

SUMMARY
Photoreceptor death is the endpoint of many blinding diseases. Identifying unifying pathogenic mechanisms in these diseases may offer global approaches for facilitating photoreceptor survival. We found that rod or cone photoreceptor-specific deletion of nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in the major NAD+ biosynthetic pathway beginning with nicotinamide, caused retinal degeneration. In both cases, we could rescue vision with nicotinamide mononucleotide (NMN). Significantly, retinal NAD+ deficiency was an early feature of multiple mouse models of retinal dysfunction, including light-induced degeneration, streptozotocin-induced diabetic retinopathy, and age-associated dysfunction. Mechanistically, NAD+ deficiency caused metabolic dysfunction and consequent photoreceptor death. We further demonstrate that the NAD+-dependent mitochondrial deacylases SIRT3/SIRT5 play important roles in retinal homeostasis and that NAD+ deficiency causes SIRT3 dysfunction. These findings demonstrate that NAD+ biosynthesis is essential for vision, provide a foundation for future work to further clarify the mechanisms involved, and identify a unifying therapeutic target for diverse blinding diseases.
Keywords: retina, neurodegeneration, photoreceptor, metabolism, NAD+, sirtuins, mitochondria
Graphical abstract

INTRODUCTION
Vision is a crucial sense that depends on photoreceptors for light transduction. Photoreceptors are divided into two classes: rods and cones. Rod photoreceptors mediate dim vision, while cone photoreceptors mediate precise central vision in ambient light. Photoreceptors make up a significant portion of the neurosensory retina, one of the most metabolically active tissues in the body (Ames et al., 1992; Kooragayala et al., 2015; Okawa et al., 2008). Although terminally differentiated and non-proliferative, photoreceptors have tremendous metabolic demands throughout their lives and experience significant light-induced oxidative stress due to their function of performing light transduction (Fu and Yau, 2007; Yau and Hardie, 2009).
Because of the critical role that photoreceptors play in light transduction, photoreceptor death leads to blindness. Despite their differing etiologies, many blinding diseases share this final pathway of photoreceptor death, which inevitably causes vision loss. For example, age-related macular degeneration (AMD) is one leading cause of blindness in older adults (Apte, 2010; Klein et al., 2004; van Leeuwen et al., 2003). Although advanced AMD takes on two forms (i.e., dry or wet AMD) (Sene and Apte, 2014), both pathways ultimately lead to photoreceptor death. Similarly, inherited retinal degenerations, including retinitis pigmentosa (RP), rod and cone dystrophies, and Leber congenital amaurosis (LCA), are caused by genetic defects in over 100 different genes that lead to photoreceptor death (Astuti et al., 2015; Hartong et al., 2006; Wright et al., 2010). Developing treatment strategies for this broad spectrum of retinal degenerative diseases is challenging given these diverse pathogenic mechanisms.
Nicotinamide adenine dinucleotide (NAD+) is both an essential coenzyme, functioning as an electron carrier in glycolysis and the Krebs cycle, and an essential cosubstrate for NAD+-consuming enzymes, including sirtuins, poly(ADP-ribose) polymerases (PARPs), mono-ADP ribosyltransferases, and cyclic ADP-ribose hydrolase (CD38) (Ying, 2006). Given these numerous functions, NAD+ has been shown to be important in many biological processes, including metabolism (Garten et al., 2009; Mouchiroud et al., 2013), circadian rhythms (Nakahata et al., 2009; Ramsey et al., 2009), and aging (Imai and Guarente, 2014). Relevant to our studies, NAD+ has also been shown to be important in neurodegeneration (Alano et al., 2010; Gerdts et al., 2015; Stein et al., 2014; Zhou et al., 2015). However, the role of NAD+ in retinal degeneration has been relatively unexplored to date.
We hypothesized that NAD+ biosynthesis plays an important role in photoreceptor function and survival. This hypothesis is supported by past studies demonstrating that LCA, the leading cause of childhood blindness from retinal disease, can be caused by mutations in nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) (Chiang et al., 2012; Falk et al., 2012; Koenekoop et al., 2012; Perrault et al., 2012), an enzyme involved in NAD+ biosynthesis. The various mutant forms of NMNAT1 associated with LCA exhibit reduced NAD+ biosynthetic capacity and/or impaired protein folding (Falk et al., 2012; Koenekoop et al., 2012; Sasaki et al., 2015) with both factors contributing to disease pathogenesis.
NAD+ can be synthesized in three ways: 1) de novo from tryptophan, 2) salvaged from nicotinamide or nicotinic acid (NAM or NA, respectively), or 3) from nicotinamide riboside (NR). In mammals, the salvage pathway beginning with NAM is the predominant NAD+ biosynthetic pathway (Imai and Yoshino, 2013; Imai and Guarente, 2014). The first step of this biosynthetic pathway is catalyzed by nicotinamide phosphoribosyltransferase (NAMPT), which combines NAM with 5-phosphoribosyl pyrophosphate (PRPP) to form nicotinamide mononucleotide (NMN). NMN is then adenylated by NMNAT1-3 to synthesize NAD+.
In this study, we focus on the role of NAMPT-mediated NAD+ biosynthesis in photoreceptor survival and vision. Our results suggest that NAD+ biosynthesis may be important not only in LCA but also in a broad spectrum of retinal degenerations and age-associated retinal dysfunction, opening up the possibility of using NAD+ intermediates to protect against retinal degeneration. Although these studies provide a strong foundation for future studies in this area, further work is needed to clarify the mechanisms involved. If successful, this therapeutic strategy would be far-reaching since it could be used for diverse retinal degenerations regardless of their etiology.
RESULTS
Loss of NAMPT-mediated NAD+ biosynthesis impairs photoreceptor survival and vision
Photoreceptors are one of the most metabolically active cells of the body with demanding energy requirements (Ames et al., 1992; Okawa et al., 2008) but limited mitochondrial reserve capacity (Kooragayala et al., 2015). Therefore, we hypothesized that photoreceptors depend on NAMPT-mediated NAD+ biosynthesis to meet their catalytic requirements. To test this hypothesis, we examined the effect of disrupting NAMPT-mediated NAD+ biosynthesis, the predominant pathway in mammals, selectively in rod photoreceptors by generating conditional knockout mice lacking Nampt in rod photoreceptors (Nampt−rod/−rod).
As expected, rod-enriched retinal isolates from Nampt−rod/−rod mice showed significant reduction in Nampt gene expression (Fig 1A). This rod photoreceptor-specific deletion of Nampt led to a ~26% reduction in retinal NAD+ levels in Nampt−rod/−rod mice compared to NamptF/F littermates at 3 weeks (Fig 1B). We also measured NAD+ levels in rod-enriched retinal isolates to more specifically characterize the magnitude of NAD+ deficiency in rod photoreceptors. We found that rod-enriched isolates from Nampt−rod/−rod mice had a ~43% reduction in NAD+ levels compared to those from NamptF/F mice (Fig 1C), suggesting that the NAD+ deficiency is predominantly specific to the rod photoreceptors.
Figure 1.
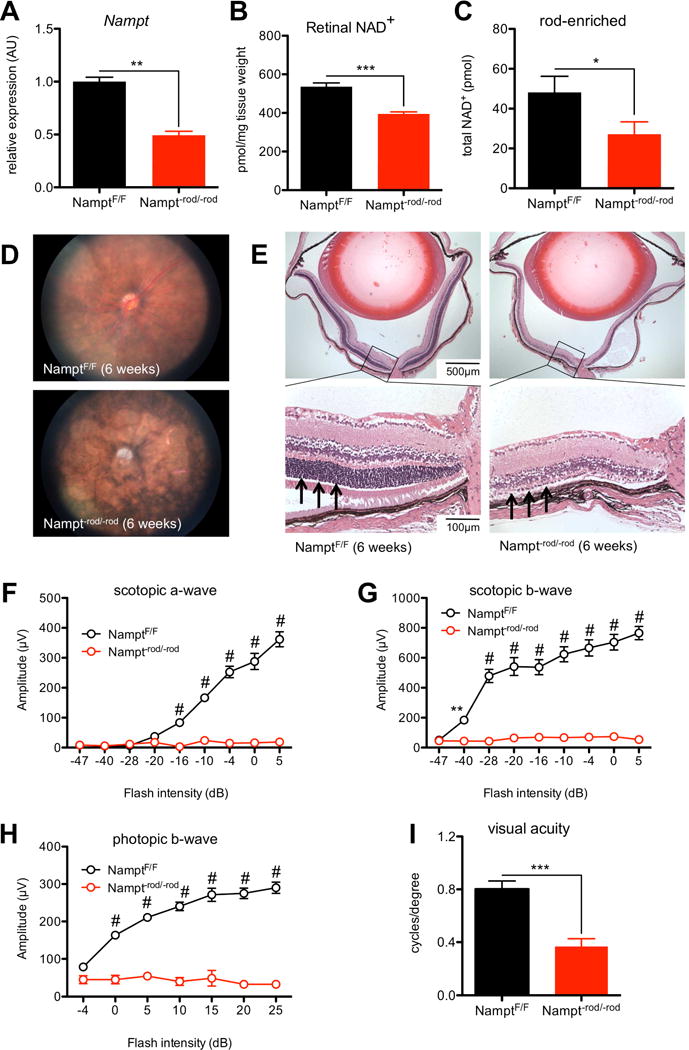
Nampt−rod/−rod mice exhibit severe retinal degeneration. (A) Rod-specific deletion of Nampt caused significant reduction in Nampt mRNA expression from rod-enriched retinal isolates (n=7 isolates/group; Mann-Whitney U test). (B) Nampt−rod/−rod retinas had lower NAD+ levels than those from NamptF/F mice (n=5–8 retinas/group; 2-tailed, unpaired t-test). (C) Rod-enriched retinal isolates from Nampt−rod/−rod mice had lower NAD+ levels than those from NamptF/F retinas (n=9–10 pooled samples/group; 1-tailed, unpaired t-test). (D) Representative fundus images from Nampt−rod/−rod mice demonstrated severe signs of retinal degeneration including vascular attenuation and optic nerve atrophy. (E) Representative retinal sections from Nampt−rod/−rod mice at 6 weeks stained with hematoxylin & eosin showed significantly reduced outer nuclear layer thickness (see arrows) with secondary retinal degeneration. (F–H) Nampt−rod/−rod mice exhibited impaired retinal function on ERG (n=5 NamptF/F mice/10 Nampt−rod/−rod mice; 2-way mixed ANOVA with Bonferroni post-hoc test), and reduced photopic visual acuity (I; n=5–6 mice/group; 2-tailed, unpaired t-test). Graphs depict mean + S.E.M. (A–C, I) or mean ± S.E.M. (F–H) (* p < .05; ** p < .01; *** p < .001; # p < .0001).
On biomicroscopy, we found that Nampt−rod/−rod mice had a degenerative phenotype that, by 6 weeks, was characterized by massive atrophy of the neurosensory retina, vascular attenuation with pigment mottling, and atrophy of the underlying retinal pigment epithelium (RPE) cells, which was not observed in NamptF/F littermates (Fig 1D). This neurosensory retinal degeneration was also associated with secondary atrophy and pallor of the optic nerve (Fig 1D). We confirmed this degeneration with histological examination of eyes from Nampt−rod/−rod mice. Although the histology was relatively normal at 2 weeks (Fig S1A), there was progressive loss of the outer nuclear layer with associated retinal degeneration, significant reduction of retinal thickness, and secondary extension to multiple retinal layers in Nampt−rod/−rod eyes that was complete by 6 weeks (Fig 1E). Of note, the outer nuclear layer (arrows; Fig 1E) appeared to be almost completely absent by 6 weeks, indicating substantial photoreceptor death.
We performed electroretinography (ERG) to confirm the functional deficits associated with this profound anatomic degeneration. By 6 weeks, Nampt−rod/−rod mice demonstrated a dramatic reduction in the amplitudes of rod-generated scotopic a-waves (Fig 1F), indicating significant impairment of rod function. This impairment in rod photoreceptor function also led to declines in the amplitudes of the scotopic b-waves (Fig 1G). Interestingly, Nampt−rod/−rod mice also exhibited deficits in cone function, as manifested by a decline in the amplitudes of the photopic b-waves (Fig 1H). Although Nampt deletion was specifically in rods, this cone dysfunction was not surprising; cone photoreceptor degeneration is often observed as a secondary effect of rod photoreceptor degeneration since rods are required for cone survival (Ait-Ali et al., 2015; Wong and Kwok, 2016). Consistently, by 6 weeks, Nampt−rod/−rod mice had significantly worse photopic visual acuity compared to age-matched NamptF/F controls (Fig 1I). As such, our findings replicate the progression of rod degeneration followed by cone degeneration that is often observed in patients with inherited retinal degenerations such as RP.
We also generated Nampt−rod/wt mice to determine the effect of partial ablation of NAMPT-mediated NAD+ biosynthesis. As expected, rod-enriched retinal isolates from Nampt−rod/wt mice exhibited partial reduction in Nampt expression compared to NamptF/wt controls (Fig S1B), which was roughly half of the reduction in Nampt−rod/−rod versus NamptF/F mice. This single-allele Nampt deletion did not cause a statistically significant reduction in retinal NAD+ levels (Fig S1C) nor did it cause significant retinal degeneration by 6 weeks as measured on ERG (Figs S1D–F), suggesting that Nampt is haplosufficient during this short time scale.
To confirm that this effect of Nampt deletion was cell autonomous, we generated mice lacking Nampt specifically from cone photoreceptors (Nampt−cone/−cone). Although cones constitute only ~3% of the photoreceptors, they are exclusively responsible for precise color and central vision. As expected, immunohistochemistry revealed reduced intracellular NAMPT staining (red) within cone photoreceptors (green) in retinal sections from Nampt−cone/−cone mice, while retinal sections from NamptF/F mice demonstrated robust cone NAMPT expression (Fig 2A).
Figure 2.

Nampt−cone/−cone mice exhibit cone-specific degeneration. (A) Cone-specific deletion of Nampt caused reduced intracellular NAMPT staining (red) in cone photoreceptors stained with cone-arrestin (green) and the nuclear DAPI stain (blue). (B) Representative fundus images from Nampt−cone/−cone mice demonstrated retinal pigment epithelial cell mottling (arrows) and optic nerve atrophy (arrowhead) consistent with mild retinal degeneration. (C–E) Nampt−cone/−cone mice also exhibited impaired retinal function on ERG (n=8–10 mice/group; 2-way mixed ANOVA with Bonferroni post-hoc test) and reduced photopic visual acuity (F; n=4 mice/group; Mann-Whitney U test). Graphs depict mean + S.E.M. (F) or mean ± S.E.M. (C–E) (* p < .05; ** p < .01; # p < .0001).
On biomicroscopy, Nampt−cone/−cone mice demonstrated changes consistent with cone degeneration, including mottling of the RPE and pallor of the optic nerve (Fig 2B). This structural degeneration also manifested in functional deficits on ERG. By 6 weeks, Nampt−cone/−cone mice exhibited mild declines in their scotopic a- and b-wave amplitudes (Figs 2C–D). More strikingly, Nampt−cone/−cone mice exhibited dramatic declines in the amplitudes of their photopic b-waves (Fig 2E), indicating predominant cone dysfunction. These structural and functional changes in Nampt−cone/−cone mice were also associated with a significant decrease in photopic visual acuity (Fig 2F). Cumulatively, these findings provide strong support for the essential role of NAMPT-mediated NAD+ biosynthesis in photoreceptor survival, as cell-specific deletion of this key enzyme led to rapid photoreceptor degeneration and vision loss.
Exogenous NMN prevents photoreceptor degeneration and restores vision
To confirm the importance of NAMPT-mediated biosynthesis in photoreceptor survival, we tested whether exogenous supplementation with NMN could rescue photoreceptor degeneration by bypassing the NAMPT-catalyzed reaction. NAMPT catalyzes the conversion of NAM to NMN and should reduce photoreceptor death by ameliorating the NAD+ deficiency. We gave Nampt−rod/−rod mice daily intraperitoneal injections of NMN (150 mg/kg) or vehicle alone beginning at P5 and ending at the time of ERG testing. Strikingly, ERG at 4 weeks in NMN-treated Nampt−rod/−rod mice revealed significant recovery of scotopic and photopic retinal function compared to vehicle-treated Nampt−rod/−rod mice (Figs 3A–C). There was no dramatic effect of NMN on the ERG of NamptF/F controls (Figs S2A–C). Consistently, retinal sections from NMN-treated Nampt−rod/−rod mice showed rescue of retinal degeneration compared to vehicle-treated Nampt−rod/−rod mice, as manifested by relative preservation of the outer nuclear layer (Fig 3D).
Figure 3.
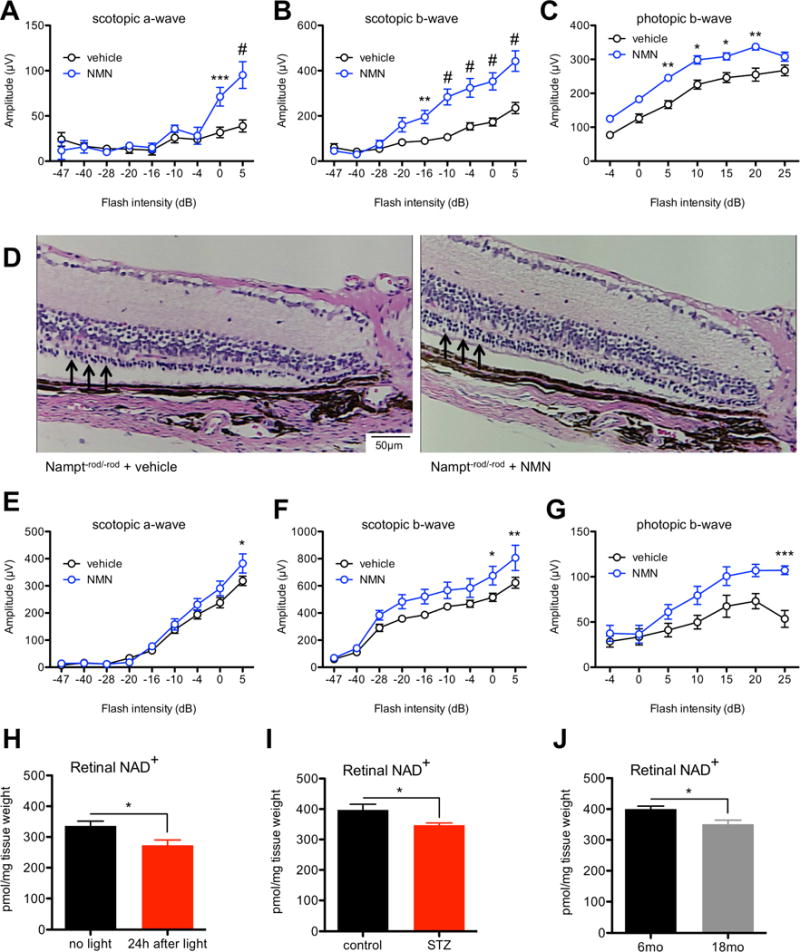
Exogenous NMN protects against retinal degeneration in mice lacking Nampt and may have efficacy against diverse retinal degenerative diseases. (A–C) Nampt−rod/−rod mice receiving daily intraperitoneal injections of 150 mg/kg NMN beginning at P5 had improved retinal function on ERG (n=12 vehicle/5 NMN; 2-way mixed ANOVA with Bonferroni post-hoc test) compared to vehicle-treated mice, consistent with relative preservation of the outer nuclear layer on histology (D; see arrows). (E–G) Nampt−cone/−cone mice receiving daily intraperitoneal injections of 150 mg/kg NMN beginning at P5 also had improved retinal function on ERG (n=9 vehicle/5 NMN; 2-way mixed ANOVA with Bonferroni post-hoc test). Retinal NAD+ deficiency is a feature of multiple mouse models of retinal dysfunction, including light-induced degeneration (H; n=10–12/group; 2-tailed, unpaired t-test), streptozotocin-induced diabetic retinopathy (I; n=5/group; 2-tailed, unpaired t-test), and age-associated retinal dysfunction (J; n=5/group; 2-tailed, unpaired t-test). Graphs depict mean + S.E.M. (H–J) or mean ± S.E.M. (A–C, E–G) (* p < .05; ** p < .01; *** p < .001; # p < .0001).
Correspondingly, daily intraperitonal NMN injections also significantly improved retinal function by ERG in Nampt−cone/−cone mice compared to vehicle-treated Nampt−cone/−cone mice (Figs 3E–G). These data clearly demonstrate that NAMPT-mediated NAD+ biosynthesis is necessary for the survival and function of both rod and cone photoreceptors, as promoting NAD+ biosynthesis in the retina with NMN supplementation can compensate for Nampt deletion, thereby reducing photoreceptor death and improving vision.
Early NAD+ deficiency is a feature of multiple mouse models of retinal dysfunction
Next, we sought to determine whether NAD+ deficiency is an early feature of other mouse models of retinal disease or dysfunction, which would support the possibility that NAD+ intermediates may have therapeutic potential against a broad spectrum of retinal degenerative diseases. Light-induced degeneration is a well-characterized model that is widely used to study mechanisms of photoreceptor death (Grimm and Reme, 2013) and is known to cause retinal dysfunction, which can be observed on histology and ERG (Schimel et al., 2011). We found that there was a significant reduction in retinal NAD+ levels as early as 24 hours following light exposure (Fig 3H). In addition, we tested whether mice with streptozotocin (STZ)-induced diabetic retinopathy, whose retinal dysfunction has been well characterized (Samuels et al., 2015), exhibit retinal NAD+ deficiency. At 3 weeks after STZ induction, hyperglycemic mice had a significant reduction in retinal NAD+ compared to non-hyperglycemic controls (Fig 3I). Finally, it is well known that there are defects in retinal structure and function in old mice, such as a decreased number of rod photoreceptors, a decreased level of total opsin in the retina, and diminished rod ERG recordings (Kolesnikov et al., 2010; Lin et al., 2016). Consistent with these past reports, we found that 18-month-old wild-type mice (C57BL/6J) had worse retinal function on ERG compared to strain-matched 6-month-old mice from the same source (Figs S2D–F). As in light-induced degeneration and STZ-induced diabetic retinopathy, this age-associated retinal dysfunction was associated with a significant decline in retinal NAD+ levels (Fig 3J). These findings support the idea that NAD+ deficiency may be a shared feature of retinal dysfunction.
NMN rescues retinal dysfunction in light-induced degeneration
Since we showed that there is NAD+ deficiency associated with the retinal dysfunction that follows light-induced degeneration, we sought to determine whether exogenous NMN supplementation to rescue NAD+ deficiency would rescue retinal dysfunction. We gave wild-type mice (129S1/SvImJ) daily intraperitoneal injections of NMN (300 mg/kg) or vehicle alone beginning 6 days prior to light exposure, on the day of light exposure, and for 3 days following light exposure (total of 10 injections/mouse). Remarkably, NMN-treated wild-type mice were more resilient against light exposure and retained improved retinal function on ERG compared to vehicle-treated mice (Figs 4A–C). These findings suggest that the NAD+ deficiency associated with retinal disease can indeed be rescued by NAD+ intermediates such as NMN, further supporting the possibility of using NAD+ intermediates to treat retinal degenerative diseases.
Figure 4.

NAD+ deficiency disrupts retinal energy homeostasis and can be rescued with exogenous NMN. (A–C) Wild-type mice (129S1/SvImJ) receiving intraperitoneal injections of 300 mg/kg NMN were more resilient against light exposure (n=8/group; 2-way mixed ANOVA with Bonferroni post-hoc test) compared to vehicle-treated mice. (D) Representative electron microscopy images of retinas from 4-week old Nampt−rod/−rod mice revealed mitochondria that are rounded, disorganized, and with loss of cristae compared to NamptF/F mice. (E) Nampt−rod/−rod retinas also exhibited significant disruption of numerous metabolic pathways on metabolite set enrichment analysis. Graphs depict mean ± S.E.M. (A–C) (* p < .05).
Loss of NAMPT-mediated NAD+ biosynthesis leads to metabolic dysfunction
To provide insight into how NAD+ deficiency leads to retinal degeneration, we performed transmission electron microscopy on Nampt−rod/−rod retinas. Although there were no obvious differences in mitochondrial ultrastructure at 3 weeks (Fig S3A), we observed profoundly dysmorphic changes in photoreceptor inner segments by 4 weeks along with disruption of the outer segments, which we did not observe in NamptF/F littermates of the same age (Fig S3B). Of interest, by 4 weeks, the mitochondria of Nampt−rod/−rod retinas were rounded and constricted with loss of cristae with a morphology that was distinctly different from that observed in NamptF/F littermates (Fig 4D). In addition, at this time point, we observed degenerative vacuoles in the cytoplasm of Nampt−rod/−rod retinas, which appeared to contain degenerated organelles including ruptured mitochondria (Fig S3C). These results suggest that loss of NAMPT-mediated NAD+ biosynthesis in photoreceptors leads to mitochondrial structural irregularities that may lead to or be associated with defects in mitochondrial function.
To characterize these possible mitochondrial defects, we performed non-biased metabolomic analysis with liquid chromatography-mass spectrometry (LC-MS) and gas chromatography-mass spectrometry (GC-MS), comparing retinas isolated from 3-week old Nampt−rod/−rod mice from those isolated from gender-matched NamptF/F littermates. We chose this time point since it would allow us to identify signs of mitochondrial dysfunction that precede gross retinal degeneration. The LC-MS results revealed that although there were no statistically significant differences, there was a trend toward accumulation of some acylcarnitine species in Nampt−rod/−rod retinas, suggesting a possible defect in Krebs cycle efficiency (Fig S3D). In addition, the GC-MS results revealed that numerous mitochondrial metabolites were significantly elevated or reduced in Nampt−rod/−rod retinas (Fig S3E). To determine whether the identities of these dysregulated metabolites were suggestive of defects in certain metabolic pathways, we performed metabolite set enrichment analysis with MetaboAnalyst 3.0 (Xia et al., 2015) using the over representation analysis algorithm. This analysis revealed that there are numerous metabolic pathways that are broadly dysregulated in Nampt−rod/−rod retinas, including protein biosynthesis, propanoate metabolism, and the citric acid cycle, among others (Fig 4E).
To confirm that these effects on mitochondrial function were indeed related to loss of NAMPT function within photoreceptors, we treated 661W cone photoreceptor-like cells with the pharmacological NAMPT inhibitor FK866 (20 μM) and measured reductive capacity to approximate metabolic activity. NAMPT inhibition caused considerable reduction of photoreceptor cell reductive capacity. By 24 hours, FK866-treated cells had roughly 40% of the reductive capacity of vehicle-treated cells (Fig 5A). By 48 hours, there was an even more dramatic effect with reductive capacity reduced down to ~20% (Fig 5B). Although there was no cell death at 24 hours (Fig 5C), this loss of reductive capacity ultimately led to cell death by 48 hours (Fig 5D). This finding highlights that metabolic dysfunction, as represented by a decrease in reductive capacity, precedes cell death.
Figure 5.

NAMPT inhibition causes metabolic dysfunction and photoreceptor death. (A–B) NAMPT inhibition with 20 μM FK866 caused loss of reductive capacity in 661W cone photoreceptor-like cells by 24 hours and 48 hours (n=15/group from three independent experiments; 1-way ANOVA with Tukey post-hoc test). (C–D) This metabolic dysfunction caused cell death by 48 hours (n=14/group from three independent experiments; 1-way ANOVA with Tukey post-hoc test). The effects of NAMPT inhibition were rescued with 100 μM NMN (A–B, D). (E) NAMPT inhibition with 20 μM FK866 led to a significant reduction in total NAD+ in photoreceptor cells by 6 hours, which was restored to near-normal levels with 100 μM NMN (n=3/group; 1-way ANOVA with Tukey post-hoc test). (F) 24 hours of 20 μM FK866 treatment led to undetectable levels of NAD+ (N.D. = not detected), which once again was restored with 100 μM NMN (n=5–7/group). (G) NAMPT inhibition also caused ATP depletion but in a delayed time frame relative to NAD+ depletion (n=4/group). (H–I) FK866-treated photoreceptor cells had reduced oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) at baseline, impaired ECAR acceleration after oligomycin treatment, and impaired OCR acceleration after FCCP treatment (n=15–16/group from representative experiment; 2-way mixed ANOVA with Bonferroni post-hoc test). 100 μM NMN restored normal metabolic responses (H–I). (J) NAD+-dependent isocitrate dehydrogenase (NAD-IDH/IDH3) activity was reduced in rods isolated from Nampt−rod/−rod mice compared to those isolated from NamptF/F mice, even when sufficient NAD+ was added to the reaction mixture (n=6/group from three independent experiments; 2-tailed, unpaired t-test) and despite similar Idh3a expression levels (K; n=11–13/group from three independent experiments; 2-tailed, unpaired t-test). (L–M) The activities of other NAD+-dependent enzymes alpha-ketoglutarate dehydrogenase (AGDH; n=5/group from three independent experiments; Mann-Whitney U test) and malate dehydrogenase (MDH; n=7/group from three independent experiments; 2-tailed, unpaired t-test) in rods from Nampt−rod/−rod mice were restored with exogenous NAD+. Graphs depict mean + S.E.M. (A–F, J–M) or mean ± S.E.M. (G–I) (* p < .05; ** p < .01; *** p < .001; # p < .0001; red: vehicle versus FK; blue: FK versus FK+NMN).
Since we observed that exogenous NMN supplementation rescued retinal function in Nampt−rod/−rod and Nampt−cone/−cone mice, we tested whether NMN supplementation could rescue the deleterious effects of NAMPT inhibition in photoreceptor cells. Our results demonstrate that providing FK866-treated photoreceptors with NMN (100 μM) could completely restore normal reductive capacities at 24 hours (Fig 5A) and at 48 hours (Fig 5B), and prevent subsequent cell death (Fig 5D). These results provide strong evidence for the importance of NAMPT-mediated NAD+ biosynthesis in photoreceptors since we demonstrate that bypassing the NAMPT-catalyzed reaction could restore normal reductive capacity and prevent photoreceptor cell death.
To determine whether these dramatic effects of NAMPT inhibition were unique to photoreceptors, we tested the effects of FK866 (20 μM) on the reductive capacity and cell survival of ARPE-19 RPE cells. Of interest, an identical dose of FK866 had no effect on the reductive capacity of RPE cells at 24 hours (Fig S4A) and only a modest albeit statistically significant effect on reductive capacity at 48 hours (Fig S4B). Remarkably, FK866 did not reduce RPE cell survival at either 24 hours (Fig S4C) or 48 hours (Fig S4D), demonstrating that RPE cells are more resilient to metabolic dysfunction and thereby protected from cell death. Ultimately, these results suggest that photoreceptors are uniquely vulnerable to impairment of NAMPT-mediated NAD+ biosynthesis and implicate this pathway as a master regulator of photoreceptor metabolism and survival.
NAMPT inhibition causes rapid NAD+ depletion, leading to an ATP crisis
To further characterize the effects of NAMPT inhibition, we measured the NAD+ levels in photoreceptor cells after treatment with FK866 (20 μM). As expected, by 6 hours, FK866-treated photoreceptor cells contained significantly less total NAD+ compared to vehicle-treated cells plated at the same density (Fig 5E). Exogenous NMN supplementation (100 μM) returned NAD+ back to near-normal levels (Fig 5E). After 24 hours, there were undetectable levels of NAD+ in FK866-treated cells compared to vehicle-treated cells (Fig 5F). Similar to the 6-hour time point, exogenous NMN supplementation for 24 hours prevented FK866-associated NAD+ depletion, bringing NAD+ back to even higher levels compared to vehicle-treated cells (Fig 5F). We interpret these results to indicate that NAMPT inhibition causes rapid NAD+ deficiency, which likely contributes to the metabolic dysfunction described above.
To further characterize the connection between NAD+ depletion and cell death, we investigated the time course of NAD+ depletion and its effect on ATP production by simultaneously measuring NAD+ and ATP content in photoreceptor cells at various times after treatment with FK866 (20 μM). As above, we found that FK866 caused rapid NAD+ deficiency. By 6 hours, NAD+ levels were down to ~50% of their original levels (Fig 5G). NAD+ depletion continued rapidly, dropping to ~15% by 12 hours and to undetectable levels by 24 hours (Fig 5G). NAMPT inhibition also caused ATP depletion, but ATP levels did not decline after 6 hours of FK866 treatment and dropped only modestly to ~70% of the original levels by 12 hours (Fig 5G). By 24 hours however, ATP levels had dropped down to ~10% of the original levels (Fig 5G). The temporal relationship between NAD+ and ATP depletion suggests that the ATP crisis is a downstream effect of NAD+ deficiency-associated metabolic dysfunction.
To confirm the connection between NAD+ availability, ATP content, metabolic function, and cell survival, we also measured the NAD+ and ATP levels in RPE cells after NAMPT inhibition. Consistent with the mild effects on metabolic function (Figs S4A–B) and the lack of effect on cell survival (Figs S4C–D), 24 hours of FK866 treatment (20 μM) caused only a ~60% reduction in total NAD+ levels (Fig S4E), a smaller effect than that observed after 24 hours of FK866 treatment in photoreceptor cells. This intermediate decrease in total NAD+ levels was accompanied by a concomitant increase in ATP levels (Fig S4F), drastically different from the severe ATP depletion exhibited by photoreceptor cells. These findings suggest that photoreceptor cells are more vulnerable to perturbations in NAD+ biosynthesis compared to other non-photoreceptor eye cells, such as RPE cells.
NAMPT inhibition impairs basal metabolism and the normal response to metabolic stress
The rapid decline in ATP in photoreceptor cells within 24 hours of FK866 treatment suggests that NAMPT inhibition causes significant metabolic dysfunction. To characterize what elements of metabolic function were impaired, we treated photoreceptor cells with FK866 (20 μM) and profiled various aspects of metabolism by measuring oxygen consumption rate (OCR) as a measure of aerobic respiration and extracellular acidification rate (ECAR) as a measure of glycolytic flux. We measured these parameters of metabolic function at baseline and under various conditions of stress induced by sequential treatments with oligomycin, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and antimycin A/rotenone.
At baseline, FK866-treated photoreceptor cells exhibited reduced OCR and ECAR (Figs 5H–I), indicating that NAMPT inhibition caused reduced basal oxidative respiration and basal glycolytic flux. While vehicle-treated cells responded appropriately to the ATP synthase inhibitor oligomycin by shifting toward glycolytic metabolism (reduced OCR; increased ECAR), FK866-treated photoreceptor cells were unable to shift toward glycolytic metabolism (Figs 5H–I). Moreover, while vehicle-treated cells responded appropriately to the ionophore FCCP by elevating their oxidative respiration (increased OCR), FK866-treated photoreceptor cells were unable to accelerate oxidative respiration in response to metabolic stress (Fig 5H). NMN supplementation restored normal baseline metabolism and the appropriate responses to stress (Figs 5H–I), confirming that these phenotypes were specific to NAD+ deficiency.
To confirm that these effects on metabolic function were specific to NAMPT inhibition, we performed Nampt knockdown in photoreceptor cells. Partial reduction of NAMPT-mediated NAD+ biosynthesis through Nampt knockdown in photoreceptor cells caused a reduction in NAD+ levels (Fig S5A) within 24 hours after knockdown. Although this intermediate reduction in NAD+ did not affect reductive capacity (Fig S5B) or cell survival at the early 48-hour time point (Fig S5C), it did impair the ability of photoreceptor cells to maximally accelerate oxidative respiration in response to FCCP (Fig S5D–E), further reinforcing the importance of NAMPT-mediated NAD+ biosynthesis for maintaining mitochondrial homeostasis in photoreceptors.
Taken together, these data confirm the importance of NAMPT-mediated NAD+ biosynthesis for maintaining photoreceptor glycolytic and mitochondrial function – both at baseline and in response to metabolic stress – and highlight a NAMPT-specific effect due to the ability of NMN to restore normal metabolic responses. Importantly, we observed these signs of metabolic dysfunction after 24 hours of FK866 treatment prior to when we observed the effects on photoreceptor cell viability. Once again, these results highlight that glycolytic and mitochondrial dysfunction precede and therefore likely cause photoreceptor cell death.
NAD+ deficiency causes NAD-IDH dysfunction
Our in vitro and in vivo results suggest that there are defects in oxidative metabolism in photoreceptors under conditions of NAD+ deficiency. To more specifically characterize these defects in oxidative metabolism, we tested the activity of three enzymes of the Krebs cycle that require NAD+ as a coenzyme: NAD+-dependent isocitrate dehydrogenase (NAD-IDH/IDH3), alpha-ketoglutarate dehydrogenase (AGDH), and malate dehydrogenase (MDH). This fact that these enzymes require NAD+ as a coenzyme provides the opportunity to determine whether their enzymatic dysfunction is caused solely by loss of NAD+ as a coenzyme. If loss of NAD+ as a coenzyme were solely responsible for enzymatic dysfunction, we would expect to be able to restore enzymatic function simply by providing sufficient NAD+ to the reaction mixture.
We found that NAD-IDH activity was significantly lower in rods isolated from Nampt−rod/−rod retinas compared to those isolated from NamptF/F retinas, even when sufficient NAD+ was supplied in the reaction (Fig 5J) and despite similar NAD-IDH expression levels (Fig 5K). These findings suggest that NAD-IDH dysfunction cannot be explained by loss of NAD+ as a coenzyme alone. Of significant interest, the rod AGDH and MDH activities were completely rescued by NAD+ (Figs 5L–M), indicating selective enzymatic dysfunction. These findings not only characterize one specific aspect of mitochondrial dysfunction but also suggest that NAD+ plays important roles in regulating metabolism in addition to its role as a coenzyme.
SIRT3 and SIRT5 play important roles in photoreceptor survival
In addition to serving as a coenzyme for crucial steps of glycolysis and the Krebs cycle, NAD+ also serves as a cosubstrate for NAD+-consuming enzymes, including the sirtuins. Of the seven sirtuin family members, three of them, SIRT3, SIRT4, and SIRT5, are known to regulate mitochondrial function (Laurent et al., 2013; Yang et al., 2007; Zhang et al., 2015). Because other sirtuin family members have been shown to play crucial roles in regulating the survival of retinal cells (Jaliffa et al., 2009; Silberman et al., 2014) and because sirtuins depend on NAD+ availability for optimal function (Satoh and Imai, 2014), we hypothesized that NAD+ deficiency may also impair mitochondrial sirtuin activity, contributing to mitochondrial dysfunction.
To determine whether the mitochondrial sirtuins are essential for photoreceptor survival, we performed individual and combined knockdown of SIRT3, SIRT4, and SIRT5 in photoreceptor cells. Individual knockdown of SIRT3 and SIRT5 caused significant reduction in reductive capacity compared to knockdown by negative control (Fig 6A). Interestingly, individual knockdown of SIRT4 did not diminish reductive capacity, highlighting the specificity of the effect of SIRT3 and SIRT5 (Fig 6A). Combined SIRT3 and SIRT5 knockdown had a synergistic effect, leading to significantly lower reductive capacity compared to either single knockdown alone (Fig 6A). Of note, NMN was not able to rescue the effect on reductive capacity in the SIRT3/SIRT5 double knockdown (Fig 6A), suggesting that exogenous NMN cannot protect against SIRT3/SIRT5 deletion.
Figure 6.

SIRT3 and SIRT5 are essential for photoreceptor survival. (A) Individual SIRT3 and SIRT5 knockdown but not SIRT4 knockdown caused significant loss of reductive capacity relative to negative control (NC); combined SIRT3/SIRT5 knockdown caused significantly more loss of reductive capacity than individual knockdowns, which could not be rescued with NMN (n=6/group from representative experiment; 1-way ANOVA with Tukey post-hoc test). (B) Individual SIRT3 and SIRT5 knockdown but not SIRT4 knockdown caused significant cell death relative to negative control (NC); combined SIRT3/SIRT5 knockdown caused significantly more cell death than individual knockdowns, which could not be rescued with NMN (n=18/group from three independent experiments; 1-way ANOVA with Tukey post-hoc test). (C) Individual and combined SIRT3/SIRT5 knockdowns recapitulated NAD-IDH dysfunction compared to negative control (NC; n=3/group from three independent experiments; 1-way ANOVA with Tukey post- hoc test). (D–F) Mice lacking SIRT3 and SIRT5 (SIRT3KOSIRT5KO) were significantly more vulnerable to light-induced degeneration (LID) compared to SIRT3hetSIRT5het mice, while SIRT3KOSIRT5het and SIRT3hetSIRT5KO mice exhibited intermediate vulnerability to LID (n=5–7 mice/group; 2-way mixed ANOVA with Bonferroni post-hoc test). Graphs depict mean + S.E.M. (A–C) or mean ± S.E.M. (D–F) (* p < .05; ** p < .01; *** p < .001; # p < .0001; ^ p < .05 relative to both SIRT3KD and SIRT5KD; red: SIRT3hetSIRT5het versus SIRT3KOSIRT5KO; brown: SIRT3KOSIRT5het versus SIRT3KOSIRT5KO; grey: SIRT3hetSIRT5KO versus SIRT3KOSIRT5KO).
This decline in reductive capacity secondary to SIRT3/SIRT5 knockdown was also accompanied by progressive cell death. Individual knockdown of SIRT3 and SIRT5 caused significant reduction in cell survival compared to knockdown by negative control (Fig 6B). Again, individual knockdown of SIRT4 had no effect on cell viability (Fig 6B), highlighting the specificity of the SIRT3/SIRT5 effect. Once again, combined SIRT3 and SIRT5 knockdown had a more profound effect on cell death compared to either individual knockdown alone (Fig 6B). Consistently, NMN supplementation did not rescue the cell death caused by combined SIRT3/SIRT5 double knockdown (Fig 6B).
To test whether loss of SIRT3 and SIRT5 causes mitochondrial dysfunction that is similar to that caused by NAD+ deficiency, we performed individual and combined SIRT3 and SIRT5 knockdowns in photoreceptors and measured the NAD-IDH activity 24 hours after transfection. We found that knocking down SIRT3, SIRT5, or both SIRT3/SIRT5 in photoreceptor cells recapitulated NAD-IDH dysfunction (Fig 6C). The fact that SIRT3 and SIRT5 knockdowns phenocopy the NAD-IDH dysfunction observed in rods isolated from Nampt−rod/−rod retinas suggests that SIRT3 and SIRT5 may play a role in governing the metabolic dysfunction associated with NAD+ deficiency.
To test the role of SIRT3 and SIRT5 in retinal function in vivo, we tested mice lacking SIRT3 and SIRT5. Both SIRT3KO and SIRT5KO mice have normal-appearing fundi on biomicroscopy (Figs S6A–B) and normal retinal function on ERG compared to strain-matched controls (Figs S6C–H). However, since many studies report that SIRT3 and SIRT5 regulate many of the same protein targets and even the same lysine residues within the same protein (Hebert et al., 2013; Park et al., 2013; Rardin et al., 2013a; Rardin et al., 2013b; Schwer et al., 2009; Sol et al., 2012; Still et al., 2013), it is possible that SIRT3 and SIRT5 can compensate for one another. To test this possibility, we also tested mice lacking multiple mitochondrial sirtuins.
Although SIRT3KOSIRT5KO mice had normal retinal function at baseline (Figs S6I–K), we could not yet conclude that SIRT3 and SIRT5 are inessential for retinal function since previous studies have reported that mice lacking mitochondrial sirtuins rarely show striking phenotypes (Lombard et al., 2007; Yu et al., 2013) until they are challenged by specific stimuli such as fasting or a high-fat diet (Hirschey et al., 2011). To this end, we tested whether SIRT3KOSIRT5KO mice are more vulnerable to retinal degeneration after exposure to light. We found that SIRT3KOSIRT5KO mice were strikingly more vulnerable to retinal degeneration upon light stress compared to SIRT3hetSIRT5het controls as manifested by retinal dysfunction on ERG (Figs 6D–F) when tested four days after light treatment. Interestingly, SIRT3hetSIRT5KO and SIRT3KOSIRT5het mice demonstrated an intermediate degenerative phenotype (Figs 6D–F), supporting the notion that SIRT3 and SIRT5 synergistically regulate mitochondrial function in photoreceptors. Together, these results provide strong evidence that SIRT3 and SIRT5 not only are important for maintaining photoreceptor survival but also have distinct, non-redundant roles in regulating retinal homeostasis.
NAMPT inhibition causes SIRT3 dysfunction
Since we found that SIRT3 and SIRT5 are essential for photoreceptor survival, we sought to determine whether NAD+ deficiency caused by NAMPT inhibition impairs SIRT3 and SIRT5 function in photoreceptor cells. Since SIRT3 regulates predominantly protein acetylation (Parihar et al., 2015) and SIRT5 regulates predominantly protein succinylation, malonylation, and glutarylation (Du et al., 2011; Nishida et al., 2015; Papanicolaou et al., 2014; Tan et al., 2014), we tested whether NAMPT inhibition modulates the acylation of mitochondrial proteins in photoreceptor cells. We found that mitochondrial lysates generated from photoreceptor cells treated with FK866 (20 μM) for 24 hours showed obvious hyperacetylation (Figs 7A–B), indicating SIRT3 dysfunction. Normal acetylation patterns were restored with NMN (Figs 7A–B), confirming that this hyperacetylation was specific to NAMPT inhibition. Although there were clear acetylation changes, these mitochondrial lysates demonstrated only modest changes in succinylation (Figs 7C–D) and no obvious differences in malonylation (Fig S7A) or glutarylation (Fig S7B), suggesting that NAD+ deficiency primarily impairs SIRT3 function.
Figure 7.

NAD+ deficiency impairs mitochondrial sirtuin function. (A–B) NAMPT inhibition caused selective mitochondrial protein hyperacetylation (n=4 biological replicates from two independent blots; 1-way ANOVA with Tukey post-hoc test). (C–D) NAMPT inhibition caused only modest changes in mitochondrial protein succinylation (n=6 biological replicates from three independent blots; 1-way ANOVA with Tukey post-hoc test). (E) NAD+ deficiency impaired SIRT3 activity (n=6/group; 2-tailed, unpaired t-test) but not SIRT5 activity (F; n=15–16/group; 2-tailed, unpaired t-test). Graphs depict mean + S.E.M. (B, D–F) (** p < .01; *** p < .001).
To confirm that these effects were caused by NAMPT inhibition rather than being secondary to metabolic dysfunction, we measured the activity of SIRT3 and SIRT5 in the mitochondrial lysates generated from FK866-treated photoreceptor cells. Consistent with our Western Blots, mitochondrial lysates generated from FK866-treated photoreceptor cells had a significant reduction in SIRT3 activity compared to mitochondrial lysates generated from vehicle-treated cells (Fig 7E). There was, however, no difference in SIRT5 activity (Fig 7F). Taken together, these results demonstrate that NAD+ deficiency leads to dysregulation of the mitochondrial acylome primarily through impairment of SIRT3 function. Since we show that SIRT3 is important for photoreceptor survival, this NAD+ deficiency-associated impairment of SIRT3 activity likely contributes to the mitochondrial dysfunction observed in photoreceptors lacking Nampt, providing additional mechanistic evidence for why NAMPT-mediated NAD+ biosynthesis is essential for mitochondrial homeostasis, photoreceptor survival, and vision.
DISCUSSION
NAD+ has numerous functions in diverse biological processes including metabolism, circadian rhythms, aging, and neurodegeneration (Alano et al., 2010; Garten et al., 2009; Gerdts et al., 2015; Imai and Yoshino, 2013; Mouchiroud et al., 2013; Nakahata et al., 2009; Ramsey et al., 2009; Stein et al., 2014; Zhou et al., 2015). In the current study, we demonstrate that NAMPT-mediated NAD+ biosynthesis is indispensable for photoreceptor survival and vision. Using loss of function approaches, we show that disrupting NAMPT-mediated NAD+ biosynthesis in rod and cone photoreceptors leads to photoreceptor death, retinal degeneration, and blindness. By testing the effects of NAMPT inhibition on non-photoreceptor cells, we confirm that photoreceptors are uniquely susceptible to perturbations in NAD+ biosynthesis.
Moreover, we demonstrate that, in photoreceptors, loss of NAMPT-mediated NAD+ biosynthesis leads to NAD+ deficiency, significant glycolytic and mitochondrial dysfunction under basal conditions, and the inability to respond appropriately to metabolic stress, which ultimately lead to photoreceptor death and retinal degeneration. Corresponding with this energetic failure, metabolic profiling of retinas from Nampt−rod/−rod mice revealed dysregulation of numerous metabolic pathways. Of interest, there was a trend (p = .0704) toward dysregulation of the citric acid cycle, a key pathway in oxidative metabolism. The trends toward accumulation of various acylcarnitine species also support a failure of Krebs cycle efficiency. These hallmarks of metabolic dysfunction can be identified prior to cell death and vision loss, supporting the possibility of probing mitochondrial function to predict subsequent photoreceptor death (Perron et al., 2013). These findings are interesting, especially considering recent studies that show that photoreceptors have limited mitochondrial reserve capacity, which may make them susceptible to defects in energy homeostasis (Kooragayala et al., 2015). We speculate that defects in the Krebs cycle cause broad energetic failure, which contributes to downstream defects in numerous metabolic pathways, such as protein biosynthesis and propanoate metabolism. Further studies are necessary to explore the precise connection between these phenomena.
Our results also identify that NAD+ deficiency leads to selective enzymatic dysfunction and that cell death is unlikely to be caused by loss of NAD+ as a coenzyme alone. When we tested the activity of the three NAD+-dependent enzymes of the Krebs cycle in rods isolated from Nampt−rod/−rod retinas, the activity of only one of the three, NAD-IDH, could not be rescued by providing sufficient NAD+ as a coenzyme, while the activity of the other NAD+-dependent Krebs cycle enzymes AGDH and MDH were restored by providing exogenous NAD+. This result stresses the importance of NAD-IDH in maintaining metabolic homeostasis in photoreceptors. In support, the retina has been shown to be highly dependent on NAD-IDH and exquisitely sensitive to defects in NAD-IDH function, such that mutations in NAD-IDH that impair its catalytic activity cause RP (Hartong et al., 2008). Of significant interest, these RP patients have normal NADP+-dependent IDH (i.e., NADP-IDH or IDH1/IDH2) activity and no other manifestations of disease despite the fact that they carry this mutation in all cells of their body (Hartong et al., 2008). This finding suggests that in contrast with most organ systems, the retina uniquely relies on the NAD+-dependent form of IDH (Hartong et al., 2008).
We further demonstrate that SIRT3 and SIRT5 both play important roles in photoreceptor survival and that NAD+ deficiency leads to predominately SIRT3 dysfunction. Our results agree with past studies reporting that other sirtuin family members, including SIRT1 (Jaliffa et al., 2009) and SIRT6 (Silberman et al., 2014), play roles in photoreceptor survival and survival of other retinal cells. Interestingly, we demonstrate that the deleterious effects of SIRT3 and SIRT5 deletion are synergistic. These findings strongly suggest that SIRT3 and SIRT5 are not redundant even though they may regulate the acylation status of the same mitochondrial proteins and even the same lysine residues within the same proteins (Hebert et al., 2013; Park et al., 2013; Rardin et al., 2013a; Rardin et al., 2013b; Schwer et al., 2009; Sol et al., 2012; Still et al., 2013).
Based on our results, we hypothesize that SIRT3 dysfunction caused by NAD+ deficiency may contribute to mitochondrial dysfunction by causing aberrant hyperacetylation of key mitochondrial proteins, such as NAD-IDH. This possibility that SIRT3 dysfunction is linked to decreased NAD-IDH activity is supported not only by our ability to recapitulate NAD-IDH dysfunction with SIRT3 knockdown but also by the fact that NAD-IDH has been identified as a target of SIRT3 (Hebert et al., 2013; Rardin et al., 2013b; Schwer et al., 2009; Sol et al., 2012; Still et al., 2013). Further studies are needed to explore this possibility and to extend these findings in vivo, perhaps using new technologies such as CRISPR/Cas9 to delete Nampt selectively from photoreceptors.
Cumulatively, these findings provide unique insights that point to the dominant mammalian NAD+ biosynthesis pathway as a master regulator of photoreceptor metabolism. Human retinal degenerations encompass a broad spectrum of diseases that include seemingly unrelated disorders such as LCA and RP, which have been associated with mutations in enzymes involved in NAD+ biosynthesis (Chiang et al., 2012; Falk et al., 2012; Koenekoop et al., 2012; Perrault et al., 2012) and the Krebs cycle (Hartong et al., 2008). Despite these genetic insights, the mechanisms underlying these conditions and what ultimately causes photoreceptor degeneration in these genotypically diverse disorders are poorly understood. We propose a model linking NAD+ biosynthesis, SIRT3/SIRT5, and metabolism in photoreceptors, which may connect these diverse retinal neurodegenerations at the molecular level.
Remarkably, our studies also demonstrate exogenous NMN supplementation to bypass the NAMPT-catalyzed reaction can restore normal NAD+ levels in photoreceptor cells despite NAMPT inhibition and reduce photoreceptor death in Nampt−rod/−rod mice. In photoreceptor cells subjected to NAMPT inhibition, NMN supplementation prevented metabolic dysfunction and cell death by restoring normal basal glycolytic and mitochondrial function and the ability to respond appropriately to metabolic stress. This therapeutic effect is likely explained by the importance of NAD+ not only to perform its coenzyme role in various steps of the Krebs cycle and glycolysis but also to maintain optimal SIRT3 activity. We show that multiple mouse models of retinal dysfunction, including light-induced degeneration, STZ-induced diabetic retinopathy, and age-associated retinal dysfunction all exhibit early retinal NAD+ deficiency. Moreover, the retinal dysfunction associated with light-induced degeneration can be partially rescued with NMN. These findings offer powerful therapeutic avenues for degenerative diseases of the eye and are supported by past studies exploring the therapeutic applications of NAD+ supplementation in light-induced degeneration (Bai and Sheline, 2013), noise-induced hearing loss (Brown et al., 2014), and high-fat diet- and age-induced metabolic complications (Canto et al., 2012; Ramsey et al., 2008; Stein and Imai, 2014; Yoshino et al., 2011).
Once successfully implemented, this treatment strategy would be far-reaching since it could be implemented for multiple diseases with diverse pathogenic mechanisms, including not only inherited forms of retinal degenerations but also other blinding diseases characterized by a final pathway of photoreceptor death such as age-related macular degeneration and diabetic retinopathy. Given the global importance of NAD+ biosynthesis and mitochondrial dysfunction in neurons, our findings may also be broadly relevant to other systemic neurodegenerative diseases such as Alzheimer’s disease where NAD+ intermediate supplementation may be neuroprotective.
EXPERIMENTAL PROCEDURES
Mice
All animal experiments were approved by the Animal Studies Committee and performed in accordance with the Washington University School of Medicine Animal Care and Use guidelines. Namptflox/flox (NamptF/F) mice were previously characterized (Rongvaux et al., 2008) and were provided by Dr. Shin-ichiro Imai. To generate mice lacking Nampt specifically from rod photoreceptors, we crossed NamptF/F mice with mice carrying one copy of the Rhodopsin-iCre75 transgene, which were provided by Dr. Ching-Kang Jason Chen and have been previously characterized (Li et al., 2005). To generate mice lacking Nampt specifically from cone photoreceptors, we crossed NamptF/F mice with mice carrying one copy of the human red/green pigment-Cre (HRGP-Cre) transgene, which were provided by Dr. Yun Le and have been previously characterized (Le et al., 2004). We received 6-month-old and 18-month-old wild-type C57BL/6J mice from the National Institutes on Aging (Bethesda MD) and purchased streptozotocin (STZ)-induced hyperglycemic mice (C57BL/6J) from Jackson Laboratories (Bar Harbor ME). We also purchased SIRT3−/−, SIRT5−/−, and the appropriate strain-matched wild-type mice (129S1/SvImJ for SIRT3−/− and B6129SF2/J for SIRT5−/−) from Jackson Laboratories.
Cells and reagents
We routinely cultured 661W cone photoreceptor-like cells, provided by Dr. Muayyad Al Ubaidi (Tan et al., 2004), in Dulbecco’s Modified Eagle Medium with 1 g/L glucose and 110 mg/L sodium pyruvate (DMEM; Thermo Fisher, Carlsbad CA) supplemented with 10% Fetal Bovine Serum (FBS; Sigma, St. Louis MO) and 1% penicillin-streptomycin (Thermo Fisher, Carlsbad CA). We prepared stock solutions of FK866 (Santa Cruz Biotechnology, Dallas TX) at 200 mM in DMSO. We purchased nicotinamide mononucleotide (NMN) from Sigma (St. Louis MO) and dissolved it directly in cell culture media, prepared as described above.
Real-time PCR in rod-enriched samples
We generated rod-enriched samples by vortexing dissected whole retinas for 60 seconds at medium speed and centrifuging the resulting supernatant at 12,800 g for 10 minutes. We extracted total RNA from the resulting pellet with the RNeasy Mini Kit (Qiagen, Valencia CA), prepared cDNA with the High Capacity cDNA Reverse Transcription Kit (Life Technologies, Grand Island NY), and performed PCR amplification of cDNA using Taqman probe-based gene expression assays (Life Technologies, Grand Island NY) as described previously (Kelly et al., 2007). We used probes for Nampt (Mm01293560_m1) and Idh3a (Mm00499674_m1) normalized to ActB (Mm00607939_s1) or Rho (Mm01184405_m1) with the ΔΔCT method.
Retinal imaging
We performed digital color fundus photography using the Micron III™ animal fundus camera (Phoenix Research Laboratories, Pleasanton CA). Prior to fundus imaging, we anesthetized mice with an intraperitoneal injection of 86.9 mg/kg ketamine and 13.4 mg/kg xylazine, and administered 1.0% tropicamide eye drops (Bausch & Lomb, Tampa FL) to dilate the pupils.
Histology and immunohistochemistry
After euthanizing the mice, we enucleated the eyes and fixed them overnight in 10% formalin. Next, we embedded the eyes in methacrylate and prepared eight to ten sections of 6–8 μm thickness cut at different planes for each eye. We stained slides with hematoxylin and eosin, rabbit anti-NAMPT(161–173) (B5812-200UL; Sigma, St. Louis MO), and rabbit anti-cone arrestin (AB15282; Millipore, Billerica MA). We acquired images with an Olympus FV1000™ upright confocal microscope with UV-sensitive (405), multi-Argon (458, 488 & 513) & Helium-Neon (543 & 633) lasers, and a Plan Apo N 60x oil objective (N.A.=1.42).
Electroretinography
We performed electroretinography (ERG) as previously described (Hennig et al., 2013) by using the UTAS-E3000 Visual Electrodiagnostic System running EM for Windows (LKC Technologies, Gaithersburg MD). We extracted quantitative measurements from the ERG waveforms using an existing Microsoft Excel macro that defines the a-wave amplitude as the difference between the average pre-trial baseline and the most negative point of the average trace and defines the b-wave amplitude as the difference between this most negative point to the highest positive point, without subtracting oscillatory potentials.
Photopic visual acuity
We measured mouse photopic visual acuity under standard photopic conditions (1.85 log cd m−2) by testing the optokinetic reflex with the OptoMotry™ System (CerebralMechanics) as described previously (Kolesnikov et al., 2011; Umino et al., 2008).
Transmission electron microscopy
To perform transmission electron microscopy, we dissected enucleated eyes to remove the cornea and lens, and fixed eye cups for 4 hours at room temperature in 2% paraformaldehyde/2.5% glutaraldehyde (Polysciences, Warrington PA) diluted in 100 mM sodium cacodylate (pH 7.2). We then washed the fixed eye cups in cacodylate buffer and post-fixed them in 1% osmium tetroxide (Polysciences) for 1 hour. We then rinsed the postfixed eye cups extensively in deionized H2O prior to en bloc staining with 1% aqueous uranyl acetate (Ted Pella, Redding CA) for 1 hour. Following several rinses, we dehydrated the stained eye cups with a graded series of ethanol solutions and embedded them in Eponate 12 resin (Ted Pella). We cut 95 nm sections with a Leica Ultracut UCT ultramicrotome (Leica Microsystems, Bannockburn IL), stained the sections with uranyl acetate and lead citrate, and imaged them on a JEOL 1200 EX transmission electron microscope (JEOL USA, Peabody MA) equipped with an AMT 8 megapixel digital camera (Advanced Microscopy Techniques, Woburn MA).
Metabolomic analysis
We isolated retinas at the same time of day after restricting oral nutrient intake for 4 hours. We pooled 6 retinas per group and snap-froze them in liquid nitrogen. We then added ammonium bicarbonate-buffered solution to the tissue pellets and transferred these tissue suspensions to tubes for subsequent metabolite extraction. We extracted intracellular metabolites and culture media samples into cold methanol:acetonitrile (ACN):water (H2O), dried them, derivatized them to their tert-butyldimethylsilyl esters (tBDMS), and then analyzed them on an Agilent 7200 GC-quadrupole-time of flight (QTOF)-MS operating in electron impact ionization mode. We dried an aliquot of each extract and dissolved it in ACN:H2O for subsequent LC-QTOF-MS analysis. We performed metabolite set enrichment analysis with MetaboAnalyst 3.0 (Xia et al., 2015) by inputting the list of dysregulated metabolites with a statistically significant difference (corrected p-value < .05) and a fold-change > 1.10. These data were processed with the over representation algorithm and the metabolic pathway-associated metabolite set library.
Reductive capacity assay
To quantify reductive capacity, we used the Cell Proliferation Reagent WST-1 (Roche Applied Science, Indianapolis IN) according to manufacturer’s instructions. In short, we washed cells once with PBS after the desired treatment and replaced the media with fresh media containing the WST-1 assay solution at a 1:10 dilution. After 2 hours of incubation at 37°C, we measured absorbance at 450 nm with the SpectraMax 190 Absorbance Microplate Reader (Molecular Devices, Sunnyvale CA).
Cell survival assay
To determine cell survival, we used the cell-permeant dye calcein AM (Thermo Fisher, Carlsbad CA) according to manufacturer’s instructions. In short, we washed cells once with PBS after the desired treatment and replaced the media with calcein AM diluted in PBS (working concentration of 2 μM). After 30 minutes at 37°C, we measured fluorescence at 485 nm excitation/520 nm emission with the Infinite 200 PRO (Tecan, Männedorf Switzerland).
Quantification of NAD+ & ATP levels with HPLC
To quantify the NAD+ and ATP levels of mouse retina, pooled rod-enriched isolates, or 661W cells, we performed reverse-phase high-performance liquid chromatography (HPLC) as described previously (Yoshino and Imai, 2013). We quantified NAD+ and ATP levels based on the peak area on the HPLC spectrogram relative to a standard curve, and normalized these values to tissue weights for retinas, number of retinas for rod-enriched isolates, or based on cell number.
OCR and ECAR measurements
We performed detailed metabolic characterization of 661W cells after NAMPT inhibition using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica MA). After the desired treatment, we washed the cells three times and left them in 180 μL of prewarmed bicarbonate-free DMEM (pH 7.4). After 30–60 minutes in a non-CO2 incubator, we simultaneously measured oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) to quantify oxidative respiration and glycolytic flux, respectively, both at baseline and after sequential treatments of 1.5 μM oligomycin, 1.0 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), and a combination of 1.0 μM antimycin A and 1.0 μM rotenone. Each 5-minute measurement period was preceded by 2 minutes of mixing and a 1-minute pause; we made 3 sequential measurements per treatment condition.
SIRT3-5 and Nampt knockdown in 661W cells
We transfected 661W cells with custom LNA longRNA GapmeRs reagents (Exiqon, Woburn MA) targeting SIRT3, SIRT4, or SIRT5 at a final concentration of 100 nM, and commercially available siRNA targeting Nampt (Mm_Pbef1_5; Qiagen, Valencia CA) at a final concentration of 50 nM. In short, we seeded 661W cells and allowed them to adhere for 30 minutes. We prepared the transfection complexes by incubating the appropriate GapmeR/siRNA with Lipofectamine RNAiMAX transfection reagent (Invitrogen, Grand Island NY) for 5 minutes at room temperature (1.0 μl or 2.4 μl of RNAiMAX per well for the 96-well and 24-well plate formats, respectively). We confirmed knockdown efficiency with real-time PCR.
Light-induced degeneration (LID)
We performed light-induced degeneration as described previously (Grimm and Reme, 2013). In short, we dilated pupils with two sequential drops of 1.0% atropine sulfate and 1.0% tropicamide (Bausch & Lomb, Tampa FL), and then exposed the mice to 13,000 lux from fluorescent lights suspended directly over the mice. We placed the mice in clear plastic cages that were surrounded on all sides with reflective aluminum foil and re-dilated pupils with two additional drops every 2 hours. We assessed retinal function 4 days following light-induced degeneration with ERG as described above. All the mice we tested carried the RPE65Leu/Leu variant, making them equally vulnerable to light-induced degeneration (Wenzel et al., 2001).
Krebs cycle enzymatic activity assays
We performed retinal dissociation by incubating dissected retinas in 12 U/ml papain (Sigma, St. Louis MO) and 5 mM L-cysteine in DMEM for 30 minutes on a small vibrating aquarium pump at room temperature. After washing away residual papain with 5 exchanges to fresh DMEM, we triturated the samples with fire-polished Pasteur pipets. We then separated rod photoreceptors with the EasySEP Mouse PE Positive Selection Kit (Stem Cell Technologies, Vancouver Canada) and PE-conjugated anti-CD73 antibodies (Miltenyi Biotec, Bergisch Gladback Germany). We measured the enzymatic activity of NAD+-dependent isocitrate dehydrogenase (NAD-IDH/IDH3), alpha-ketoglutarate dehydrogenase (AGDH), and malate dehydrogenase (MDH) in these rods with assay kits from Sigma (St. Louis MO) according to the manufacturer’s instructions. We normalized enzymatic activity to total protein and/or viable rod cell number.
Western Blotting for mitochondrial protein acylation
We isolated mitochondria from 661W cells as described previously (Frezza et al., 2007). We denatured mitochondrial lysates in NuPAGE LDS Sample Buffer and NuPAGE Sample Reducing Agent (Thermo Fisher, Carlsbad CA) for 10 minutes at 70°C, loaded them into 4–12% Novex Bis-Tris Protein Gels, and then ran them at 50 V for 15 minutes followed by 150 V for 60 minutes. We transferred the gel to a 0.2 μm nitrocellulose membrane (Bio-Rad, Hercules CA) in transfer buffer (192 mM glycine, 25 nM Tris-base, 10% methanol) for 60 minutes at a constant current of 400 mA. We blocked the membranes for 60 minutes at room temperature with 5% (w/v) bovine serum albumin (BSA; Sigma) in TBS with 0.05% Tween-20 (0.05% TBST). We then stained the membranes overnight at 4°C with 1:1,000 anti-acetyllysine (#9441; Cell Signaling Technology, Danvers MA), 1:1,000 anti-succinyllysine (PTM-401; PTM BioLabs, Chicago IL), 1:1,000 anti-malonyllysine (PTM-901; PTM BioLabs), or 1:1,000 anti-glutaryllysine (PTM-1151; PTM BioLabs) diluted in blocking buffer. We then washed the membranes and incubated them for 60 minutes at room temperature with the appropriate secondary antibody conjugated to either IRDye 800CW or IRDye 680LT (LI-COR, Lincoln NE) diluted 1:5,000 in blocking buffer. We detected proteins and analyzed the band densities with the Odyssey Infrared Imaging System (LI-COR). We normalized protein loading per lane with the Pierce BCA Protein Assay Kit (Thermo Fisher, Carlsbad CA) and used anti-COX IV (4D11-B3-E8) antibody (#11967; Cell Signaling Technology) as the loading control.
SIRT3 and SIRT5 activity assays
We measured the SIRT3 activity and SIRT5 activity of mitochondrial isolates from 661W cells with commercially available kits (Abcam, Milton Cambridge; Enzo Life Sciences, Farmingdale NY; respectively) according to manufacturer’s instructions. We did not add exogenous SIRT3/5 or NAD+ to the reaction mixture to allow us to accurately quantify native deacylase activity.
Statistics
We performed statistical testing with GraphPad Prism (Version 6.0), using the appropriate test for each data set. Prior to all data analysis, we assessed the normality of the data graphically and with the Kolmogorov-Smirnov test and, when necessary, used appropriate non-parametric alternatives. We defined statistical significance as a p-value < 0.05.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants R01EY019287 (R.S.A.), AG024150 (S.I.), AG037457 (S.I.), KL2TR000450 (J.Y.), P30DK56341 (J.Y.), P30DK020579 (J.Y.), DK104995 (J.Y.), and P30EY02687 (Vision Core Grant); the Carl Marshall Reeves and Mildred Almen Reeves Foundation (R.S.A.); a Physician-Scientist Award from Research to Prevent Blindness (R.S.A.); the Hope Center (R.S.A., S.I.), the Lacy Foundation (A. Sene, S.K.); the American Federation for Aging Research (R.S.A.); the Schulak Family Gift Fund for Retinal Research (R.S.A.); the Jeffrey Fort Innovation Fund (R.S.A.); Hope For Vision (R.S.A.); the Robert Machemer Foundation (S.K.); and the Central Society for Clinical and Translational Research (J.Y.). Additional funding comes from an unrestricted grant to the Department of Ophthalmology and Visual Sciences of Washington University School of Medicine from Research to Prevent Blindness. J.B.L. was supported by the Washington University in St. Louis Medical Scientist Training Program (NIH Grant T32GM007200), the Washington University in St. Louis Institute of Clinical and Translational Sciences (NIH Grants UL1TR000448, TL1TR000449), the Washington University Diabetic Cardiovascular Disease Center, the American Federation for Aging Research, and the VitreoRetinal Surgery Foundation. The authors acknowledge Douglas Cox for technical assistance, Dr. David Beebe for advice regarding Seahorse experiments, Dr. Kevin Yarasheski at the Washington University Metabolomics Core for assistance with analyzing retinal samples, and Dr. Raul Mostoslavsky for providing resources. R.S.A. and S.I. are co-founders of Metro Midwest Biotech, which is developing NMN-based therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
J.B.L. performed most of the experiments related to Figures 3H–J, 4A–C/E, 5, 6, and 7; S.K. performed most of the experiments related to Figures 1, 2, 3A–G, and 4D. Conceptualization: R.S.A., S.I., K.T.; Investigation: J.B.L., S.K., N.B., M.Y., A. Santeford, A. Sene, R.N., N.Z., M.K., J.Y.; Writing – Original Draft: J.B.L., S.K.; Writing – Review & Editing: J.B.L., R.S.A., S.I., A. Sene; Supervision: R.S.A., S.I., K.T.; Funding Acquisition: R.S.A., S.I., K.T.
References
- Ait-Ali N, Fridlich R, Millet-Puel G, Clerin E, Delalande F, Jaillard C, Blond F, Perrocheau L, Reichman S, Byrne LC, et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell. 2015;161:817–832. doi: 10.1016/j.cell.2015.03.023. [DOI] [PubMed] [Google Scholar]
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames A, 3rd, Li YY, Heher EC, Kimble CR. Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1992;12:840–853. doi: 10.1523/JNEUROSCI.12-03-00840.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte RS. Regulation of angiogenesis by macrophages. Adv Exp Med Biol. 2010;664:15–19. doi: 10.1007/978-1-4419-1399-9_2. [DOI] [PubMed] [Google Scholar]
- Astuti GD, Bertelsen M, Preising MN, Ajmal M, Lorenz B, Faradz SM, Qamar R, Collin RW, Rosenberg T, Cremers FP. Comprehensive genotyping reveals RPE65 as the most frequently mutated gene in Leber congenital amaurosis in Denmark. European journal of human genetics: EJHG. 2015 doi: 10.1038/ejhg.2015.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai S, Sheline CT. NAD(+) maintenance attenuates light induced photoreceptor degeneration. Experimental eye research. 2013;108:76–83. doi: 10.1016/j.exer.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KD, Maqsood S, Huang JY, Pan Y, Harkcom W, Li W, Sauve A, Verdin E, Jaffrey SR. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell metabolism. 2014;20:1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell metabolism. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PW, Wang J, Chen Y, Fu Q, Zhong J, Yi X, Wu R, Gan H, Shi Y, Barnett C, et al. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat Genet. 2012;44:972–974. doi: 10.1038/ng.2370. [DOI] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, Zhang Q, Nakamaru-Ogiso E, Kannabiran C, Fonseca-Kelly Z, Chakarova C, Audo I, Mackay DS, Zeitz C, Borman AD, et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet. 2012;44:1040–1045. doi: 10.1038/ng.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nature protocols. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Fu Y, Yau KW. Phototransduction in mouse rods and cones. Pflugers Arch. 2007;454:805–819. doi: 10.1007/s00424-006-0194-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten A, Petzold S, Korner A, Imai S, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009;20:130–138. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science (New York, NY) 2015;348:453–457. doi: 10.1126/science.1258366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm C, Reme CE. Light damage as a model of retinal degeneration. Methods Mol Biol. 2013;935:87–97. doi: 10.1007/978-1-62703-080-9_6. [DOI] [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, Colman RF. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet. 2008;40:1230–1234. doi: 10.1038/ng.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Molecular cell. 2013;49:186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig AK, Peng GH, Chen S. Transcription coactivators p300 and CBP are necessary for photoreceptor-specific chromatin organization and gene expression. PLoS One. 2013;8:e69721. doi: 10.1371/journal.pone.0069721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Molecular cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab. 2013;15(Suppl 3):26–33. doi: 10.1111/dom.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai SI, Guarente L. NAD and sirtuins in aging and disease. Trends Cell Biol. 2014 doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaliffa C, Ameqrane I, Dansault A, Leemput J, Vieira V, Lacassagne E, Provost A, Bigot K, Masson C, Menasche M, et al. Sirt1 involvement in rd10 mouse retinal degeneration. Investigative ophthalmology & visual science. 2009;50:3562–3572. doi: 10.1167/iovs.08-2817. [DOI] [PubMed] [Google Scholar]
- Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest. 2007;117:3421–3426. doi: 10.1172/JCI32430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Peto T, Bird A, Vannewkirk MR. The epidemiology of age-related macular degeneration. American journal of ophthalmology. 2004;137:486–495. doi: 10.1016/j.ajo.2003.11.069. [DOI] [PubMed] [Google Scholar]
- Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, Chen Y, Li Y, Fishman GA, Genead M, et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012;44:1035–1039. doi: 10.1038/ng.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikov AV, Fan J, Crouch RK, Kefalov VJ. Age-related deterioration of rod vision in mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:11222–11231. doi: 10.1523/JNEUROSCI.4239-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikov AV, Tang PH, Parker RO, Crouch RK, Kefalov VJ. The mammalian cone visual cycle promotes rapid M/L-cone pigment regeneration independently of the interphotoreceptor retinoid-binding protein. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:7900–7909. doi: 10.1523/JNEUROSCI.0438-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooragayala K, Gotoh N, Cogliati T, Nellissery J, Kaden TR, French S, Balaban R, Li W, Covian R, Swaroop A. Quantification of Oxygen Consumption in Retina Ex Vivo Demonstrates Limited Reserve Capacity of Photoreceptor Mitochondria. Investigative ophthalmology & visual science. 2015;56:8428–8436. doi: 10.1167/iovs.15-17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent G, German NJ, Saha AK, de Boer VC, Davies M, Koves TR, Dephoure N, Fischer F, Boanca G, Vaitheesvaran B, et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Molecular cell. 2013;50:686–698. doi: 10.1016/j.molcel.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le YZ, Ash JD, Al-Ubaidi MR, Chen Y, Ma JX, Anderson RE. Targeted expression of Cre recombinase to cone photoreceptors in transgenic mice. Mol Vis. 2004;10:1011–1018. [PubMed] [Google Scholar]
- Li S, Chen D, Sauve Y, McCandless J, Chen YJ, Chen CK. Rhodopsin-iCre transgenic mouse line for Cre-mediated rod-specific gene targeting. Genesis. 2005;41:73–80. doi: 10.1002/gene.20097. [DOI] [PubMed] [Google Scholar]
- Lin JB, Tsubota K, Apte RS. A glimpse at the aging eye. Npj Aging And Mechanisms Of Disease. 2016;2:16003. doi: 10.1038/npjamd.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Molecular and cellular biology. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW, et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Molecular cell. 2015;59:321–332. doi: 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa H, Sampath AP, Laughlin SB, Fain GL. ATP consumption by mammalian rod photoreceptors in darkness and in light. Current biology: CB. 2008;18:1917–1921. doi: 10.1016/j.cub.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou KN, O’Rourke B, Foster DB. Metabolism leaves its mark on the powerhouse: recent progress in post-translational modifications of lysine in mitochondria. Front Physiol. 2014;5:301. doi: 10.3389/fphys.2014.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar P, Solanki I, Mansuri ML, Parihar MS. Mitochondrial sirtuins: emerging roles in metabolic regulations, energy homeostasis and diseases. Experimental gerontology. 2015;61:130–141. doi: 10.1016/j.exger.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, et al. SIRT5–mediated lysine desuccinylation impacts diverse metabolic pathways. Molecular cell. 2013;50:919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrault I, Hanein S, Zanlonghi X, Serre V, Nicouleau M, Defoort-Delhemmes S, Delphin N, Fares-Taie L, Gerber S, Xerri O, et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat Genet. 2012;44:975–977. doi: 10.1038/ng.2357. [DOI] [PubMed] [Google Scholar]
- Perron NR, Beeson C, Rohrer B. Early alterations in mitochondrial reserve capacity; a means to predict subsequent photoreceptor cell death. Journal of bioenergetics and biomembranes. 2013;45:101–109. doi: 10.1007/s10863-012-9477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell metabolism. 2013a;18:920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proceedings of the National Academy of Sciences of the United States of America. 2013b;110:6601–6606. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Galli M, Denanglaire S, Van Gool F, Dreze PL, Szpirer C, Bureau F, Andris F, Leo O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J Immunol. 2008;181:4685–4695. doi: 10.4049/jimmunol.181.7.4685. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Bell BA, Pereira A, Saxon J, Peachey NS. Early retinal pigment epithelium dysfunction is concomitant with hyperglycemia in mouse models of type 1 and type 2 diabetes. Journal of neurophysiology. 2015;113:1085–1099. doi: 10.1152/jn.00761.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Margolin Z, Borgo B, Havranek JJ, Milbrandt J. Characterization of Leber Congenital Amaurosis-associated NMNAT1 Mutants. The Journal of biological chemistry. 2015;290:17228–17238. doi: 10.1074/jbc.M115.637850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh A, Imai S. Systemic regulation of mammalian ageing and longevity by brain sirtuins. Nat Commun. 2014;5:4211. doi: 10.1038/ncomms5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimel AM, Abraham L, Cox D, Sene A, Kraus C, Dace DS, Ercal N, Apte RS. N-acetylcysteine amide (NACA) prevents retinal degeneration by up-regulating reduced glutathione production and reversing lipid peroxidation. The American journal of pathology. 2011;178:2032–2043. doi: 10.1016/j.ajpath.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW, Lombard DB. Calorie restriction alters mitochondrial protein acetylation. Aging cell. 2009;8:604–606. doi: 10.1111/j.1474-9726.2009.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sene A, Apte RS. Eyeballing cholesterol efflux and macrophage function in disease pathogenesis. Trends Endocrinol Metab. 2014;25:107–114. doi: 10.1016/j.tem.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman DM, Ross K, Sande PH, Kubota S, Ramaswamy S, Apte RS, Mostoslavsky R. SIRT6 Is Required for Normal Retinal Function. PloS one. 2014;9:e98831. doi: 10.1371/journal.pone.0098831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sol EM, Wagner SA, Weinert BT, Kumar A, Kim HS, Deng CX, Choudhary C. Proteomic investigations of lysine acetylation identify diverse substrates of mitochondrial deacetylase sirt3. PLoS One. 2012;7:e50545. doi: 10.1371/journal.pone.0050545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Imai S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. The EMBO journal. 2014;33:1321–1340. doi: 10.1002/embj.201386917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Wozniak DF, Dearborn JT, Kubota S, Apte RS, Izumi Y, Zorumski CF, Imai S. Expression of Nampt in hippocampal and cortical excitatory neurons is critical for cognitive function. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:5800–5815. doi: 10.1523/JNEUROSCI.4730-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still AJ, Floyd BJ, Hebert AS, Bingman CA, Carson JJ, Gunderson DR, Dolan BK, Grimsrud PA, Dittenhafer-Reed KE, Stapleton DS, et al. Quantification of mitochondrial acetylation dynamics highlights prominent sites of metabolic regulation. The Journal of biological chemistry. 2013;288:26209–26219. doi: 10.1074/jbc.M113.483396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E, Ding XQ, Saadi A, Agarwal N, Naash MI, Al-Ubaidi MR. Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Investigative ophthalmology & visual science. 2004;45:764–768. doi: 10.1167/iovs.03-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell metabolism. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umino Y, Solessio E, Barlow RB. Speed, spatial, and temporal tuning of rod and cone vision in mouse. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:189–198. doi: 10.1523/JNEUROSCI.3551-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen R, Klaver CC, Vingerling JR, Hofman A, de Jong PT. Epidemiology of age-related maculopathy: a review. European journal of epidemiology. 2003;18:845–854. doi: 10.1023/a:1025643303914. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2001;21:53–58. doi: 10.1523/JNEUROSCI.21-01-00053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F, Kwok SY. The Survival of Cone Photoreceptors in Retinitis Pigmentosa. JAMA ophthalmology. 2016:1–2. doi: 10.1001/jamaophthalmol.2015.5490. [DOI] [PubMed] [Google Scholar]
- Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nature reviews Genetics. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic acids research. 2015;43:W251–257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau KW, Hardie RC. Phototransduction motifs and variations. Cell. 2009;139:246–264. doi: 10.1016/j.cell.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W. NAD+ and NADH in cellular functions and cell death. Frontiers in bioscience: a journal and virtual library. 2006;11:3129–3148. doi: 10.2741/2038. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Imai S. Accurate measurement of nicotinamide adenine dinucleotide (NAD(+)) with high-performance liquid chromatography. Methods Mol Biol. 2013;1077:203–215. doi: 10.1007/978-1-62703-637-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell metabolism. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Sadhukhan S, Noriega LG, Moullan N, He B, Weiss RS, Lin H, Schoonjans K, Auwerx J. Metabolic characterization of a Sirt5 deficient mouse model. Scientific reports. 2013;3:2806. doi: 10.1038/srep02806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Bharathi SS, Rardin MJ, Uppala R, Verdin E, Gibson BW, Goetzman ES. SIRT3 and SIRT5 regulate the enzyme activity and cardiolipin binding of very long-chain acyl-CoA dehydrogenase. PLoS One. 2015;10:e0122297. doi: 10.1371/journal.pone.0122297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Ottenberg G, Sferrazza GF, Hubbs C, Fallahi M, Rumbaugh G, Brantley AF, Lasmezas CI. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain: a journal of neurology. 2015;138:992–1008. doi: 10.1093/brain/awv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.