

Abstract
Mitochondrial dysfunction is associated with pathogenic mitochondrial (mt)DNA mutations. The majority of mtDNA point mutations have a heteroplasmic status, which is defined as the coexistence of wild-type and mutated DNA within a cell or tissue. Previous findings demonstrated that certain mtDNA heteroplasmic mutations contribute to widely spread chronic diseases, including cancer, and alterations in the heteroplasmy level are associated with the clinical phenotype and severity of cancer. In the present study, the proportions of mutant mtDNA 10398G were assessed using amplification-refractory mutation system-quantitative polymerase chain reaction (PCR) assay in 129 non-small cell lung cancer (NSCLC) tissue samples. Wild-type and mutant sequences were separately amplified using allele-specific primers and, subsequently, the PCR products containing the mtDNA 10398 site were ligated into vectors to construct a standard plasmid DNA construct. The association between mtDNA A10398G and the prognosis of patients was analyzed by survival analysis and Cox proportional hazards model. For the patient cohort, the median follow-up time and overall survival time were 20.6 and 26.3 months, respectively. The ratios of mutant heteroplasmy ranged between 0.31 and 97.04%. Patients with a high degree of mutant mtDNA 10398G had a significantly longer overall survival time compared with those with a low degree of mutant mtDNA 10398G (28.7 vs. 22.5 months, respectively; P<0.05). In addition, multivariate analysis demonstrated that epidermal growth factor receptor mutation status, tumor stage and the possession of a low degree of mutant 10398G were the three most independent prognostic factors. In conclusion, the present study suggests that, among NSCLC patients, there are large shifts in mutant mtDNA 10398G heteroplasmy and a low degree of mutant mtDNA 10398G heteroplasmy may be a marker of poor prognosis in patients with NSCLC.
Keywords: heteroplasmy, mitochondrial DNA 10398, mutation, non-small cell lung cancer, prognosis
Introduction
Mitochondria are important in cell and energy metabolism, and are required for cell apoptosis. Mitochondrial dysfunction has been associated with a wide range of degenerative and metabolic diseases, including ageing and cancer (1). According to the Warburg hypothesis (2), dysfunction of mitochondria has been postulated to render cancer cells resistant to apoptosis.
Mitochondrial DNA (mtDNA) possesses several unique characteristics (3–5), such as non-Mendelian maternal inheritance, a mutation rate that is 10–20 times higher than nuclear DNA, the threshold effect and heteroplasmy (6). In addition, there are 100–1,000 mitochondria per cell and 2–10 copies of circular double-stranded mitochondrial genomes per mitochondrion (6). When a mutation arises in mtDNA, it creates a coexistence of wild-type and mutant mtDNA, resulting in heteroplasmy. When a heteroplasmic cell divides, the two variants of mtDNA are randomly distributed into the daughter cells, resulting in genetic drift; thus, the proportions of mutant mtDNA in these daughter cells ranges between 1 and 100% (4,7). As the percentage of mutant mtDNA increases to a threshold value, tissue dysfunction and clinical symptoms appear in the individual, and apoptosis or necrosis may be initiated (8,9). Therefore, the proportion of mutant mtDNA may contribute to the clinical symptoms of mitochondrial disease, including cancer (9).
Previously, somatic mtDNA mutations, including point mutations, insertions and deletions, were identified in various types of human cancer, including non-small cell lung cancer (NSCLC) (10). Furthermore, mtDNA 8701 and 10398, which code for ATPase 6 and NADH dehydrogenase 3, respectively, were identified as mutational hotspots in the mitochondrial genome of patients with lung cancer (11). African American women carrying wild-type mtDNA 10398A were observed to have an increased risk of invasive breast cancer (12). In cancer, it has been hypothesized that these somatic mtDNA alterations may lead to a switch in the energy supply between mitochondrial oxidative phosphorylation and aerobic glycolysis, leading to cancer progression (13). However, few studies have investigated the prognostic value of heteroplasmy of mutant mtDNA 10398G in patients with NSCLC. The proportion of mutant mtDNA must reach the threshold to cause a clinical phenotype (14); therefore, in order to predict the clinical outcome of a patient, the proportion of mutant DNA in the affected or relevant tissue should be determined.
In the present study, the heteroplasmy of mtDNA A10398G was assessed in tumor tissues from 129 patients with NSCLC using an amplification refractory mutation system-quantitative polymerase chain reaction (ARMS-qPCR) assay. A previous study demonstrated that the introduction of one or two mismatched nucleotides immediately 5′ to the mutation site markedly increases the binding specificity of the allele-specific modified primers toward the wild-type or mutant sequence targets (15). In addition, the present study analyzed the prognostic value of mtDNA A10398G. The present results demonstrated that the proportion of mutant heteroplasmy provides a novel biomarker in predicting the outcome of patients with NSCLC. Thus, intervention of heteroplasmy using mtDNA A10398G may become a novel approach for cancer treatment.
Materials and methods
Patient eligibility and clinical data
A total of 129 NSCLC tissue specimens were collected from patients that underwent surgical resection, including lymph node biopsy, at Zhongnan Hospital of Wuhan University (Wuhan, China) between July 2006 and July 2011. None of patients had received radiotherapy or chemotherapy prior to surgery. Clinical staging was assessed according to the American Joint Committee on Cancer (AJCC, seventh edition; https://cancerstaging.org/Pages/default.aspx). Formalin-fixed and paraffin-embedded surgical tissue samples collected from the Department of Pathology at the Zhongnan Hospital of Wuhan University were examined according to the World Health Organization 2004 classification system (16). The patients consisted of 96 men and 33 women (age range, 20–80 years). All participants provided prior written informed consent. The study protocol was reviewed and approved by the Institutional Review Board of Zhongnan Hospital of Wuhan University. Clinical information, including age, gender, pathological type, tumor-node-metastasis stage, smoking status, epidermal growth factor receptor (EGFR) mutation status and clinical follow-up data, was recorded prospectively (Table I). Patients were followed up at 3-month intervals for ≤2 years, then every 6 months for 5 years, and annually thereafter until the final follow-up on November 26, 2012. The median follow-up time and overall survival (OS) time were 20.6 and 26.3 months respectively; 69 patients (53.5%) succumbed during this period, 46 patients (35.7%) survived and 14 patients (10.9%) were lost to follow-up.
Table I.
Patient characteristics of 129 patients with non-small cell lung cancer.
| Variable | Value |
|---|---|
| Gender, n (%) | |
| Male | 96 (74.4) |
| Female | 33 (25.6) |
| Age, years | |
| Mean | 61.2 |
| Range | 20–80 |
| Smoking status, n (%) | |
| No | 50 (38.8) |
| Yes | 79 (61.2) |
| Histological status, n (%)a | |
| AC | 54 (41.9) |
| SCC | 52 (40.3) |
| ASC | 15 (11.6) |
| Other | 8 (6.2) |
| EGFR mutation, n (%) | |
| No | 115 (89.1) |
| Yes | 14 (10.9) |
| Stage, n (%)b | |
| I | 23 (17.9) |
| II | 27 (20.9) |
| III | 63 (48.8) |
| IV | 16 (12.4) |
| Follow-up status, n (%) | |
| Survival | 46 (35.7) |
| Mortality | 69 (53.5) |
| Lost to follow-up | 14 (10.9) |
World Health Organization
American Joint Committee on Cancer. AC, adenocarcinoma; SCC, squamous cell carcinoma; ASC, adenosquamous carcinoma; Other, large cell carcinoma and anaplastic carcinoma; EGFR, epidermal growth factor receptor.
Tissue genomic DNA isolation
Total DNA was extracted from formalin-fixed, paraffin-embedded tissues using the E.Z.N.A.® FFPE DNA kit (Omega Bio-Tek, Inc., Norcross, GA, USA), according to the manufacturer's protocol. The isolated DNA was eluted in 100 µl Tris-EDTA buffer included in the E.Z.N.A.® FFPE DNA kit and stored at −20°C until required. The quality of the isolated DNA was assessed by NanoDrop spectrophotometer (NanoDrop Technologies, Thermo Fisher Scientific, Inc., Wilmington, DE, USA). The optical density values of all samples ranged from 1.8 to 2.0.
Primers for ARMS-qPCR
The forward primers for ARMS-qPCR analysis of the mtDNA A10398G mutation are listed in Table II. The primers were designed by Primer Premier 6.0 (Premier Biosoft International, Palo Alto, CA, USA) and generated by Sangon Biotech Co., Ltd. (Shanghai, China). The numbers in Table I correspond to the nucleotide positions in GenBank (accession no. NC012920). Two mismatches were introduced at the penultimate and antepenultimate nucleotide positions of the mutation site in the forward primers (Table II) to increase the specificity of the ARMS reaction, as described by Newton et al (15). At the mutation site, the wild-type primer containing an A would perfectly match the wild-type target sequence, but would be a weak AC mismatch with the mutant target sequence. Similarly, the mutant primer containing a G would be a perfect match with the mutant target sequence, but would be a weak GT mismatch with the wild-type target sequence. Therefore, two clear TT and CC mismatches at the two nucleotides immediately 5′ to the mutation site were introduced, which were hypothesized to increase the primer specificity (15). The reverse primer was at nucleotide positions 10753–10776 for the amplification of the mtDNA A10398G mutation. However, the wild-type primer produced two PCR products of different sizes, indicating that the specificity of the wild-type primer was not sufficient. Therefore, two and one nucleotides were added to the forward and reverse primers, respectively, at each 5 terminal (Table II), which improved the specificity, since only one PCR product was generated upon addition of the above nucleotides. PCR products amplified by the two pairs of primers were identified by agarose gel electrophoresis and sequencing, as follows.
Table II.
Primers used in amplification refractory mutation system-quantitative polymerase chain reaction.
| Primer | Sequence, 5′-3′ |
|---|---|
| Forward (nucleotide position 10373–10398) | |
| Wild-type sequence | GT GACTACAAAAAGGATTAGACT g a A |
| Wild-type 10398A primer | GT GACTACAAAAAGGATTAGACT c t A |
| Mutated 10398G primer | GACTACAAAAAGGATTAGACT c t G |
| Reverse (nucleotide position 10753–10777) | |
| Wild-type sequence | TACTCCAATGCTAAAACTAATCGT C |
| Antisense strand sequence | G ACGATTAGTTTTAGCATTGGAGTA |
| Wild-type 10398A primer | G ACGATTAGTTTTAGCATTGGAGTA |
| Mutated 10398G primer | ACGATTAGTTTTAGCATTGGAGTA |
Underlined lowercase bases indicate the mismatches.
ARMS-qPCR
qPCR was performed using a 20-µl PCR reaction mixture, containing 100 ng DNA template (2 µl), 10 µl QuantiTect SYBR-Green PCR Master Mix (Takara Bio, Inc., Otsu, Japan), 200 nM of each primer (Table II), 0.4 µl ROX Reference Dye II (Takara Bio, Inc.) and 7.2 µl ddH2O, according to the manufacture's protocol. As reference gene, s a single copy region of the nuclear gene β2 microglobulin (β2M), was used, which was amplified with the following primers: β2M forward, 5′-GCTGGGTAGCTCTAAACAATGTATTCA-3′ and reverse, 5′-CCATGTACTAACAAATGTCTAAAATGGT-3′ (93 bp product). The PCR cycling conditions were as follows: 2 min at 50°C and 10 min at 95°C; followed by 40 cycles of denaturation for 15 sec at 95°C and annealing/extension for 60 sec at 60°C. The fluorescent signal intensities were recorded and analyzed using an ABI Prism 7300 Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Dissociation curves were generated following each run to confirm that the increased fluorescence intensities were not associated with primer-dimer formation. The quantification cycle (Cq) values (17) within the linear exponential increase phase were used to construct the calibration curve and to measure the original DNA template content, as well as to calculate the mutant heteroplasmy of each sample.
Preparation of standard curves
The fragments of wild-type and mutant target sequences were amplified by the specific primers presented in Table II. Subsequently, the products were analyzed by electrophoresis on a 2% agarose gel. Bands of interest were cut from the gel and purified using a commercial kit (E.Z.N.A Gel Extraction kit; Omega Bio-Tek, Inc.). Plasmids were constructed by cloning the purified PCR products into the pGEM®-T Easy Vector (Promega Corporation, Madison, WI, USA), according to the manufacturer's protocol, and subsequently transformed into competent JM109 cells (Beijing Transgen Biotech Co., Ltd., Beijing, China) by the heat shock method. The clones were identified by digesting the plasmid DNA with NotI (Fermentas; Thermo Fisher Scientific, Inc.; Pittsburgh, PA, USA). The positive plasmids were sequenced and their nucleotide sequences were analyzed using Lasergene version 7.1 software (DNASTAR, Inc., Madison, WI, USA). The copy numbers were calculated based on the size and molecular weight of the plasmid DNA. Five 10-fold serial dilutions of plasmid DNA between 105 and 109 molecules were prepared, and ARMS-qPCR reactions were performed to construct the calibration curves for the wild-type and mutant mtDNA 10398 sequences. The slope of the curves was determined and the corresponding qPCR efficiency for the specific amplification was calculated according to the following equation (18): E = 10(−1 / slope) - 1.
Measurement of mutant heteroplasmy
The ARMS-qPCR assay was performed three times for each wild-type and mutant target sequence of each sample by relative quantification to detect the heteroplasmy of mtDNA A10398G. The proportion of the mutant mtDNA that was used to assess the degree of mutant heteroplasmy was calculated from ΔCq (Cqwild-type - Cqmutant), using the formula: Proportion of mutant (%) = 1 / (1+1/2ΔCq) × 100% (18).
Detection of mtDNA copy number and EGFR mutations
Quantification of a unique fragment in the human mitochondrial genome (accession no. NC012920) relative to a single copy region of the nuclear gene β2M was performed using a qPCR assay to assess mtDNA content (19), using reagents purchased from Sangon Biotech Co., Ltd.
PCR-restriction fragment length polymorphism was employed for mutation analysis of EGFR exons 18, 19 and 21 (19), using reagents purchased from Sangon Biotech Co., Ltd. The mutant allele of exons 18 and 21 were not digested by the restriction enzymes ApaI and MscI (New England BioLabs, Inc., Ipswich, MA, USA), respectively. Exon 19 deletion was distinguished by the generation of PCR products of different sizes.
Statistical analysis
Statistical analyses were performed with SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA). The association between heteroplasmy of mtDNA A10398G and survival time was analyzed using the χ2 test. Kaplan-Meier survival curves and the log-rank test were used to analyze OS time. Univariate and multivariate Cox proportional hazard models were used to assess the effects of all clinical variables on patient survival (Table I). The parameters were then tested using the multivariate Cox proportional hazards model, which was performed to identify the independent variables for predicting survival. Two-sided P<0.05 was considered to indicate a statistically significant difference.
Results
Assay specificity, standard amplification curves and amplification efficiency
The ARMS-qPCR primer system, for which the primers were specifically designed, exhibited high specificity and qPCR efficiency. The wild-type primer only amplified the wild-type target sequence, but did not amplify the mutant target sequence. Similarly, the mutant primer only targeted the mutant target sequence, but did not target the wild-type target sequence (Fig. 1).
Figure 1.

Analysis of primer specificity. (A) Gel electrophoresis of the PCR products. Lane M, 50 bp DNA ladder; lane 1, negative controls of wild-type primers (ddH2O instead of DNA template was added to the PCR); lane 2, negative control of mutation patterns (ddH2O instead of DNA template was added to the PCR); lanes 3 and 4, plasmid with wild-type sequence amplified using wild-type (lane 3) and mutant (lane 4) primers; lanes 5 and 6, plasmid with mutant and wild-type sequences amplified using wild-type (lane 5) and mutant (lane 6) primers; lanes 7 and 8, plasmid with mutant sequence amplified using wild-type (lane 7) and mutant (lane 8) primers. (B and C) Reverse sequence (detecting the sequence of the complementary strand) results of PCR products extended using two pairs of primers: (B) Wild-type (TA) and (C) mutant (CG). Results were unimodal, which indicated high specificity of the primers. Subsequently, these primers were used to amplify reconstructed plasmids. PCR, polymerase chain reaction; dd, double distilled.
Calibration curves were generated from duplicate samples with high linearity. Quantitative equations were as follows: Wild-type, y = −3.3517x + 28.52 (R2=0.9964); and mutant, y = −3.4723x + 28.676 (R2=0.9977) where × and y represent the Cq value of qPCR and the logarithmic value of the DNA copy numbers, respectively. The corresponding amplification efficiencies that were calculated from the slopes of two equations, Ewild-type=98.77% and Emutant=94%, were in the optimum range, since both efficiency values were >90% and the difference between them was <5% (Fig. 2).
Figure 2.

Amplification plots obtained using amplification refractory mutation system-quantitative polymerase chain reaction for the respective (A) wild-type and (B) mutant primer systems. Fluorescence is presented as fluorescence intensity over background. Standard calibration curves were generated by plotting the Cq values versus the logarithm of diluted standard for (C) wild-type and (D) mutant DNA copy number. Amplification efficiency was calculated from the slope of the curve. Rn, normalized reporter.
Prognostic significance of mtDNA A10398G heteroplasmy
The mean Cq values for wild-type and mutant sequences in NSCLC tissues ranged between 22.11 and 35.71, and between 22.10 and 36.06, respectively. The proportion of mutant heteroplasmy in the NSCLC tissue samples ranged between 0.31 and 97.04%. The mtDNA content of the NSCLC tissue samples ranged between 23.8 and 1,833.0 copy numbers (data not shown).
There are few studies published at present regarding heteroplasmic mutations, and none regarding mtDNA 0398 heteroplasmic mutation. In the present study, it was observed that heteroplasmic mutations were not normally distributed. therefore, they were divided into low proportion (≤ median) and high proportion (> median) on the basis of their median mtDNA content, as previously analyzed (20). A low proportion of mutant mtDNA 10398G was observed marginally less often in adenocarcinoma compared with other histological types of NSCLC tissue (32.8 vs. 67.2%, respectively; χ2=3.567; P=0.050). There was no association between proportion of mutant mtDNA 10398G, and gender, age, smoking status, EGFR mutation status or tumor stage (P>0.05; Table III). Subsequently, mutant mtDNA 10398G heteroplasmy was subjected to survival analysis. Patients with a low percentage of mutant mtDNA 10398G heteroplasmy had a significantly shorter survival time compared with patients with a high proportion of mutant mtDNA 10398G heteroplasmy (median survival time 22.5 vs. 28.7 months, respectively; χ2=5.656; P=0.017; Fig. 3). All the analyzed clinical parameters (gender, age, smoking status, histology, EGFR mutation status and tumor stage) and mtDNA (mutant mtDNA A10398G heteroplasmy and mtDNA content) were entered into a Cox proportional hazards regression analysis. The univariate analysis revealed that the EGFR mutation status (P<0.001), stage of tumor (P=0.044) and mutant mtDNA A10398G heteroplasmy (P=0.038) were significantly associated with prognosis. The prognosis exhibited no significant difference for age (P=0.354), gender (P=0.405), smoking status (P=0.319), tumor histology (P=0.463) or mtDNA content (P=0.764). In the multivariate statistical analysis, the Cox regression analysis demonstrated that EGFR mutation status and possessing a low degree of mutant mtDNA A10398G were two independent prognostic factors in patients with NSCLC (χ2=24.595, P<0.001 and χ2= 0.557, P=0.022, respectively) (Table IV). Forward: Wald, P=0.05, entry and P=0.10, removal (Wald probability and statistical method, forward by stepwise selection of variables).
Table III.
Associations between mutant heteroplasmy proportions of mtDNA 10398 and clinical parameters in 129 patients with non-small cell lung cancer.
| Variable | Low mutant proportion, n (%) | High mutant proportion, n (%) | χ2 | P-value | OR (95% CI) |
|---|---|---|---|---|---|
| Total | 64 (49.6) | 65 (50.4) | |||
| Gender | 0.000 | 1.000 | 1.063 (0.482–2.344) | ||
| Male | 48 (50.0) | 48 (50.0) | |||
| Female | 16 (48.5) | 17 (51.5) | |||
| Age, years | 0.916 | 0.321 | 0.542 (0.199–1.481) | ||
| <50 | 7 (36.8) | 12 (63.2) | |||
| ≥50 | 57 (51.8) | 53 (48.2) | |||
| Smoking status | 0.063 | 0.720 | 1.169 (0.575–2.375) | ||
| No | 26 (52.0) | 24 (48.0) | |||
| Yes | 38 (48.1) | 41 (51.9) | |||
| Histology | 3.567 | 0.050 | 0.474 (0.232–0.966) | ||
| AC | 21 (38.9) | 33 (61.1) | |||
| Other | 43 (57.3) | 32 (42.7) | |||
| EGFR mutation | 0.064 | 0.778 | 1.357 (0.443–4.158) | ||
| No | 58 (50.4) | 57 (49.6) | |||
| Yes | 6 (42.9) | 8 (57.1) | |||
| Stage | 0.012 | 0.857 | 0.900 (0.443–1.828) | ||
| I + II | 24 (48.0) | 26 (52.0) | |||
| III + IV | 40 (50.6) | 39 (49.4) | |||
χ2 tests were two-sided. Staging was assessed according to the American Joint Committee on Cancer. mtDNA, mitochondrial DNA; AC, adenocarcinoma; EGFR, epidermal growth factor receptor; OR, odds ratio; CI, confidence interval.
Figure 3.
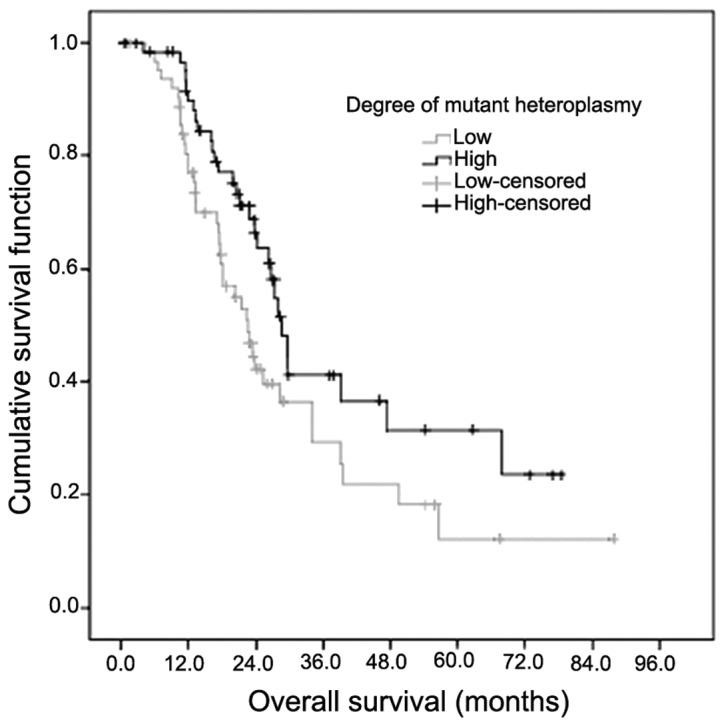
Survival analysis for mutant heteroplasmy. Overall survival time in patients with non-small cell lung cancer was significantly decreased in patients with a low mutant load of mitochondrial DNA (mtDNA) 10398 compared with patients with a high mutant load of mtDNA 10398. Median survival time: 22.5 vs. 28.7 months; χ2=5.656; P=0.017 (Kaplan-Meier analysis and log-rank test).
Table IV.
Multivariate analysis of overall survival.
| Variables | Wald | P-value | HR | 95% CI |
|---|---|---|---|---|
| EGFR mutation | 24.595 | <0.001 | 5.662 | (2.854, 11.235) |
| Heteroplasmy | 5.257 | 0.022 | 0.557 | (0.338, 0.919) |
Cox proportional hazards regression model was used. HR, hazard ratio; CI, confidence interval; EGFR, epidermal growth factor receptor.
Discussion
Mitochondria are semi-autonomous cytoplasmic organelles that are involved in the production of energy for cell metabolism. Each mitochondria contains 2–10 copies of its genome. Double-stranded circular mtDNA is unique genetic material located outside of the cell nucleus and has a higher mutation rate compared with nuclear DNA (6). mtDNA is key in maintaining organelles that are functionally competent and an accumulation of somatic mtDNA mutations may affect energy production; therefore, it is crucial in cancer pathogenesis and progression (21).
mtDNA mutations have been increasingly observed to be potentially involved in cancer. mtDNA 8701 and 10398, which code for ATPase6 and NADH dehydrogenase 3 respectively, were reported to have the highest mutation frequencies in the mitochondrial genome in lung cancer. The A-to-G point mutation of mtDNA 10398 (A10398G) leads to the conversion of Ala to Thr, resulting in an alteration in the structure of complex I in the mitochondrial electron transport chain, which is an important site of free radical production. This polymorphism is associated with several neurodegenerative disorders. For example, a previous study demonstrated that African-American female patients with the wild-type mtDNA 10398A allele had a significantly increased risk of invasive breast cancer (20). In addition, the wild-type mtDNA 10398A variant in African American women with breast cancer provides resistance to cell apoptosis and promotes metastasis in mice (22). In NSCLC, patients with a high mtDNA content in combination with the mutant mtDNA 10398G allele had a significantly increased overall survival time compared with those that had a low mtDNA content and the wild-type mtDNA 10398A allele (19).
Presently, few studies have reported the heteroplasmy of wild-type mtDNA 10398A. However, the potential involvement of mutant heteroplasmy and tumor biological behavior has been indicated by studies at other positions; for example, the proportion of mutant mtDNA 3243 in patients with mitochondrial disease was detected at between 0.02 and 100% (5). However, the proportion of mutant mitochondria must reach the threshold to cause a clinical phenotype. Depending on the proportion of mutant heteroplasmy and the affected tissue, patients with the mtDNA A3243G mutation may be completely unaffected, or present with a broad spectrum of diseases, including diabetes, deafness or a MELAS phenotype (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes) (23). In addition, mutation copy number determined using ARMS-qPCR was associated with the degree of deafness in a small number of patients with heteroplasmic mutations of mtDNA A1555G (14). Another study reported five family pedigrees with multiple members who had the mtDNA A1555G mutation, and exhibited diverse clinical manifestations and various heteroplasmy levels; the risk of deafness rose with the increase in heteroplasmy level (24). Warowicka et al (25) identified that mtDNA copy numbers with a 4.977 bp deletion were significantly different at various stages of carcinogenesis in control, low-grade squamous intraepithelial lesions (L-SIL), high grade-SIL (H-SIL) and cervical cancer samples. In L-SIL cells, only one third of the mtDNA copies did not contain a deletion, while in H-SIL and cervical cancer cells the proportion that contained a deletion reduced to almost half (24). However, to the best of our knowledge, the association between the mtDNA G10398A polymorphism and NSCLC prognosis has not been reported. In the present study, the overall survival time increased by 27.6% and the mortality risk decreased in NSCLC patients with a high proportion of mutant mtDNA 1039 compared with a low proportion of mutant mtDNA 10398G (median survival time, 28.7 vs. 22.5 months, respectively; χ2=5.656; P=0.017).
The present study confirmed that the ARMS-qPCR assay is a rapid, sensitive, reliable and cost-effective one-step qPCR method for the quantification of mutant mtDNA heteroplasmy. This method may be used for the quantification of heteroplasmy for any mtDNA point mutation (5,23). However, it is challenging to identify the optimal allele-specific primers and mode for qPCR to ensure correct hybridization of allele-specific primers (26). The present study designed several pairs of primers, as reported in previous studies (14,15,18), and each amplified PCR product was identified by sequencing and electrophoresis to ensure allele specificity. Subsequently, the primers with strict specificity were selected for use inARMS-qPCR. The mtDNA 10400 was included in the sequence amplified by the specific primers with presence of mutation, which caused the difference in the site located two bases prior to mtDNA 10398 (T and C, Fig. 1B and C). However, the site was located downstream of mtDNA 10398, not in the primer binding region, and therefore, did not influence the study of mtDNA 10398 mutation heteroplasmy.
In conclusion, the present study investigated the association between the heteroplasmic mtDNA A10398G mutation and the prognosis of patients with NSCLC. There was no association between the proportion of mutant mtDNA, and gender, smoking status, tumor stage, EGFR mutation status or mtDNA content. Furthermore, the present study confirmed that a low proportion of mutant mtDNA 10398G may be a marker of poor prognosis in patients with NSCLC. However, the mtDNA A10398G mutation and the accumulation of this mutation may result in mitochondrial dysfunction, sequentially affecting biological behaviors and the sensitivity to anticancer treatment, leading to an alteration in the prognosis of patients. The small size of the patient cohort in the present study was also a limitation of the study. Therefore, additional studies are required to verify the prognostic value of mtDNA A10398G heteroplasmy in various types of cancer, and identify the threshold value that causes a clinical phenotype.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (Beijing, China; grant no. 81472798).
References
- 1.Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanchez-Aragó M, Chamorro M, Cuezva JM. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis. 2010;31:567–576. doi: 10.1093/carcin/bgq012. [DOI] [PubMed] [Google Scholar]
- 3.Johns DR. Seminars in medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA and disease. N Engl J Med. 1995;333:638–644. doi: 10.1056/NEJM199509073331007. [DOI] [PubMed] [Google Scholar]
- 4.Schon EA. Mitochondrial genetics and disease. Trends Biochem Sci. 2000;25:555–560. doi: 10.1016/S0968-0004(00)01688-1. [DOI] [PubMed] [Google Scholar]
- 5.Bai RK, Wong LJ. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: A single-step approach. Clin Chem. 2004;50:996–1001. doi: 10.1373/clinchem.2004.031153. [DOI] [PubMed] [Google Scholar]
- 6.Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: Similar pathways? Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Rozwodowska M, Drewa G, Zbytniewski Z, Woźniak A, Krzyzyńska-Malinowska E, Maciak R. Mitochondrial diseases. Med Sci Monit. 2000;6:817–822. [PubMed] [Google Scholar]
- 8.Wallace DC. The mitochondrial genome in human adaptive radiation and disease: On the road to therapeutics and performance enhancement. Gene. 2005;354:169–180. doi: 10.1016/j.gene.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- 10.Lin CS, Wang LS, Tsai CM, Wei YH. Low copy number and low oxidative damage of mitochondrial DNA are associated with tumor progression in lung cancer tissues after neoadjuvant chemotherapy. Interact Cardiovasc Thorac Surg. 2008;7:954–958. doi: 10.1510/icvts.2008.177006. [DOI] [PubMed] [Google Scholar]
- 11.Choi SJ, Kim SH, Kang HY, Lee J, Bhak JH, Sohn I, Jung SH, Choi YS, Kim HK, Han J, et al. Mutational hotspots in the mitochondrial genome of lung cancer. Biochem Biophys Res Commun. 2011;407:23–27. doi: 10.1016/j.bbrc.2011.02.078. [DOI] [PubMed] [Google Scholar]
- 12.Pezzotti A, Kraft P, Hankinson SE, Hunter DJ, Buring J, Cox DG. The mitochondrial A10398G polymorphism, interaction with alcohol consumption and breast cancer risk. PLoS One. 2009;4:e5356. doi: 10.1371/journal.pone.0005356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin X, Zhang J, Gao Y, Ding K, Wang N, Zhou D, Jen J, Cheng S. Relationship between mitochondrial DNA mutations and clinical characteristics in human lung cancer. Mitochondrion. 2007;7:347–353. doi: 10.1016/j.mito.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Zu-Jian C, Bin Y, Qi-Cai L, Lin J, Jing C, Qi-Shui O. Quantification of mitochondrial DNA with the A1555G mutation in deaf patients using real-time amplification refractory mutation system-quantitative PCR. J Mol Diagn. 2009 Dec 3; doi: 10.2353/jmoldx.2010.090065. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 15.Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, Markham AF. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS) Nucleic Acids Res. 1989;17:2503–2516. doi: 10.1093/nar/17.7.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beasley MB, Brambilla E, Travis WD. The 2004 World Health Organization classification of lung tumors. Semin Roentgenol. 2005;40:90–97. doi: 10.1053/j.ro.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Venegas V, Halberg MC. Quantification of mtDNA mutation heteroplasmy (ARMS qPCR) Methods Mol Biol. 2012;837:313–326. doi: 10.1007/978-1-61779-504-6_21. [DOI] [PubMed] [Google Scholar]
- 19.Kulawiec M, Owens KM, Singh KK. mtDNA G10398A variant in African-American women with breast cancer provides resistance to apoptosis and promotes metastasis in mice. J Hum Genet. 2009;54:647–654. doi: 10.1038/jhg.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canter JA, Kallianpur AR, Parl FF, Millikan RC. Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women. Cancer Res. 2005;65:8028–8033. doi: 10.1158/0008-5472.CAN-05-1428. [DOI] [PubMed] [Google Scholar]
- 21.Lee HC, Chang CM, Chi CW. Somatic mutations of mitochondrial DNA in aging and cancer progression. Ageing Res Rev. 2010;9(Suppl 1):S47–S58. doi: 10.1016/j.arr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Xu H, He W, Jiang HG, Zhao H, Peng XH, Wei YH, Wei JN, Xie CH, Liang C, Zhong YH, et al. Prognostic value of mitochondrial DNA content and G10398A polymorphism in non-small cell lung cancer. Oncol Rep. 2013;30:3006–3012. doi: 10.3892/or.2013.2783. [DOI] [PubMed] [Google Scholar]
- 23.Wong LJ, Bai RK. Real-time quantitative polymerase chain reaction analysis of mitochondrial DNA point mutation. Methods Mol Biol. 2006;335:187–200. doi: 10.1385/1-59745-069-3:187. [DOI] [PubMed] [Google Scholar]
- 24.Zhu Y, Huang S, Kang D, Han M, Wang G, Yuan Y, Su Y, Yuan H, Zhai S, Dai P. Analysis of the heteroplasmy level and transmitted features in hearing-loss pedigrees with mitochondrial 12S rRNA A1555G mutation. BMC Genet. 2014;15:26. doi: 10.1186/1471-2156-15-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warowicka A, Kwasniewska A, Gozdzicka-Jozefiak A. Alterations in mtDNA: A qualitative and quantitative study associated with cervical cancer development. Gynecol Oncol. 2013;129:193–198. doi: 10.1016/j.ygyno.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Sobenin IA, Mitrofanov KY, Zhelankin AV, Sazonova MA, Postnov AY, Revin VV, Bobryshev YV, Orekhov AN. Quantitative assessment of heteroplasmy of mitochondrial genome: Perspectives in diagnostics and methodological pitfalls. Biomed Res Int. 2014;2014:292017. doi: 10.1155/2014/292017. [DOI] [PMC free article] [PubMed] [Google Scholar]