

Abstract
Cisplatin is commonly recognized as a DNA-damaging drug; however, its versatile antitumor effects have been demonstrated to extend beyond this narrow functional attribute. The present study determined how cisplatin regulates alternative pathways and transcription factors to exert its additional antitumor actions. Cisplatin was observed to be able to trigger an endoplasmic reticulum stress response through aggravated nitrosative stress coupled to perturbed mitochondrial calcium (Ca2+) homeostasis, which substantially downregulated glucose-regulated protein (GRP) 78 expression by suppressing the cleavage of activating transcription factor (ATF) 6α (90 kDa) to its active 50 kDa subunit. Concomitantly, the ATF4-ATF3-C/emopamil binding protein homologous protein axis was activated by cisplatin, which triggered cellular glutathione (GSH) depletion by strongly inhibiting γ-glutamylcysteine synthetase heavy chain (γ-GCSh), a key enzyme in GSH biosynthesis. The present study also demonstrated that cisplatin substantially inhibited β-catenin, causing a marked downregulation of survivin and B-cell lymphoma (Bcl)-2. Taken together, the present results uncovered a novel mechanism of cisplatin that could simultaneously trigger the inhibition of three prominent antiapoptotic effector molecules (Bcl-2, survivin and GRP78) and effectively promote GSH depletion by inhibiting γ-GCSh. These newly discovered functional attributes of cisplatin can provide an avenue for novel combined therapeutic strategies to kill hepatocellular carcinoma cells effectively.
Keywords: cisplatin, GSH depletion, ER stress, GRP78, survivin, Bcl-2
Introduction
Among men, hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide and the second leading cause of cancer-associated mortalities annually (1). Current curative treatments, including surgical resection and liver transplantation, are generally ineffective, and can be applied only during early-stage HCC (2,3). In addition, only 10–15% of patients are qualified to be treated with curative surgery (2,3). Thus, the majority of HCC patients must be considered for systemic chemotherapies or supportive therapies (3). However, the majority of chemotherapeutic agents exhibit poor effectiveness and can only marginally improve patient survival rates (3).
Cisplatin is one of the most widely used chemotherapeutic agents for treating various types of malignancies (4). Cisplatin is primarily considered as a DNA-damaging agent that forms various types of bifunctional adducts upon reacting with cellular DNA (4). The final cellular outcome of DNA adduct formation is generally apoptotic cell death, which is considered to occur through the disruption of cellular processes such as the deregulation of signal transduction pathways involved in growth, differentiation and stress responses (5). Although DNA damage is widely considered to be responsible for cisplatin-induced apoptosis, the molecular mechanisms that establish the formation of DNA adducts in response to cisplatin-induced apoptosis have not been identified conclusively (6,7). This research gap raises the question of whether alternative and/or additional mechanisms are involved. Accumulating evidence has revealed that cisplatin-induced apoptosis could occur independently of DNA damage through oxidative stress in various cell types (8–10). Therefore, alternative pathways affected by cisplatin should be further explored, since they could yield critical insights for improving the current cisplatin-based regimens. Despite the fact that the current postulated mechanisms remain controversial, there is a leading theory that involves the ability of cisplatin to inhibit sublethal damage repair in resistant tumor cells (5,6). Thus, further characterization of the possible underlying mechanism(s) associated with the sensitization engendered by cisplatin in HCC cells is justified.
With these premises in mind, the aim of the present study was to identify the molecular signatures of selected HCC cell lines and to provide mechanistic insight into apoptotic pathways alternatively activated by cisplatin to elucidate the fundamental causes of sensitization during endoplasmic reticulum (ER) stress. In the present study, two p53-dysfunctional HCC models, Hep3B and Mahlavu (11), were selected, of which, the former was more differentiated than the latter. It was noticed that Mahlavu cells were distinctly more sensitive to cisplatin than Hep3B cells. Our results also revealed a novel functional attribute of cisplatin involving the robust suppression of two prominent pathways that regulate the expression of B-cell lymphoma (Bcl)-2, survivin and glucose-regulated protein (GRP) 78 independently of DNA damage and repair mechanisms. Thus, these findings provide a crucial missing link in the understanding of the mechanism of cisplatin-induced ER stress. Critically, this novel functional attribute of cisplatin may also provide an avenue for novel therapeutic strategies against chemotherapy-resistant HCC cells.
Materials and methods
Cell culture and cell viability assay
The two HCC cell lines used in the present study, Hep3B cells purchased from Bioresource Collection and Research Center (Hsinchu, Taiwan) and Mahlavu cells provided by Professor K.H. Lin (Chang Gung University, Taiwan), were grown in Dulbecco's modified Eagle's medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum (Biological Industries, Cromwell, CT, USA) in 5% CO2, in a humidified incubator (37°C). The cells were treated with cisplatin (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) at various concentrations for 24 or 72 h, and cell viability was measured using a sulforhodamine B (SRB) assay, as previously described (12). Briefly, cells were fixed using trichloroacetic acid and then incubated with SRB (Sigma-Aldrich; Merck Millipore) upon being washed and air-dried. The precipitate was dissolved in Trizma® base solution (Sigma-Aldrich; Merck Millipore). The absorbance was measured at 540 nm by using a microplate reader.
Western blot analysis
A total of 5×105 cells were seeded for 24 h and treated with cisplatin for different periods. Cells were lysed in radioimmunoprecipitation assay buffer (Sigma-Aldrich; Merck Millipore), and protein concentrations were assayed using the Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). In total, 40 µg of proteins was separated on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred electrophoretically to a polyvinylidene difluoride membrane. Upon blocking, the membrane was hybridized at 4°C overnight with one of the following primary antibodies: Anti-GRP78 (catalog no. sc-13968; dilution, 1:500), anti-activating transcription factor (ATF) 3 (catalog no. sc-188; dilution, 1:500), anti-ATF4 (catalog no. sc-200; dilution, 1:500), anti-ATF6 (50 kDa; catalog no. sc-22799; dilution, 1:500), anti-C/emopamil binding protein homologous protein (CHOP; (catalog no. sc-7351; dilution, 1:500), anti-γ-glutamylcysteine synthetase heavy chain (γ-GCSh; catalog no. sc-100747; dilution, 1:500), anti-Bcl-2 (ctalog no. sc-7382; dilution, 1:500), anti-survivin (catalog no. sc-10811; dilution, 1:500) (all Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-β-catenin (catalog no. 06-734; dilution, 1:2,500; Upstate Biotechnology, Inc., Lake Placid, NY, USA), anti-poly(adenosine diphosphate-ribose) polymerase 1 (PARP-1; 1074-1; dilution, 1:1,000; Epitomics, Burlingame, CA, USA) and anti-β-actin (catalog no. A5441; dilution, 1:10,000; Sigma-Aldrich; Merck Millipore) antibodies, followed by incubation with secondary antibodies conjugated to horseradish peroxidase. The antigen-antibody complexes were detected using a Pierce enhanced chemiluminescence detection system (Thermo Fisher Scientific, Inc.).
Flow cytometric measurement of intracellular nitric oxide (NO) and cellular glutathione (GSH) depletion
All fluorescence reagents were purchased from Sigma-Aldrich (Merck Millipore), unless otherwise specified. Intracellular NO production was measured using 4,5-diaminofluorescein (DAF-2), as described previously (13). Briefly, cells were treated with cisplatin, washed with phosphate-buffered saline (PBS) and then incubated with 1 µM DAF-2 for 10 min in the dark. The cells were then washed with PBS, detached by trypsinization, collected by centrifugation at 0.2 × g at room temperature and resuspended in PBS. To measure cellular GSH depletion, cisplatin-treated cells were incubated with 25 µM 5-chloromethylfluorescein (CMF) diacetate for 20 min at 5% CO2 in a 37°C incubator. The CMF fluorescence intensity was measured using a BD FACSCalibur™ flow cytometer (version 3.3; BD Biosciences, San Jose, CA, USA) and analyzed using BD CellQuest Pro software (BD Biosciences).
Detecting intracellular NO using confocal microscopy
To measure the production of intracellular NO, cisplatin- and sham-treated HCC cells were cultured in poly-L-lysine-coated slides. All fluorescence reagents were purchased from Sigma-Aldrich (Merck Millipore). Upon reaching 80% cell density, the cells were incubated with 2 µM DAF-2′,7′-difluorofluorescein diacetate in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid for 30 min in the dark and imaged using a Leica TCS SP2 laser scanning confocal microscope (Leica Microsystems GmbH, Wetzlar, Germany).
Confocal microscopic detection of mitochondrial and cytosolic calcium (Ca2+)
Cells were stained with 1.5 µM Rhod-2 and 2 mM Fluo-4 (Invitrogen; Thermo Fisher Scientific, Inc.) to detect mitochondrial and cytosolic Ca2+, respectively. Fluorescent images of Rhod-2 and Fluo-4 were obtained prior and subsequent to treating the cells with cisplatin. At the end of each experimental period, cells were mounted on the stage of a Leica TCS SP2 laser scanning confocal microscope. Then, cells were randomly selected and imaged.
Terminal deoxynucleotidyl transferase 2′-deoxyuridine 5′-triphosphate nick end labeling (TUNEL) assay
DNA fragmentation was detected using a TUNEL assay and an Apo-BrdU In Situ DNA Fragmentation Assay kit (BioVision, Inc., Milpitas, CA, USA), according to the manufacturer's protocol. A BD FACSCalibur™ was used to perform the analyses. DNA content was quantified using ModFit LT™ software (version 3.0; Verity Software House, Inc., Topsham, ME, USA).
Statistical analysis
All data are presented as the mean ± standard deviation from ≥3 independent experiments, and were analyzed using the Student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
HCC cells exhibit distinct disparity in response to chemotherapeutic drugs
Two chemotherapeutic drugs, cisplatin and doxorubicin, were selected to test their cytotoxicity effect on two HCC cell lines, Mahlavu (a poorly differentiated and highly Bcl-2-expressing cell line with a p53 mutation at codon 249) and Hep3B (a well-differentiated and low Bcl-2-expressing subline formed by p53-null cells) (13–15). As indicated in Fig. 1A, upon treatment with cisplatin, cell viability decreased to 5% in the Mahlavu cells, whereas 28% of the Hep3B cells remained viable. However, there was no significant difference between the Mahlavu and Hep3B cells upon treatment with doxorubicin (P=0.086). By contrast, the Mahlavu cells were observed to be distinctly more sensitive to cisplatin than Hep3B cells. Next, cisplatin-induced apoptosis was investigated by a TUNEL assay. Cisplatin induced cell apoptotic death in a concentration-dependent manner in the Mahlavu cells (Fig. 1B), and it also induced the cleavage of PARP-1 (a 116-kDa nuclear enzyme downstream of caspase 3) to produce an 85-kDa fragment (Fig. 1C). These data clearly demonstrated that the cell death associated with cisplatin treatment was apoptotic in nature.
Figure 1.

Distinct disparity of sensitivity of hepatocellular carcinoma cells in response to chemotherapeutic drugs. (A) Mahlavu and Hep3B cells were treated with cisplatin or doxorubicin for 72 h. Cell viability was determined using a sulforhodamine B assay. The results are expressed as the mean ± standard deviation. Each point represents an average of ≥3 determinations. ***P<0.05 vs. control. (B) Evaluation of cisplatin-induced apoptosis in Mahlavu cells. Control (untreated) cells and cells treated with 5, 10 or 25 µg/ml cisplatin for 24 h were subjected to terminal deoxynucleotidyl transferase 2′-deoxyuridine 5′-triphosphate assay. The representative plots depict DNA content on the X axis and 5-bromo-2′-deoxyuridine-fluorescein isothiocyanate-labeled apoptotic DNA strand breaks on the Y axis. The data represent the percentage of apoptotic cells in the upper boxes. (C) The cleavage of PARP-1 (a 116-kDa nuclear enzyme downstream of caspase 3) into an 85-kDa fragment by cisplatin-treated cells provides further evidence of the occurrence of apoptosis through the activation of caspase 3. UN, untreated; PARP-1, poly(adenosine diphosphate-ribose) polymerase 1.
Cisplatin provokes NO production and perturbs Ca2+ homeostasis
Accumulating evidence indicates that cisplatin-induced apoptosis could occur independently of DNA damage through oxidative stress in various cell types (5–7). Therefore, it was examined whether cisplatin regulates ER stress through aggravated nitrosative stress coupled to perturbed mitochondrial Ca2+ homeostasis. As represented in Fig. 2A, when Mahlavu cells were treated with cisplatin, a stimulated overproduction of NO was observed, according to the data obtained using flow cytometry. Furthermore, cisplatin triggered an increased influx of cytosolic Ca2+ into the mitochondria and caused a Ca2+ overload in these organelles, as indicated by the red fluorescence of Rhod-2 (Fig. 2B). Thus, cisplatin was demonstrated to induce ER stress response through aggravated nitrosative stress coupled to perturbed mitochondrial Ca2+ homeostasis.
Figure 2.
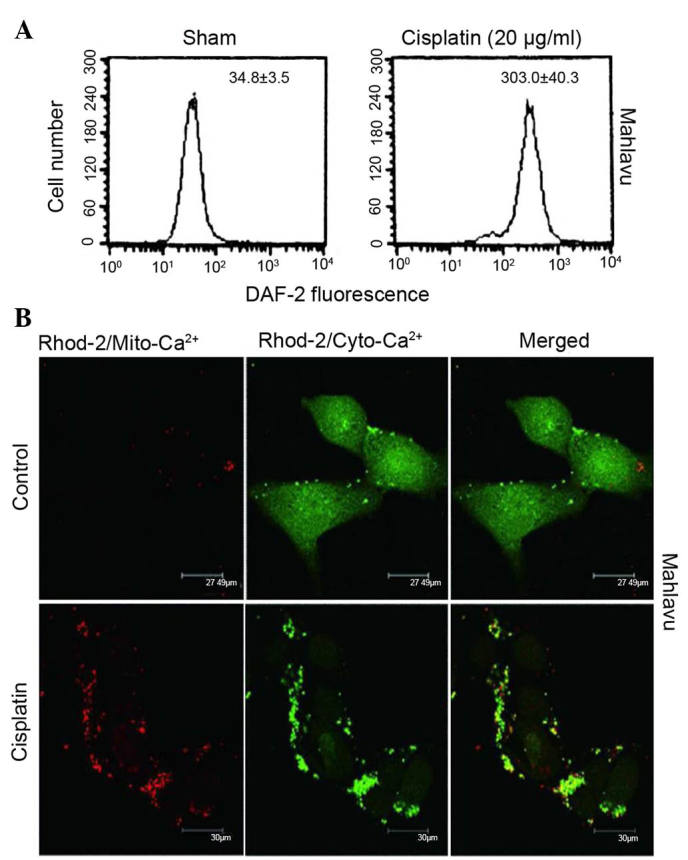
Cisplatin could provoke increased production of NO and perturbed Ca2+ homeostasis. (A) A nearly 10-fold increase in NO production was observed when Mahlavu cells were treated with 20 µg/ml cisplatin, as demonstrated by flow cytometry using DAF-2 as a probe. (B) At resting state, Mahlavu cells exhibited relatively low levels of mitochondrial Ca2+ (Rhod-2/sham) (×400 magnification). Upon cisplatin treatment (25 µg/ml), significantly increased Rhod-2 fluorescence (mito-Ca2+) (red color) was detected, accompanied by a concomitantly reduced Fluo-4 fluorescence (cyto-Ca2+) (green color), indicating that mitochondrial Ca2+ overload had occurred. DAF-2, 4,5-diaminofluorescein; NO, nitric oxide; Ca2+, calcium; Mito, mitochondrial; Cyto, cytosolic.
Cisplatin-induced ER stress response activates the ATF4- ATF3-CHOP axis
The present study demonstrated that cisplatin-induced ER stress response could also regulate the ATF4-ATF3-CHOP axis and its downstream molecules. In Mahlavu cells treated with cisplatin for 24 h, the ATF4-ATF3-CHOP axis was concomitantly activated following treatment (Fig. 3A). The CHOP-mediated downstream molecule GSH may be critical for cell apoptosis (16). In the present study, a concentration-dependent depletion of intracellular GSH was identified using flow cytometry (Fig. 3B). Additionally, the current results revealed the first evidence that CHOP-mediated GSH depletion is at least in part due to the strong inhibition by cisplatin of γ-GCSh, a rate-limiting enzyme responsible for cellular GSH biosynthesis (Fig. 3C) (17).
Figure 3.

(A) Cisplatin could trigger an ER stress response in Mahlavu cells by activating the pro-apoptotic ATF4-ATF3-CHOP pathway. (B) The cisplatin-induced ER stress response concomitantly caused cellular GSH depletion in a concentration-dependent manner. (C) The cisplatin-induced cellular GSH depletion was demonstrated to be due to a strong inhibition of γ-GCSh, an enzyme involved in the regulation of GSH biosynthesis. ER, endoplasmic reticulum; ATF, activating transcription factor; CHOP, C/emopamil binding protein homologous protein; CMF, 5-chloromethylfluorescein; GSH, glutathione; γ-GCSh, γ-glutamylcysteine synthetase heavy chain.
Cisplatin severely downregulates GRP78 via suppressed ATF6α-p50 expression
ER stress can induce transcriptional factor activation; therefore, the present study sought to determine whether cisplatin-induced ER stress regulates the activation of the transcriptional factor ATF6α. At resting state, Mahlavu cells were observed to be a high GRP78-expressing HCC subline, due to its constitutive high levels of active ATF6α-p50. However, upon cisplatin treatment, strong suppression of the active ATF6α-p50 form and downregulation of GRP78 expression was observed (Fig. 4A and B).
Figure 4.

Inhibition of three prominent anti-apoptotic effectors by cisplatin and the underlying mechanisms involved. (A and B) Cisplatin concomitantly inhibited GRP78 and ATF6α (50 kDa) in Mahlavu cells in a (A) concentration- and (B) time-dependent manner. (C and D) Cisplatin-treated Mahlavu cells exhibited suppressed expression of β-catenin and survivin, two major modifying molecules regulating anti-apoptotic signaling pathways, in a (C) concentration-and (D) time-dependent manner. Thus, (A and B) the blockade of ATF6α (50 kDa) expression could suppress GRP78 production, whereas the suppression of β-catenin expression could perturb the Wnt signaling pathway and render (C and D) survivin and (E and F) Bcl-2 downregulation in a (E) concentration- and (F) time-dependent manner. ATF, activating transcription factor; GRP, glucose-regulated protein; Bcl-2, B-cell lymphoma 2.
Cisplatin induces survivin and Bcl-2 downregulation through inhibiting β-catenin
Since the availability of β-catenin and its nuclear translocation is key in controlling the transcription of downstream genes of the Wnt signaling pathway (18,19), the current study investigated whether cisplatin could also downregulate the expression of survivin and Bcl-2 through inhibiting β-catenin. The results revealed that the expression of survivin (Fig. 4C and D) and Bcl-2 (Fig. 4E and F) in the Mahlavu cells was substantially inhibited by cisplatin in a time- and concentration-dependent manner. This phenomenon correlated closely with the inhibition of β-catenin, suggesting that the expression of survivin and Bcl-2 through the Wnt signaling pathway was largely blocked.
Discussion
The present study identified a crucial missing link in the understanding of the mechanism associated with cisplatin-induced ER stress. Two HCC model cell lines were selected, Mahlavu and Hep3B, with different p53 dysfunctions. Generally, HCC lines were subjected to higher doses of cisplatin (25–200 µM) in previous platinum-based drug studies (20,21), according to the literature. For example, Hep3B was observed to be resistant to p53-mediated growth arrest and apoptosis (22), and received a high dose of cisplatin (60 µM for 24 h) in a previous study (20). Notably, when comparing the two HCC cell lines used in the present study, the Mahlavu cells were observed to be considerably more sensitive to cisplatin than the Hep3B cells (Fig. 1). This result may reflect differences in their p53 and molecule expression status (14,15). The cisplatin response has been reported to involve ER stress (8,23,24). The ER is the most critical protein-folding compartment and an intracellular Ca2+ storage organelle in cells (25). Thus, accumulating reactive nitrogen species could modify proteins and disrupt Ca2+ homeostasis, which may trigger the induction of cisplatin-induced ER stress (26,27). Upon cisplatin treatment, intracellular NO levels increased nearly by 10-fold in the Mahlavu cells (Fig. 2A). This increased NO production could have caused ER Ca2+ depletion and subsequently increased the influx of cytosolic Ca2+ into the mitochondria, which could eventually trigger mitochondrial Ca2+overload (Fig. 2B). These findings support the notion that NO-dependent mitochondrial disruption can be coupled to the ER stress response (14,15,20,21). The present study revealed that cisplatin-induced ER stress response simultaneously provoked two prominent pathways that may induce cells apoptosis. The key effectors of ER stress-signaling pathways may modulate cisplatin-induced cell death (27) through GRP78 (24) or β-catenin (28). Therefore, identifying those molecules that can be used to predict cisplatin treatment response would enable therapy to be tailored on an individual HCC patient basis. GRP78 is an ER-resident protein responsible for protein folding and assembly, and its expression level has been correlated with the acquisition of resistance by certain malignancies against diverse anticancer drugs (29,30). Since the upregulation of GRP78 was frequently observed in HCC cells (15), the present study investigated how cisplatin affects the expression of GRP78 in association with the possible nitrosative stress-mediated mechanism in the Mahlavu cell model. The uniqueness of the Mahlavu cells was that they constitutively overexpressed GRP78, likely through a mild, endogenously generated NO-mediated increase in the active p50 form of the transcription factor ATF6 (ATF6α-p50), which, by nuclear translocation, could release GRP78 from its conjugated form (18,26,27,31). In the present study, cisplatin was observed to suppress GRP78 through a mechanism involving the strong inhibition of the expression of ATF6α-p50. In this situation, the ability of the cells to withstand ER stress-induced apoptosis could then be considerably weakened. The present results also revealed that ATF4, ATF3 and CHOP were all highly induced by cisplatin (Fig. 3A). Cisplatin-induced CHOP expression is mediated through the cooperative interaction of ATF4 and ATF3 (Fig. 3A). Since ATF3 has been reported to be induced by a variety of stress-causing agents, including DNA damages (25), it is expected that cisplatin, a chemotherapeutic agent regarded as a typical DNA-damaging agent (Fig. 3A), could induce ATF3 expression. The upregulation of ATF3 has been demonstrated to play a pivotal role in the apoptosis induced by various histone deacetylase inhibitors and certain chemotherapeutic agents such as cisplatin (32,33). Based on these observations, it can be concluded that the suppressed active ATF6α-p50 formed by cisplatin can render GRP78 downregulation. In this situation, the induction of CHOP mediated through the cross-talk of ATF4 and ATF3 can be triggered (31). Consistent with the findings reported in the literature, the present authors previously observed that cisplatin-induced ER stress response and CHOP expression could trigger GSH depletion (31,34). Whether cisplatin-induced CHOP induction can also be triggered by a similar mechanism is currently under investigation by the present authors. Furthermore, the mechanism by which GSH depletion occurs during ER stress-mediated CHOP induction has not previously been addressed. The present study provides the first evidence that CHOP-mediated cellular GSH depletion is primarily due to the robust inhibition by cisplatin of γ-GCSh, a rate-limiting enzyme responsible for the synthesis of cellular GSH (Fig. 2C) (17).
The activation of the Wnt/β-catenin pathway has been reported in HCC cells (18,19). β-catenin, a central effector molecule, works with the T-cell factor family of transcription factors to activate the expression of specific oncogenes, including cyclin D1, c-Myc, vascular endothelial growth factor, Bcl-2 and survivin (35–37). Mechanistically, determining the extent to which cisplatin is involved in modulating the transcriptional activity of the Wnt/β-catenin signaling pathway that could eventually lead to the downregulation of survivin is warranted. Using confocal microscopy imaging, the present study determined that the majority of β-catenin was localized in the nucleus, thus leading to an increased expression of survivin (data not shown). Since β-catenin was overexpressed in Mahlavu cells, the present study observed that, upon cisplatin treatment, the β-catenin expression of the Mahlavu cells was substantially suppressed. This suppression could in turn markedly reduce the nuclear translocation of β-catenin, thus leading to the suppressed oncogenic expression of survivin (Fig. 4C and D) and Bcl-2 (Fig. 4E and F).
In conclusion, the present study provides the first evidence that the cisplatin-triggered apoptosis of HCC Mahlavu cells involves the robust inhibition of three prominent effector molecules, including GRP78, Bcl-2 and survivin, through the dualistic modulation of nitrosative stress-mediated ER stress response via activation of the ATF4-ATF3-CHOP axis and the downregulation of the Wnt/β-catenin signaling pathway (Fig. 5). The current results suggest that cisplatin could be clinically useful in eradicating cancer cells that are resistant to chemotherapeutic agents due to the enhanced expression of GRP78, Bcl-2 or survivin.
Figure 5.
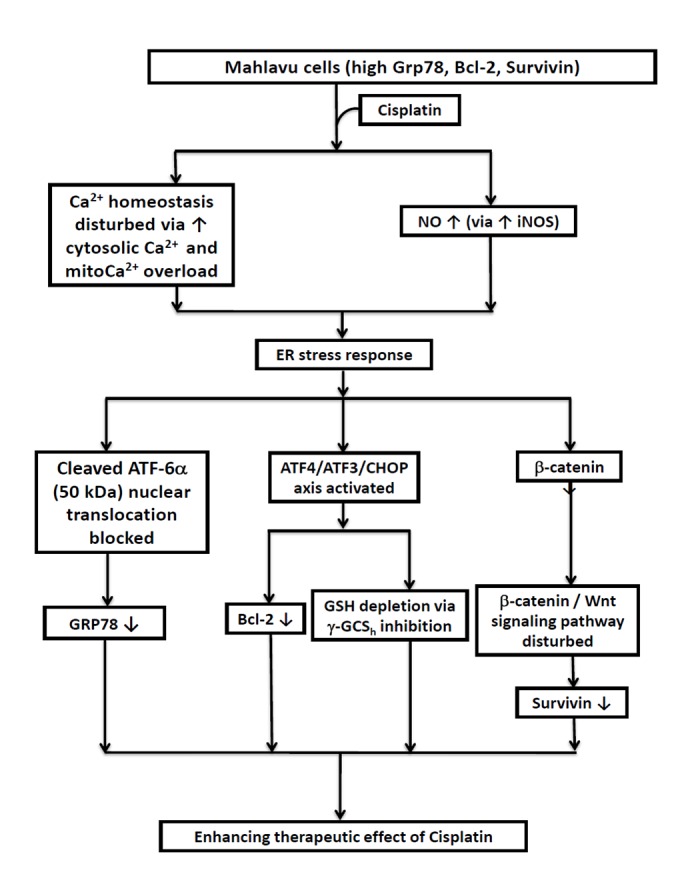
Diagrammatic illustration of the three critical cascades of events that negatively modulate the expression of three prominent cisplatin-associated anti-apoptotic effectors of hepatocellular carcinoma Mahlavu cells. RT, radiotherapy; Ca2+, calcium; NO, nitric oxide; iNOS, inducible nitric oxide synthase; ER, endoplasmic reticulum; ATF, activating transcription factor; CHOP, C/emopamil binding protein homologous protein; CMF, 5-chloromethylfluorescein; GSH, glutathione; γ-GCSh, γ-glutamylcysteine synthetase heavy chain; GRP, glucose-regulated protein; Bcl-2, B-cell lymphoma 2.
Acknowledgements
The present study was supported by grants from the Taiwan National Science Council (Taipei, Taiwan; grant no. NSC 99-2314-B-038-026-MY3), the Ministry of Science and Technology of the R.O.C (Tapei, Taiwan; grant no. MOST 103-2314-B-038-058) and a Chi Mei Medical Center (CM) - Taipei Medical University (TMU) joint grant (Taipei, Taiwan; grant no. 98 CM-TMU 01-02).
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Lim YS, Han S, Heo NY, Shim JH, Lee HC, Suh DJ. Mortality, liver transplantation, and hepatocellular carcinoma among patients with chronic hepatitis B treated with entecavir vs lamivudine. Gastroenterology. 2014;147:152–161. doi: 10.1053/j.gastro.2014.02.033. [DOI] [PubMed] [Google Scholar]
- 3.Tabrizian P, Roayaie S, Schwartz ME. Current management of hepatocellular carcinoma. World J Gastroenterol. 2014;20:10223–10237. doi: 10.3748/wjg.v20.i30.10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 5.Stewart DJ. Mechanisms of resistance to cisplatin and carboplatin. Crit Rev Oncol Hematol. 2007;63:12–31. doi: 10.1016/j.critrevonc.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Vaisman A, Varchenko M, Said I, Chaney SG. Cell cycle changes associated with formation of Pt-DNA adducts in human ovarian carcinoma cells with different cisplatin sensitivity. Cytometry. 1997;27:54–64. doi: 10.1002/(SICI)1097-0320(19970101)27:1<54::AID-CYTO7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 7.Burger H, Nooter K, Boersma AW, Kortland CJ, Stoter G. Lack of correlation between cisplatin-induced apoptosis, p53 status and expression of Bcl-2 family proteins in testicular germ cell tumour cell lines. Int J Cancer. 1997;73:592–599. doi: 10.1002/(SICI)1097-0215(19971114)73:4<592::AID-IJC22>3.3.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 8.Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003;278:9100–9106. doi: 10.1074/jbc.M210284200. [DOI] [PubMed] [Google Scholar]
- 9.Baek SM, Kwon CH, Kim JH, Woo JS, Jung JS, Kim YK. Differential roles of hydrogen peroxide and hydroxyl radical in cisplatin-induced cell death in renal proximal tubular epithelial cells. J Lab Clin Med. 2003;142:178–186. doi: 10.1016/S0022-2143(03)00111-2. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Zhou S, Qui L, Wu J, Xu C. Effects of zinc gluconate on nephrotoxicity and glutathione metabolism disorder induced by cis-platin in mice. Drug Metabol Drug Interact. 1997;14:41–46. doi: 10.1515/DMDI.1997.14.1.41. [DOI] [PubMed] [Google Scholar]
- 11.Kuo TC, Chang PY, Huang SF, Chou CK, Chao CC. Knockdown of HURP inhibits the proliferation of hepacellular carcinoma cells via downregulation of gankyrin and accumulation of p53. Biochem Pharmacol. 2012;83:758–768. doi: 10.1016/j.bcp.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 12.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 13.Chen CY, Liu TZ, Liu YW, Tseng WC, Liu RH, Lu FJ, Lin YS, Kuo SH, Chen CH. 6-shogaol (alkanone from ginger) induces apoptotic cell death of human hepatoma p53 mutant Mahlavu subline via an oxidative stress-mediated caspase-dependent mechanism. J Agric Food Chem. 2007;55:948–954. doi: 10.1021/jf0624594. [DOI] [PubMed] [Google Scholar]
- 14.Matouk IJ, Mezan S, Mizrahi A, Ohana P, Abu-Lail R, Fellig Y, Degroot N, Galun E, Hochberg A. The oncofetal H19 RNA connection: Hypoxia, p53 and cancer. Biochim Biophys Acta. 2010;1803:443–451. doi: 10.1016/j.bbamcr.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 15.Wu CH, Uen YH, Ho CT, Tseng YT, Liu TZ, Chiou JF, Leung SW. Constitutive overexpression of Bcl-2, Survivin and ER stress chaperone GRP-78 confers intrinsic radioresistance in human hepatocellular carcinoma cells: Insight into the mechanistic pathways involved*. J Cancer Ther. 2013;4:399. doi: 10.4236/jct.2013.42A048. [DOI] [Google Scholar]
- 16.Hsu HC, Chiou JF, Wang YH, Chen CH, Mau SY, Ho CT, Change PJ, Liu TZ, Chen CH. Folate deficiency triggers an oxidative-nitrosative stress-mediated apoptotic cell death and impedes insulin biosynthesis in RINm5F pancreatic islet beta-cells: Relevant to the pathogenesis of diabetes. PLoS One. 2013;8:e77931. doi: 10.1371/journal.pone.0077931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffith OW, Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine) J Biol Chem. 1979;254:7558–7560. [PubMed] [Google Scholar]
- 18.Lee HC, Kim M, Wands JR. Wnt/Frizzled signaling in hepatocellular carcinoma. Front Biosci. 2006;11:1901–1915. doi: 10.2741/1933. [DOI] [PubMed] [Google Scholar]
- 19.Takigawa Y, Brown AM. Wnt signaling in liver cancer. Curr Drug Targets. 2008;9:1013–1024. doi: 10.2174/138945008786786127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim Y, Jang M, Lim S, Won H, Yoon KS, Park JH, Kim HJ, Kim BH, Park WS, Ha J, Kim SS. Role of cyclophilin B in tumorigenesis and cisplatin resistance in hepatocellular carcinoma in humans. Hepatology. 2011;54:1661–1678. doi: 10.1002/hep.24539. [DOI] [PubMed] [Google Scholar]
- 21.Lim SC, Choi JE, Kang HS, Han SI. Ursodeoxycholic acid switches oxaliplatin-induced necrosis to apoptosis by inhibiting reactive oxygen species production and activating p53-caspase 8 pathway in HepG2 hepatocellular carcinoma. Int J Cancer. 2010;126:1582–1595. doi: 10.1002/ijc.24853. [DOI] [PubMed] [Google Scholar]
- 22.Friedman SL, Shaulian E, Littlewood T, Resnitzky D, Oren M. Resistance to p53-mediated growth arrest and apoptosis in Hep 3B hepatoma cells. Oncogene. 1997;15:63–70. doi: 10.1038/sj.onc.1201149. [DOI] [PubMed] [Google Scholar]
- 23.Rabik CA, Fishel ML, Holleran JL, Kasza K, Kelley MR, Egorin MJ, Dolan ME. Enhancement of cisplatin [cis-diammine dichloroplatinum (II)] cytotoxicity by O6-benzylguanine involves endoplasmic reticulum stress. J Pharmacol Exp Ther. 2008;327:442–452. doi: 10.1124/jpet.108.141291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmad M, Hahn IF, Chatterjee S. GRP78 up-regulation leads to hypersensitization to cisplatin in A549 lung cancer cells. Anticancer Res. 2014;34:3493–3500. [PubMed] [Google Scholar]
- 25.Xu W, Liu L, Charles IG, Moncada S. Nitric oxide induces coupling of mitochondrial signalling with the endoplasmic reticulum stress response. Nat Cell Biol. 2004;6:1129–1134. doi: 10.1038/ncb1188. [DOI] [PubMed] [Google Scholar]
- 26.Zhao S, Xiong Z, Mao X, Meng D, Lei Q, Li Y, Deng P, Chen M, Tu M, Lu X, et al. Atmospheric pressure room temperature plasma jets facilitate oxidative and nitrative stress and lead to endoplasmic reticulum stress dependent apoptosis in HepG2 cells. PLoS One. 2013;8:e73665. doi: 10.1371/journal.pone.0073665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Y, Wang C, Li Z. A new strategy of promoting cisplatin chemotherapeutic efficiency by targeting endoplasmic reticulum stress. Mol Clin Oncol. 2014;2:3–7. doi: 10.3892/mco.2013.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verras M, Papandreou I, Lim AL, Denko NC. Tumor hypoxia blocks Wnt processing and secretion through the induction of endoplasmic reticulum stress. Mol Cell Biol. 2008;28:7212–7224. doi: 10.1128/MCB.00947-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiu CC, Lee LY, Li YC, Chen YJ, Lu YC, Li YL, Wang HM, Chang JT, Cheng AJ. Grp78 as a therapeutic target for refractory head-neck cancer with CD24(−)CD44(+) stemness phenotype. Cancer Gene Ther. 2013;20:606–615. doi: 10.1038/cgt.2013.64. [DOI] [PubMed] [Google Scholar]
- 30.Lin JA, Fang SU, Su CL, Hsiao CJ, Chang CC, Lin YF, Cheng CW. Silencing glucose-regulated protein 78 induced renal cell carcinoma cell line G1 cell-cycle arrest and resistance to conventional chemotherapy. Urol Oncol. 2014;32:29.e1–e11. doi: 10.1016/j.urolonc.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 31.St Germain C, O'Brien A, Dimitroulakos J. Activating Transcription Factor 3 regulates in part the enhanced tumour cell cytotoxicity of the histone deacetylase inhibitor M344 and cisplatin in combination. Cancer Cell Int. 2010;10:32. doi: 10.1186/1475-2867-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang N, Zhang H, Si-Ma H, Fu Y, Zhao W, Li D, Yang G. Dexamethasone decreases hepatocellular carcinoma cell sensitivity to cisplatin-induced apoptosis. Hepatogastroenterology. 2011;58:1730–1735. doi: 10.5754/hge11153. [DOI] [PubMed] [Google Scholar]
- 33.Liu G, Su L, Hao X, Zhong N, Zhong D, Singhal S, Liu X. Salermide up-regulates death receptor 5 expression through the ATF4-ATF3-CHOP axis and leads to apoptosis in human cancer cells. J Cell Mol Med. 2012;16:1618–1628. doi: 10.1111/j.1582-4934.2011.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, Mao C, Ye R, Wang M, Pen L, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 35.Altieri DC. Molecular circuits of apoptosis regulation and cell division control: The survivin paradigm. J Cell Biochem. 2004;92:656–663. doi: 10.1002/jcb.20140. [DOI] [PubMed] [Google Scholar]
- 36.Tien LT, Ito M, Nakao M, Niino D, Serik M, Nakashima M, Wen CY, Yatsuhashi H, Ishibashi H. Expression of beta-catenin in hepatocellular carcinoma. World J Gastroenterol. 2005;11:2398–2401. doi: 10.3748/wjg.v11.i16.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson MD, Monga SP. WNT/beta-catenin signaling in liver health and disease. Hepatology. 2007;45:1298–1305. doi: 10.1002/hep.21651. [DOI] [PubMed] [Google Scholar]