

Abstract
By analyzing the exomes of 12,332 unrelated Swedish individuals – including 4,877 affected with schizophrenia – in ways informed by exome sequences from 45,376 other individuals, we identified 244,246 coding-sequence and splice-site ultra-rare variants (URVs) that were unique to individual Swedes. We found that gene-disruptive and putatively protein-damaging URVs (but not synonymous URVs) were more abundant in schizophrenia cases than controls (P = 1.3 × 10−10). This elevation of protein-compromising URVs was several times larger than an analogously elevated rate for de novo mutations, suggesting that most rare-variant effects on schizophrenia risk are inherited. Among individuals with schizophrenia, the elevated frequency of protein-compromising URVs was concentrated in brain-expressed genes, particularly in neuronally expressed genes; most of this genetic signal arose from large sets of genes whose RNAs have been found to interact with synaptically localized proteins. Our results suggest that synaptic dysfunction may mediate a large fraction of strong, individually rare genetic influences on schizophrenia risk.
Schizophrenia is a psychiatric disorder with a lifetime risk of about 0.7%1 and a heritability of 60–80%2,3 despite greatly reduced reproductive fecundity4,5. Because individuals affected with schizophrenia have fewer offspring, purifying selection is expected to prevent high-risk alleles from reaching even modest allele frequencies6. Indeed, estimates of selection (when based only on the reproductive costs of schizophrenia) may underestimate the actual selective pressure against such alleles given emerging evidence that such alleles have multiple adverse effects: for example, rare copy number variations (CNVs), implicated with penetrances ranging from 2–30% (the latter observed for 22q11.2 deletions), negatively impact cognition and fecundity even in their more-typical presentation without schizophrenia7. To the extent that such observations condition expectations for rare single-nucleotide variants, variants with a large effect on schizophrenia risk are likely to be rare in populations, requiring sequencing to find them.
Distinguishing those variants that are extremely rare from variants that are segregating in a population is ideally informed by sequencing very-large numbers of individuals from the same population. We thus analyzed the sequences of 12,332 unrelated individuals (4,946 affected with schizophrenia, 6,242 unaffected controls, and 1,144 with other psychiatric illnesses whose analysis is beyond the scope of the current study) from Sweden (Online Methods). We further informed this analysis with a much larger set of exome sequencing data from 45,376 individuals from multiple non-psychiatric cohorts ascertained by the Exome Aggregation Consortium8. This made it possible to identify among the Swedish research participants 244,246 coding-sequence and splice-site ultra-rare variants (URVs) that were present in single individuals – a set of variants that is greatly enriched for recent mutations, relative to the vastly larger fraction of heterozygosity that is due to less-rare variants (Fig. 1a). This large set of variants made it possible to identify broad biological patterns among an excess of more than 1,000 protein-damaging URVs that we found in the exomes of 4,946 individuals affected with schizophrenia.
Figure 1. Ultra-rare variants distribution and association with schizophrenia.

(a-b) Counts across coding-sequence and splice-site rare variants stratified by minor allele count across exome sequencing data from 12,332 individuals indicating (a) how many variants were observed in the ExAC cohort and (b) how many variants were classified as disruptive, damaging, missense non-damaging, and synonymous. (c) Observed enrichment in schizophrenia cases compared to controls for coding-sequence and splice-site URVs across the main four annotation types. Enrichment and P values were computed using a linear regression model (left panel) and a logistic regression model (right panel). Horizontal bars indicate 95% confidence intervals.
RESULTS
Exome-wide enrichment of ultra-rare variants
We analyzed the protein-coding sequences (the exomes) of 12,332 unrelated Swedish individuals, including 4,946 affected with schizophrenia (2,951 males and 1,995 females), 6,242 unaffected controls (3,182 males and 3,060 females), and 1,144 affected with other disorders (443 males and 701 females, used for population genetic analyses but not as cases or controls). After removing 119 individuals for quality control reasons (mostly due to divergent ancestry, Online Methods), we identified 244,246 coding-sequence and splice-site URVs (among 4,877 schizophrenia cases and 6,203 controls) that were present in only one of the 12,332 unrelated Swedish exomes analyzed and never seen in the Exome Aggregation Consortium (ExAC) cohort (which numbered 45,376 individuals after excluding the subjects from this cohort and other subjects ascertained for psychiatric disorders).
We focused on URVs in most analyses because such variants – although comprising a tiny fraction (less than 0.2%) of the heterozygous sites in an individual – will be greatly enriched for recent mutations and thus have been exposed to fewer generations of purifying selection. The size of the Swedish cohort analyzed, and the additional sequence data for the Exome Aggregation Consortium, allowed us to greatly refine the identification of URVs; for example, among 5,092 (of the 12,332) individuals who were also part of an earlier sequencing study9, the additional data allowed us to re-classify ~66% of variants that had been “singletons” as segregating variants (not URVs in the current analysis). This may have been particularly helpful for refining analyses of challenging-to-interpret missense variants, as we describe below.
We classified coding-sequence and splice-site variants into four groups (Fig. 1b):
synonymous: exonic variants not predicted to change the encoded protein (63,230 URVs);
missense non-damaging: missense variants not predicted to damage protein function (by the criteria below) (134,100 URVs);
damaging: missense variants predicted to compromise protein function per an algorithm (Online Methods), in-frame indels, or variants affecting protein-protein-contact domains (27,390 URVs); and
disruptive: variants that truncate or abrogate the encoded protein in a way that is readily classified as loss-of-function10 or as triggering nonsense mediated decay (NMD)11. These included nonsense, frameshift, splice-site, and very rarely, read-through variants. (19,526 URVs).
The terms protein-“damaging” and gene-“disruptive” refer to predicted effects on individual gene copies and the encoded proteins, rather than effects on phenotypes; effects on phenotypes can be inferred only from association analysis.
Missense damaging URVs accounted for approximately 15% of all missense URVs (Supplementary Fig. 1). There was a median of 2 disruptive and 2 damaging URVs per individual (4 total) (Supplementary Fig. 2).
To assess whether schizophrenia was associated with an increased number of coding-sequence and splice-site URVs (in specific genes, across the exome, or in sets of genes), we used a linear regression model to control for possible confounding variables, including each individual’s overall number of detected URVs (including non-coding URVs), sex, birth year, the hybrid selection kit used for exome enrichment, and the first 20 principal components estimated from exome-wide SNP and indel genotypes (Supplementary Table 1).
An important negative control – to address the possibility that analyses could be affected by population structure, differences in average relatedness within the case and control groups, or by technical variation – is to ask whether functionally neutral forms of variation show any apparent differences in frequency between case and control groups. We did not observe a significant difference in the rate of synonymous URVs between schizophrenia cases and controls (Fig. 1c). We also did not observe a significant difference for non-coding URVs (Supplementary Fig. 3a).
In contrast, we observed significant case-control differences in the rates of disruptive URVs (a difference of 0.12 variants/person; 95% confidence interval [CI]=0.07–0.17; P = 1.8 × 10−6) and damaging URVs (0.12 variants/person; 95% CI=0.07–0.18; P = 3.4 × 10−5). (P values determined by permuting the phenotype data 10 million times agreed with P values from the linear regression analysis, and were: P = 0.81 for synonymous, P = 0.045 for missense non-damaging, P = 3.5 × 10−5 for damaging, and P = 1.8 × 10−6 for disruptive; this suggests that the P values from the regression model are well-calibrated) Damaging and disruptive URVs showed similarly elevated frequencies in cases and were thus combined into a single category termed dURVs (disruptive and damaging ultra-rare variants) for subsequent analyses.
Adjusting for covariates, there were 7% more dURVs in affected individuals than in controls (odds ratios [OR]=1.07; 95% CI=1.05-1.09; P = 1.5 × 10−10), as the case-associated elevation in dURVs (of about 0.25 variants/patient; 95% CI=0.17-0.32) occurred on a background of about 4 dURVs per patient. The elevated frequency of dURVs among individuals affected with schizophrenia appeared to arise from multiple types of dURVs, including in-frame indels, protein-protein-contact, splice-acceptor, splice-donor, stop-gained, and frame-shift variants (Supplementary Fig. 3b).
To assure that this result was not the result of population stratification within Sweden, we further estimated the enrichment in a more genetically homogeneous subset of the Swedish cohort (3,554 schizophrenia cases and 5,164 controls) that excluded individuals with significant amounts of Finnish or Northern Sweden ancestry. Individuals with schizophrenia showed a similar dURV excess in this more genetically homogeneous group (excess of 0.25 dURVs/case, 95% CI 0.16-0.34; P = 2.2 × 10−8).
We next estimated the extent to which dURVs tend to be inherited or de novo. While parental DNA would be necessary to directly ascertain which specific dURVs are de novo mutations (DNMs), we can compare the schizophrenia-associated elevation in dURVs (~0.25 per exome) to an analogous elevation in DNMs detected in earlier studies of 617 affected and 1911 unaffected father-mother-offspring trios12,13. Using data from the trios, we estimated the frequencies of DNMs that were protein-damaging or -disruptive (dDNMs) by the same criteria we used to identify dURVs (including restricting to variants not previously observed in ExAC8). These data yielded an elevation of about 0.03 such DNMs per exome, based on the difference between rates of 0.185 (95% CI 0.151-0.219) for individuals with schizophrenia12 and 0.156 (95% CI 0.139-0.174) for unaffected individuals13. This estimate (0.03 per exome) was several times smaller than the elevation of dURVs in affected individuals in our population-based study (0.25 per exome). (We note that such a comparison requires the imperfect assumption of uniform technical ascertainment across the sequencing studies; even under plausible relaxations of this assumption, the dURV excess greatly exceeds the dDNM excess. Also, when estimated by this same approach, rates of synonymous and non-damaging DNMs were similar – 0.475 in affected and 0.459 in unaffected individuals – suggesting that the analysis is well-calibrated.) We conclude that the great majority of the dURVs driving the elevated rates in schizophrenia were inherited rather than de novo, though the very-low allele frequency of these variants suggests that they are on average just a few generations old.
Although the elevated frequency of dURVs among affected individuals was statistically significant (P = 1.4 × 10−10), it was still only a modest increase of 0.25 dURVs on a background of about 4 dURVs per individual. This excess could in principle be concentrated in individual genes or in sets of functionally related genes, possibilities we address below.
Single gene burden analysis
Joint analysis of many rare variants that affect the same gene or sets of genes can increase power to identify genes whose disruption increases the risk of schizophrenia. To find individual genes that had significantly more rare variants in cases or controls, we performed a burden test using SKAT14 adjusting for previously defined covariates (Online Methods). We tested for (a) disruptive, (b) damaging, (c) disruptive and damaging, and (d) missense variants that were either (i) ultra-rare, (ii) singletons in the Sweden cohort, (iii) had a minor allele count ≤5 (minor allele frequency <0.02%), (iv) had a minor allele count ≤10 (minor allele frequency <0.05%), (v) had a minor allele frequency <0.1%, or (vi) had a minor allele frequency <0.5%.
Given the sample size, our analysis would have >90% power (at α = 2.5 × 10−6) to detect any gene for which rare, disruptive and damaging variants were present in 1% of schizophrenia cases, even if such variants had only a relatively modest effect size15 (odds ratio of at least 3, i.e. about 2% penetrance), and still greater power if effect sizes were larger. No individual gene surpassed exome-wide significance in this analysis (Supplementary Fig. 4), suggesting that no one gene is likely to have rare variants that explain even 1% of schizophrenia cases. The individual gene with the strongest enrichment was KL (klotho) (Supplementary Table 2), in which we found eight different dURVs in cases and none in controls (P = 3.7 × 10−4), but this result was not significant given the number of genes tested. Other models, based on higher levels of polygenicity, therefore appear to be more plausible: in a model in which a hypothetical gene is affected in 0.1% of schizophrenia cases, we would have only ~4% power to conclusively find this effect at exome-wide significance, and a far-larger sample would be required. The finding that no individual gene surpassed exome-wide significance in this analysis (Supplementary Fig. 4), suggests that no one gene is likely to have rare variants that explain even 1% of schizophrenia cases.
Among genes previously reported to have potential connections between rare variants and schizophrenia, we identified an ultra-rare splice donor variant in TAF1312, an ultra-rare nonsense variant in SETD1A16,17, and a single ultra-rare nonsense variant in NRXN1, a gene in which exonic deletions associate with schizophrenia18. We did not find any evidence of enrichment of dURVs in DPYD19 (in which we found two dURVs in cases and six in controls), nor in DISC120 (one dURV among cases and two in controls) (Supplementary Table 3).
With this high level of polygenicity foreshadowed by earlier results9,16, it appears that definitive implication of individual genes will require sequencing still-larger numbers of exomes or whole genomes6. We therefore focused on sets of genes with plausibly overlapping biological functions as a way of concentrating a diffuse genetic signal.
Enrichment of variants from cases in constrained genes
We tested gene sets for an enrichment of dURVs (in cases relative to controls) by comparing each gene set’s enrichment level to that of the average gene (Online Methods). We made this stringent correction to account for the fact that any large gene set is more likely to encompass the exome-wide excess of dURVs we see in schizophrenia cases. Our practice greatly deflates the resulting P values.
Subsets of human genes have been previously identified as “missense constrained” (based on a lack of functional coding variation in controls) or “loss-of-function intolerant” (based on a smaller-than-expected number of loss-of-function mutations in population-scale data)13,21. Similar to recent findings in autism, we observed a significant enrichment (in cases relative to controls) of dURVs in missense constrained genes22 (OR=1.28; 95% CI=1.20–1.37; P = 3.2 × 10−8) and loss-of-function intolerant genes8 (OR=1.17; 95% CI=1.12-1.21; P = 1.7 × 10−8) (Fig. 2). Both missense constrained and loss-of-function intolerant genes were enriched for disruptive variants relative to damaging variants (Supplementary Fig. 5); for the latter set, this may reflect these genes having been ascertained specifically for intolerance to disruptive mutations.
Figure 2. dURVs enrichement in schizophrenia cases across selected gene sets.

Excess per case and odds ratios for dURVs across loss-of-function intolerant (LoF-intolerant) genes, missense constrained genes, protein complexes genes, genes associated through common variants, predicted microRNA-137 targets, and intellectual disability genes. Enrichment and P values were computed using a linear regression model (left panel) and a logistic regression model (right panel) using exome-wide dURV count as a covariate to correct for average exome-wide burden (dot-dashed line). Horizontal bars indicate 95% confidence intervals.
In contrast, genes not meeting earlier criteria for loss-of-function intolerance or missense constraint were much less enriched for dURVs (Supplementary Fig. 6). This important negative control confirms that the schizophrenia-associated elevation we observe (for constrained genes) is not due to false positives disproportionally represented across disruptive and damaging variants in cases. The observed enrichment was consistent across data from previously analyzed exomes9 and newly generated data (Supplementary Fig. 5); as the previously analyzed exomes were sequenced across randomized batches with equal number of cases and controls in each batch, this provides additional evidence that the enrichment is not due to technical effects.
Tissues and cell types
The excess of dURVs could in principle be concentrated in genes expressed within specific tissues. Distinct tissues have both shared and tissue-specific sets of expressed genes. We found that a set of 2,647 genes expressed specifically in brain tissue23 was strongly enriched for dURVs (OR=1.17; 95% CI=1.11–1.23; P = 1.2 × 10−4), whereas sets of genes with expression specific to other tissues (including immune cells) were not (Fig. 3a and Supplementary Fig. 7). (At the same time, the “brain-specific” genes explained only part of this signal, while a larger set of brain-expressed genes explained most of it, suggesting that much of the signal may come from genes that are expressed in brain as well as other tissues, Fig. 3a). This aligns with earlier findings that SNP haplotypes implicated in schizophrenia genome-wide association studies (GWAS) tend to overlap (to a non-random degree) with sequences identified as putative enhancers in chromatin-profiling experiments on brain tissue24,25.
Figure 3. dURVs enrichment in schizophrenia cases across tissue, brain cell type, and synaptic gene sets.

Excess per case and odds ratios for dURVs across genes with higher expression in a given tissue (a), genes with higher expression in a given cell type (b), and genes expected to localize to synapses (c). Enrichment and P values were computed using a linear regression model (left panels) and a logistic regression model (right panels) using exome-wide dURV count as a covariate to correct for average exome-wide burden (dot-dashed line). Horizontal bars indicate 95% confidence intervals.
The brain contains a complex mixture of cell types, each of which expresses different, only partially overlapping sets of genes. To identify cell types through which rare variants might act to affect risk of schizophrenia, we evaluated (for enrichment of dURVs in affected relative to unaffected individuals) sets of genes identified as specific to neurons, astrocytes, and oligodendrocytes by earlier cell sorting and transcriptional profiling experiments26. A set of 3,388 neuron-specific genes had a strong enrichment of mutations in schizophrenia cases (OR=1.17; 95% CI=1.12–1.22; P = 1.9 × 10−7), comparable to that observed for genes specific to brain tissue itself. Genes specifically expressed in other brain cell types, such as astrocytes and oligodendrocytes, were no more enriched than the average gene (Fig. 3b and Supplementary Fig. 8a). These results nominate neurons as the central nervous system (CNS) cell type in which genetic perturbations most affect schizophrenia risk, though they do not exclude more-modest contributions from other CNS cell types.
Neurons are broadly classified into excitatory and inhibitory classes. The case-control excess of dURVs showed a similar degree of concentration into genes expressed in excitatory and inhibitory neurons (Supplementary Fig. 8b). The small number of genes that were specific to excitatory or inhibitory neurons (relative to the other class) were insufficient to concentrate this genetic signal, which appeared to reside primarily in genes that were expressed in both neuronal classes (Supplementary Fig. 8b).
Synaptic mRNAs
A strong and consistent finding in exome-sequencing studies of schizophrenia involves an excess of variants in genes whose mRNAs are bound by the fragile X mental retardation protein (FMRP)9,12,27. The large excess of dURVs ascertained in the current set of schizophrenia cases elevated evidence for this relationship (OR=1.23; 95% CI=1.17–1.30; P = 8.2 × 10−9).
The enrichment of dURVs among genes that encode FMRP-bound transcripts has multiple potential biological explanations. One potential explanation could involve the translational-inhibition capacity of FMRP, as implicit in the common description of such genes as FMRP “targets”. Another potential interpretation is that it is in fact the localization of these RNAs to neuronal processes and synapses by FMRP – its shuttling activity – that defines the important biological commonality among these genes. Yet a third possibility is that FMRP-binding experiments have simply been effective ways of ascertaining neuronally expressed genes.
To evaluate these possibilities, we first considered a different set of genes whose mRNAs are carried to synapses by a different shuttling protein, CELF428. The genes encoding CELF4-bound mRNAs also showed an enrichment of dURVs in schizophrenia cases; this enrichment (OR=1.14; 95% CI=1.09–1.19; P = 6.6 × 10−4) was greater than that of the average gene though less strong than that of genes encoding FMRP-bound RNAs.
We also investigated whether genes encoding mRNAs that are bound by RBFOX splicing factors, known to regulate synaptic genes29 and also previously observed at synapses30, could explain a substantial fraction of the dURVs. Earlier experimental work (based on the HITS-CLIP technique for identifying RNAs bound to proteins of interest) has defined constellations of genes whose RNAs are bound by RBFOX1, RBFOX2, or RBFOX3. (We considered RBFOX1 and RBFOX3 together below due to their largely overlapping sets of bound genes31). Genes whose transcripts are bound by RBFOX1 or RBFOX3 were enriched in dURVs (OR=1.16; 95% CI=1.11–1.21; P = 6.7 × 10−7). A somewhat stronger enrichment was apparent for genes whose RNAs are bound by RBFOX2 (OR=1.21; 95% CI=1.16–1.26; P = 6.3 × 10−12).
We also observed enrichment in synaptic genes as defined by the SynaptomeDB32 (OR=1.14; 95% CI=1.09–1.20; P = 0.0022), though this smaller set of genes explained a smaller fraction of the case-control difference in dURVs (Fig. 3c).
We were concerned that the enrichment for dURVs in genes with synaptically localized transcripts could, in principle, be simply due to these experiments having been highly effective at isolating transcripts that are present in neurons (which strongly express FMR1, CELF4, and RBFOX1/2/3); in this case, the importance of synaptic localization would be uncertain. To address this possibility, we identified, from earlier experimental data, sets of genes expressed in brain tissue23, neurons26, excitatory neurons, and inhibitory neurons33. Within each set, we defined a gene as “potentially synaptic” if it was in any of the previously constructed FMRP, CELF4, RBFOX2, or SynaptomeDB gene sets, then stratified each of the neuronal/brain expression gene sets based on whether or not the genes were potentially synaptic (Fig. 4). No matter how we defined neuronally expressed genes, we observed that this tendency to contain an excess of dURVs in schizophrenia cases distinguished the potentially synaptic genes (which showed elevated rates of dURVs in schizophrenia) from other neuronally expressed genes (which did not) (Fig. 4).
Figure 4. dURVs enrichment in schizophrenia cases across brain cell type gene sets stratified by synaptic localization.
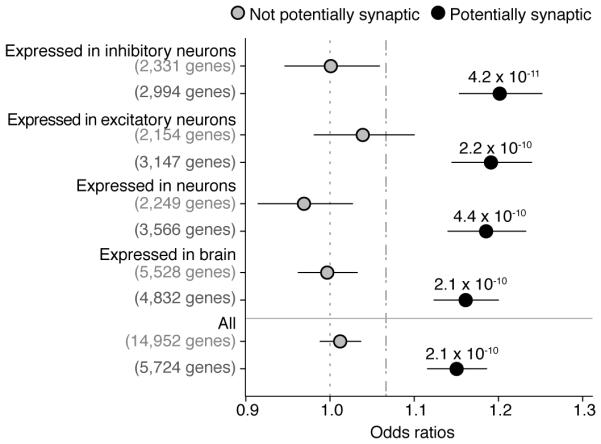
Odds ratios for enrichment of dURVs across genes expressed in brain tissue, neuronal cells, inhibitory neurons, and excitatory neurons, stratified between genes recognized as synaptic and genes recognized as non-synaptic. Synaptic genes were defined as genes part of either the FMRP, RBFOX2, CELF4, or SynaptomeDB gene sets. Enrichment and P values were computed using a logistic regression model using exome-wide dURV count as a covariate to correct for average exome-wide burden (dot-dashed line). Horizontal bars indicate 95% confidence intervals. Across each gene set, synaptic genes are clearly more enriched for variants in schizophrenia cases than non-synaptic genes.
These large constellations of potentially synaptic genes appeared to explain a large fraction (collectively more than 70%) of the exome-wide enrichment in dURVs (Fig. 4).
Protein complexes
Protein complexes have been used to define sets of genes with aligned activities, offering potentially meaningful ways to group genes for genetic analysis. We focused on genes encoding proteins that have been detected at synaptic complexes by co-immunoprecipitation with known synaptic components followed by mass spectrometric proteomic analyses. These gene sets have been the source of primary enrichment results in earlier studies of CNVs and rare and de novo SNVs in schizophrenia patients9,12,34. We observed case-control enrichment of dURVs among genes thus defined as encoding interactors with PSD-95 (OR=1.52; 95% CI 1.21–1.90; P = 0.0017), ARC and N-methyl-D-aspartate receptors (NMDAR)35 (OR=1.55; 95% CI=1.21–1.98; P = 0.0028) (Fig. 2). Despite these elevated levels of enrichment, these smaller gene sets explained much smaller fractions (collectively 4-12%) of the case-control enrichment in dURVs, perhaps reflecting that these gene sets include just a fraction of the proteins that are present at synapses.
More-complete ascertainment of the protein components of synaptic structures is an important future research direction that might advance functional analysis and interpretation of larger constellations of rare variants.
Overlap with GWAS genes and intellectual disability
We tested whether genes within the 108 GWAS loci recently identified in schizophrenia also contain an excess of dURVs. We observed a nominally significant enrichment in genes overlapping regions near common variants associated with schizophrenia24 (OR=1.37; 95% CI=1.09–1.73; P = 0.027) (Fig. 2) hinting at some degree of convergence. This overlap was greater than could be explained by any individual gene or small set of genes. Predicted targets of microRNA-13736, previously identified as localized near common SNPs associated with schizophrenia37, were also significantly enriched for dURVs (OR=1.13; 95% CI=1.09–1.18; P = 6.8 × 10−4) (Fig. 2).
Mutations associated with intellectual disability and developmental disorders are often also substantial risk factors for syndromic forms of autism and perhaps schizophrenia38–40,17. We did observe the dURV elevation to be concentrated in X-linked intellectual disability (XLID) genes41,42 (OR=1.88; 95% CI=1.34–2.64; P = 9.5 × 10−4) and in developmental disorder (DD) genes43 (OR=1.67; 95% CI=1.31-2.13; P = 1.6 × 10−4) (Online Methods). Of potential interest, we identified four dURVs in schizophrenia cases (and none in controls) in XLID gene KDM5C, an H3K4 methylation eraser gene44, 11 dURVs in cases (and 2 in controls) in DD gene KDM5B, another H3K4 methylation eraser gene45, and 11 dURVs in cases (and 3 in controls) in DD gene ITPR1, which encodes an inositol triphosphate receptor46 (Supplementary Tables 2 and 3). The enrichment of XLID variants was not different between female and male cases (Supplementary Fig. 9).
Ovrelap with de novo mutations ascertained in trios
We further tested for enrichment of dURVs in (i) genes overlapping de novo copy number variants (CNVs) previously found in individuals with schizophrenia, bipolar disorder, and autism (Supplementary Tables 4 and 5, see Online Methods), and (ii) genes in which de novo non-synonymous mutations were previously ascertained in individuals with autism, congenital heart disease, epilepsy, intellectual disability, and schizophrenia (Supplementary Tables 6 and 7, see Online Methods). Because de novo non-synonymous mutations have been ascertained in such a large number of genes, we sought to increase specificity by restricting this analysis to loss-of-function intolerant (LoF-intolerant) genes, as previously defined8. We observed a significant enrichment in genes within de novo deletions previously ascertained in schizophrenia cases (OR=1.34; 95% CI=1.13–1.59; P = 0.0052) (Fig. 5a), as well as an enrichment in loss-of-function intolerant genes with de novo non-synonymous mutations in schizophrenia cases (OR=1.41; 95% CI=1.25–1.60; P = 0.0011) (Fig. 5b).
Figure 5. dURVs enrichment in schizophrenia cases across genes previously observed as affected by de novo mutations.

Odds ratios for enrichment of dURVs across (a) genes overlapping de novo deletions and duplications in schizophrenia, bipolar disorder, and autism trios, and across (b) loss-of-function intolerant (LoF-intolerant) genes with observed de novo mutations in schizophrenia, intellectual disability, congenital heart disease, epilepsy, and autism trios. Enrichment and P values were computed using a logistic regression model using exome-wide dURV count (a) and dURV count across LoF-intolerant genes (b) as a covariate to correct for average burden (dot-dashed line). Horizontal bars indicate 95% confidence intervals.
DISCUSSION
By sequencing the exomes of 12,332 unrelated individuals from Sweden, including 4,946 affected with schizophrenia, we observed an exome-wide burden of dURVs in schizophrenia cases. This excess rare-variant burden – approximately 0.25 such variants per person (on a background of 4 such variants) – was several times greater than the schizophrenia-associated elevation in rates of gene-disruptive and protein-damaging de novo mutations, suggesting that the observed excess arose mostly from inherited variants. For less-rare (segregating) variants of even modest allele frequencies, we were unable to detect any excess in affected relative to unaffected individuals, consistent with a previous analysis of non-ultra-rare exonic variants in other cohorts47.
The excess of dURVs in schizophrenia cases largely resided in brain-expressed genes, and more specifically in genes that are expressed in neurons, rather than in other CNS cell types (Fig. 6). It is possible that earlier associations to small, protein-interaction-defined gene sets (such as PSD-95, NMDAR and ARC9,12,34), which appear to explain combined a much-smaller fraction of the exome-wide dURV burden in schizophrenia (collectively 4-12%), have been proxies for a far-wider set of rare-variant effects at synapses.
Figure 6. Dissection of the dURVs enrichment in schizophrenia cases.

An enrichment of URVs in the exomes of individuals affected with schizophrenia (relative to variants in control exomes) is observed exclusively in dURVs. After correcting for exome-wide dURV count, this enrichment is observed as concentrated in brain-specific genes while not in other tissue-specific genes, in neuron-specific genes while not in other brain-cell type-specific genes, and finally in potentially synaptic genes while not in other neuronally expressed genes.
Most of the excess of dURVs in affected individuals’ exomes appeared to be concentrated in a larger set of genes encoding potentially synaptic proteins. Genes whose transcripts are bound by FMRP or CELF4 – which transport a subset of neuronal RNAs to neuronal processes and synapses – or RBFOX2 – which regulates many synaptic RNAs and has been observed at synapses – explained considerably larger fractions (collectively more than 70%) of the global rare-variant enrichment observed in cases. Genes encoding RNAs bound by FMRP or RBFOX proteins have previously been shown to be enriched for mutations in subjects with autism and/or schizophrenia48,9,12,13,49, though it has been unclear whether such potential effects are a small or a large fraction of strongly risk-increasing variants. While it is tempting to attribute the association of schizophrenia with dURVs in FMRP–associated, CELF4–associated, and RBFOX2–associated genes to the specific biological activities of these proteins, we propose that their association may simply reflect the synaptic localization and function of the transcripts and proteins encoded by these genes.
We observed a significant overlap of the dURV excess with genes in which de novo non-synonymous mutations and deletions have been found in schizophrenia cases. We also observed a significant enrichment across intellectual disability genes on the X chromosome and in developmental disorder genes. This enrichment is compatible with observations of the role of intellectual disability genes in some cases of autism38–40 and schizophrenia17, though the penetrance of such mutations for schizophrenia may be much less than their penetrance for intellectual disability, and they may reside primarily in syndromic cases in which schizophrenia is preceded by other developmental disorders17.
The fact that an analysis of the current scale (4,877 cases, 6,203 controls, and 45,376 other genomes used to help identify ultra-rare variants) did not implicate individual genes of large effect in an unbiased exome-wide search – while documenting a very clear exome-wide elevation of hundreds of pathogenic variants across 4,877 individuals affected with schizophrenia relative to controls – lends further support to the emerging impression that the high polygenicity of schizophrenia extends to rare as well as common variants9,16. Because of the rareness of these variants and the infrequency with which any individual gene is affected by them – even among schizophrenia cases – the sequencing of much larger cohorts will be needed to identify the specific individual genes in which rare variants shape risk for schizophrenia.
ONLINE METHODS
Sample collection and sequencing
A total of 12,384 blood-derived DNA samples from Swedish research participants were collected from 2005 to 2013. Psychiatric cases with a diagnosis of schizophrenia or bipolar disorder were ascertained from the Swedish National Hospital Discharge Register as described in previous studies9,50, which captures all inpatient hospitalizations. Controls were randomly selected from population registers. Excluding subjects with bipolar disorder, age information at the time of DNA sampling was available for each individual. All subjects provided informed consent; institutional human subject committees approved the research (UNC IRB # 04-1465). All procedures were approved by the ethical committees in Sweden and in the United States.
The 12,384 samples collected were sequenced in twelve separate waves. The first wave employed an earlier version of the hybrid-capture procedure (Agilent SureSelect Human All Exon Kit), which targets ~28 million base pairs of the human genome, partitioned in ~160,000 intervals, whereas the samples from the other waves used a newer version (Agilent SureSelect Human All Exon v.2 Kit), which targets ~32 million base pairs of the human genome, partitioned in ~190,000 intervals. The first wave was sequenced using Illumina GAII instruments and the remaining waves were sequenced using Illumina HiSeq 2000 and HiSeq 2500 instruments, with pair ended sequencing reads of 76 base pairs across all waves. Sequencing was performed at the Broad Institute of MIT and Harvard across the period of time from 2010 to 2013. With the exception of the first wave, we did not observe significant differences across waves and cases status beyond what could be explained by ancestry (Supplementary Fig. 10).
This cohort has been previously analyzed in relation to schizophrenia for common variants51,37,52,50,24 and copy number variants52,53, and in relation to somatic mosaic mutations54. Exome sequence data for approximately half of the individuals in the cohort had already been analyzed in relation to schizophrenia phenotype in a previous study9 and in a more recent study17.
Preliminary quality control for individuals
Exome sequence data from 12,384 samples was aligned against the GRCh37 human genome reference with bwa aln 0.5.955 and further processed using the GATK framework56. Genotype calls were generated using GATK Haplotype Caller version 3.1-144-g00f68a3 and best practices57,58. Variants filtered out by the GATK Variant Quality Score Recalibration (VQSR) tool were excluded. Genotypes over sites with less than 10x sequencing coverage were set to missing. We identified and removed 4 duplicate individuals and 48 individuals with a first degree relationship (Supplementary Fig. 11) with other individuals in the cohort using the plink59,60 software. We then computed the number of ultra-rare single nucleotide polymorphysms (SNPs) and indels never observed in ExAC8 for each of the remaining 12,332 samples and identified one individual with 1,757 ultra-rare SNPs and 22 ultra-rare indels from the sixth sequencing wave (see Supplementary Table S5F from Genovese et al.54), four individuals with between 92 and 127 ultra-rare indels from the first sequencing wave, five individuals from waves 11 and 12 with between 410 and 496 ultra-rare SNPs due to African ancestry. These 10 individuals were excluded from further analysis. The resulting individuals had a range of 5-259 ultra-rare SNPs and 0-19 ultra-rare indels (Supplementary Fig. 12a). We further removed 15 individuals for whom the reported sex and the inferred sex from inbreeding coefficients on the X chromosome mismatched (Supplementary Fig. 13), including 7 individuals with 47, XXY karyotype (Klinefelter syndrome), and 94 individuals with more than 100 URVs.
Association with common variants
A logistic regression model was used to estimate association between single variants and schizophrenia phenotype correcting for sex and the first five principal components using plink59,60. We identified two loci (Supplementary Fig. 14) with statistically significant associations (p<10−6) replicating a couple of common variants associations previously observed for this cohort50: a single variant rs281766 on chromosome 2 in the UTR5 of genes TYW5 and C2orf47, a variant in strong linkage disequilibrium with the seventh strongest independently associated variant in the largest meta-analysis for schizophrenia24, and seven variants in the MHC region around the HLA genes, also a region with extensive linkage to known causal variants associated with schizophrenia61.
Variant annotation
We annotated all genotyped variants with SnpEff 4.2 (build 2015-12-05)62 using Ensembl gene models from database GRCh37.75. We further annotated variants with SnpSift 4.2 (build 2015-12-05)63 using annotations from database dbNSFP 2.964,65. Variants identified within transcripts from UCSC known genes66 were further classified into four groups:
synonymous: whenever classified with synonymous effect by SnpEff
missense non-damaging: whenever classified with missense effect but not classified as damaging (by the criteria below)
putatively protein-damaging: whenever classified with MODERATE impact by SnpEff and further predicted as damaging by each among SIFT67, PolyPhen-268, LRT69, Mutation Taster70, Mutation Assessor71, and PROVEAN72 algorithms or classified as either in-frame indels or protein-protein-contact variants73
gene disruptive: whenever classified with HIGH impact by SnpEff with the exclusion of protein-protein-contact variants
Notice that FATHMM74 predictions included in dbNSFP were not used due to poor performance with respect to minor allele count (Supplementary Fig. 1) and a small number of variants defined as damaging by the predictor (Supplementary Fig. 3b). The final predictor performed better than all other individual predictors (Supplementary Fig. 3b) but it was not overfit as to be the best predictor for this cohort (Supplementary Fig. 15).
Estimation of principal components
Out of a total of 1,753,312 variants passing VQSR filters, 66,874 were identified as in common with variants from the 1000 Genomes project phase 1 dataset75 and included as part of the Omni2.5 genotype array. We used this subset of highly confident variants to estimate population stratification. We selected exclusively Omni2.5 polymorphic sites because more robust in the 1000 Genomes dataset to artifacts due to the heterogeneity of the sequencing technologies used within the 1000 Genomes project. We then further restricted to variants with minor allele frequency larger than 1% in both the Sweden and the 1000 Genomes dataset and we pruned for variants in linkage disequilibrium using plink59,60 (with command line '--indep 50 5 2'). We then merged the Sweden and the 1000 Genomes dataset and computed principal components using plink and GCTA76 (Supplementary Fig. 12c-d). Estimated 3rd and 5th principal components corresponded to previously observed Finnish and Northern-Southern Sweden clines12 (Supplementary Fig. 12d), while 1st, 2nd, and 4th principal components corresponded to the three main principal components in the 1000 Genomes project phase 1 distinguishing African, East Asian, and Native American ancestry. While principal components did correlate with overall amounts of URVs (Supplementary Fig. 12e-f), rather than removing individuals with exotic ancestry based on principal components loading, we simply removed individuals with more than 100 URVs and we included sex, year of birth (Supplementary Fig. 12b), exome capturing kit, the first 20 principal component loadings, and the total number of URVs for each individual as covariates in all statistical analysis involving URVs and dURVs.
Quality control for common variants
Variants were excluded whether failing the GATK VQSR tool (117,629 variants), having inbreeding coefficient less than zero (that is, more observed heterozygotes than expected) while at the same time failing a Hardy-Weinberg disequilibrium test with a false discovery rate of 10−6 (8,306 additional variants), or whether associating with any of the 146 batches among which the cohort was split for sequencing in the sequencing facility at the Broad Institute (3,700 additional variants). Due to prevalent population stratification within batches, we estimated unusual associations with a logistic regression model including sex and the first 20 principal components loadings as covariates.
Excess of dURVs
For each gene set, to estimate the excess of dURVs in cases with schizophrenia (or the odds ratios for schizophrenia phenotype) we used a linear (or logistic) regression model correcting for: (i) sex; (ii) overall URV count; (iii) birth year; (iv) the hybrid selection kit used to enrich for exome sequence; and (v) the first twenty principal components estimated from exome-wide SNP genotypes. When estimating P values, to estimate the importance of each gene set with respect to the observed exome-wide enrichment, we further corrected for exome-wide dURV count. This expedient allows to better estimate the importance of each gene set irrespectively of its size and to answer the more precise question of whether a gene set concentrates the exome-wide dURV enrichment better than the average gene.
Construction of gene sets
We used different resources to build the gene sets for which the burden of dURVs was computed:
For missense constrained genes we used genes from supplementary table 2 of Samocha et al.22.
For loss-of-function intolerant (LoF-intolerant) genes we used genes from Lek et al.8 available online (ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.3/functional_gene_constraint/).
For genes with expression specific to the brain, we used expression table from supplementary data set 1 of Fagerberg et al.23 and we selected genes for which expression in brain was four times higher than the median expression across all 27 different tissues (Supplementary Fig. 7).
For brain genes with expression specific to neurons, we used expression table from supplementary table S3b of Cahoy et al.26 and we selected genes for which log-expression in Neurons P7n cell type was 0.5 greater than the median log-expression across 11 central nervous system cell types (Supplementary Fig. 8a).
For RBFOX2 and RBFOX1/3 gene sets we selected genes from supplementary table S1 of Weyn-Vanhentenryck et al.31 for which at least, respectively, one Rbfox2 tag count was measured greater than or equal to 4, and one of the sum of Rbfox1 and Rbfox3 tag counts was greater than or equal to 12. A single gene set was generated for RBFOX1 and RBFOX3 due to high correlation between tag counts for the two genes.
Instead of using the classical FMRP Darnell gene set of 842 mouse genes from supplementary table S2A of Darnell et al.27 including all genes with FDR<0.01, we used a larger gene set of 1,285 mouse genes from supplementary table S2C of Darnell et al.27 including genes with FDR<0.1.
For CELF4 we used genes with “iCLIP occupancy” greater than 0.2 from supplementary table S4 of Wagnon et al.28.
To create a gene set with synaptic genes we included 1,887 genes from the SynaptomeDB32 from the presynaptic proteins, presynaptic activezone, synaptic vesicles, and postsynaptic density categories.
To create a set of genes expressed in brain, we used expression table from supplementary data set 1 of Fagerberg et al.23 and we selected genes for which fragments per kilobase of transcript per million (FPKM) in brain was larger than 5.
To create a set of genes expressed in neurons, we used expression table from supplementary table S3b of Cahoy et al.26 and we selected genes for which log-expression in Neurons P7n cell type was larger than 9.
To create sets of genes expressed in excitatory and inhibitory neurons, we used expression table from Table S2 of Mo et al.33 and we selected genes for which the average transcripts per million (TPM) of, respectively, excitatory pyramidal neurons and inhibitory neurons, the latter including parvalbumin (PV)-expressing fast-spiking or vasoactive intestinal peptide (VIP)-expressing interneurons, was larger than 50. Similarly for sets of genes specific for each neuron type, we selected genes expressed more than 5 times the minimum expression observed across all types (Supplementary Fig. 8b).
To generate a list of predicted targets of microRNA-137, we used human targets with good mirSVR score from Betel at al.36 available online (http://www.microrna.org/).
To generate PSD-95 complex gene sets, we used a gene list generated from human cortex biopsy data35 available online (http://www.genes2cognition.org/db/GeneList/L00000049).
To compute a combined NMDAR and ARC complexes gene set, we used genes from Table S9 of Kirov et al.34.
For genes implicated in common variant association studies, we used genes overlapping 62 regions from the 108 regions known to be associated with schizophrenia24, for which the overlap yielded at most four genes.
To generate genes involved in X-linked intellectual disability (XLID) we used gene lists available online (see next section).
To generate genes involved in developmental disorder, we selected genes from supplementary table 3 of McRae et al.43.
For genes implicated in de novo CNV studies, we used genes overlapping de novo deletions and duplications identified in autism77–84 and identified in bipolar disorder and schizophrenia34,85–88 cases (Supplementary Tables 4 and 5).
For genes implicated in de novo nonsynonymous mutations from exome sequencing studies, we used genes identified as mutated in autism13,49,89,90, epilepsy91,92, congenital heart disease93, intellectual disability94–97, and schizophrenia98,19,99,100,12 (Supplementary Tables 6 and 7).
Enrichment in X-linked intellectual disability genes
We used three different resources available online to define X-linked intellectual disability (XLID) genes:
XLID OMIM genes were defined as those genes causing mental retardation phenotype in the OMIM database101 (http://omim.org/geneMap/X)
XLID GCC genes were defined as those genes tested by the Greenwood Genetic Center102 (http://www.ggc.org/diagnostic/tests-costs/test-finder/test-finder.html?id=242).
XLID Chicago genes were those tested by the Genetic Services Laboratories of the university of Chicago103,41,42,104 (http://dnatesting.uchicago.edu/tests/x-linked-non-specific-intellectual-disability-sequencing-panel).
We tested for enrichment of dURVs in the above gene sets and in genes including all of the above gene sets, and seperately genes believed to escape and not escape X-inactivation in humans105, genes from OMIM including autosomal genes causing intellectual disability, and genes involved in developmental disorders through de novo mutations43. We tested for enrichment separately in males and females, as well as combined (Supplementary Fig. 9).
While XLID genes and developmental disorders genes were strongly enriched in schizophrenia cases, autosomally linked intellectually disability genes were not. This discrepancy might reflect a better characterization of intellectual disability genes on the X chromosome due to a more straightforward study design for how these genes where discovered. We also observed that XLID genes which escape X-inactivation were more enriched than other XLID genes. This might reflect a disproportionate contribution to intellectual disability and psychosis from dosage sensitive brain-related genes on the X chromosome106–108. We did not observe a difference in effect sizes between males and females.
Similarly to rare variants enrichment, common variants associated with schizophrenia are localized near XLID genes CNKSR2 and NLGN4X, both of which escape X inactivation, as well as non-XLID gene PJA124.
No detectable enrichment of less-rare variants
Given the strong case-control enrichment of dURVs in potentially synaptic genes, we used these genes to perform a sensitive evaluation of whether we could observe an increased burden of less-rare disruptive and damaging variants in the same set. Using a standard burden test for non-ultra-rare variants with minor allele count 10 or less and controlling for covariates, we observed no statistically significant enrichment of disruptive and damaging variants in schizophrenia cases compared to controls (P = 0.59), wheareas the same test was highly significant when restricted to URVs (P = 1.7 × 10−19, without controlling for exome-wide enrichment) (Supplementary Table 8).
Variance explained
While a predictor based on common variants24,109 explained 15% of the variance in schizophrenia liability in this cohort, a predictor based on the cumulative burden of dURVs in all genes explained only 0.48% (P = 1.5 × 10−10), and a similar predictor in potentially synaptic genes explained only 0.92% (Nagelkerke's coefficient of determination) (P = 6.3 × 10−19). We also attempted to generate a polygenic score based on the cumulative number of dURVs in genes that had burden of dURVs in cases greater than or equal to controls. Using a leave-one-out strategy, the resulting predictor explained 0.47% (P = 2.3 × 10−10) of the phenotypic variability. These estimates are naturally lower bounds on the effect of rare variants; knowledge of the correct effect size of each variant would significantly increase the predictive value of dURVs, though obtaining such knowledge will require sequencing a vastly larger number of exomes.
Supplementary Material
ACKNOWLEDGEMENTS
We thank C. Usher for comments on the manuscript and work on the figures. This study was supported by grants from the National Human Genome Research Institute (U54 HG003067, R01 HG006855 to S.A.M.), the National Institute of Mental Health (R01 MH077139 to P.F.S. and RC2 MH089905), the Stanley Center for Psychiatric Research, the Alexander and Margaret Stewart Trust, and the Sylvan C. Herman Foundation.
Footnotes
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
Accession codes dbGaP: phs000473.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
G.G. and S.A.M. designed the analyses and wrote early drafts of the manuscript. G.G. performed the analyses. M.F. contributed to analyses of de novo mutated genes, D.M.R and E.A.S. contributed with the specific design of the analyses. K.C. contributed with sample processing and data management, M.L., J.L.M., S.M.P., P.S., P.F.S., and C.M.H. contributed with sample and phenotpye collection. All authors contributed to interpretation of the findings and revisions of the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Data availability
Scripts used to perform all of the analyses are available at https://github.com/freeseek/gwaspipeline and data are available through dbGAP at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000473
References
- 1.McGrath J, Saha S, Chant D, Welham J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 2008;30:67–76. doi: 10.1093/epirev/mxn001. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry. 2003;60:1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 3.Lichtenstein P, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet Lond. Engl. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bundy H, Stahl D, MacCabe JH. A systematic review and meta-analysis of the fertility of patients with schizophrenia and their unaffected relatives. Acta Psychiatr. Scand. 2011;123:98–106. doi: 10.1111/j.1600-0447.2010.01623.x. [DOI] [PubMed] [Google Scholar]
- 5.Power RA, et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiatry. 2013;70:22–30. doi: 10.1001/jamapsychiatry.2013.268. [DOI] [PubMed] [Google Scholar]
- 6.Zuk O, et al. Searching for missing heritability: designing rare variant association studies. Proc. Natl. Acad. Sci. U. S. A. 2014;111:E455–464. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stefansson H, et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505:361–366. doi: 10.1038/nature12818. [DOI] [PubMed] [Google Scholar]
- 8.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purcell SM, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacArthur DG, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 12.Fromer M, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iossifov I, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am. J. Hum. Genet. 2013;92:841–853. doi: 10.1016/j.ajhg.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinforma. Oxf. Engl. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 16.Takata A, et al. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron. 2014;82:773–780. doi: 10.1016/j.neuron.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh T, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 2016;19:571–577. doi: 10.1038/nn.4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rujescu D, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 2009;18:988–996. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu B, et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 2012;44:1365–1369. doi: 10.1038/ng.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Millar JK, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- 21.Pinto D, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014;94:677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samocha KE, et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014;46:944–950. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fagerberg L, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics MCP. 2014;13:397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finucane HK, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. Off. J. Soc. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darnell JC, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagnon JL, et al. CELF4 regulates translation and local abundance of a vast set of mRNAs, including genes associated with regulation of synaptic function. PLoS Genet. 2012;8:e1003067. doi: 10.1371/journal.pgen.1003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J-A, et al. Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron. 2016;89:113–128. doi: 10.1016/j.neuron.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamada N, et al. Biochemical and morphological characterization of A2BP1 in neuronal tissue. J. Neurosci. Res. 2013;91:1303–1311. doi: 10.1002/jnr.23266. [DOI] [PubMed] [Google Scholar]
- 31.Weyn-Vanhentenryck SM, et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014;6:1139–1152. doi: 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pirooznia M, et al. SynaptomeDB: an ontology-based knowledgebase for synaptic genes. Bioinforma. Oxf. Engl. 2012;28:897–899. doi: 10.1093/bioinformatics/bts040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mo A, et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron. 2015;86:1369–1384. doi: 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirov G, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bayés A, et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011;14:19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson EB, et al. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc. Natl. Acad. Sci. U. S. A. 2014;111:15161–15165. doi: 10.1073/pnas.1409204111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson EB, Neale BM, Hyman SE. Genetic research in autism spectrum disorders. Curr. Opin. Pediatr. 2015;27:685–691. doi: 10.1097/MOP.0000000000000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson EB, et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat. Genet. 2016;48:552–555. doi: 10.1038/ng.3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moeschler JB. Genetic evaluation of intellectual disabilities. Semin. Pediatr. Neurol. 2008;15:2–9. doi: 10.1016/j.spen.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 42.Gécz J, Shoubridge C, Corbett M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. TIG. 2009;25:308–316. doi: 10.1016/j.tig.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 43.McRae JF, et al. Prevalence, phenotype and architecture of developmental disorders caused by de novo mutation. bioRxiv. 2016:49056. [Google Scholar]
- 44.Jensen LR, et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiang Y, et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 2007;104:19226–19231. doi: 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsumoto M, et al. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- 47.Richards AL, et al. Exome arrays capture polygenic rare variant contributions to schizophrenia. Hum. Mol. Genet. 2016;25:1001–1007. doi: 10.1093/hmg/ddv620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Rubeis S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ripke S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.International Schizophrenia Consortium et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergen SE, et al. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol. Psychiatry. 2012;17:880–886. doi: 10.1038/mp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szatkiewicz JP, et al. Copy number variation in schizophrenia in Sweden. Mol. Psychiatry. 2014;19:762–773. doi: 10.1038/mp.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKenna A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. Ed. Board Andreas Baxevanis Al. 2013;11:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sekar A, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cingolani P, et al. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012;3:35. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013;34:E2393–2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu F, et al. The UCSC Known Genes. Bioinforma. Oxf. Engl. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 67.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 68.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 71.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PloS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shihab HA, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Autism Genome Project Consortium et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marshall CR, et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Itsara A, et al. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Levy D, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 84.Gilman SR, et al. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 86.Malhotra D, et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72:951–963. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Noor A, et al. Copy number variant study of bipolar disorder in Canadian and UK populations implicates synaptic genes. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2014;165B:303–313. doi: 10.1002/ajmg.b.32232. [DOI] [PubMed] [Google Scholar]
- 88.Georgieva L, et al. De novo CNVs in bipolar affective disorder and schizophrenia. Hum. Mol. Genet. 2014;23:6677–6683. doi: 10.1093/hmg/ddu379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang Y, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 2013;93:249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Epi4K Consortium et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.EuroEPINOMICS-RES Consortium. Epilepsy Phenome/Genome Project. Epi4K Consortium De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 2014;95:360–370. doi: 10.1016/j.ajhg.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zaidi S, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de Ligt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 95.Rauch A, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 96.Gilissen C, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–347. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- 97.Hamdan FF, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10:e1004772. doi: 10.1371/journal.pgen.1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Girard SL, et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 2011;43:860–863. doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- 99.Gulsuner S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McCarthy SE, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry. 2014;19:652–658. doi: 10.1038/mp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stevenson RE, Schwartz CE, Rogers RC, Rogers RC. Atlas of X-linked intellectual disability syndromes. Oxford University Press; 2012. [Google Scholar]
- 103.Moeschler JB, Shevell M, American Academy of Pediatrics Committee on Genetics Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics. 2006;117:2304–2316. doi: 10.1542/peds.2006-1006. [DOI] [PubMed] [Google Scholar]
- 104.Rauch A, et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am. J. Med. Genet. A. 2006;140:2063–2074. doi: 10.1002/ajmg.a.31416. [DOI] [PubMed] [Google Scholar]
- 105.Cotton AM, et al. Analysis of expressed SNPs identifies variable extents of expression from the human inactive X chromosome. Genome Biol. 2013;14:R122. doi: 10.1186/gb-2013-14-11-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Crow TJ. The XY gene hypothesis of psychosis: origins and current status. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2013;162B:800–824. doi: 10.1002/ajmg.b.32202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ji B, Higa KK, Kelsoe JR, Zhou X. Over-expression of XIST, the Master Gene for X Chromosome Inactivation, in Females With Major Affective Disorders. EBioMedicine. 2015;2:907–916. doi: 10.1016/j.ebiom.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Crow TJ. Is Psychosis a Disorder of XY Epigenetics? EBioMedicine. 2015;2:792–793. doi: 10.1016/j.ebiom.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vilhjálmsson BJ, et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am. J. Hum. Genet. 2015;97:576–592. doi: 10.1016/j.ajhg.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.