

Abstract
T follicular helper (Tfh) cells are a subset of CD4+ T lymphocytes that promote the development of humoral immunity. While the triggers required for the differentiation of the other major T helper subsets are well defined, those responsible for Tfh responses are still poorly understood. We determined that mice immunized with either peptide or protein antigens emulsified in incomplete Freund’s adjuvant or related water-in-oil adjuvants develop a highly polarized response in which the majority of the antigen-specific CD4+ T cells are germinal center-homing CXCR5+Bcl6+ Tfh. Despite the absence of exogenous microbial PAMPs, the Tfh responses observed were partially MyD88 dependent. Importantly, in addition to IL-6, T cell intrinsic type I IFN signaling is also required for optimal Tfh polarization. These findings suggest that water-in-oil adjuvants promote Tfh dominated responses by triggering endogenous alarm signals that in turn induce type I IFN-dependent differentiation pathway functioning in T cells.
Introduction
A major challenge of vaccine research is to induce high titer durable antibody responses against protein antigens that represent major targets for disease prevention. T follicular helper (Tfh) CD4+ T lymphocytes are well recognized as specialized providers of T cell help to B cells within germinal centers of secondary lymphoid organs. B lymphocyte responses are dramatically impaired in the absence of Tfh cells (1) and boosting Tfh function has been shown to promote long-lived humoral immunity by amplifying antibody production and class switch recombination (2). Therefore, an understanding of the mechanisms underlying the induction and regulation of Tfh cells could provide important clues for the rational design and development of more durable and effective antibody-based vaccines.
In comparison with other helper T cell subsets, Tfh differentiation in vivo is poorly understood and importantly cannot be fully recapitulated in vitro. Tfh cells express the lineage defining transcription repressor Bcl6 (3-5), which is expressed by naive T cells early after encounter with antigen-presenting dendritic cells (6, 7) in the presence of IL-6 (1). This leads to the up-regulation of the chemokine receptor CXCR5 and cell migration from the cortex to the B cell follicles of secondary lymphoid organs. At the T-B border, pre-Tfh cells interact with cognate B cells, resulting in their differentiation into either short-lived extrafollicular plasmablasts or their co-migration with pre-Tfh cells into B cell follicles to form germinal centers. The proliferation and B cell helper activities of Tfh are in turn controlled by a subset of Foxp3+ T follicular regulatory cells (Tfr) that also express Bcl6 and CXCR5 and arise either from thymically-derived natural Treg cells or from naïve cells in the periphery (8, 9).
Numerous studies have investigated Tfh differentiation in the context of infection and/or by the use of adoptive transfer experiments with TCR transgenic T cells. In an attempt to better characterize the minimal endogenous signals required for Tfh differentiation, we utilized a basic model of immunization with peptide or protein antigen emulsified in water-in-oil (W/O) adjuvants in the absence of microbial immunostimulants. The primary adjuvant chosen for these studies was incomplete Freund’s adjuvant (IFA), which consists of paraffin oil and mannide mono-oleate as a surfactant. Although less immunostimulatory than complete Freund’s adjuvant (CFA), which is supplemented with heat killed Mycobacterium tuberculosis extracts, IFA has previously been shown to promote substantial T cell-dependent antibody production associated with Tfh activity (10). In performing these studies, we also compared IFA with other more chemically defined, clinically acceptable, W/O adjuvants.
As reported here, we found that in contrast to other adjuvant preparations, W/O only adjuvants appear to selectively trigger the polarization of naïve T cells into germinal center (GC)-associated Tfh. In addition, we uncovered unexpected roles for MyD88 and Type I IFN signaling in driving this preferential Tfh response. Together, these findings establish a convenient model for studying the signals involved in Tfh differentiation and support a major role for non-microbial stimuli in the development of adjuvants designed to promote Tfh activity.
Material and Methods
Mice
Male or female (7-14 weeks old) C57BL/6 (CD45.2/2), B6.SJL-Cd45a(Ly5a)/Nai (CD45.1/1) and C57BL/6J x B6.SJL-CD45a(Ly5a)/Nai F1 (CD45.1/2) mice were used alternatively as wild type (WT) animals and supplied by Taconic Farms via a contract with the National Institute of Allergy and Infectious Diseases (NIAID). Il12p40−/−, Il1r1−/−, Tlr3−/−, Ifnar−/−, Ifih1−/− (MDA5), muMT−/− and Tcra−/− mice were also obtained from Taconic Farms. Il18−/− (stock #004130) and Sting−/− (Tmem−/−, strain #17537) were from the Jackson Laboratory. Myd88−/− and Trif−/− mice, backcrossed to B6 for 10 generations, were originally obtained from S. Akira (Osaka University, Osaka, Japan). Stat1fl/fl CD4 CRE and Stat3fl/fl CD4 CRE mice were a gift from J. O’Shea (NIAMS, NIH), Tlr4−/−, Tlr5−/−, Tlr9−/− mice from G. Trinchieri (NCI, NIH) and Unc92b−/− animals from E. Latz and D. Golenbock (University of Massachusetts, Worcester, MA). All mice were maintained in an AALAC-accredited animal facility at the NIAID, National Institutes of Health (Bethesda, MD). Mice were used according to animal study proposals approved by the NIAID Animal Care and Use Committee.
Bone marrow reconstitutions and chimeras
Congenically-marked recipient mice were lethally irradiated (950 rad) and on the same day reconstituted with a total of 107 donor bone-marrow (BM) cells isolated from WT, Myd88−/− or Ifnar−/− animals or with a 50/50 mixture of congenically marked BM cells from either WT and Myd88−/− or WT and Ifnar−/− animals. Mice were allowed to reconstitute for 8-10 weeks before immunizations.
Adjuvants
Alum hydroxide (Alhydrogel 2%) was from Brenntag (Baltimore, MD), IFA and CFA were from Sigma-Aldrich (St. Louis, MO) and Montanide ISA 51VG and Montanide ISA 720VG from Seppic (Fairfield, NJ). The adjuvant/antigen emulsions were created using two silicone and latex free syringes (Air-Tite, Virginia Beach, VA) connected with a double female luer lock adapter (Air-Tite, Virginia Beach, VA) and mixed for up to 8 minutes.
Antigens
AS15 (AVEIHRPVPGTAPPS), ESAT6 (QQWNFAGIEAAASA) or Ag85b (FQDAYNAAGGHNAVF) peptides were provided by Jan Lukszo at the Research Technologies Branch, NIAID. Recombinant Ag85b protein from M. tuberculosis was obtained from Biodefense and Emerging Infections Research Resources Repository (BEI Resources, Manassas, VA, USA) and from Aeras (Rockville, MD).
Immunization and harvest
Unless otherwise indicated, immunizations were performed by subcutaneous (s.c) injection at four sites along the back (50 μl/site) with Alum, IFA or CFA containing a total of 100μg or less if indicated of AS15, ESAT6 or Ag85b peptides or a total of 4μg of Ag85b protein. Unless otherwise stated, the draining (inguinal, axillary, and brachial) lymph nodes (LNs) were harvested 10-14 days after immunization. LNs were dissociated through 100-μm cell strainers, and 2×106 LN cells were plated in a round-bottom 96-well plate and stained for flow cytometry. To measure antibody responses, WT and Tcra−/− mice were immunized twice with Ag85b in IFA, first with s.c. priming immunization and then boosted 21 days later by i.p. immunization.
Flow cytometry
I-A(b) T. gondii TGME49_012300 605-619 AVEIHRPVPGTAPPS (AS15), I-A(b) Mtb ESAT6 4-17 QQWNFAGIEAAASA and I-A(b) Mtb Ag85b 280-294 FQDAYNAAGGHNAVF tetramers were obtained from the NIH Tetramer core facility. Tetramer staining (15-30 μg/ml) was performed at 37°C for one hour. Antibodies against the following molecules were purchased from eBioscience (San Diego, CA), BioLegend (San Diego, CA), or BD Biosciences (San Jose, CA): CD4 (RM4-4), CD45.1 (A20), CD45.2 (104), CD44 (IM7), CD62L (MEL-14), SLAM, (TC15-12F12.2), CXCR5 (SPRCL5), PD-1 (29F.1A12), ICOS (7E.17G9), GL7 (GL7), Foxp3 (FJK-16s), Tbet (4B10), RORgt (B2D), Bcl6 (K112-91). Fixable Viability Dye eFluor 780 was from eBioscience (San Diego, CA). All samples were acquired on an LSR Fortessa flow cytometer (BD Biosciences, (San Jose, CA)) and analyzed using FlowJo software (Tree Star).
Immunofluorescence and confocal microscopy
dLNs were harvested and fixed with PLP buffer (0.05 M phosphate buffer containing 0.1 M L-lysine [pH 7.4], 2 mg/ml NaIO4, and 10 mg/ml paraformaldehyde) for 12 hours. Following fixation, dLNs were incubated in 30% sucrose for 6 hours before embedding in OCT compound (Tissue-Tek). 30 μm sections were cut on a CM3050S cryostat (Leica) and adhered to Super Frost Plus Gold slides (Electron Microscopy Services). Frozen sections were permeabilized and blocked for 1-2 hours in PBS containing 0.3% Triton X-100 (Sigma, St. Louis, MO), 1% normal mouse serum, 1% bovine serum albumin, and 10% normal goat serum. Sections were stained for a minimum of 12 hours at 4°C with the following directly conjugated antibodies from BioLegend (San Diego, CA) unless stated otherwise: anti-CD4 (GK1.5), anti-B220 (RA3-6B2), anti-IgD (11-26c.2a), anti-PD-1 (29F.1A12), and anti-Bcl6 (K112-91, BD Biosciences, San Jose, CA). Stained slides were mounted with Fluoromount G (eBioscience, San Jose, CA) and sealed with a glass coverslip. Each section was visually inspected by epifluorescent light microscopy and several representative sections from different dLNs were acquired using a SP8 confocal microscope (Leica), objectives with 40X magnification (NA 1.3) or 63X magnification (NA 1.4). Fluorophore emission was collected on separate detectors with sequential laser excitation used to minimize spectral spillover. To calculate the total number of GCs per dLN, whole dLNs were serially sectioned and GCs were scored for each section. GC area was calculated by using the surface creation module in Imaris (Bitplane). Graph plots depict the widest area for each GC present in the dLNs examined.
ELISA
Total anti-Ag85b specific IgM, IgA and IgG antibodies were detected in the serum. In brief, 96-well plates were coated with 1μg/ml Ag85b, incubated with three fold serial dilutions of the sera followed by an incubation with HRP-coupled anti IgM, IgA and IgG antibodies. ABTS was added as a substrate and optical density measured at 405nm.
Quantitative PCR
Total RNA from draining lymph nodes was isolated using MagMAX™-96 Total RNA Isolation Kit (Thermo Fisher, Waltham, MA) according to the manufacturer’s protocol and cDNA was reverse transcribed with Superscript III reverse transcriptase and random primers (Thermo Fisher, Waltham, MA). Quantitative PCR was performed on an ABI Prism 7900 HT Sequence Detection System using Power SYBR Green Master Mix (Applied Biosystems/Life Technologies) for detection. Fold induction of gene expression was calculated by normalizing mRNA levels for each sample to levels of hypoxanthine guanine phosphoribosyltransferase. The following primer pairs were used: Hprt F, 5′-GCCCTTGACTATAATGAGTACTTCAGG-3’ and R, 5′-TTCAACTTGCGCTCATCTTAGG-3’, pan-ifna F 5′-SAWCYCTCCYAGACTCMTTCTGCA-3’ and R 5’-TATDTCCTCACAGCCAGCAG-3’, ifnb F 5′- CCCTATGGAGATGACGGAGA-3’ and R 5′-ACCCAGTGCTGGAGAAATTG-3’
In vivo treatment
In vivo cytokine blockade was performed by i.p. injections of 500μg lg MP5-20F3 mAb (anti-mIL-6R, BioXcell, West Lebanon, NH) or Ig MAR1-5A3 mAb (anti-mIFNAR-1, BioXcell, West Lebanon, NH) for three consecutive days at the time of immunization followed by 250μg once every three days for the remainder time of the experiment. Mice were partially depleted of gut microbiota by providing antibiotic-treated water (containing ampicillin (1g/l), vancomycin (500mg/l), neomycin trisulfate (1g/l), and metronidazole (1g/l)) (Sigma, St. Louis, MO) ad libitum to WT animals for a month prior to immunization.
Statistical analysis
The statistical significance of differences between groups was analyzed via unpaired Mann-Whitney t test or paired t test (when comparing groups in the mixed bone marrow chimera settings) using GraphPad Prism software version 5.0c (GraphPad). The p values are shown as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Water-in-oil adjuvants in the absence of microbial components promote Tfh polarization in response to peptide immunization
IFA and CFA contain non-metabolizable paraffin oil, resulting in W/O emulsions and form long-lasting depots at sites of injection. CFA is IFA supplemented with heat-killed Mycobacterium tuberculosis and induces a strong pro-inflammatory response associated with Th1, Th17 and Tfh development along with B cell maturation (10). However, oil and surfactant-only adjuvants such as IFA are also known to promote antibody responses (11). This led us to address the question of whether these adjuvants promote Tfh responses in the total absence of additional microbial stimuli.
We first performed kinetic studies comparing the responses of mice following subcutaneous immunization with antigen either admixed with alum or emulsified in IFA or CFA. The immunogen chosen for our initial studies was a 15mer peptide (AS15) representing a highly immunodominant T cell epitope present in an unidentified protein from Toxoplama gondii (12). We found that IFA was able to promote a substantial AS15-specific CD4+ T cell response in draining lymph nodes (supplementary Figure 1 A and 1B) that includes Tfh cells as defined by the co-expression of CXCR5 and Bcl6 (supplementary Figure 1C and 1D). At the peak of the response at approximately day 12 post immunization, both the frequency (Figure 1A and Figure 1B) and the total number of antigen-specific CD4+ T cells (Figure 1C) were significantly increased after IFA as compared with alum immunization, although this response was lower than that observed with CFA emulsified Ag. Interestingly, a significantly higher percentage of antigen-specific Tfh cells were induced following IFA compared to CFA immunization (Figure 1A and 1D), although the overall number of antigen-specific Tfh cells was lower (Figure 1E). In contrast, Ag incorporated in alum induced only low-level Tfh responses in this model (Figure 1A, 1D and 1E). The high frequency of Tfh observed following immunization with IFA was not due to the choice of Ag, as similar results were seen following administration of immunogenic peptides from Mycobacterium tuberculosis proteins (Supplementary Figure 1E-H).
Figure 1. IFA promotes Tfh polarization in response to peptide or protein immunization.

Mice were immunized with AS15 peptide in PBS, in Alhydrogel (AH) or emulsified in incomplete (IFA) or complete (CFA) Freund’s adjuvants and draining lymph nodes harvested approximately 12 days after immunization. (A) Representative flow cytometry plots, (B) percentage and (C) total number of AS15 MHCII tetramer+ cells within the CD4+ Foxp3− CD44hi population. (D) Percentage and (E) total number of CXCR5+ Bcl6+ Tfh cells within the tetramer+ population cells. (F) Pie charts displaying the lineage-defining transcription factor profile among CD4+ Foxp3− tetramer+ cells. (G) Total number of T-bet+, RORγT+ or Bcl6+ cells within the tetramer+ populations. (H) Mean fluorescent intensities of several markers on antigen specific Tfh cells (top row) versus other antigen-specific cells (bottom row) following IFA and CFA Immunization. Data shown are a pool of more than five experiments where each dot represents an individual mouse. Data were analysed using the non-parametric Mann–Whitney test.; **P<0.01; ***P<0.001.
Importantly in terms of the total response, IFA immunization resulted in the selective polarization of CD4+ T cells towards the Tfh subset, with nearly half of the tetramer positive cells expressing the Tfh lineage-defining transcription factor Bcl6 versus a total of only 14% expressing the T-bet or RORγt canonical transcription factors for Th1 and Th17 subset (Figure 1F). In contrast, CFA immunization induced a less Tfh-polarized T lymphocyte response with higher percentages of both Th1 and Th17 cells. This dominance of Tfh over Th1 and Th17 following IFA immunization was also apparent when the total numbers of each subset were calculated (Figure 1G). The Tfh cells induced by both adjuvants were indistinguishable in their increased expression of CXCR5 and PD1 and reduced expression of SLAM (Figure 1H), arguing that both protocols trigger Tfh cells that are at the same state of differentiation.
It has been reported that the antigen dose can critically impact the extent of Th1 versus Tfh differentiation (13). However, under the experimental conditions we employed, no significant dose-dependent differences were observed in either antigen-specific T cell expansion (Supplementary Figure 2A) or the frequency of Tfh cells within the antigen specific population (Supplementary Figure 2B).
Although IFA is not approved for human use, other better defined oil- and surfactant-only containing adjuvants have been utilized clinically (14). To determine whether the ability to induce polarized Tfh responses is shared by these adjuvants, we tested two of them, Montanide ISA 51 VG™ (ISA51) and Montanide ISA 720 VG™ (ISA720), which have been both used in numerous clinical trials related to therapeutic vaccines (14, 15). Montanide ISA 51 VG™, similar to IFA, contains a non-metabolizable mineral oil as its main component whereas Montanide ISA 720 VG™ incorporates squalene as a metabolizable oil. We found that ISA51 and ISA720, like IFA, were able to drive AS15-specific T cell expansion (Figure 2A) and promote the generation of high percentages of tetramer-positive CXCR5+ Bcl6+ Tfh cells (Figure 2B and 2C). Similarly to IFA, immunization with ISA51 or ISA720 resulted in a Tfh subset dominated profile (Figure 2D).
Figure 2. Tfh polarization in a common feature of oil and surfactant only adjuvants.
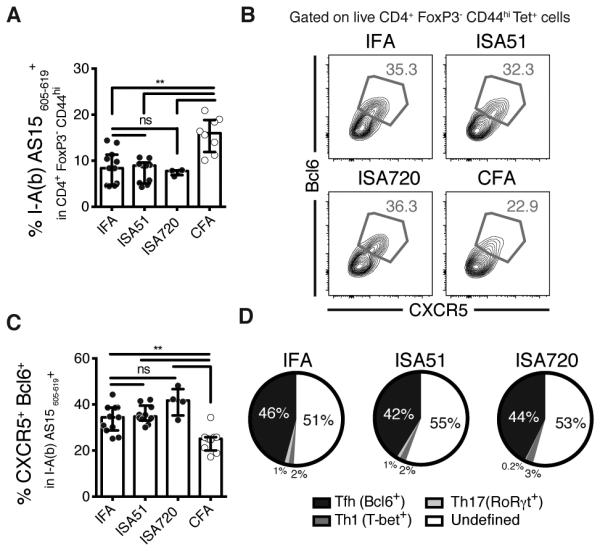
Mice were immunized with AS15 peptide emulsified in the water-in-oil (W/O) and surfactant only adjuvants IFA, Montanide ISA51 VG, Montanide ISA720 VG or CFA. (A) Frequencies of AS15 MHCII tetramer+ cells within the CD4+ Foxp3− CD44hi population. (B) Representative flow cytometry plots and (C) frequencies of CXCR5+ Bcl6+ Tfh cells within the tetramer+ population cells. (D) Pie charts displaying the lineage-defining transcription factor profile among CD4+ Foxp3− tetramer+ cells. Data shown are a pool of two separated experiments where each dot represents an individual mouse. Data were analysed using the non-parametric Mann–Whitney test.; ns: not significant, **P<0.01.
Having shown that W/O adjuvants are preferential stimulators of Tfh responses, we performed imaging studies to visualize the location of these Tfh cells within draining lymph nodes. We observed equivalent total numbers of germinal centers (GCs), defined as IgD− Bcl6+ structures, in naïve and immunized mice maintained in specific-pathogen free conditions (Figure 3A). This background level of GC likely reflects the steady-state response to commensals and/or self-antigens. In contrast, the administration of AS15 peptide in IFA or CFA induced a significant increase in the GC size (area) (Figure 3B). As expected, GC formation in mice immunized with IFA in the absence of peptide antigen was comparable to that observed in naïve animals. Importantly, the vast majority of the CD4+PD1+Bcl6+ Tfh induced by peptide immunization in IFA were localized within these structures and hence were considered as classical GC Tfh (Figure 3C). Together the above experiments performed with peptide immunization models indicated that W/O only adjuvants preferentially trigger antigen-specific GC Tfh cells.
Figure 3. IFA immunization induces GC-homing Tfh.

Mice were immunized with AS15 peptide emulsified in IFA or CFA. dLNs were collected after 12 days, sectioned, stained by immunofluorescence and analysed by confocal microscopy. (A) Total number of GC per lymph node. (B) Area of the GCs in age-matched naïve mice or following immunization with IFA only (no antigen) or AS15 peptide emulsified in IFA or CFA. (C) Representative images from naïve, IFA, or CFA immunized dLNs. (D) Magnification of a representative GC after IFA immunization. CD4+ cells are shown in blue, IgD+ naïve B cells in green, PD-1 in white and Bcl6+ GC B cells/Tfh cells in red. Data shown are representative of two separate experiments. Data were analysed using the non-parametric Mann–Whitney test.; ns: not significant, *P<0.05.
Water-in-oil only adjuvants also promote Tfh dominated T cell responses and antibody production during immunization with protein antigen
We next asked whether proteins and not just peptide antigens also trigger polarized Tfh responses when emulsified in oil and surfactant-only adjuvants. For these experiments we utilized antigen 85b (Ag85b), a mycobacterial protein for which class II tetramers are available. We found that when administered in IFA, ISA51 or ISA720, Ag85b stimulates an antigen-specific T cell response (Supplementary Figure 2C and 2D) containing a high proportion of CXCR5+ Bcl6+ Tfh cells (Supplementary Figure 2C and 2E). As predicted from previous studies, protein immunization in IFA in this setting resulted in the production of a substantial Ag85b-specific Ab response in C57BL6 but not C57BL6 TCRA−/− mice arguing that the antigen-specific Tfh induced in our IFA immunization protocol have functional B cell helper activity (Supplementary Figure 2F).
The Tfh response induced by immunization with antigen in IFA depends on both B cells and IL-6 upstream of STAT1 and STAT3 signaling
The differentiation of conventional GC Tfh cells is known to require antigen presentation and co-stimulation from B cells (1). Consistent with this property, B cell-deficient μMT mice immunized with peptide in IFA displayed a profound impairment in total CD4+ T cell responses (Supplementary figure 3A) and more importantly in the antigen-specific Tfh population (Supplementary figure 3B).
IL-6 has also been shown to play a role in Tfh differentiation by triggering both Bcl6 (4) and CXCR5 (7, 16) expression in the precursors of these cells. We confirmed that the Tfh induced following peptide immunization in IFA display a similar IL-6 dependency. Thus, immunized Il6−/− mice, while possessing indistinguishable levels of AS15 tetramer-positive lymphocytes as compared to immunized wild-type mice (Supplementary Figure 3C), exhibited a significant reduction in the frequency of Tfh cells within this antigen-specific population (Supplementary Figure 3D). Although IL-12 has also been reported to be required for human (17) and in some cases murine Tfh (18) differentiation in vitro, no reduction in the generation of these cells was observed when Il12p40−/− mice were immunized with peptide in IFA (Supplementary Figure 3E and 3F).
IL-6 is thought to mediate its effects on infection-induced Tfh differentiation through STAT1 and STAT3 (19). Consistent with these findings, when immunized with peptide in IFA, mice with a T cell-intrinsic deficiency in STAT1 or STAT3 display reduced frequencies of antigen-specific Tfh (Supplementary Figure 3G) while exhibiting normal levels of tetramer positive cells (Supplementary Figure 3H) after immunization with peptide in IFA. Nevertheless, the reduction in the Tfh response was not as profound as that observed in the prior studies involving lymphocytic choriomeningitis virus (LCMV) infection (20).
IFA-induced Tfh responses are dependent on MyD88
MyD88 is a major downstream signaling adaptor for most TLR as well as IL-1 and IL-18 receptors. We and others have previously shown that MyD88 is critical for the Th1 and Th17 responses triggered by either the heat-killed Mycobacterium tuberculosis in CFA (21) or by an analog of the mycobacterial cord factor trehalose-6,6-dimycolate (22). Similarly, we found that MyD88-deficient mice immunized with peptide in CFA display reduced frequencies (Figure 4A) and total numbers of antigen-specific cells (Supplementary Figure 3I). Interestingly, while the total number of tetramer+ Tfh cells was also reduced in these animals (Supplementary Figure 3J), their frequency was not (Figure 4B and 4C).
Figure 4. IFA-induced Tfh polarization requires MyD88 signaling.

WT and Myd88−/− mice were immunized with AS15 peptide emulsified in IFA or CFA and dLNs were harvested at either D6 (E) or D12 (A-G) post-immunization. (A) Frequencies of AS15 MHCII tetramer+ cells within the CD4+ Foxp3− CD44hi population. (B) Representative flow cytometry plots and (C) frequencies of CXCR5+ Bcl6+ Tfh cells within the tetramer+ population cells. (D) Representative flow cytometry plots and (E) frequencies of Gl7+ Bcl6+ GC B cells. (F) Schematic of the experiments using irradiated WT or Myd88−/− mice reconstituted with WT or Myd88−/− bone marrow cells and their relative frequencies of CXCR5+ Bcl6+ Tfh cells within the tetramer+ population cells after IFA immunization. (G) Schematic of mixed bone marrow chimera experiments using WT irradiated mice reconstituted with a 50/50 mixture of congenically-marked WT/Myd88−/− bone marrow cells. The frequencies of WT (white dots) and Myd88−/− (grey dots) CXCR5+ Bcl6+ Tfh cells within the tetramer+ population cells after IFA immunization are shown. Each line represents a single recipient animal. Data shown are representative of 2-5 experiments where each dot represents an individual mouse. Data were analysed using the non-parametric Mann–Whitney test.; ns: not significant, *P<0.05; **P<0.01; ***P<0.001.
Unexpectedly, we observed that the response to peptide immunization in IFA, which does not carry exogenous Pathogen-Associated Molecular Patterns (PAMPs), also requires MyD88 signaling. We found that MyD88-deficient mice display significant decreases in both the percentages (Figure 4A) and total numbers (Supplementary Figure 3I) of antigen-specific cells as well as in the frequency of Tfh cells within the antigen specific population (Figure 4B and 4C and Supplementary Figure 3J). Of note, the requirement for MyD88 appears early since a significant defect in the frequency of antigen-specific Tfh cells was already present as early as 6 days post IFA immunization (Supplementary Figure 3K). Importantly, the effect of MyD88 deficiency on IFA adjuvanted Tfh responses was also observed in mice immunized with protein antigen (Ag85b) (Supplementary Figure 3L). In addition to dampening Tfh responses, the absence of MyD88 also impaired the frequency of GC B cells (Figure 4D and 4E).
We next used reciprocal bone marrow chimeric mice to address the cellular basis of the role of MyD88 in Tfh generation in IFA plus peptide-immunized mice. These experiments showed that the requirement for MyD88 expression is restricted to the hematopoietic compartment (Figure 4F) but is not T cell intrinsic, as no difference was observed in MyD88-sufficient versus MyD88-deficient T cells found in the same irradiated host following reconstitution with a 50/50 mixture of WT and MyD88−/− bone marrow cells (Figure 4G).
One hypothesis to explain the unexpected involvement of MyD88 in the IFA-induced Tfh response is that the oil component and/or the surfactant contained in IFA leads to local cell death and/or tissue damage that could provide endogenous danger signals for MyD88-dependent triggering of Tfh differentiation (23). However, no difference in the frequency of Tfh cells within the antigen-specific population was observed in the absence of TLR2, TLR4 or IL-1R (Supplementary Figure 3M), three receptors which have been implicated in danger-induced signaling. In related experiments, normal Tfh responses were also observed in IFA immunized IL-18-deficient mice (Supplementary Figure 3M). A second hypothesis we considered is that the requirement for MyD88 reflects the involvement of microbiota-derived signals. In this regard, it has been shown recently that the sensing of the gut microbiota through TLR5 is necessary for the antibody response observed after subcutaneous immunization with an unadjuvanted influenza vaccine (24). Nevertheless, no impairment of the Tfh response was observed in IFA-immunized Tlr5−/− mice or mice treated with antibiotics using the identical protocol employed in the latter study (Supplementary Figure 3M). Similarly, no reduction in Tfh response was observed in the absence of the microbial (or self) DNA sensor TLR9 (Supplementary Figure 3M). Together, these experiments demonstrated a contribution for MyD88-dependent signaling in Tfh induction, but did not identify an individual TLR or IL-1R family member responsible for this effect.
Optimal water-in-oil adjuvant Tfh induction depends on T cell intrinsic type I IFN signaling
Type I IFN has been reported to either promote (18, 25) or inhibit (20) Tfh responses. We observed that WT, as well as MyD88 deficient mice, IFA-immunized mice displayed a marked upregulation of both Ifna and Ifnb mRNA in total lymph nodes as compared to unimmunized animals (Supplementary Figure 4A). To examine the possible function of this IFN production in Tfh induction, we compared the responses of WT versus IFN receptor deficient mice (Ifnar−/−) following IFA or CFA immunization. IFNAR-deficient mice displayed unaltered levels of antigen-specific cells (Figure 5A) but had significantly reduced frequencies of Tfh cells within this population at D12 (Figure 5B and 5C and Supplementary figure 4B) but not at D6 post-immunization (Supplementary figure 4B). In addition, the absence of IFNAR also led to a decreased frequency of GC B cells (Figure 5D and 5E). Importantly, a role for type I IFN signaling in Tfh induction was also observed with two other non-microbial containing W/O only containing adjuvants, Montanide ISA51™ and Montanide ISA720™ (Figure 5F).
Figure 5. Water-in-oil and surfactant only adjuvants induced tfh polarization involves type I IFN signaling on T cells.

(A-H) WT and type I IFN receptor (Ifnar)−/− mice were immunized with AS15 peptide emulsified in IFA or CFA and dLNs were harvested at either D6 (E) or D12 (A-I) post-immunization. (A) Frequencies of AS15 MHCII tetramer+ cells within the CD4+ Foxp3− CD44hi population. (B) Representative flow cytometry plots and (C) frequencies of CXCR5+ Bcl6+ Tfh cells within the tetramer+ population cells comparing IFA with CFA (D) Representative flow cytometry plots and (E) frequencies of Gl7+ Bcl6+ GC B cells. (F) Frequencies of CXCR5+ Bcl6+ Tfh cells within the tetramer+ population cells comparing the W/O and surfactant only adjuvants IFA, Montanide ISA51 VG, Montanide ISA720 VG. (G) Schematic of mixed bone marrow chimera experiments using WT irradiated mice reconstituted with a 50/50 mixture of congenically marked WT/Ifnar−/− bone marrow cells. The frequencies of WT (white dots) and Ifnar−/− (grey dots) CXCR5+ Bcl6+ Tfh cells within the tetramer+ population cells after IFA immunization are shown, where each line represents a single animal. (H-I) WT and Unc93b1−/− mice were immunized with IFA and AS15 peptide. (H) Frequencies of AS15 MHCII tetramer+ cells within the CD4+ Foxp3− CD44hi population and (I) frequencies of CXCR5+ Bcl6+ Tfh cells within the tetramer+ population cells. Data shown are representative of 2-6 experiments where each dot represents an individual mouse. Data were analysed using the non-parametric Mann–Whitney test.; ns: not significant *P<0.05; ***P<0.001.
To examine the cellular basis of the contribution of Type I IFN signaling to Tfh differentiation, we utilized mixed bone chimera experiments in which sub-lethally irradiated mice were reconstituted with equivalent numbers of CD45.1 WT and CD45.2 IFNAR-deficient bone marrow cells and the animals were then immunized with IFA plus AS15 peptide eight weeks after reconstitution (Figure 5G). In this system, WT and IFNAR-deficient T cells are readily distinguishable by their different congenic marker expression. Since each population receives the same environmental signals the intrinsic requirement of IFNAR can be addressed. While the WT and IFNAR-deficient CD4+ T lymphocytes recovered from the draining LN contained comparable frequencies of activated CD44hi cells (Supplementary Figure 4C), T cell-intrinsic IFNAR expression was required to obtain full expansion of tetramer positive cells (Supplementary Figure 4D) as well as the Tfh within this population (Figure 5G). In these IFA-immunized animals, the decreased frequency of Tfh developing in the absence of IFNAR was not associated with a concomitant increase in the other T cell subsets (data not shown).
Since W/O only adjuvants preferentially induce Tfh cells, it was possible that the role of type I IFN in this system reflects a general effect not restricted to that subset. To address this question, we immunized the same mixed bone marrow chimeric mice with peptide and CFA, a protocol that simultaneously induces Th1/Th17 and Tfh subsets (Figure 1F). With this type of immunization, the absence of type I IFN receptor did not alter the overall frequency of antigen specific-cells (Supplementary Figure 4E) but again resulted in a marked reduction in the Tfh response (Supplementary Figure 4F) with an accompanying increase in the percentages of T-bet- and RORγT-expressing cells (Supplementary Figure 4G).
Type I IFN is induced through a variety of innate receptors that include the cytoplasmic and endosomal sensors STING, MDA5, AIM2, TLR3, TLR7/8, and TLR9. The experiments previously described above argued against a role for TLR9 in Tfh induction (Supplementary Figure 3M) and parallel experiments involving TLR3-, STING- and MDA5-deficient mice failed to reveal a function for these PRRs or the TRIF adaptor molecule in IFA-induced Tfh responses (Supplementary Figure 4H). However, we found that IFA immunized UNC93b1-deficient mice, in which all endosomal TLRs including the type I IFN inducing receptors TLR3, TLR7/8, and TLR9 are non-functional, exhibit a significant impairment in Tfh response (Figure 5H and 5I). Taken together with the other data on PRR involvement shown above, this finding suggests a possible role for TLR7/8 or a combination of multiple endosomal TLRs in W/O adjuvant-promoted Tfh induction. In conclusion, the experiments described above identified important roles for both MyD88 and type I IFN receptor signaling in the Tfh polarized responses promoted by W/O adjuvants, with IFNAR but not MyD88 having a T cell intrinsic function.
The roles of MyD88 and IFNAR in Tfh generation are IL-6 independent
As discussed above, IL-6 in both our experiments and in other studies (4) has been shown to play a major function in Tfh generation. To investigate whether the influence of MyD88 and IFNAR on Tfh responses in IFA-immunized mice relates to their involvement in IL-6 induction and/or signaling, we simultaneously treated MyD88- or IFNAR-deficient mice with an anti-IL-6 monoclonal antibody. Interestingly, IL-6 neutralization in either of these deficient settings caused a marked reduction in frequencies (Figure 6A) as well as in total numbers (Figure 6B) of antigen-specific Tfh cells that exceeded in magnitude the decreased response seen in anti-IL-6 treated WT mice. In contrast, treatment of immunized Myd88−/− mice with an IFNAR blocking monoclonal antibody caused only a minor, statistically insignificant decrease in the magnitude of the Tfh response (Figure 6B). Together, these findings are consistent with the existence of a common pathway for MyD88 and IFNAR in the Tfh response, which is likely to be unrelated to that involved to IL-6 induction and function.
Figure 6. Type I IFN and IL-6 independently promote Tfh differentiation.
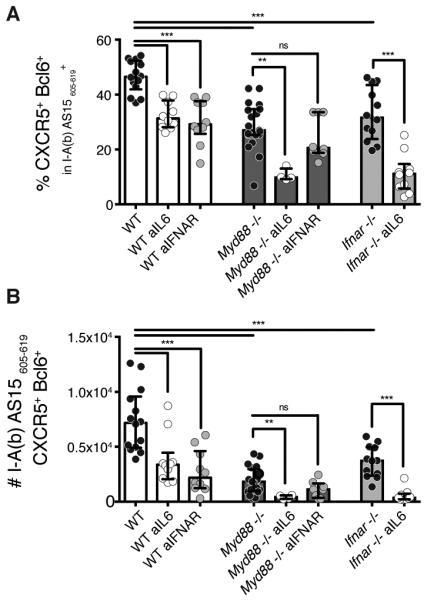
Frequencies of antigen specific cells (A) and of CXCR5+ Bcl6+ Tfh cells within the tetramer+ population (B) of IFA immunized WT, Myd88−/− or Ifnar−/− mice treated or not with anti-IL6 antibody (clone MP5-20F3, 250/500μg by i.p. injections on day -1,1,3,6 and 9) or with anti-IFNAR-1 antibody (clone BE0241, 250/500μg by i.p. injections on day 0,1,3,6 and 9). Data shown are representative of 2-3 experiments where each dot represents an individual mouse. Data were analysed using the non-parametric Mann–Whitney test.; ns: not significant **P<0.01; ***P<0.001.
Discussion
The stimulation of Tfh development and function is a logical approach toward the development of improved vaccines based on humoral immunity. Nevertheless, Tfh cells are distinct from other CD4+ T cell subsets in that relatively little is known about the signals that guide their selective differentiation. In this report, we have described a model employing W/O adjuvants that can be utilized to study the polarization and immunologic activity of Tfh cells in vivo and employed this system to reveal MyD88 and type I IFN signaling as two innate determinants involved in their induction.
Most previous studies on the mechanism of Tfh differentiation have utilized either infectious challenge and/or adoptive transfer of transgenic T cells to dissect this process. In contrast, the IFA immunization model employed here offers the ability to study Tfh induction endogenously and in the absence of added microbial stimuli. We chose initially to examine the response to an immunogenic peptide (AS15) with a high precursor frequency (26) that is readily detected by tetramer staining. This allowed us to focus on endogenous Tfh development driven solely by a single epitope. Although a previous study in an infection model had indicated that the choice of peptide epitope can influence the degree of Tfh polarization, we confirmed that the Tfh induction observed following immunization with AS15 in water-in-oil adjuvant can be reproduced with other structurally unrelated peptides. Similarly, in contrast to what has been observed in other models (13) the capacity of IFA immunization to trigger polarized Tfh responses appeared to be independent of the dose of peptide employed.
In the present study, immunization with each of the three W/O only adjuvants tested consistently resulted in strong Tfh polarization. The oils incorporated in each adjuvant differ in their degree of refinement and biodegradability. However, all three share the same surfactant, mannide mono-oleate, at different degrees of purity, raising the question of its direct contribution to Tfh induction. Since the presence of a surfactant is essential for the generation of W/O emulsions, this possibility cannot be formally ruled out. Nevertheless, immunization with peptide plus mannide mono-oleate (the same preparation used in the manufacture of IFA) in PBS in the absence of oil induced the same basal Tfh response observed with peptide alone (data not shown). Regardless, the presence of microbial PAMPs in the adjuvant(s) was clearly not required for Tfh induction nor was the response altered in antibiotic-treated animals. The latter observation argues against a significant role for the endogenous microbiota as has been documented previously for the B cell response observed following immunization with a non-adjuvanted flu vaccine (24).
W/O only adjuvants induced a frequency of Tfh that was substantially higher than that promoted by either alum or CFA although the latter adjuvant stimulated a greater total number of these cells. This finding is consistent with the differences in antibody response typically triggered by above adjuvants (11). Indeed, when compared to CFA, W/O only adjuvants barely stimulated Th1, Th17, and Th2 (data not shown) induction, supporting their preferential capacity to trigger Tfh response and, as also shown here, germinal center formation.
The central question raised by the data presented here concerns the mechanism underlying the Tfh polarization observed following immunization with W/O only adjuvants. Since these formulations do not contain any microbial components able to drive the differentiation of other CD4+ T cell subsets (e.g. Th1 or Th17), one possibility is that the Tfh polarization occurring in this system represents a default pathway seen when T cell responses are induced in the absence of exogenous PAMPs. This explanation seems unlikely, however, given the failure of αCD3/CD28 activated purified CD4+ T cells to default to a CXCR5- and Bcl6-expressing Tfh-like phenotype even in the presence of IL-6 or IL-21 (27). Nevertheless, in contrast to the in vitro situation, Bcl6-expressing CD4+ T cells are readily induced during early immune responses in vivo (6, 28) and appear to be either followed by a second wave of differentiated Tfh cells or replaced by T cells that belong to other subsets depending on whether the appropriate transcriptional activators are present. Thus, Tfh response induction may require both a positive differentiation signal that promotes the development of early Bcl6 and CXCR5 cells into mature Tfh as well as the concomitant absence of polarizing stimuli for the other major CD4+ T cell subsets.
In the experiments reported here on W/O only adjuvant immunization, we were able to identify two innate immune system components (in addition to IL-6) that influence Tfh polarization and which may provide clues as to the nature of the positive differentiation signal referred to above. The first, the adaptor protein MyD88, has already been extensively studied in the context of antibody responses where its role is controversial (11, 29). In the case of immunization with protein antigens in non-PAMP-containing oil adjuvants, MyD88-deficient mice have been observed to display either normal or defective antibody production (11, 30). Although the effects of MyD88 deficiency on Tfh induction have previously been reported (24, 31), these have involved experimental models in which direct microbial stimulation from the gut flora was proposed to play a major contribution. In the studies reported here, MyD88 dependence was observed in a system in which exogenous microbial PAMPs should be absent and oral antibiotic treatment failed to impair the response. Although we were unable to formally identify the ligands and host receptors involved, the reduction in Tfh response seen in UNC93b1-deficient mice implicates a role for TLR3/7 or 9 or a combination of these receptors in this pathway. Moreover, based on our bone marrow reconstitution experiments, these receptors appear to be functioning exclusively in the hematopoietic compartment.
In addition to MyD88, type I IFN signaling was implicated in our experiments as an innate immune component participating in adjuvant-induced Tfh induction. The role of this pathway has been investigated previously but with differing outcomes, either promoting (18, 25) or inhibiting (20) Tfh responses depending on the system studied. In our model, which does not utilize adoptive transfer and/or viral infection, we observed significant IFNAR involvement in the Tfh responses following immunization with all three W/O only adjuvants tested. Additionally, our mixed bone marrow chimera experiments revealed a major CD4+ T cell-intrinsic component for this function. Further analysis suggested that MyD88 and type I IFN signaling may be part of a common pathway, since in contrast to IL-6 blockade, antibody-mediated IFNAR blockade failed to significantly diminish the Tfh responses observed in immunized MyD88-deficient mice. However, at day 10 post-immunization and in total lymph nodes, we found a similar upregulation of Ifna and Ifnb mRNA in both WT and Myd88−/− mice, revealing a more complex relationship between MyD88 and type I IFN for which in-depth kinetic and anatomic (e.g. lymph nodes versus immunization sites) studies would be required. Interestingly, whereas the requirement for MyD88 to generate antigen-specific Tfh cells appears already early after immunization (day 6), presumably acting during the APC activation and/or T cell priming steps, that of IFNAR is important between D6 and D12 post-immunization when GC B cells play a prominent role in the persistence of Tfh cells. Of note, both Myd88−/− and Ifnar−/− displayed a significantly lower frequency of GC B cells at day 12 post-immunization, but not at day 6, as compared to WT mice. Based on the above series of observations, we speculate that the adjuvant-stimulated Tfh polarization occurring in our model may involve the production of type I IFN from a hematopoietically-derived cell population (e.g. B lymphocytes, plasmacytoid DC, myeloid cells) that in turn triggers the induction of a T cell-intrinsic type I IFN-dependent signal important for the differentiation and persistence of this subset. Further work is necessary to formally confirm the central features and in particular the role of MyD88 in this pathway.
The finding that W/O only adjuvants preferentially drive Tfh responses in the absence of exogenous microbial PAMPs suggests an important role for endogenous danger signals or DAMPs in the mechanism underlying this process. While the ligands involved were not identified in the present study, the dual dependency on MyD88 and type I IFN signaling raises the question of a possible role for self nucleic acids derived from stressed or damaged cells (32).
Once delineated, these signals could be potentially utilized to engineer enhanced vaccine-induced antibody production. Nevertheless, the simple induction of higher frequencies of Tfh may not necessarily lead to improved B cell maturation and humoral immunity. In this regard, it is becoming clear that Foxp3+ T follicular regulatory cells, which balance the function of Tfh in germinal centers, can also strongly influence the outcome of immunization. Based on previous studies (9) and our own preliminary observations (data not shown), major increases in this cell population appear to be strongly promoted by W/O only adjuvants and could contribute positively or negatively to the overall functional outcome on humoral immunity. Further studies on the role of adjuvants in regulating the Tfh/Tfr balance offer an approach for addressing this question.
Supplementary Material
Acknowledgments
We are grateful to the NIAID Flow Cytometry and Tetramer Facilities and Animal Care Branch for major contributions to this work. We thank Daniel L. Barber (NIAID, NIH) helpful discussions and Ronald N. Germain (NIAID, NIH) for his thoughtful input and support. We also acknowledge Marie-Eve Koziol (SEPPIC Inc. / Air Liquide Healthcare North America) for the generous gift of Montanide ISA 51VG and Montanide ISA 720VG adjuvants and Paul Herzog (Hudson Institute of Medical Research) for interferon primer sequences.
Abbreviations
- GC
Germinal center
- PAMPs
Pathogen-Associated Molecular Patterns
- Tfh
T follicular helper
- W/O adjuvant
Water-in-oil adjuvant
Footnotes
This work was supported by the Intramural Research and the Intramural Vaccine Adjuvant Programs of the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH).
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 3.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, Dong C. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, Ellyard JI, Parish IA, Ma CS, Li QJ, Parish CR, Mackay CR, Vinuesa CG. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Baumjohann D, Okada T, Ansel KM. Cutting Edge: Distinct waves of BCL6 expression during T follicular helper cell development. J Immunol. 2011;187:2089–2092. doi: 10.4049/jimmunol.1101393. [DOI] [PubMed] [Google Scholar]
- 7.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular Helper T Cells. Annu Rev Immunol. 2016 doi: 10.1146/annurev-immunol-041015-055605. [DOI] [PubMed] [Google Scholar]
- 9.Aloulou M, Carr EJ, Gador M, Bignon A, Liblau RS, Fazilleau N, Linterman MA. Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells. Nat Commun. 2016;7:10579. doi: 10.1038/ncomms10579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang YH, Watowich SS, Jetten AM, Tian Q, Dong C. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, Nemazee D. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science. 2006;314:1936–1938. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grover HS, Blanchard N, Gonzalez F, Chan S, Robey EA, Shastri N. The Toxoplasma gondii peptide AS15 elicits CD4 T cells that can control parasite burden. Infect Immun. 2012;80:3279–3288. doi: 10.1128/IAI.00425-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, Sallusto F. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38:596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 14.Aucouturier J, Dupuis L, Deville S, Ascarateil S, Ganne V. Montanide ISA 720 and 51: a new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev Vaccines. 2002;1:111–118. doi: 10.1586/14760584.1.1.111. [DOI] [PubMed] [Google Scholar]
- 15.van Doorn E, Liu H, Huckriede A, Hak E. Safety and tolerability evaluation of the use of Montanide ISA51 as vaccine adjuvant: A systematic review. Hum Vaccin Immunother. 2016;12:159–169. doi: 10.1080/21645515.2015.1071455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35:583–595. doi: 10.1016/j.immuni.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitt N, Liu Y, Bentebibel SE, Munagala I, Bourdery L, Venuprasad K, Banchereau J, Ueno H. The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol. 2014;15:856–865. doi: 10.1038/ni.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakayamada S, Poholek AC, Lu KT, Takahashi H, Kato M, Iwata S, Hirahara K, Cannons JL, Schwartzberg PL, Vahedi G, Sun HW, Kanno Y, O'Shea JJ. Type I IFN induces binding of STAT1 to Bcl6: divergent roles of STAT family transcription factors in the T follicular helper cell genetic program. J Immunol. 2014;192:2156–2166. doi: 10.4049/jimmunol.1300675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J Immunol. 2013;190:3049–3053. doi: 10.4049/jimmunol.1203032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity. 2014;40:367–377. doi: 10.1016/j.immuni.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shenderov K, Barber DL, Mayer-Barber KD, Gurcha SS, Jankovic D, Feng CG, Oland S, Hieny S, Caspar P, Yamasaki S, Lin X, Ting JP, Trinchieri G, Besra GS, Cerundolo V, Sher A. Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through mincle/CARD9 signaling and the inflammasome. J Immunol. 2013;190:5722–5730. doi: 10.4049/jimmunol.1203343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desel C, Werninghaus K, Ritter M, Jozefowski K, Wenzel J, Russkamp N, Schleicher U, Christensen D, Wirtz S, Kirschning C, Agger EM, Prazeres da Costa C, Lang R. The Mincle-activating adjuvant TDB induces MyD88-dependent Th1 and Th17 responses through IL-1R signaling. PLoS One. 2013;8:e53531. doi: 10.1371/journal.pone.0053531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–119. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 24.Oh JZ, Ravindran R, Chassaing B, Carvalho FA, Maddur MS, Bower M, Hakimpour P, Gill KP, Nakaya HI, Yarovinsky F, Sartor RB, Gewirtz AT, Pulendran B. TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity. 2014;41:478–492. doi: 10.1016/j.immuni.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cucak H, Yrlid U, Reizis B, Kalinke U, Johansson-Lindbom B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity. 2009;31:491–501. doi: 10.1016/j.immuni.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Nelson RW, Beisang D, Tubo NJ, Dileepan T, Wiesner DL, Nielsen K, Wuthrich M, Klein BS, Kotov DI, Spanier JA, Fife BT, Moon JJ, Jenkins MK. T cell receptor cross-reactivity between similar foreign and self peptides influences naive cell population size and autoimmunity. Immunity. 2015;42:95–107. doi: 10.1016/j.immuni.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, Yusuf I, Crotty S. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One. 2011;6:e17739. doi: 10.1371/journal.pone.0017739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, Sun HW, Vahedi G, Hakim O, Handon R, Schwartzberg PL, Hager GL, O'Shea JJ. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity. 2011;35:919–931. doi: 10.1016/j.immuni.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 30.Seubert A, Calabro S, Santini L, Galli B, Genovese A, Valentini S, Aprea S, Colaprico A, D'Oro U, Giuliani MM, Pallaoro M, Pizza M, O'Hagan DT, Wack A, Rappuoli R, De Gregorio E. Adjuvanticity of the oil-in-water emulsion MF59 is independent of Nlrp3 inflammasome but requires the adaptor protein MyD88. Proc Natl Acad Sci U S A. 2011;108:11169–11174. doi: 10.1073/pnas.1107941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O'Connell RM, Round JL. MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe. 2015;17:153–163. doi: 10.1016/j.chom.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat Immunol. 2007;8:11–13. doi: 10.1038/ni0107-11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.