

SUMMARY
Przewalski’s horses (PHs, Equus ferus ssp. przewalskii) were discovered in the Asian steppes in the 1870s and represent the last remaining true wild horses. PHs became extinct in the wild in the 1960s but survived in captivity, thanks to major conservation efforts. The current population is still endangered, with just 2,109 individuals, one-quarter of which are in Chinese and Mongolian reintroduction reserves [1]. These horses descend from a founding population of 12 wild-caught PHs and possibly up to four domesticated individuals [2–4]. With a stocky build, an erect mane, and stripped and short legs, they are phenotypically and behaviorally distinct from domesticated horses (DHs, Equus caballus). Here, we sequenced the complete genomes of 11 PHs, representing all founding lineages, and five historical specimens dated to 1878–1929 CE, including the Holotype. These were compared to the hitherto-most-extensive genome dataset characterized for horses, comprising 21 new genomes. We found that loci showing the most genetic differentiation with DHs were enriched in genes involved in metabolism, cardiac disorders, muscle contraction, reproduction, behavior, and signaling pathways. We also show that DH and PH populations split ~45,000 years ago and have remained connected by gene-flow thereafter. Finally, we monitor the genomic impact of ~110 years of captivity, revealing reduced heterozygosity, increased inbreeding, and variable introgression of domestic alleles, ranging from non-detectable to as much as 31.1%. This, together with the identification of ancestry informative markers and corrections to the International Studbook, establishes a framework for evaluating the persistence of genetic variation in future reintroduced populations.
Graphical abstract
RESULTS AND DISCUSSION
Despite their additional chromosomal pair (2n = 66 versus 2n = 64 [5]), PHs can successfully reproduce with DHs, resulting in fully viable and fertile offspring. The extent to which those horses admixed in the past is, however, contentious. Mitochondrial DNA (mtDNA) [6–8], the X [9–11] and Y [11–13] chromosomes, and a limited number of autosomal SNPs [14] have supported admixture, with PHs appearing within the genetic variability of DHs, in line with possible domestic contributions listed in the International Studbook [2–4]. Conversely, ~54,000 SNPs [15] and one complete PH genome [16] indicated that PH and DH populations probably diverged ~38,000–72,000 years ago [17], with no sign of admixture [16].
To elucidate the evolutionary history of PHs, we used the Illumina paired-end technology and generated whole-genome sequences of 33 living horses, including 11 PHs representing all pedigree founding lineages (18.0–23.4× average depth-of-coverage), one F1 PH × DH hybrid (22.8×), and 21 horses from five domestic breeds (7.7–22.6×). These complement an existing dataset of seven DH and three PH genomes [16, 18]. We also sequenced the genomes of five historical specimens: two PH pedigree founders from the early 1900s (SB11/Ewld1_Bijsk1 and SB12/Ewld2_Bijsk2, each at 0.9×), one hybrid (SB56/Ewld5_Theodor, 4.3×), as well as the PH Holotype (1.4×) and one Paratype specimen (3.7×), both from the late 19th century (Table S1). DNA fragmentation and nucleotide mis-incorporation patterns, indicative of post-mortem damage [17, 19], supported the authenticity of our ancient genomes (Figure S1). We limited the impact of nucleotide mis-incorporations on downstream analyses by enzymatically treating ancient DNA extracts [20] and/or by downscaling base quality scores at damaged positions [14]. The genomes of the Holotype, Para-type, and SB11/Ewld1_Bijsk1 specimens showed error rates lower than those of the other historical horses (0.099%–0.258% versus 0.463%–0.622% errors/base) but similar to those of Late Pleistocene horses (0.103%–0.207% [21]). In comparison, error rates in modern horses were 0.018%–0.040% in DHs and 0.036%–0.051% in PHs (Table S1).
Phylogenetic analyses identified three main mtDNA lineages and two Y chromosome haplotypes in PHs. PHs represent a paraphyletic assemblage within DHs in the mtDNA tree [6–8] and exhibit Y chromosome haplotypes not found in any other domesticated stallion, nor in a ~16,000-year-old horse [21] (Figures S2A and S2B). The maternal and paternal lineages are in agreement with the recorded PH pedigree, except for SB528 and SB285, respectively. An exome-based maximum-likelihood (ML) phylogeny revealed DHs and PHs as two reciprocally monophyletic clades distinct from Late Pleistocene horses (Figure 1A) [21], in accordance with the population tree reconstructed in TreeMix using 198,932 SNPs (Figure S2C). These results are consistent with Pairwise Sequential Markovian Coalescent (PSMC) demographic trajectories ([21]; Figure 1B), indicating an early divergence (~150,000 years ago) for the population of Late Pleistocene horses and synchronized DH and PH profiles until their most recent history. This also reflects the clusters identified in the STRUCTURE analyses of 12 autosomal STRs (Figure S2D) and principal-component analyses based on genome-wide SNP calls or genotype likelihoods (Figure 1C, S2E, and S2F). The latter show no overlap for historical and modern PHs along the first principal component, potentially as a consequence of captivity. Altogether, our results support the two Late Pleistocene horses analyzed, DHs, and PHs as three populations that diverged ~150,000 years ago [16, 21]. The discrepancy between the phylogenies inferred from autosomes, the Y chromosome, and mtDNA could reflect the recruitment of only few stallions during the domestication process, the co-segregation of mtDNA haplogroups in the ancestral populations of DHs and PHs, and/or maternally driven gene-flow post-divergence [10–12, 22, 23].
Figure 1. Genomic Structure among Late Pleistocene Horses in Blue, DHs in Red, and PHs in Green.
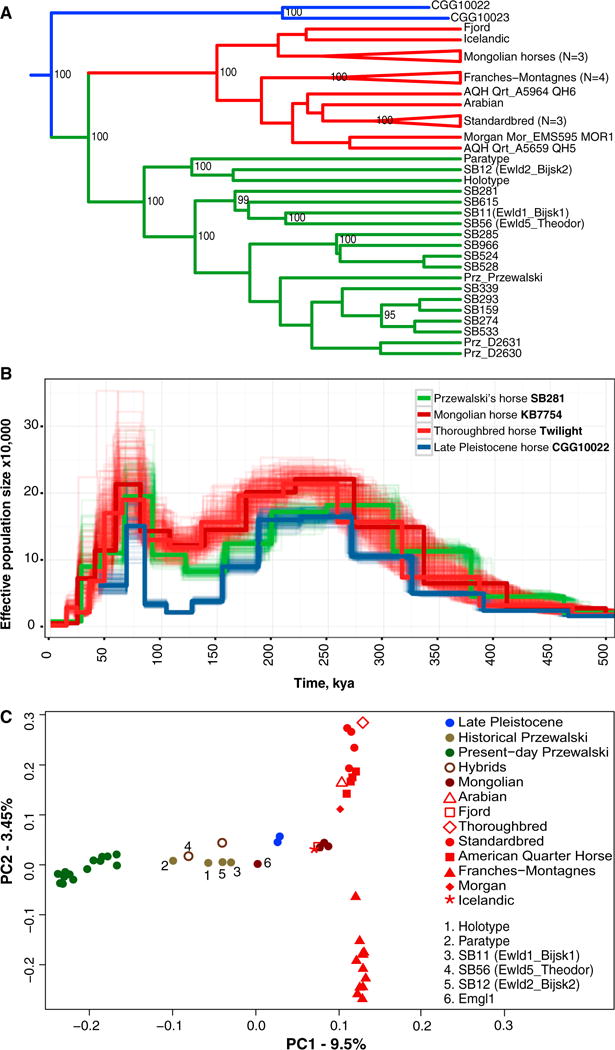
(A) Exome-based ML tree. Nodes show bootstrap support ≥ 95%. AQH, American Quarter Horse.
(B) PSMC profiles. Thin lines represent 100 bootstrap replicates. kya, thousand years ago.
(C) PCA based on genotype likelihoods.
See also Figures S1 and S2 and Tables S1 and S2.
We then aimed at identifying the genetic variants underlying the striking phenotypical differences between DHs and PHs. We found 509 copy-number variants (CNVs) in PHs, encompassing a total number of 1.65 Mb, 219 kb of which were previously reported in [24]. These contained 233 genes, including genes encoding proteins associated with glycolsphingolipid metabolic process, scarring, decreased corneal thickness, and the sulfuric ester hydrolase activity, which is key for the bone and cartilage matrix composition [25]. We also extracted 101 exonic ancestry informative markers for PHs, of which 53 represent non-synonymous mutations in genes involved in cardiac activity (CACNA1D, ITGA10), protein digestion, and absorption (COL18A1, COL15A1); disorders in muscles, ligaments, and the tissues surrounding muscles, blood vessels, and nerves (PALMD, MCPH1); diseases of the sebaceous glands (PALMD, MCPH1); and musculoskeletal and craniofacial abnormalities (MCPH1, SETBP1) (Table S2). We further sought for regions maximizing the genetic differentiation between DHs and PHs, using the FST fixation index. These contained 874 genes showing significant enrichment in signaling pathways (chemokine, kit receptor, and the inhibitor of DNA binding), in the glycogene and glycerophospholipid metabolisms, and in pathways involved in striated muscle contraction, heart and metabolic diseases, and mood disorders (Table S2). We finally used SweeD [26] to identify 76 protein-coding candidates for selective sweeps within PHs, including MCR2, which encodes one receptor of the ACTH stress hormone. Candidate genes showed enrichment for energetic and metabolic pathways, the cardiovascular system, muscular contraction, and immunity. Two genes, CACNA1D and PLA2G1B, supported a significant enrichment for the gonadotropin-releasing hormone signaling pathway, which is essential in sexual behavior and aggressiveness [27], possibly in relation to the temper of PHs (Table S2). Overall, our analyses unveil some potential drivers of the marked anatomical, physiological, and behavioral differences between DHs and PHs, some of which might have been enhanced during domestication.
We next reconstructed the past population history of DHs and PHs. Using dadi [28], our best-fit comparison consistently supported models involving asymmetrical gene flow after divergence (~35,000–54,000 years ago, in agreement with [16, 21]), almost entirely from PHs into DHs (Figure 2A; Table S3). We also noticed that the fit could be substantially improved when considering that migration stopped within recent times, i.e., as early as the last 200 years, suggesting that genetic restocking from wild animals was likely common until recently (Figure 2B; Table S3). Using genome projections [29], the best model was recovered by accounting for a recent 4% migration pulse from DHs and two epochs with asymmetrical gene flow, first from DHs into PHs, then reversed (Figure 2C; Table S3). The best fit was obtained for migration changing during the Last Glacial Maximum (~23,000 years ago; Figure 2D), when rates decreased ~31- to 160-fold. This change in the direction and magnitude of migration could reflect a combination of sexual behavior and demographic factors, as the detected contribution of DHs was higher when populations were much larger (Figure 1B). Altogether, our analyses suggest connection by gene flow after divergence and prior to horse domestication.
Figure 2. DH and PH Demographic Models Explored in Dadi and Projection Analyses.
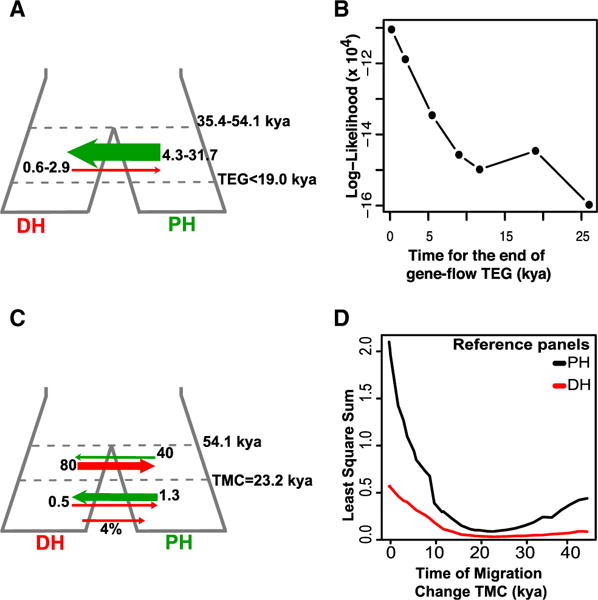
(A) Best two-population model supported by dadi.
(B) Dadi model Log(likelihood) for varying starting dates of isolation between DHs (Franches-Montagnes) and PHs (TEG).
(C) Best two-population model supported by projection analyses.
(D) Model LSS when considering varying dates for the change in migration dynamics (TMC) for the genome projections based on the DH (Franches-Montagnes, red) and PH (black) reference panels.
See also Table S3.
We then evaluated the genetic impact of the bottleneck associated with ~110 years of captivity in PHs. Their average heterozygosity (0.39–0.59 heterozygous sites per kilobase) was comparable to that in other endangered mammalian species (e.g., 0.12–0.65 in [30, 31]) but lower than in DHs (0.40–0.98) and historical PHs (0.74–2.35; Mann-Whitney test, p ≤ 1.88 × 10−3; Figure 3A). Another consequence of the bottleneck is the higher inbreeding coverage, i.e., the proportion of mostly homozygous blocks [21, 32], in PHs (0.052–0.388) than in modern DHs (0.006–0.285; two-sample t test, p = 6.81 × 10−3). Additionally, the inbreeding coefficient of historical PHs, as estimated using a novel method accommodating data uncertainty (FHMM, Supplemental Experimental Procedures), was lower than in modern PHs (p = 4.43 × 10−2) (Figure 3B; Table S1). Altogether, this demonstrates that captivity has significantly impacted the genetic diversity of PHs, reducing heterozygosity and increasing inbreeding.
Figure 3. Heterozygosity and Inbreeding Estimates.
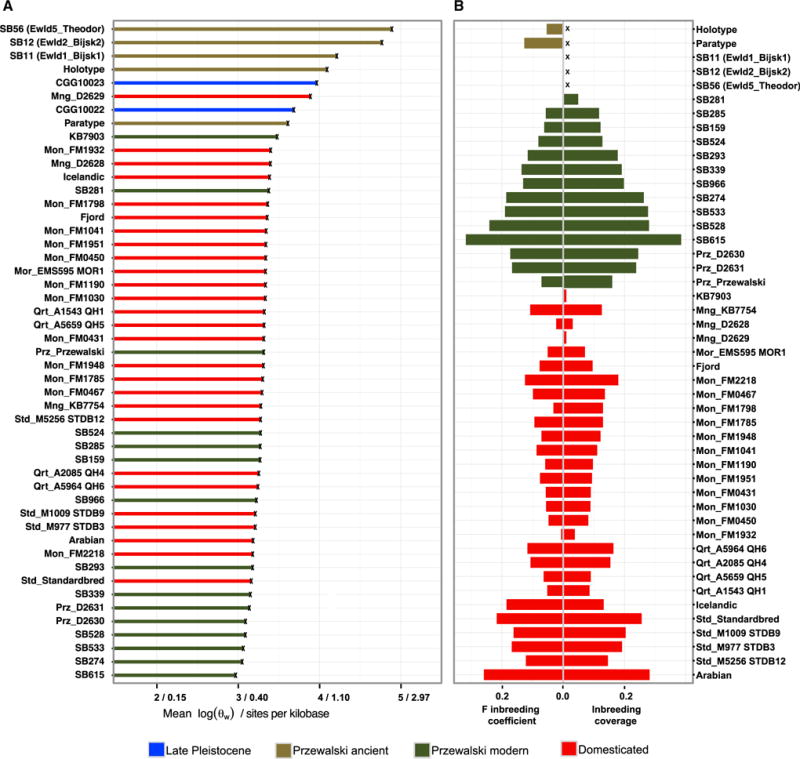
(A) Average genome-wide heterozygosity for autosomes, disregarding transitions.
(B) FHMM inbreeding coefficient and inbreeding coverage across modern and historical PHs and DHs. X denotes not estimated.
We investigated admixture with DHs as another potential consequence of captivity. Using NGSAdmix [33], eight clusters recapitulated DH breeds and their differentiation from PHs (Figures 4A and S3A). Although inconsistent with the pedigree describing the historical specimen SB56/Ewld5_Theodor as the F1 offspring of pure PH (SB11/Ewld1_Bjisk1) and DH (Emgl1) parents, this horse displayed, respectively, 79% and 21% of PH and DH components, providing clear evidence of introgression into the early descent of the PH founders.
Figure 4. DH Introgression into PHs.
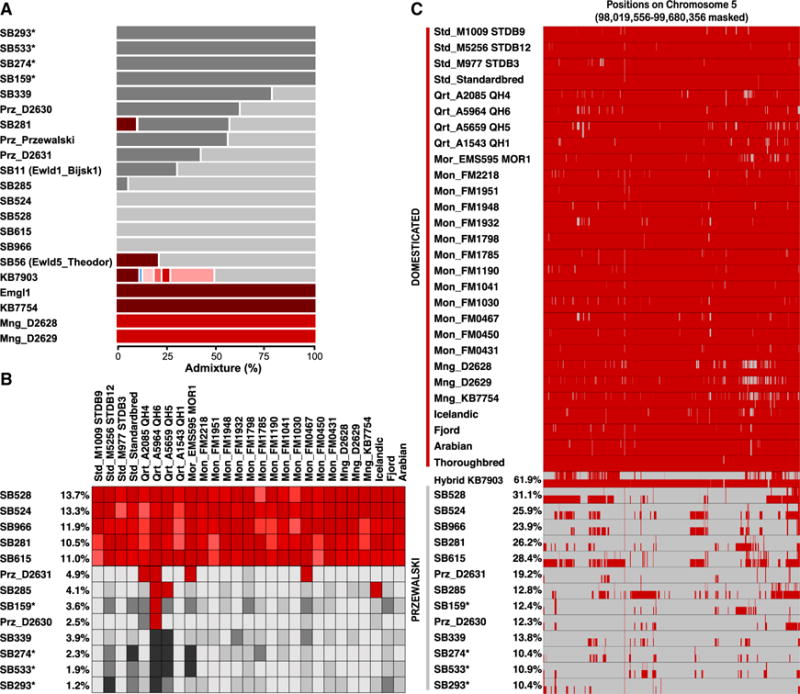
(A) Genetic components estimated in NGSAdmix (eight clusters). The gray and red gradients represent the PH and DH components, respectively.
(B) D statistic heatmaps. The tree topology (Outgroup,(PH,(DH,Paratype))) is tested for each DH and PH combination, disregarding transitions. Darker red colors indicate the admixture component in significant tests; darker gray shades indicate non-significant tests. The proportion of DH introgression within each PH genome, estimated by the average of f4 ratios involving Franches-Montagnes horses, is indicated next to the PH sample labels.
(C) Chromosome painting of DH (red) and PH (gray) ancestry blocks identified along chromosome 5.
Asterisk denotes A-line PHs. See also Figure S3 and Tables S4 and S5.
We sought further admixture evidence using the f3 statistics in (PH,DH;PH) tests [34] and found significant evidence of DH ancestry within modern PHs (Table S4). This was confirmed using D statistics [35] (Figures S3B–S3D), as topologies in the form of (Outgroup,(DH,(PH,PH))) were rejected in a large proportion of the tests. Taking advantage of the genome sequence of the Paratype specimen and (Outgroup,(DH,(PH,Paratype))) trees, we identified three groups of PHs with increasing levels of domestic ancestry (Figure 4B; Table S4): (1) SB274, SB293, SB339, and SB533 were found to be no closer to DHs than the Paratype; (2) SB281, SB524, SB528, SB615, and SB966 showed a significant genetic contribution from all the DHs tested (representing 10.5%–13.7% of their genome); (3) whereas SB159, SB285, Prz_D2630, Prz_D2631, and Prz_Przewalski showed a significant genetic contribution (2.5%–4.9%) from a minority of the DHs tested.
We next mapped admixture blocks along the genome using LAMP [36] and the whole panel of DH and modern PH genomes (except Prz_Przewalski, showing coverage artifacts confounding LAMP). A first group (SB281, SB524, SB528, SB615, and SB966), showed LAMP-admixture proportions of 23.9%–31.1%, with introgression tracts unevenly scattered across the genome. The remaining PHs exhibited 10.4%–19.2% admixture, corresponding to shorter tracts and earlier admixture events (Figure 4C). Interestingly, we found a derived allele associated with increasing wither height at ZFAT in SB524, SB528, and SB966, in a region likely introgressed from DHs (Table S5). Overall, these results confirm the DH introgression depicted in the International Studbook, with individuals tested for the A line (SB159, SB274, SB293, and SB533) considered as the pedigree’s purest line, virtually devoid of admixture [37]. We should, however, caution that, despite being significantly correlated (Pearson’s correlation coefficients ≥ 0.98, p ≤ 7.94 × 10−6), admixture coefficients estimated using LAMP and D statistics were likely overestimated. The DH ancestry for the KB7903 hybrid is indeed ~62% in LAMP, instead of the expected ~50%, and the higher error rate of the Paratype genome inflates allele sharing—hence, D statistics—between modern PHs and DHs.
In addition, to provide the most extensive genomic resource for horses classified as “endangered” on the IUCN Red List [1], our study fulfills the goal of the IUCN/SSC Equid Specialist Group conservation program [38] by identifying hybrids, AIMs, and candidate alleles maximizing the adaptation of PHs to their native environment. In the future, cost-effective genome-wide analyses targeting these markers should help minimize the contribution of domestic ancestry and limit loss of genetic diversity in a closed breeding program, with the ultimate objective of maintaining a viable population, once reintroduced in the wild.
Supplementary Material
In Brief.
Der Sarkissian et al. characterized complete genomes of modern and historical Przewalski’s horses, a wild and endangered population distinct from domesticated horses. Comparative analyses revealed specific signatures of selection, increased inbreeding, and variable introgression from domesticated horses in the last 110 years in captivity.
Highlights.
Complete genomes were sequenced for 21 domesticated and 17 Przewalski’s horses
Selection signatures and variants specific to Przewalski’s horses were detected
Domesticated and Przewalski’s horse ancestors mixed post-divergence ~45,000 years ago
Captivity increased inbreeding and domestic introgression in Przewalski’s horses
Acknowledgments
We thank the staff of the Danish National High-Throughput DNA Sequencing Center for technical assistance. This work was supported by the Danish Council for Independent Research, Natural Sciences (FNU-4002-00152B); the Danish National Research Foundation (DNFR94); the Villum Fonden Blokstipendium (2014); the Lundbeck Foundation (R52-A5062); the Israel Science Foundation (1365/10); the German Research Council (DFG-LU852/7-4); the NIH (R01-GM40282); the Caesar Kleberg Foundation for Wildlife Conservation; the John and Beverly Stauffer Foundation; FP7 European Marie-Curie programs (CIG-293845, ITN-290344, IEF-328024, IEF-299176, and IEF-302617); a National Science Foundation Graduate Research Fellowship; and the Human Frontier Science Program (LT000320/2014).
Footnotes
EXPERIMENTAL PROCEDURES
Samples and methods are described in Supplemental Experimental Procedures. Material was shipped in compliance with CITES regulation for endangered species, following Materials Transfer Agreements between Copenhagen and San Diego (Biomaterial Request BR2013045) and Certificate of Scientific Exchange (COSE) in place at both institutions (12US50818A/9 San Diego; DK003 Natural History Museum of Denmark, Copenhagen). An extended Supplementary Material can be found at: http://geogenetics.ku.dk/publications/si-przewalski.
ACCESSION NUMBERS
The accession number for the sequences reported in this paper is European Nucleotide Archive: PRJEB10098.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, three figures, and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.cub.2015.08.032.
AUTHOR CONTRIBUTIONS
W.Z., O.A.R., and L.O. conceived the project, and L.O. designed the study. N.I.A., R.S., A.T., O.A.R., M.M., S.R., and T.L. provided samples. C.D., L.E., G.K.B., S.P., C.S., M.N., V.J., M.M., and L.O. performed laboratory analyses. C.D., L.E., M. Schubert, M.A.Y., P.L., M.F., H.J., G.K.B., A.A., F.G.V., B.P., A.G., A.F., B.L.-G., T.M.B., M. Slatkin, and L.O. analyzed genomic data. G.K.B., A.S.O., K.M., C.F., C.L.G., A.L., T.S.P., E.W., T.M.B., O.A.R., M.M., S.R., T.L., and L.O. provided reagents and material. L.O. wrote the paper with input from all co-authors. C.D., P.L., M. Schubert, M.F., M.A.Y., and L.O. wrote the supplementary information, with input from C.G.
References
- 1.Boyd L, King S. Equus ferus ssp przewalskii. The IUCN Red List of Threatened Species. 2011 http://www.iucnredlist.org.
- 2.Mohr E. Das Urwildpferd (Equus przewalski Poljakoff 1881) Mit 87 Abbildungen Die Neue Brehm-Bücherei 249. Wittenberg Lutherstadt: 1959. [Google Scholar]
- 3.Volf J, Kus E, Prokopova L. General Studbook of the Przewalski Horse. Zoological Garden Prague: 1991. [Google Scholar]
- 4.Zimmermann W. International Przewalski’s Horse Studbook (Equus ferus przewalskii) Cologne Zoo: 2014. [Google Scholar]
- 5.Benirschke K, Malouf N, Low RJ, Heck H. Chromosome complement: differences between Equus caballus and Equus przewalskii, Poliakoff. Science. 1965;148:382–383. doi: 10.1126/science.148.3668.382. [DOI] [PubMed] [Google Scholar]
- 6.Ishida N, Oyunsuren T, Mashima S, Mukoyama H, Saitou N. Mitochondrial DNA sequences of various species of the genus Equus with special reference to the phylogenetic relationship between Przewalskii’s wild horse and domestic horse. J Mol Evol. 1995;41:180–188. doi: 10.1007/BF00170671. [DOI] [PubMed] [Google Scholar]
- 7.Oakenfull EA, Ryder OA. Mitochondrial control region and 12S rRNA variation in Przewalski’s horse (Equus przewalskii) Anim Genet. 1998;29:456–459. doi: 10.1046/j.1365-2052.1998.296380.x. [DOI] [PubMed] [Google Scholar]
- 8.Goto H, Ryder OA, Fisher AR, Schultz B, Kosakovsky Pond SL, Nekrutenko A, Makova KD. A massively parallel sequencing approach uncovers ancient origins and high genetic variability of endangered Przewalski’s horses. Genome Biol Evol. 2011;3:1096–1106. doi: 10.1093/gbe/evr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowling AT, Zimmermann W, Ryder O, Penado C, Peto S, Chemnick L, Yasinetskaya N, Zharkikh T. Genetic variation in Przewalski’s horses, with special focus on the last wild caught mare, 231 Orlitza III. Cytogenet Genome Res. 2003;102:226–234. doi: 10.1159/000075754. [DOI] [PubMed] [Google Scholar]
- 10.Wallner B, Brem G, Müller M, Achmann R. Fixed nucleotide differences on the Y chromosome indicate clear divergence between Equus przewalskii and Equus caballus. Anim Genet. 2003;34:453–456. doi: 10.1046/j.0268-9146.2003.01044.x. [DOI] [PubMed] [Google Scholar]
- 11.Lau AN, Peng L, Goto H, Chemnick L, Ryder OA, Makova KD. Horse domestication and conservation genetics of Przewalski’s horse inferred from sex chromosomal and autosomal sequences. Mol Biol Evol. 2009;26:199–208. doi: 10.1093/molbev/msn239. [DOI] [PubMed] [Google Scholar]
- 12.Lindgren G, Backström N, Swinburne J, Hellborg L, Einarsson A, Sandberg K, Cothran G, Vilà C, Binns M, Ellegren H. Limited number of patrilines in horse domestication. Nat Genet. 2004;36:335–336. doi: 10.1038/ng1326. [DOI] [PubMed] [Google Scholar]
- 13.Wallner B, Vogl C, Shukla P, Burgstaller JP, Druml T, Brem G. Identification of genetic variation on the horse y chromosome and the tracing of male founder lineages in modern breeds. PLoS ONE. 2013;8:e60015. doi: 10.1371/journal.pone.0060015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wade CM, Giulotto E, Sigurdsson S, Zoli M, Gnerre S, Imsland F, Lear TL, Adelson DL, Bailey E, Bellone RR, et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science. 2009;326:865–867. doi: 10.1126/science.1178158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCue ME, Bannasch DL, Petersen JL, Gurr J, Bailey E, Binns MM, Distl O, Guérin G, Hasegawa T, Hill EW, et al. A high density SNP array for the domestic horse and extant Perissodactyla: utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet. 2012;8:e1002451. doi: 10.1371/journal.pgen.1002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orlando L, Ginolhac A, Zhang G, Froese D, Albrechtsen A, Stiller M, Schubert M, Cappellini E, Petersen B, Moltke I, et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature. 2013;499:74–78. doi: 10.1038/nature12323. [DOI] [PubMed] [Google Scholar]
- 17.Briggs AW, Stenzel U, Johnson PLF, Green RE, Kelso J, Prüfer K, Meyer M, Krause J, Ronan MT, Lachmann M, Pääbo S. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci USA. 2007;104:14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Do KT, Kong HS, Lee JH, Lee HK, Cho BW, Kim HS, Ahn K, Park KD. Genomic characterization of the Przewalski׳s horse inhabiting Mongolian steppe by whole genome re-sequencing. Livest Sci. 2014;167:86–91. [Google Scholar]
- 19.Sawyer S, Krause J, Guschanski K, Savolainen V, Pääbo S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE. 2012;7:e34131. doi: 10.1371/journal.pone.0034131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Briggs AW, Stenzel U, Meyer M, Krause J, Kircher M, Pääbo S. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010;38:e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schubert M, Jónsson H, Chang D, Der Sarkissian C, Ermini L, Ginolhac A, Albrechtsen A, Dupanloup I, Foucal A, Petersen B, et al. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc Natl Acad Sci USA. 2014;111:E5661–E5669. doi: 10.1073/pnas.1416991111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallner B, Piumi F, Brem G, Müller M, Achmann R. Isolation of Y chromosome-specific microsatellites in the horse and cross-species amplification in the genus Equus. J Hered. 2004;95:158–164. doi: 10.1093/jhered/esh020. [DOI] [PubMed] [Google Scholar]
- 23.Lippold S, Knapp M, Kuznetsova T, Leonard JA, Benecke N, Ludwig A, Rasmussen M, Cooper A, Weinstock J, Willerslev E, et al. Discovery of lost diversity of paternal horse lineages using ancient DNA. Nat Commun. 2011;2:450. doi: 10.1038/ncomms1447. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh S, Qu Z, Das PJ, Fang E, Juras R, Cothran EG, McDonell S, Kenney DG, Lear TL, Adelson DL, et al. Copy number variation in the horse genome. PLoS Genet. 2014;10:e1004712. doi: 10.1371/journal.pgen.1004712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soong BW, Casamassima AC, Fink JK, Constantopoulos G, Horwitz AL. Multiple sulfatase deficiency. Neurology. 1988;38:1273–1275. doi: 10.1212/wnl.38.8.1273. [DOI] [PubMed] [Google Scholar]
- 26.Pavlidis P, Zivkovic D, Stamatakis A, Alachiotis N. SweeD: likelihood-based detection of selective sweeps in thousands of genomes. Mol Biol Evol. 2013;30:2224–2234. doi: 10.1093/molbev/mst112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ransom JI, Powers JG, Garbe HM, Oehler MW, Nett TM, Baker DL. Behavior of feral horses in response to culling and GnRH immunocontraception. Appl Anim Behav Sci. 2014;157:81–92. [Google Scholar]
- 28.Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009;5:e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang MA, Harris K, Slatkin M. The projection of a test genome onto a reference population and applications to humans and archaic hominins. Genetics. 2014;198:1655–1670. doi: 10.1534/genetics.112.145359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho YS, Hu L, Hou H, Lee H, Xu J, Kwon S, Oh S, Kim HM, Jho S, Kim S, et al. The tiger genome and comparative analysis with lion and snow leopard genomes. Nat Commun. 2013;4:2433. doi: 10.1038/ncomms3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cahill JA, Green RE, Fulton TL, Stiller M, Jay F, Ovsyanikov N, Salamzade R, St John J, Stirling I, Slatkin M, Shapiro B. Genomic evidence for island population conversion resolves conflicting theories of polar bear evolution. PLoS Genet. 2013;9:e1003345. doi: 10.1371/journal.pgen.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505:43–49. doi: 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skotte L, Korneliussen TS, Albrechtsen A. Estimating individual admixture proportions from next generation sequencing data. Genetics. 2013;195:693–702. doi: 10.1534/genetics.113.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, Genschoreck T, Webster T, Reich D. Ancient admixture in human history. Genetics. 2012;192:1065–1093. doi: 10.1534/genetics.112.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28:2239–2252. doi: 10.1093/molbev/msr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pasaniuc B, Sankararaman S, Kimmel G, Halperin E. Inference of locus-specific ancestry in closely related populations. Bioinformatics. 2009;25:i213–i221. doi: 10.1093/bioinformatics/btp197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seal U, Lacy R, Zimmermann W, Ryder O, Princee F. Przewalski’s Horse, Equus przewalskii, Global Conservation Plan Draft: Captive Breeding Specialist Group (SSC/IUCN) 1990 [Google Scholar]
- 38.Moehlman PDR, et al. Equids: Zebras, Asses, and Horses: Status Survey and Conservation Action Plan (IUCN) 2002 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.