contribution of transporting activity changes by SLCO1B1*15. Pharmacogenet Genomics. 2008; 18( 5): 424– 433. [DOI] [PubMed] [Google Scholar]
- 108. Kameyama Y, Yamashita K, Kobayashi K. et al. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics. 2005; 15( 7): 513– 522. [DOI] [PubMed] [Google Scholar]
- 109. Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics. 2006; 16( 12): 873– 879. [DOI] [PubMed] [Google Scholar]
- 110. Hsiang B, Zhu Y, Wang Z. et al. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J Biol Chem. 1999; 274( 52): 37161– 37168. [DOI] [PubMed] [Google Scholar]
- 111. Chen C, Mireles RJ, Campbell SD. et al. Differential interaction of 3-hydroxy-3-methylglutaryl-coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos. 2005; 33( 4): 537– 546. [DOI] [PubMed] [Google Scholar]
- 112. Tirona RG, Leake BF, Wolkoff AW, Kim RB. Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J Pharmacol Exp Ther. 2003; 304( 1): 223– 228. [DOI] [PubMed] [Google Scholar]
- 113. Vavricka SR, Van Montfoort J, Ha HR. et al. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002; 36( 1): 164– 172. [DOI] [PubMed] [Google Scholar]
- 114. Lau YY, Huang Y, Frassetto L, Benet LZ. effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007; 81( 2): 194– 204. [DOI] [PubMed] [Google Scholar]
- 115. Asberg A, Hartmann A, Fjeldsa E. et al. Bilateral pharmacokinetic interaction between cyclosporine A and atorvastatin in renal transplant recipients. Am J Transplant. 2001; 1( 4): 382– 386. [DOI] [PubMed] [Google Scholar]
- 116. Hermann M, Asberg A, Christensen H. et al. Substantially elevated levels of atorvastatin and metabolites in cyclosporine-treated renal transplant recipients. Clin Pharmacol Ther. 2004; 76( 4): 388– 391. [DOI] [PubMed] [Google Scholar]
- 117. Lemahieu WP, Hermann M, Asberg A. et al. Combined therapy with atorvastatin and calcineurin inhibitors: no interactions with tacrolimus. Am J Transplant. 2005; 5( 9): 2236– 2243. [DOI] [PubMed] [Google Scholar]
- 118. Park JW, Siekmeier R, Lattke P. et al. Pharmacokinetics and pharmacodynamics of fluvastatin in heart transplant recipients taking cyclosporine A. J Cardiovasc Pharmacol Ther. 2001; 6( 4): 351– 361. [DOI] [PubMed] [Google Scholar]
- 119. Olbricht C, Wanner C, Eisenhauer T. et al. Accumulation of lovastatin, but not pravastatin, in the blood of cyclosporine-treated kidney graft patients after multiple doses. Clin Pharmacol Ther. 1997; 62( 3): 311– 321. [DOI] [PubMed] [Google Scholar]
- 120. Regazzi MB, Iacona I, Campana C. et al. Altered disposition of pravastatin following concomitant drug therapy with cyclosporin A in transplant recipients. Transplant Proc. 1993; 25( 4): 2732– 2734. [PubMed] [Google Scholar]
- 121. Simonson SG, Raza A, Martin PD. et al. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther. 2004; 76( 2): 167– 177. [DOI] [PubMed] [Google Scholar]
- 122. Arnadottir M, Eriksson LO, Thysell H, Karkas JD. Plasma concentration profiles of simvastatin 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitory activity in kidney transplant recipients with and without ciclosporin. Nephron. 1993; 65( 3): 410– 413. [DOI] [PubMed] [Google Scholar]
- 123. Ichimaru N, Takahara S, Kokado Y. et al. Changes in lipid metabolism and effect of simvastatin in renal transplant recipients induced by cyclosporine or tacrolimus. Atherosclerosis. 2001; 158( 2): 417– 423. [DOI] [PubMed] [Google Scholar]
- 124. Martin PD, Warwick MJ, Dane AL. et al. Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin Ther. 2003; 25( 11): 2822– 2835. [DOI] [PubMed] [Google Scholar]
- 125. Fujino H, Yamada I, Shimada S. et al. Metabolic fate of pitavastatin (NK-104), a new inhibitor of 3-hydroxy-3-methyl-glutaryl coenzyme A reductase. Effects on drug-metabolizing systems in rats and humans. Arzneimittelforschung. 2002; 52( 10): 745– 753. [DOI] [PubMed] [Google Scholar]
- 126. Neuvonen PJ, Jalava KM. Itraconazole drastically increases plasma concentrations of lovastatin and lovastatin acid. Clin Pharmacol Ther. 1996; 60( 1): 54– 61. [DOI] [PubMed] [Google Scholar]
- 127. Neuvonen PJ, Kantola T, Kivisto KT. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol Ther. 1998; 63( 3): 332– 341. [DOI] [PubMed] [Google Scholar]
- 128. Kivisto KT, Kantola T, Neuvonen PJ. Different effects of itraconazole on the pharmacokinetics of fluvastatin and lovastatin. Br J Clin Pharmacol. 1998; 46( 1): 49– 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Backman JT, Luurila H, Neuvonen M, Neuvonen PJ. Rifampin markedly decreases and gemfibrozil increases the plasma concentrations of atorvastatin and its metabolites. Clin Pharmacol Ther. 2005; 78( 2): 154– 167. [DOI] [PubMed] [Google Scholar]
- 130. Kyrklund C, Backman JT, Kivisto KT. et al. Plasma concentrations of active lovastatin acid are markedly increased by gemfibrozil but not by bezafibrate. Clin Pharmacol Ther. 2001; 69( 5): 340– 345. [DOI] [PubMed] [Google Scholar]
- 131. Kyrklund C, Backman JT, Neuvonen M, Neuvonen PJ. Gemfibrozil increases plasma pravastatin concentrations and reduces pravastatin renal clearance. Clin Pharmacol Ther. 2003; 73( 6): 538– 544. [DOI] [PubMed] [Google Scholar]
- 132. Schneck DW, Birmingham BK, Zalikowski JA. et al. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther. 2004; 75( 5): 455– 463. [DOI] [PubMed] [Google Scholar]
- 133. Backman JT, Kyrklund C, Kivisto KT. et al. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin Pharmacol Ther. 2000; 68( 2): 122– 129. [DOI] [PubMed] [Google Scholar]
- 134. Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004; 447( 5): 653– 665. [DOI] [PubMed] [Google Scholar]
- 135. Ogawa K, Hasegawa S, Udaka Y. et al. Individual difference in the pharmacokinetics of a drug, pravastatin, in healthy subjects. J Clin Pharmacol. 2003; 43( 11): 1268– 1273. [DOI] [PubMed] [Google Scholar]
- 136. Nishizato Y, Ieiri I, Suzuk iH. et al. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: consequences for pravastatin pharmacokinetics. Clin. Pharmacol. Ther. 2003; 73: 554– 565. [DOI] [PubMed] [Google Scholar]
- 137. Niemi M, Schaeffeler E, Lang T. et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SL-CO1B1). Pharmacogenetics. 2004; 14( 7): 429– 440. [DOI] [PubMed] [Google Scholar]
- 138. He YJ, Zhang W, Chen Y. et al. Rifampicin alters atorvastatin plasma concentration on the basis of SLCO1B1 521T>C polymorphism. Clin Chim Acta. 2009; 405( 1–2): 49– 52. [DOI] [PubMed] [Google Scholar]
- 139. SEARCH, Collaborative, Group, . et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med. 2008; 359: 789– 799. [DOI] [PubMed] [Google Scholar]
- 140. Pan HY, Waclawski AP, Funke PT, Whigan D. Pharmacokinetics of pravastatin in elderly versus young men and women. Ann Pharmacother. 1993; 27( 9): 1029– 1033. [DOI] [PubMed] [Google Scholar]
- 141. Tse FL, Nickerson DF, Yardley WS. Binding of fluvastatin to blood cells and plasma proteins. J Pharm Sci. 1993; 82( 9): 942– 947. [DOI] [PubMed] [Google Scholar]
- 142. Botti RE, Triscari J, Pan HY, Zayat J. Concentrations of pravastatin and lovastatin in cerebrospinal fluid in healthy subjects. Clin Neuropharmacol. 1991; 14( 3): 256– 261. [DOI] [PubMed] [Google Scholar]
- 143. Johnson-Anuna LN, Eckert GP, Keller JH. et al. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J Pharmacol Exp Ther. 2005; 312( 2): 786– 793. [DOI] [PubMed] [Google Scholar]
- 144. Pierno S, De Luca A, Tricarico D. et al. Potential risk of myopathy by HMG-CoA reductase inhibitors: a comparison of pravastatin and simvastatin effects on membrane electrical properties of rat skeletal muscle fibers. J Pharmacol Exp Ther. 1995; 275( 3): 1490– 1496. [PubMed] [Google Scholar]
- 145. Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther. 2005; 19( 6): 403– 414. [DOI] [PubMed] [Google Scholar]
- 146. Masters BA, Palmoski MJ, Flint OP. et al. In vitro myotoxicity of the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, pravastatin, lovastatin, and simvastatin, using neonatal rat skeletal myocytes. Toxicol Appl Pharmacol. 1995; 131( 1): 163– 174. [DOI] [PubMed] [Google Scholar]
- 147. Nakahara K, Kuriyama M, Sonoda Y. et al. Myopathy induced by HMG-CoA reductase inhibitors in rabbits: a pathological, electrophysiological, and biochemical study. Toxicol Appl Pharmacol. 1998; 152( 1): 99– 106. [DOI] [PubMed] [Google Scholar]
- 148. Gadbut AP, Caruso AP, Galper JB. Differential sensitivity of C2-C12 striated muscle cells to lovastatin and pravastatin. J Mol Cell Cardiol. 1995; 27( 10): 2397– 2402. [DOI] [PubMed] [Google Scholar]
- 149. Reijneveld JC, Koot RW, Bredman JJ. et al. Differential effects of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitors on the development of myopathy in young rats. Pediatr Res. 1996; 39( 6): 1028– 1035. [DOI] [PubMed] [Google Scholar]
- 150. Prueksaritanont T, Ma B, Yu N. The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br J Clin Pharmacol. 2003; 56( 1): 120– 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol. 2004; 94( 9): 1140– 1146. [DOI] [PubMed] [Google Scholar]
- 152. Prueksaritanont T, Gorham LM, Ma B. et al. In vitro metabolism of simvastatin in humans: Identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab Dispos. 1997; 25( 10): 1191– 1199. [PubMed] [Google Scholar]
- 153. Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006; 80( 6): 565– 581. [DOI] [PubMed] [Google Scholar]
- 154. Scripture CD, Pieper JA. Clinical pharmacokinetics of fluvastatin. Clin Pharmacokinet. 2001; 40( 4): 263– 281. [DOI] [PubMed] [Google Scholar]
- 155. Transon C, Leemann T, Dayer P. In vitro comparative inhibition profiles of major human drug metabolising cytochrome P450 isozymes (CYP2C9, CYP2D6 and CYP3A4) by HMG-CoA reductase inhibitors. Eur J Clin Pharmacol. 1996; 50( 3): 209– 215. [DOI] [PubMed] [Google Scholar]
- 156. Transon C, Leemann T, Vogt N, Dayer P. In vivo inhibition profile of cytochrome P450TB (CYP2C9) by (+/−)-fluvastatin. Clin Pharmacol Ther. 1995; 58( 4): 412– 417. [DOI] [PubMed] [Google Scholar]
- 157. Lamba JK, Lin YS, Schuetz EG, Thummel KE. Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev. 2002; 54( 10): 1271– 1294. [DOI] [PubMed] [Google Scholar]
- 158. Wang D, Guo Y, Wrighton SA. et al. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 2010; 11( 4): 274– 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Elens L, Becker ML, Haufroid V. et al. Novel CYP3A4 intron 6 single nucleotide polymorphism is associated with simvastatin-mediated cholesterol reduction in the Rotterdam study. Pharmacogenet Genomics. 2011. 21( 12): 861– 866. [DOI] [PubMed] [Google Scholar]
- 160. Kirchheiner J, Kudlicz D, Meisel C. et al. Influence of CYP2C9 polymorphisms on the pharmacokinetics and cholesterol-lowering activity of (−)-3S,5R-fluvastatin and (+)-3R,5S-fluvastatin in healthy volunteers. Clin Pharmacol Ther. 2003; 74( 2): 186– 194. [DOI] [PubMed] [Google Scholar]
- 161. Fujino H, Yamada I, Shimada S, Yoneda M, Kojima J. Metabolic fate of pitavastatin, a new inhibitor of HMG-CoA reductase: human UDP-glucuronosyltransferase enzymes involved in lactonization. Xenobiotica. 2003; 33( 1): 27– 41. [DOI] [PubMed] [Google Scholar]
- 162. Prueksaritanont T, Subramanian R, Fang X. et al. Glucuronidation of statins in animals and humans: a novel mechanism of statin lactonization. Drug Metab Dispos. 2002; 30( 5): 505– 512. [DOI] [PubMed] [Google Scholar]
- 163. Fujino H, Saito T, Tsunenari Y, Kojima J, Sakaeda T. Metabolic properties of the acid and lactone forms of HMG-CoA reductase inhibitors. Xenobiotica. 2004; 34( 11–12): 961– 971. [DOI] [PubMed] [Google Scholar]
- 164. Riedmaier S, Klein K, Hofmann U. et al. UDP-glucuronosyltransferase (UGT) polymorphisms affect atorvastatin lactonization in vitro and in vivo. Clin Pharmacol Ther. 2010; 87( 1): 65– 73. [DOI] [PubMed] [Google Scholar]
- 165. Cho SK, Oh ES, Park K. et al. The UGT1A3*2 polymorphism affects atorvastatin lactonization and lipid-lowering effect in healthy volunteers. Pharmacogenet Genomics. 2012; 22( 8): 598– 605. [DOI] [PubMed] [Google Scholar]
- 166. Leschziner GD, Andrew T, Pirmohamed M, Johnson MR. ABCB1 genotype and PGP expression, function and therapeutic drug response: a critical review and recommendations for future research. Pharmacogenomics J. 2007; 7( 3): 154– 179. [DOI] [PubMed] [Google Scholar]
- 167. Keskitalo JE, Kurkinen KJ, Neuvonen M. et al. No significant effect of ABCB1 haplotypes on the pharmacokinetics of fluvastatin, pravastatin, lovastatin, and rosuvastatin. Br J Clin Pharmacol. 2009; 68( 2): 207– 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Keskitalo JE, Kurkinen KJ, Neuvoneni PJ, Niemi M. ABCB1 haplotypes differentially affect the pharmacokinetics of the acid and lactone forms of simvastatin and atorvastatin. Clin Pharmacol Ther. 2008; 84( 4): 457– 461. [DOI] [PubMed] [Google Scholar]
- 169. Imai Y, Nakane M, Kage K. et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther. 2002; 1( 8): 611– 616. [PubMed] [Google Scholar]
- 170. Keskitalo JE, Pasanen MK, Neuvonen PJ, Niemi M. Different effects of the ABCG2 c.421C>A SNP on the pharmacokinetics of fluvastatin, pravastatin and simvastatin. Pharmacogenomics. 2009; 10( 10): 1617– 1624. [DOI] [PubMed] [Google Scholar]
- 171. Hirano M, Maeda K, Hayashi H. et al. Bile salt export pump (BSEP/ABCB11) can transport a nonbile acid substrate, pravastatin. J Pharmacol Exp Ther. 2005; 314( 2): 876– 882. [DOI] [PubMed] [Google Scholar]
- 172. Stern RH, Yang BB, Horton M. et al. Renal dysfunction does not alter the pharmacokinetics or LDL-cholesterol reduction of atorvastatin. J Clin Pharmacol. 1997; 37( 9): 816– 819. [DOI] [PubMed] [Google Scholar]
- 173. Halstenson CE, Triscari J, DeVault A. et al. Single-dose pharmacokinetics of pravastatin and metabolites in patients with renal impairment. J Clin Pharmacol. 1992; 32( 2): 124– 132. [DOI] [PubMed] [Google Scholar]
- 174. Hasegawa M, Kusuhara H, Sugiyama D. et al. Functional involvement of rat organic anion transporter 3 (rOat3; Slc22a8) in the renal uptake of organic anions. J Pharmacol Exp Ther. 2002; 300( 3): 746– 753. [DOI] [PubMed] [Google Scholar]
- 175. Takeda M, Noshiro R, Onozato ML. et al. Evidence for a role of human organic anion transporters in the muscular side effects of HMG-CoA reductase inhibitors. Eur J Pharmacol. 2004; 483( 2–3): 133– 138. [DOI] [PubMed] [Google Scholar]
- 176. Nakagomi-Hagihara R, Nakai D, Tokui T. Inhibition of human organic anion transporter 3 mediated pravastatin transport by gemfibrozil and the metabolites in humans. Xenobiotica. 2007; 37( 4): 416– 426. [DOI] [PubMed] [Google Scholar]
- 177. Hebbel RP. Special issue of Microcirculation: examination of the vascular pathobiology of sickle cell anemia. Foreword. Microcirculation. 2004; 11( 2): 99– 100. [PubMed] [Google Scholar]
- 178. Hoppe C, Kuypers F, Larkin S. et al. A pilot study of the short-term use of simvastatin in sickle cell disease: effects on markers of vascular dysfunction. Br J Haematol. 2011. 153( 5): 655– 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Mahle WT, Vincent RN, Berg AM, Kanter KR. Pravastatin therapy is associated with reduction in coronary allograft vasculopathy in pediatric heart transplantation. J. Heart Lung Transplant. 2005; 24: 63066. [DOI] [PubMed] [Google Scholar]
- 180. Singh TP, Naftel DC, Webber S. et al. Hyperlipidemia in children after heart transplantation. J. Heart Lung Transplant. 2006; 25: 1199– 1205. [DOI] [PubMed] [Google Scholar]
- 181. Azuma RW, Suzuki J, Ogawa M. et al. HMG-CoA reductase inhibitor attenuates experimental autoimmune myocarditis through inhibition of T cell activation. Cardiovasc Res. 2004; 64( 3): 412– 420. [DOI] [PubMed] [Google Scholar]
- 182. Tang Q, Huang J, Qian H. et al. Antiarrhythmic effect of atorvastatin on autoimmune myocarditis is mediated by improving myocardial repolarization. Life Sci. 2007; 80( 7): 601– 608. [DOI] [PubMed] [Google Scholar]
- 183. Zembala M, Wojnicz R, Zakliczynski M. et al. Cellular adhesion molecules changes in myocardium during first year post heart transplant. Ann Transplant. 1997; 2( 2): 16– 19. [PubMed] [Google Scholar]
- 184. Stojanovic I, Vrtovec B, Radovancevic B. et al. Survival, graft atherosclerosis, and rejection incidence in heart transplant recipients treated with statins: 5-year follow-up. J Heart Lung Transplant. 2005; 24( 9): 1235– 1238. [DOI] [PubMed] [Google Scholar]
- 185. Wenke K, Meiser B, Thiery J, Reichart B. Impact of simvastatin therapy after heart transplantation an 11-year prospective evaluation. Herz. 2005; 30( 5): 431– 432. [DOI] [PubMed] [Google Scholar]
- 186. Fildes JE, Shaw SM, Mitsidou A. et al. HMG-CoA reductase inhibitors deplete circulating classical and non-classical monocytes following human heart transplantation. Transpl Immunol. 2008; 19( 2): 152– 157. [DOI] [PubMed] [Google Scholar]
- 187. Grigioni F, Carigi S, Potena L. et al. Long-term safety and effectiveness of statins for heart transplant recipients in routine clinical practice. Transplant Proc. 2006; 38( 5): 1507– 1510. [DOI] [PubMed] [Google Scholar]
- 188. Kobashigawa JA, Katznelson S, Laks H. et al. Effect of pravastatin on outcomes after cardiac transplantation. N Engl J Med. 1995; 333( 10): 621– 627. [DOI] [PubMed] [Google Scholar]
- 189. Mahle WT, Vincent RN, Berg AM, Kanter KR. Pravastatin therapy is associated with reduction in coronary allograft vasculopathy in pediatric heart transplantation. J Heart Lung Transplant. 2005; 24( 1): 63– 66. [DOI] [PubMed] [Google Scholar]
- 190. Singh TP, Naftel DC, Webber S. et al. Hyperlipidemia in children after heart transplantation. J Heart Lung Transplant. 2006; 25( 10): 1199– 1205. [DOI] [PubMed] [Google Scholar]
- 191. Frohlich GM, Rufibach K, Enseleit F. et al. Statins and the risk of cancer after heart transplantation. Circulation. 2012; 126( 4): 440– 447. [DOI] [PubMed] [Google Scholar]
- 192. Murtola TJ, Tammela TL, Maattanen L. et al. Prostate cancer and PSA among statin users in the Finnish prostate cancer screening trial. Int J Cancer. 2010; 127( 7): 1650– 1659. [DOI] [PubMed] [Google Scholar]
- 193. Cho SJ, Kim JS, Kim JM. et al. Simvastatin induces apoptosis in human colon cancer cells and in tumor xenografts, and attenuates colitis-associated colon cancer in mice. Int J Cancer. 2008; 123( 4): 951– 957. [DOI] [PubMed] [Google Scholar]
- 194. Spampanato C, De Maria S, Sarnataro M. et al. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of Bax and down-regulation of BCL-2 gene expression. Int J Oncol. 2012; 40( 4): 935– 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Denoyelle C, Vasse M, Korner M. et al. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis. 2001; 22( 8): 1139– 1148. [DOI] [PubMed] [Google Scholar]
- 196. Sanguankeo A, Upala S, Cheungpasitporn W. et al. Effects of statins on renal outcome in chronic kidney disease patients: a systematic review and meta-analysis. PLoS One. 2015; 10( 7): e0132970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Athyros VG, Katsiki N, Karagiannis A, Mikhailidis DP. Statins can improve proteinuria and glomerular filtration rate loss in chronic kidney disease patients, further reducing cardiovascular risk. Fact or fiction? Expert Opin Pharmacother. 2015; 16( 10): 1449– 1461. [DOI] [PubMed] [Google Scholar]
- 198. Yan YL, Qiu B, Wang J. et al. High-intensity statin therapy in patients with chronic kidney disease: a systematic review and meta-analysis. BMJ Open. 2015; 5( 5): e006886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Mundy G, Garrett R, Harris S. et al. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999; 286( 5446): 1946– 1949. [DOI] [PubMed] [Google Scholar]
- 200. Park YS, David AE, Park KM. et al. Controlled release of simvastatin from in situ forming hydrogel triggers bone formation in MC3T3-E1 cells. AAPS J. 2013; 15( 2): 367– 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Majima T, Shimatsu A, Komatsu Y. et al. Short-term effects of pitavastatin on biochemical markers of bone turnover in patients with hypercholesterolemia. Intern Med. 2007; 46( 24): 1967– 1973. [DOI] [PubMed] [Google Scholar]
- 202. Safaei H, Janghorbani M, Aminorroaya A, Amini M. Lovastatin effects on bone mineral density in postmenopausal women with type 2 diabetes mellitus. Acta Diabetol. 2007; 44( 2): 76– 82. [DOI] [PubMed] [Google Scholar]
- 203. Perez-Castrillon JL, Vega G, Abad L. et al. Effect of the TNFalpha-308 G/A polymorphism on the changes produced by atorvastatin in bone mineral density in patients with acute coronary syndrome. Ann Nutr Metab. 2008; 53( 2): 117– 121. [DOI] [PubMed] [Google Scholar]
- 204. Kanazawa I, Yamaguchi T, Yamauchi M, Sugimoto T. Rosuvastatin increased serum osteocalcin levels independent of its serum cholesterol-lowering effect in patients with type 2 diabetes and hypercholesterolemia. Intern Med. 2009; 48( 21): 1869– 1873. [DOI] [PubMed] [Google Scholar]
- 205. Baker JV, Huppler Hullsiek K, Prosser R. et al. Angiotensin converting enzyme inhibitor and HMG-CoA reductase inhibitor as adjunct treatment for persons with HIV infection: a feasibility randomized trial. PLoS One. 2012; 7( 10): e46894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Boccara F, Simon T, Lacombe K. et al. Influence of pravastatin on carotid artery structure and function in dyslipidemic HIV-infected patients receiving antiretroviral therapy. AIDS. 2006; 20( 18): 2395– 2398. [DOI] [PubMed] [Google Scholar]
- 207. Hurlimann D, Chenevard R, Ruschitzka F. et al. Effects of statins on endothelial function and lipid profile in HIV infected persons receiving protease inhibitor-containing anti-retroviral combination therapy: a randomised double blind crossover trial. Heart. 2006; 92( 1): 110– 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Ridker PM, Danielson E, Fonseca FA. et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009; 373( 9670): 1175– 1182. [DOI] [PubMed] [Google Scholar]
- 209. Calza L, Vanino E, Salvadori C. et al. Tenofovir/emtricitabine/efavirenz plus rosuvastatin decrease serum levels of inflammatory markers more than antiretroviral drugs alone in antiretroviral therapy-naive HIV-infected patients. HIV Clin Trials. 2014; 15( 1): 1– 13. [DOI] [PubMed] [Google Scholar]
- 210. Funderburg NT, Jiang Y, Debanne SM. et al. Rosuvastatin treatment reduces markers of monocyte activation in HIV-infected subjects on antiretroviral therapy. Clin Infect Dis. 2014; 58( 4): 588– 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Rasmussen LD, Kronborg G, Larsen CS. et al. Statin therapy and mortality in HIV-infected individuals; a Danish nationwide population-based cohort study. PLoS One. 2013; 8( 3): e52828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Amuro H, Ito T, Miyamoto R. et al. Statins, inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase, function as inhibitors of cellular and molecular components involved in type I interferon production. Arthritis Rheum. 2010; 62( 7): 2073– 2085. [DOI] [PubMed] [Google Scholar]
- 213. Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005; 4( 12): 977– 987. [DOI] [PubMed] [Google Scholar]
- 214. Mira E, Manes S. Immunomodulatory and anti-inflammatory activities of statins. Endocr Metab Immune Disord Drug Targets. 2009; 9( 3): 237– 247. [DOI] [PubMed] [Google Scholar]
- 215. Yasuda H, Yuen PS, Hu X. et al. Simvastatin improves sepsis-induced mortality and acute kidney injury via renal vascular effects. Kidney Int. 2006; 69( 9): 1535– 1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Tleyjeh IM, Kashour T, Hakim FA. et al. Statins for the prevention and treatment of infections: a systematic review and meta-analysis. Arch Intern Med. 2009; 169( 18): 1658– 1667. [DOI] [PubMed] [Google Scholar]
- 217. Fedson DS. Confronting an influenza pandemic with inexpensive generic agents: can it be done? Lancet Infect Dis. 2008; 8( 9): 571– 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Thomsen RW, Riis A, Kornum JB. et al. Pre-admission use of statins and outcomes after hospitalization with pneumonia: population-based cohort study of 29,900 patients. Arch Intern Med. 2008; 168( 19): 2081– 2087. [DOI] [PubMed] [Google Scholar]
- 219. Schanberg LE, Sandborg C, Barnhart HX. et al. Use of atorvastatin in systemic lupus erythematosus in children and adolescents. Arthritis Rheum. 2012; 64( 1): 285– 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Mok CC, Wong CK, To CH. et al. Effects of rosuvastatin on vascular biomarkers and carotid atherosclerosis in lupus: a randomized, double-blind, placebo-controlled trial. Arthritis Care Res (Hoboken). 2011; 63( 6): 875– 883. [DOI] [PubMed] [Google Scholar]
- 221. Ferreira GA, Navarro TP, Telles RW. et al. Atorvastatin therapy improves endothelial-dependent vasodilation in patients with systemic lupus erythematosus: an 8 weeks controlled trial. Rheumatology (Oxford). 2007; 46( 10): 1560– 1565. [DOI] [PubMed] [Google Scholar]
- 222. Ferreira GA, Teixeira AL, Sato EI. Atorvastatin therapy reduces interferon-regulated chemokine CXCL9 plasma levels in patients with systemic lupus erythematosus. Lupus. 2010; 19( 8): 927– 934. [DOI] [PubMed] [Google Scholar]
- 223. Sathyapalan T, Kilpatrick ES, Coady AM, Atkin SL. The effect of atorvastatin in patients with polycystic ovary syndrome: a randomized double-blind placebo-controlled study. J Clin Endocrinol Metab. 2009; 94( 1): 103– 108. [DOI] [PubMed] [Google Scholar]
- 224. Banaszewska B, Pawelczyk L, Spaczynski RZ. et al. Effects of simvastatin and oral contraceptive agent on polycystic ovary syndrome: prospective, randomized, crossover trial. J Clin Endocrinol Metab. 2007; 92( 2): 456– 461. [DOI] [PubMed] [Google Scholar]
- 225. Gao L, Zhao FL, Li SC. Statin is a reasonable treatment option for patients with polycystic ovary syndrome: a meta-analysis of randomized controlled trials. Exp Clin Endocrinol Diabetes. 2012; 120( 6): 367– 375. [DOI] [PubMed] [Google Scholar]
- 226. Lv S, Liu Y, Zou Z. et al. The impact of statins therapy on disease activity and inflammatory factor in patients with rheumatoid arthritis: a meta-analysis. Clin Exp Rheumatol. 2015; 33( 1): 69– 76. [PubMed] [Google Scholar]
- 227. Schoenfeld SR, Lu L, Rai SK. et al. Statin use and mortality in rheumatoid arthritis: a general population-based cohort study. Ann Rheum Dis. 2016; 75( 7): 1315– 1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Xing B, Yin YF, Zhao LD. et al. Effect of 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitor on disease activity in patients with rheumatoid arthritis: a meta-analysis. Medicine (Baltimore). 2015; 94( 8): e572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Pereira MC, Cardoso PR, Da Rocha LF., Jr et al. Simvastatin inhibits cytokines in a dose response in patients with rheumatoid arthritis. Inflamm Res. 2014; 63( 4): 309– 315. [DOI] [PubMed] [Google Scholar]
- 230. Lin H, Xiao Y, Chen G. et al. HMG-CoA reductase inhibitor simvastatin suppresses Toll-like receptor 2 ligand-induced activation of nuclear factor kappa B by preventing RhoA activation in monocytes from rheumatoid arthritis patients. Rheumatol Int. 2011; 31( 11): 1451– 1458. [DOI] [PubMed] [Google Scholar]
- 231. McCarey DW, McInnes IB, Madhok R. et al. Trial of Atorvastatin in Rheumatoid Arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet. 2004; 363( 9426): 2015– 2021. [DOI] [PubMed] [Google Scholar]
- 232. Dolkart O, Liron T, Chechik O. et al. Statins enhance rotator cuff healing by stimulating the COX2/PGE2/EP4 pathway: an in vivo and in vitro study. Am J Sports Med. 2014; 42( 12): 2869– 2876. [DOI] [PubMed] [Google Scholar]
- 233. Lin TT, Lin CH, Chang CL. et al. The effect of diabetes, hyperlipidemia, and statins on the development of rotator cuff disease: a nationwide, 11-year, longitudinal, population-based follow-up study. Am J Sports Med. 2015; 43( 9): 2126– 2132. [DOI] [PubMed] [Google Scholar]
- 234. Joukhadar C, Klein N, Prinz M. et al. Similar effects of atorvastatin, simvastatin and pravastatin on thrombogenic and inflammatory parameters in patients with hypercholesterolemia. Thromb Haemost. 2001; 85( 1): 47– 51. [PubMed] [Google Scholar]
- 235. Nomura S, Shouzu A, Omoto S. et al. Effects of losartan and simvastatin on monocyte-derived microparticles in hypertensive patients with and without type 2 diabetes mellitus. Clin Appl Thromb Hemost. 2004; 10( 2): 133– 141. [DOI] [PubMed] [Google Scholar]
- 236. Wagner AH, Kohler T, Ruckschloss U. et al. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol. 2000; 20( 1): 61– 69. [DOI] [PubMed] [Google Scholar]
- 237. Couture P, Brun LD, Szots F. et al. Association of specific LDL receptor gene mutations with differential plasma lipoprotein response to simvastatin in young French Canadians with heterozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1998; 18( 6): 1007– 1012. [DOI] [PubMed] [Google Scholar]
- 238. Vohl MC, Szots F, Lelievre M. et al. Influence of LDL receptor gene mutation and apo E polymorphism on lipoprotein response to simvastatin treatment among adolescents with heterozygous familial hypercholesterolemia. Atherosclerosis. 2002; 160( 2): 361– 368. [DOI] [PubMed] [Google Scholar]
- 239. Koeijvoets KC, Rodenburg J, Hutten BA. et al. Low-density lipoprotein receptor genotype and response to pravastatin in children with familial hypercholesterolemia: sub-study of an intima-media thickness trial. Circulation. 15 2005; 112( 20): 3168– 3173. [DOI] [PubMed] [Google Scholar]
- 240. Krauss RM, Mangravite LM, Smith JD. et al. Variation in the 3-hydroxyl-3-methylglutaryl coenzyme a reductase gene is associated with racial differences in low-density lipoprotein cholesterol response to simvastatin treatment. Circulation. 2008; 117( 12): 1537– 1544. [DOI] [PubMed] [Google Scholar]
- 241. Medina MW, Gao F, Ruan W. et al. Alternative splicing of 3-hydroxy-3-methylglutaryl coenzyme A reductase is associated with plasma low-density lipoprotein cholesterol response to simvastatin. Circulation. 2008; 118( 4): 355– 362. [DOI] [PMC free article] [PubMed] [Google Scholar]