

Abstract
Emodin, an anthraquinone derivative from the root and rhizome of Rheum palmatum L., was found to have antitumor effects in different types of cancer by regulating multi-molecular targets. The aim of the present study was to explore the effect of emodin on the migration and invasion of MHCC-97H human hepatocellular carcinoma cells and the underlying molecular mechanisms. Firstly, it was demonstrated that emodin can inhibit cell proliferation and induce apoptosis of cells in a time- and dose-dependent manner, using a MTT assay and flow cytometry, respectively. However, when emodin concentration was <50 µmol/l, it had little effect on the inhibition of proliferation or the induction of apoptosis. Then, it was observed that emodin can significantly suppress cell migration and invasion with a treatment dose <50 µmol/l compared with the control (P<0.05), which was not attributed to a decrease in cell number. Further study demonstrated that emodin significantly suppressed the expression levels of matrix metalloproteinase (MMP)-2 and MMP-9 compared with the control, which may be mediated by the activation of the p38 mitogen-activated protein kinases (MAPK) signaling pathway and suppression of extracellular signal regulated kinase (ERK)/MAPK and phosphatidylinositol 3-kinase/Akt signaling pathways. Therefore, the present study, for the first time, used MHCC-97H cells, which have the high potential of malignant invasion, to demonstrate that emodin may inhibit cell migration and invasion.
Keywords: emodin, human hepatocellular carcinoma, migration, invasion, molecular mechanisms
Introduction
Hepatocellular carcinoma (HCC) is one of the most frequent malignancies worldwide, representing the fifth most common cause of cancer in men and the seventh in women, and the third most frequent cause of cancer-related mortality (1–3). Globally, there are ~750,000 new cases of liver cancer reported per year (4). Although surgical resection provides better results in patients with HCC in early stages, the long-term prognosis remains unsatisfactory because many patients are diagnosed at the advanced stage, and tumors have an inherent capacity for invasiveness and metastasis (5,6). At present, chemotherapy is commonly used in the treatment of metastatic HCC. However, this treatment remains ineffective due to its toxicity to normal cells and the rapid development of resistance. Therefore, to further explore the biology of metastasis and develop novel therapeutics that specifically target metastasis and metastatic progression are required (7,8).
Increasing attention has been paid to drugs among traditional Chinese medicines, since these materials typically share high safety profiles and can effectively prevent and control metastasis (9,10). Rheum palmatum L. is a plant that has been widely used in Chinese medicine as a laxative for thousands of years. Emodin is an anthraquinone derivative from its root and rhizome (11). It has been reported that emodin possesses a number of biological properties such as anti-inflammatory, antiviral and anti-proliferative effects (12). In the context of cancer, several studies have indicated that emodin exerts anticancer effects on various cell lines derived from the pancreas (13), colorectum (14) and breast (15). However, so far there is little evidence revealing the possible effect of emodin on tumor metastasis in HCC.
Herein, in the present study, the inhibitory effect of migration and invasion by emodin are investigated using the HCC cell line MHCC-97H, and the molecular mechanisms underlying these actions are explored.
Materials and methods
Cell culture and reagents
The MHCC-97H cell line was purchased from the American Type Culture Collection (Manassas, VA, USA), and was cultured in high-glucose Dulbeccos modified Eagles medium with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) in a humidified atmosphere with 5% CO2 at 37°C. Emodin (1,3,8-trihydroxy-6-methyl-anthraquinine) was purchased from Sigma-Aldrich (Merck Millipore, Darmstadt, Germany). Its molecular formula is C15H10O5 and its molecular weight is 270.24 g/mol. It was dissolved at a concentration of 100 mmol/l in 100% dimethylsulfoxide (DMSO; Sigma-Aldrich; Merck Millipore) as a stock solution, stored at −20°C, and the following concentrations were prepared: 2.5, 5, 12.5, 25, 50 and 100 mmol/l. DMSO (0.2%) was used as vehicle control for all the experiments. MTT was purchased from Sigma-Aldrich (Merck Millipore). The Annexin V-FITC apoptosis detection kit (cat. no. KGA108) was purchased from KeyGen Bio-Tech Co., Ltd., Nanjing, China. Rabbit phosphorylated (p)-Akt (cat. no. 4060S) and total (t)-Akt (cat. no. 4691S) polyclonal antibody, rabbit p-extracellular-signal-regulated kinase (ERK)1/2 (cat. no. 4370S) and t-ERK1/2 (cat. no. 4695S) monoclonal antibody, rabbit p-p38 (cat. no. 4511S) and t-p38 (cat. no. 9212S) monoclonal antibody and mouse β-actin (cat. no. 3700) monoclonal antibody were all purchased from Cell Signaling Technology, Inc. (Boston, MA, USA). Rabbit matrix metalloproteinase (MMP)-2 (cat. no. BS1236) and MMP-9 (cat. no. BS1241) polyclonal antibody were purchased from Bioworld Technology, Inc. (St. Louis Park, MN, USA).
MTT assay for cell viability
Cell viability was determined by MTT assay. Briefly, Cells were seeded into a 96-well plate at a density of 1×104 cells/well and cultured for 24 h. Emodin was then added to the wells with final concentrations of 0, 5, 10, 25, 50, 100 and 200 µmol/l and incubated for 12, 24, 48 and 72 h at 37°C. DMSO (0.2%) was used as a negative control. Then, 20 µl MTT solution (5 mg/ml) was added to each well and incubated for an additional 4 h at 37°C. The medium was carefully removed and 150 µl DMSO was added to each well to dissolve the formazan crystals and the absorbance was detected at 570 nm using a multiskan spectrum microplate reader (Thermo Fisher Scientific, Inc.).
Flow cytometricanalysis of apoptosis
Cell apoptosis was detected using the Annexin V-FITC apoptosis detection kit according to the manufacturers instructions. In brief, a total of 3×105 MHCC-97H cells were seeded into each well of the 6-well plates and various concentrations (10, 25, 50,100 µmol/l) of emodin were added to the wells after 24 h of incubation at 37°C. The control group was treated with the equivalent quantity of DMSO (0.2%). Cells of each sample were harvested after an additional 12 or 24 h and suspended in 500 µl of Annexin V binding buffer (1X). Annexin V-FiTC (5 µl) and 5 µl of propidium iodide (PI) were added and incubated for 15 min in dark. The stained cells were analyzed by flow cytometry using a FACS Calibur (BD Biosciences, San Jose, CA, USA).
Migration and invasion assay
Cell migration was analyzed with the aid of a Transwell chamber (Corning Incorporated, Corning, NY, USA) with 8-µm pores and a cell invasion assay was performed using a Corning® Matrigel® invasion chamber (Corning Incorporated). Cells suspended in 200 µl serum-free medium were seeded onto the upper chambers (1×105 cells/chamber for migration and 2×105 cells/chamber for invasion) and incubated with different concentrations of emodin (10, 25 and 50 µmol/l), and the chambers were placed into 24-well plates with medium containing 10% serum. DMSO (0.2%) was used as a negative control. After 24 h, the cells remaining in the upper chamber were removed and migrated or invaded cells on the lower membrane surface were fixed with 4% formaldehyde polymerisatum followed by staining with 0.1% crystal violet for 20 min. The migrated or invaded cells in six visual fields (magnification, ×200) selected randomly were counted in each Transwell chamber under a phase-contrast microscope.
Western blot analysis
After treatment with 50 µM emodin and DMSO (0.2%) as a negative control, the cells were washed twice using ice-cold phosphate-buffered saline (pH 7.4) and lysed in radioimmunoprecipitation assay protein lysis buffer containing 1 mM PMSF on ice. Total proteins were extracted by centrifuging the cell lysates at 12,000 × g for 15 min at 4°C and the protein concentration was determined using a BCA assay kit (cat. no. P0010) Beyotime Institute of Biotechnology, Shanghai, China). A total of 30 µg protein from every sample was separated using a 10% SDS-PAGE gel and transferred to a polyvinylidene fluoride (PVDF) membrane. After blocking with Tris-buffered saline and Tween 20 (TBST) buffer containing 5% skimmed milk for 1 h at room temperature, the PVDF membrane was incubated with appropriate concentrations of primary antibodies (dilution, 1:1,000) at 4°C overnight. After washing the membrane with TBST three times for 15 min, the membrane was incubated with corresponding secondary antibody labeled with horseradish peroxidase-conjugated (goat anti-mouse, 1:5,000; goat anti-rabbit, 1:2,000) secondary antibody for 2 h at room temperature. Following three washes with TBST for 15 min, the immunoreactive bands were detected using an enhanced chemiluminescence detection kit (Sigma-Aldrich; Merck Millipore). β-actin was used as the internal control and the relative values of target protein were corrected in accordance with the absorbency of the internal control.
Statistical analysis
All results were expressed as the mean ± standard deviation of at least three independent experiments and were analyzed using SPSS version 13.0 software (SPSS, Inc., Chicago, IL, USA). The statistical analysis was performed using a one-way analysis of variance and Dunnetts test. P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of emodin on cell viability
In order to investigate the antiproliferative effect of emodin on MHCC-97H cells, MTT assay was used to quantify the effect following treatment with emodin at different concentrations (5, 10, 25, 50, 100 and 200 µmol/l) for 12, 24, 48 and 72 h. As shown in Fig. 1, when the treatment concentration was <100 µmol/l and the treatment time was <24 h, the viability of the cells changed very little. With increases in the emodin concentration and treatment time, cell viability decrease evidently in a concentration- and time-dependent manner.
Figure 1.
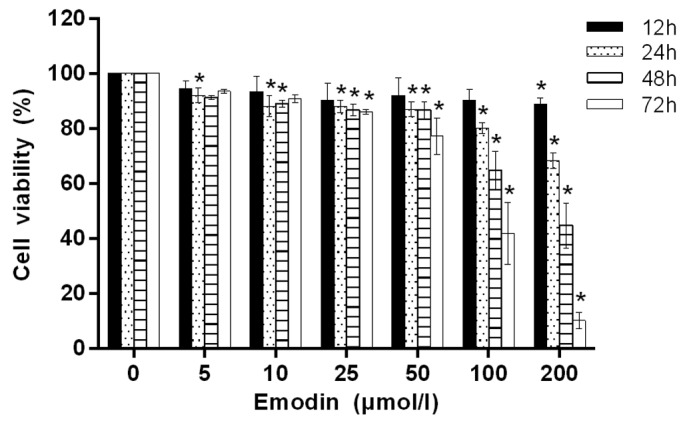
Effect of emodin on the cell viability of MHCC-97H cells. Cell proliferation and viability were determined by an MTT assay. Cells were treated with 5, 10, 25, 50, 100 and 200 µmol/l emodin for 12, 24, 48 and 72 h. Data are expressed as mean ± standard deviation and all experiments were repeated three times. *P<0.05 vs. the control group (0 µmol/l emodin).
Effect of low dose emodin on mild cells apoptosis
To determine the effect of emodin on apoptosis induction in MHCC-97H cells, flow cytometry was used to assess the cell apoptosis rate. After treatment with various concentrations of emodin (0 µmol/l for the control, 10, 25, 50 and 100 µmol/l for the experimental groups) for 24 h, MHCC-97H cells were stained with FITC-Annexin V/PI. As shown in Fig. 2, the apoptosis rate of MHCC-97H cells was shown to gradually increase as the emodin concentration increased (1.73±1.53, 6.31±0.59, 10.16±2.21, 12.99±3.77 and 53.68±8.32%, respectively). These results suggest that emodin was able to induce apoptosis in MHCC-97H cells, but the change was mild when the emodin concentration was <50 µmol/l.
Figure 2.

Low dose emodin induced mild cells apoptosis. MHCC-97H cells treated with various concentrations of emodin for 24 h were stained with FITC-Annexin V/PI and analyzed by flow cytometry. The gate setting distinguished between living (lower left), necrotic (upper left), early apoptotic (lower right) and late apoptotic (upper right) cells. (A) Control group (0 µmol/l emodin); (B) 10 µmol/l emodin; (C) 25 µmol/l emodin; (D) 50 µmol/l emodin; (E) 100 µmol/l emodin; (F) The statistical analysis of the apoptosis rate, including the combination of early apoptotic and late apoptotic MHCC-97H cells. Data are presented as mean ± standard deviation (n=3). *P<0.05 vs. the control group (0 µmol/l emodin). FITC, fluorescein isothiocyanate; PI< propidium iodide.
Effect of emodin on cell migration and invasion
To determine the effect of emodin on cell migration and invasion, MHCC-97H cells treated with 0, 10, 25 and 50 µmol/l emodin were induced to migrate through the Transwell membranes and invade in Matrigel-coated Transwells for 24 h. The number of cells that migrated was reduced by emodin in a dose-dependent manner (Fig. 3). In the same manner, following treatment with emodin at concentration of 0, 10, 25 and 50 µmol/l, the numberof invasive cells was 26.67±3.51, 18.34±4.16, 13.33±1.53 and 7.33±1.53, respectively. These data clearly demonstrate that emodin treatment significantly inhibits the migration and invasion of MHCC-97H cells in a dose-dependent manner.
Figure 3.

Effect of emodin on MHCC-97H cell migration and invasion. Cells treated with 0, 10, 25 and 50 µmol/l emodin were induced to migrate through the Transwell membranes or invade through the matrigel-coated Transwell membranes. (A) Microscopic observation of MHCC-97H cells that migrated through the Transwell chamber (magnification, ×200). (B) Quantification of MHCC-97H cells passing through the membranes. (C) Microscopic observation of MHCC-97H cells that invaded through the matrigel-coated Transwell membranes (magnification, ×200). (D) Quantification of MHCC-97H cells invading through the membranes. Data are presented as the mean ± standard deviation (n=3). *P<0.05 vs. the control group (0 µmol/l emodin).
Effect of emodin on the expression of proteins associated with tumor invasion
In order to investigate the probable mechanism of the inhibition of migration and invasion induced by emodin, the activation of metastasis-related signal pathways, such as mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways, were detected by western blotting. The results demonstrated that following treatment with 50 µM emodin, the phosphorylated (p)-extracellular signal regulated kinase (ERK1)/2 expression levels were significantly decreased in a time-dependent manner until 60 min, at which point the levels began to increase again. In addition, p-Akt expression levels were decreased in a time-dependent manner, whereas the tendency of p-p38 expression levels were inverse. However, the ERK1/2, p38 and Akt expression levels remained essentially unchanged, except for a mild decrease at 60 min in ERK1/2 expression. In addition, MHCC-97H cells were treated with 50 µmol/l emodin for 0, 3, 6, 9 and 12 h, and it was observed that the MMP-2 and MMP-9 expression levels decreased in a time-dependent manner (Fig. 4).
Figure 4.

Effect of emodin on the expression of proteins associated with tumor invasion. MHCC-97H cells were treated with 50 µmol/l emodin for the indicated times and the whole protein extracts were then analyzed by western blotting. (A) Representative western blot showing the protein expression of p-ERK, ERK, p-p38, p38, p-AKT and AKT. (B) Quantification of the relative protein expression levels were normalized against the value of β-actin protein expression. (C) Representative western blot showing the protein expression of MMP-2 and MMP-9. (D) Quantification of the relative protein expression levels normalized against β-actin protein expression. Data are presented as the mean ± standard deviation (n=3). *P<0.05 vs. the control group (0 min). p, phosphorylated; ERK1/2, extracellular signal regulated kinase; MMP, matrix metalloproteinase.
Discussion
Distant metastasis is a common occurrence in patients with HCC and is the primary hindrance in improving overall survival of HCC. Therefore, novel innovative therapeutic drugs are required to improve HCC prognosis. Emodin, an anthraquinone derivative from the root and rhizome of Rheum palmatum L., was found to have antitumor effects in several types of cancer by regulating multi-molecular targets involved in tumor growth, apoptosis and angiogenesis (16). Recent research demonstrated that emodin may inhibit growth and induce apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of signal transducer and activator of transcription 3 (17). It was also reported that emodin may suppresses migration and invasion through the modulation of C-X-C chemokine receptor type 4 expression in an orthotopic model of HCC (18). In the present study, the inhibitory effects of emodin on the migration and invasion of HCC in vitro are investigated, using MHCC-97H cell line as a model due to its high potential of malignant invasion. Furthermore, the molecular mechanisms underlying these effects are investigated.
Initially, we used a MTT assay to investigate the antiproliferative effect of emodin in MHCC-97H cells and found that emodin could suppress cell proliferation in a dose-and time-dependent manner, particularly when the treatment concentration was >100 µmol/l and the treatment time was >24 h. Then, we used flow cytometry to identify the effects of emodin on the induction of apoptosis with <100 µmol/l emodin for 24 h and found that its induction effect on apoptosis was mild when emodin was <50 µmol/l. Therefore, in order to eliminate the effect of antiproliferation and apoptosis, 0, 10, 25 and 50 µmol/l emodin was chosen to treat MHCC-97H cells for 24 h and investigate the cell migration and invasion ability with the aid of a Transwell chamber. The result indicated that emodin treatment inhibited the migration and invasion of MHCC-97H cells in a dose-dependent manner.
The establishment and progression of metastases is a complex, multi-step process involving the modulation of the cell phenotype, detachment from the primary tumor, invasion through the extracellular matrix and transportation to other normal organs (9,19). MMPs are a class of proteolytic enzymes which serve important roles in extracellular matrix degradation and their abnormal expression may promote tumor invasion and metastasis (20). MMP-2 and MMP-9 are two members of the MMP family and they are the primary enzymes that degrade diffusely basal membrane type IV collagen (21). Early studies have demonstrated that MMP-2 and MMP-9 exhibited increased expression in patients with HCC, which may be a prediction of tumor recurrence and survival in patients with HCC following surgical resection (22,23). In the current study, the effects of emodin on MMP-2 and MMP-9 expression in cells were investigated and the results suggest that emodin significantly downregulated the expression of MMP-2 and MMP-9, indicating that MMP-2 and MMP-9 were involved in the inhibitory impact of emodin.
MAPKs are serine-threonine protein kinases that mediate a wide variety of cellular behaviors, including proliferation, differentiation, apoptosis, migration and invasion (24). In mammals, MAPKs include ERK, p38 MAPK and c-Jun NH2-terminal kinase. Typically, ERK is present in its non-activated forms in the cytoplasm. When exposed to inflammatory cytokines and other factors, the ERK MAPK pathway may be activated and mediates the cancer processes. Lin et al (25) found that mammalian sterile-20-like kinase 4 (MST4) promotes HCC metastasis via the activation of the p-ERK pathway, and the combination of MST4 and p-ERK has better power to predict the outcomes of HCC. Thus, the effects of emodin on the expression of total and phosphorylated ERK1/2 was investigated and the results indicated that following exposure to 50 µmol/l emodin, the level of p-ERK1/2 was downregulated in a time-dependent manner while t-ERK1/2 remained unchanged. Thus, emodin may inhibit MHCC-97H cell metastasis through the regulation of the ERK1/2 MAPK signaling pathway. p38 MAPK is an additional important signaling pathway that regulates the proliferation of malignant tumor cells (26). The p38 MAPK signaling pathway can be activated in response to various environmental and cellular stresses, and a number of studies have shown that activation of p38-MAPK signaling pathways may produce anti-apoptotic and proliferative effects (27). However, previous studies have suggested an opposite role of p38 MAPK in mediating cell apoptosis and growth inhibition, which indicated that p38 MAPK serves a dual role as a regulator of cell behavior (28,29). In the present study, it was observed that emodin could promote the phosphorylation of p38, indicating that the p38 MAPK signaling pathway may exert a function in response to the type of stimulus and in a cell type specific manner. Further investigation regarding the specific molecular mechanism involved is required.
Akt is a serine-threonine kinase that is a primary effector of PI3K (30). Increased PI3K/Akt activation is suggested to change the migration and invasion characteristics of HCC cells, and is an independent prognostic index for patients with HCC (31,32). MMP-2 and MMP-9 expression in HCC cells could be enhanced through the activation of the PI3K/Akt signaling pathway, and as a result further regulate HCC cell invasion and metastasis (33). The observations in the present study indicated that emodin treatment inhibited the expression levels of p-Akt in the MHCC-97H cell line in a time-dependent manner, while no significant change existed in t-Akt expression levels. Since Akt is a downstream target of the PI3K signaling pathway, the inhibition of Akt phosphorylation revealed that emodin treatment inhibited the PI3K/Akt signaling pathway. This phenomenon may associated with the inhibition of MMP-2 and MMP-9 expression.
In conclusion, the current study is the first to use MHCC-97H cells, which have a high malignant and invasion potential, to demonstrate that emodin can suppress the migration and invasion at concentrations that have little effects on proliferation inhibition or apoptosis induction. Furthermore, emodin suppressed the MMP-2 and MMP-9 expression, which are closely associated with the metastasic characteristics of malignant tumors. It can be speculated that these effects were mediated by the activation of the p38 MAPK signaling pathway and the suppression of the ERK/MAPK and PI3K/Akt signaling pathways. However, further studies are required to explore and elucidate the specific molecular mechanism by which emodin inhibits the invasion and metastasis of HCC.
Acknowledgements
The present study was supported by the National Nature Science Foundation of China (grant no. 30901986), the Foundation for the Author of National Excellent Doctoral Dissertation of China (grant no. 201366) and the Shanghai Municipal Natural Science Foundation (grant no. 13ZR1448900).
Glossary
Abbreviations
- MMP
matrix metalloproteinase
- MAPK
mitogen-activated protein kinases
- ERK
extracellular signal regulated kinase
- PI3K
phosphatidylinositol 3-kinase
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 3.Bosetti C, Turati F, La Vecchia C. Hepatocellular carcinoma epidemiology. Best Pract Res Clin Gastroenterol. 2014;28:753–770. doi: 10.1016/j.bpg.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012;62:394–399. doi: 10.3322/caac.21161. [DOI] [PubMed] [Google Scholar]
- 5.Forner A, Reig ME, de Lope CR, Bruix J. Current strategy for staging and treatment: The BCLC update and future prospects. Semin Liver Dis. 2010;30:61–74. doi: 10.1055/s-0030-1247133. [DOI] [PubMed] [Google Scholar]
- 6.Baffy G. Decoding multifocal hepatocellular carcinoma: An opportune pursuit. Hepatobiliary Surg Nutr. 2015;4:206–210. doi: 10.3978/j.issn.2304-3881.2014.12.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lui GY, Kovacevic Z, Richardson V, Merlot AM, Kalinowski DS, Richardson DR. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget. 2015;6:18748–18779. doi: 10.18632/oncotarget.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling CQ, Yue XQ, Ling C. Three advantages of using traditional Chinese medicine to prevent and treat tumor. J Integr Med. 2014;12:331–335. doi: 10.1016/S2095-4964(14)60038-8. [DOI] [PubMed] [Google Scholar]
- 9.Li F, Zhang W. Role of traditional Chinese medicine and its chemical components in anti-tumor metastasis. J Cancer Res Ther. 2014;10(Suppl 1):S20–S26. doi: 10.4103/0973-1482.139748. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Wang N, Cheung F, Lao L, Li C, Feng Y. Chinese medicines for prevention and treatment of human hepatocellular carcinoma: Current progress on pharmacological actions and mechanisms. J Integr Med. 2015;13:142–164. doi: 10.1016/S2095-4964(15)60171-6. [DOI] [PubMed] [Google Scholar]
- 11.Sun ZH, Bu P. Downregulation of phosphatase of regenerating liver-3 is involved in the inhibition of proliferation and apoptosis induced by emodin in the SGC-7901 human gastric carcinoma cell line. Exp Ther Med. 2012;3:1077–1081. doi: 10.3892/etm.2012.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shrimali D, Shanmugam MK, Kumar AP, Zhang J, Tan BK, Ahn KS, Sethi G. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 2013;341:139–149. doi: 10.1016/j.canlet.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, Chen L, Bu HQ, Yu QJ, Jiang DD, Pan FP, Wang Y, Liu DL, Lin SZ. Effects of emodinon the demethylation of tumor-suppressor genes in pancreatic cancer PANC-1 cells. Oncol Rep. 2015;33:3015–3023. doi: 10.3892/or.2015.3914. [DOI] [PubMed] [Google Scholar]
- 14.Pooja T, Karunagaran D. Emodin suppresses Wnt signaling in human colorectal cancer cells SW480 and SW620. Eur J Pharmacol. 2014;742:55–64. doi: 10.1016/j.ejphar.2014.08.028. [DOI] [PubMed] [Google Scholar]
- 15.Ma J, Lu H, Wang S, Chen B, Liu Z, Ke X, Liu T, Fu J. The anthraquinone derivative Emodin inhibits angiogenesis and metastasis through downregulating Runx2 activity in breast cancer. Int J Oncol. 2015;46:1619–1628. doi: 10.3892/ijo.2015.2888. [DOI] [PubMed] [Google Scholar]
- 16.Wei WT, Lin SZ, Liu DL, Wang ZH. The distinct mechanisms of the antitumor activity of emodin in different types of cancer (Review) Oncol Rep. 2013;30:2555–2562. doi: 10.3892/or.2013.2741. [DOI] [PubMed] [Google Scholar]
- 17.Subramaniam A, Shanmugam MK, Ong TH, Li F, Perumal E, Chen L, Vali S, Abbasi T, Kapoor S, Ahn KS, et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br J Pharmacol. 2013;170:807–821. doi: 10.1111/bph.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manu KA, Shanmugam MK, Ong TH, Subramaniam A, Siveen KS, Perumal E, Samy RP, Bist P, Lim LH, Kumar AP, et al. Emodin suppresses migration and invasion through the modulation of CXCR4 expression in an orthotopic model of human hepatocellular carcinoma. PloS One. 2013;8:e57015. doi: 10.1371/journal.pone.0057015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polacheck WJ, Zervantonakis IK, Kamm RD. Tumor cell migration in complex microenvironments. Cell Mol Life Sci. 2013;70:1335–1356. doi: 10.1007/s00018-012-1115-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deryugina EI, Quigley JP. Tumor angiogenesis: MMP-mediated induction of intravasation-and metastasis-sustaining neovasculature. Matrix Biol 44–46. 2015:94–112. doi: 10.1016/j.matbio.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kessenbrock K, Wang CY, Werb Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol 44–46. 2015:184–190. doi: 10.1016/j.matbio.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daniele A, Divella R, Quaranta M, Mattioli V, Casamassima P, Paradiso A, Garrisi VM, Gadaleta CD, Gadaleta-Caldarola G, Savino E, et al. Clinical and prognostic role of circulating MMP-2 and its inhibitor TIMP-2 in HCC patients prior to and after trans-hepatic arterial chemo-embolization. Clin Biochem. 2014;47:184–190. doi: 10.1016/j.clinbiochem.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 23.Chen R, Cui J, Xu C, Xue T, Guo K, Gao D, Liu Y, Ye S, Ren Z. The significance of MMP-9 over MMP-2 in HCC invasiveness and recurrence of hepatocellular carcinoma after curative resection. Ann Surg Oncol. 2012;19(Suppl 3):S375–S384. doi: 10.1245/s10434-011-1836-7. [DOI] [PubMed] [Google Scholar]
- 24.Peti W, Page R. Molecular basis of MAP kinase regulation. Protein Sci. 2013;22:1698–1710. doi: 10.1002/pro.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin ZH, Wang L, Zhang JB, Liu Y, Li XQ, Guo L, Zhang B, Zhu WW, Ye QH. MST4 promotes hepatocellular carcinoma epithelial-mesenchymal transition and metastasis via activation of the p-ERK pathway. Int J Oncol. 2014;45:629–640. doi: 10.3892/ijo.2014.2455. [DOI] [PubMed] [Google Scholar]
- 26.Grossi V, Peserico A, Tezil T, Simone C. p38α MAPK pathway: A key factor in colorectal cancer therapy and chemoresistance. World J Gastroenterol. 2014;20:9744–9758. doi: 10.3748/wjg.v20.i29.9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim EK, Choi EJ. Compromised MAPK signaling in human diseases: An update. Arch Toxicol. 2015;89:867–882. doi: 10.1007/s00204-015-1472-2. [DOI] [PubMed] [Google Scholar]
- 28.Olson JM, Hallahan AR. p38 MAP kinase: A convergence point in cancer therapy. Trends Mol Med. 2004;10:125–129. doi: 10.1016/j.molmed.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Koul HK, Pal M, Koul S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer. 2013;4:342–359. doi: 10.1177/1947601913507951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engelman JA. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 31.Lin JJ, Su JH, Tsai CC, Chen YJ, Liao MH, Wu YJ. 11-epi-Sinulariolide acetate reduces cell migration and invasion of human hepatocellular carcinoma by reducing the activation of ERK1/2, p38MAPK and FAK/PI3K/AKT/mTOR signaling pathways. Mar Drug. 2014;12:4783–4798. doi: 10.3390/md12094783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitz KJ, Wohlschlaeger J, Lang H, Sotiropoulos GC, Malago M, Steveling K, Reis H, Cicinnati VR, Schmid KW, Baba HA. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J Hepatol. 2008;48:83–90. doi: 10.1016/j.jhep.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 33.Wu YJ, Neoh CA, Tsao CY, Su JH, Li HH. Sinulariolide suppresses human hepatocellular carcinoma cell migration and invasion by inhibiting matrix metalloproteinase-2/-9 through MAPKs and PI3K/Akt signaling pathways. Int J Mol Sci. 2015;16:16469–16482. doi: 10.3390/ijms160716469. [DOI] [PMC free article] [PubMed] [Google Scholar]