

Abstract
Bone morphogenetic protein 9 (BMP9) is a circulating growth factor that is part of the TGFβ superfamily, and an essential regulator of vascular endothelial homeostasis. Previous studies have suggested a role for BMP9 signalling in leukocyte recruitment to the endothelium, but the directionality of this effect and underlying mechanisms have not been elucidated. Here we report that BMP9 upregulates toll-like receptor 4 (TLR4) expression in human endothelial cells and that BMP9 pre-treatment synergistically increases human neutrophil recruitment to LPS-stimulated human endothelial monolayers in an in vitro flow adhesion assay. BMP9 alone did not induce neutrophil recruitment to the endothelium. We also show that E-selectin and VCAM-1, but not ICAM-1 are upregulated in response to BMP9 in LPS-stimulated human endothelial cells. siRNA knockdown of ALK1 inhibited the BMP9-induced expression of TLR4 and VCAM-1 and inhibited BMP9-induced human neutrophil recruitment to LPS-stimulated human endothelial cells. BMP9 treatment also increased leukocyte recruitment within the pulmonary circulation in a mouse acute endotoxemia model. These results demonstrate that whilst BMP9 alone does not influence leukocyte recruitment, it primes the vascular endothelium to mount a more intense response when challenged with LPS, through an increase in TLR4, E-selectin and VCAM-1 and ultimately through enhanced leukocyte recruitment.
Introduction
BMP9 is a receptor ligand belonging to the TGFβ superfamily and involved in many cellular and physiological processes. BMP9 circulates at constitutively active concentrations and acts as an endothelial quiescence factor, potentially maintaining the stability of the adult vasculature (1). On ECs, BMP9 signals primarily through receptor complexes comprising the high affinity type 1 receptor, ALK1 and the lower affinity type II receptors, BMP receptor-II (BMPR-II) and activin-receptor type IIA (ACTR-IIA) (2–4). Accordingly, deficiencies in BMP9, ALK1 and BMPR-II underlie several vascular pathologies associated with postnatal vascular instability, including hereditary haemorrhagic telangiectasia (HHT) (5–7) and pulmonary arterial hypertension (PAH) (8, 9).
The BMP/TGFβ serine/threonine kinase type I receptors (ALK1-7) and type II receptors (BMPRII, ActRIIA, ActRIIB, TGFβRII and anti-Mullerian hormone receptor type 2, AMHRII) form unique heteromeric complexes that define ligand specificity (10). BMP9 binds ALK1 with the highest affinity, but can also bind to ALK2 (11, 12). These receptors then integrate BMP9 signalling via the type II receptors BMPRII, ACTR-IIA ACTR-IIB and the BMP co-receptor endoglin (3), all of which are expressed on ECs. Upon ligand binding, a signalling cascade is initiated, which involves the C-terminal phosphorylation of the receptor Smads (R-Smads), Smad1, Smad5 and Smad8 (10, 13–15). The R-Smads then bind with Smad4, and the R-Smad-Smad4 complex translocates to the nucleus and activates transcription factors that regulate the expression of specific genes (16–19).
BMP9 signalling has been shown to be essential to endothelial homeostasis in several settings including neonatal retinal angiogenesis (20) and lymphangiogenesis (21), and BMP9 has recently been shown to protect against LPS-induced endothelial permeability (22). Moreover, the vascular endothelium plays a key role in the molecular and cellular processes that initiate infection and tissue injury within the body (23), and acts as a barrier to regulate the exposure of the underlying tissue to circulating factors, including inflammatory cells and cytokines. There is compelling evidence that BMP9 signalling is involved in regulating the barrier function of the vascular endothelium. In human pulmonary arterial endothelial cells (HPAECs), BMP9 can induce expression of E-selectin (which is involved in the initial capture of leukocytes), and the inflammatory cytokines, IL-8 and IL-6 (4). Deficiency in endoglin (the BMP co-receptor essential for BMP9 signalling), has been implicated in impaired leukocyte recruitment and endothelial mediated inflammation (24, 25), further implicating BMP9 signalling in the process of leukocyte recruitment.
TLRs are pattern recognition receptors, and 11 family members have currently been identified (26–28). Several of these recognise bacterial products including TLR4, which recognises endotoxin (LPS), a component of the outer membrane of gram-negative bacteria (29–31). Endothelial-expressed TLR4 is essential for LPS-induced neutrophil sequestration to the lung (32, 33).
The aim of this study was to investigate the role of BMP9 in leukocyte recruitment to the vascular endothelium using an in vitro flow adhesion assay and an acute endotoxemia mouse model. Our results demonstrate that while BMP9 alone does not induce leukocyte recruitment, it acts in a concentration dependent way to prime the vascular endothelium for enhanced leukocyte recruitment following LPS. This occurs through increased expression of TLR4, E-selectin and VCAM-1, and ultimately through increased leukocyte recruitment in an ALK1/2-dependent manner.
Materials and Methods
Antibodies, primers and reagents
Mouse monoclonal antibodies for flow cytometry: Anti-hE-selectin Fluorescein Conjugated mouse IgG1 (anti-human E-selectin-FITC; R&D Systems), Allophycocyanin (APC) mouse anti-human CD54 (anti-human ICAM-1-APC; BD Pharmingen), and PE/Cy5 anti-human CD106 (anti-human VCAM-1-PECy5; BioLegend). Isotype control antibodies used for flow cytometry: APC-mouse IgG1 (BD Pharmingen), Mouse IgG1 Isotype control Fluorescein (R&D Systems) and PE/Cy5 mouse IgG1 Isotype (BioLegend). Antibodies for histology: Polyclonal rabbit anti-human myeloperoxidase (MPO) antibody (A0398; DAKO). General reagents: LPS Escherichia coli 0111:B4 (Sigma-Aldrich), BMP9 (R&D Systems), Sodium Citrate (Martindale Pharma). Cell culture reagents: EGM-2 BulletKit (Lonza), Hyclone (GE Healthcare), Ficoll-Paque PLUS (GE Healthcare), Fetal bovine serum (FBS) (Sigma-Aldrich) Histopaque 1077 (Sigma-Aldrich), Histopaque 1119 (Sigma-Aldrich), Dulbecco’s phosphate buffered saline (PBS) with Ca2+, Mg2+ (Sigma-Aldrich). Albumin Bovine Fraction (BSA) V Solution (7.5%) (Sigma-Aldrich). Quantitative polymerase chain reaction (qPCR) reagents: QuantiTect Primer Assays (Qiagen), Hs_ACVR1_1_SG (ALK2), Hs-ACVRL1_1_SG (ALK1) and Hs_TLR4_2_SG. Primer sequences, Smad1 forward 5’-TAGAAAGCCCTGTACTTCCTC-3’, Smad1 reverse 5’-GGTTGCTGGAAAGAATCTGG-3’, Smad5 forward 5’-GAGAGTCCAGTCTTACCTCC-3’, Smad5 reverse 5’-GGAAAGAATCTGGAAACGTG-3’, B2M forward 5’-CTCGCGCTACTCTCTCTTTC-3’, B2M reverse 5’-CATTCTCTGCTGGATGACGTG-3. Rox reference dye (Invitrogen), SYBRGreen JumpStart Taq ReadyMix (Sigma-Aldrich). Transfection reagents: DharmaFECT 1 Transfection Reagent (Dharmacon) ON-TARGETplus siRNA (Dharmacon), siACVRL1 (ALK1), siACVR1 (ALK2), siSmad1, siSmad5 and non-targeting siRNA Pool (siCP).
Endothelial cell culture
Human pulmonary arterial endothelial cells (HPAECs; passage 4-7) were purchased from Promocell and cultured in EGM-2 media (Lonza) with 2% FBS. Blood outgrowth endothelial cells (BOECs) were isolated as previously described (34) from peripheral venous blood (40-60 ml) collected into sodium citrate-containing tubes, obtained from healthy adult donors. Blood was layered over a Ficoll-Paque PLUS density gradient and BOEC colonies appeared between 2-4 weeks of culture in EGM-2, without heparin, supplemented with 20% Hyclone. BOECs were used at passage 5-7. ECs were cultured at 37°C in a 5% CO2 humidified atmosphere. ECs were treated with indicated concentrations of BMP6 and BMP9 for 16 hours prior a 4 hours LPS (100 ng/ml) stimulation.
Neutrophil isolation from peripheral blood
Blood (10-40 ml) from healthy adult volunteers was collected into EDTA-coated tubes (Sarstedt), and neutrophils were isolated as previously described, using 2-step density gradients of Histopaque 1119 and 1077 (Sigma) (35). Neutrophils were washed once in PBS (with Ca2+, Mg2+ and 0.15% BSA) and resuspended at a cell density of 1 x 106 cells/ml in PBS (with Ca2+, Mg2+ and 0.15% BSA). Cell counts were performed using a haemocytometer.
Assessing endothelial:leukocyte interactions under flow
Endothelial:neutrophil interactions were assessed in a flow adhesion assay as previously described (36). Microslides (µ-Slide VI0.4; Ibidi), containing confluent endothelial monolayers were mounted onto the stage of a phase contrast microscope. The slides were connected to cell and wash reservoirs by silicon tubes at one end and to a withdrawal syringe pump at the other end. ECs were washed for 2 min with PBS (with Ca2+, Mg2+ and 0.15% BSA), prior to perfusion with neutrophils at 1 x 106 cells/ml for 4 min at a wall shear stress of 0.1 Pa. Endothelial:neutrophil interactions were captured using time lapse imaging, 6 min post the initial cell bolus; quantification of neutrophil behaviour including rolling, arrest and transmigration was performed offline using ImagePro software. All flow-based adhesion assays were performed within a Perspex environmental chamber at 37°C.
Flow cytometric analysis of cell surface proteins
Trypsinised ECs were collected into a 96 well U-bottom plate and centrifuged at 400g for 5 min at room temperature. Cells were resuspended in blocking buffer (5% FBS in 0.5% BSA) for ≥10 min at 4°C, and incubated at 4°C in the dark with conjugated antibodies (anti-human E-selectin- FITC; anti-human VCAM-1-PE-Cy5; anti-human ICAM-APC) for 30 min. Conjugated isotype control antibodies and unstained cells were used as negative controls. Stained cells were centrifuged at 400g for 5 min at room temperature, and fixed in 1% paraformaldehyde (PFA) in PBS in the dark at room temperature for 10 min. Fixed cells were centrifuged at 400g for 5 min at room temperature, and resuspended in 400 μl staining buffer (1% FBS and 0.09% sodium azide in PBS). Detection of cell-bound conjugated antibodies was performed using a BD FACSCanto™ II (BD Bioscences) and quantification performed using FlowJo data analysis software.
siRNA transfection
ECs were transfected with siRNAs using the DharmaFECT 1 transfection reagent as per the manufacturer’s instructions, 48 h before their use in cell culture experiments.
Quantitative polymerase chain reaction (qPCR)
Total RNA was isolated from endothelial cells using a RNAeasy Mini kit (Qiagen), and mRNA expression of target genes was assessed using qPCR in a 384 well QuantStudio 6 Flex (Applied Biosystems by Life Technologies). QPCR 10 μl reactions were set up using SYBRGreen Jumpstart Taq ReadyMix, Rox Reference Dye and primers, (either Quantitech Primer Assays or in-house designed primers). Quantification was calculated using the ΔΔCt method.
Mice
Wild type male C57BL/6 mice, aged approximately 10 weeks and weighing between 20-25g were used in this study. All animal studies were approved by the local animal care committee and performed to the respective guidelines. BMP9 (50 ng/mouse) and LPS (3 mg/kg) were administered by i.p. injection. Mice were killed humanely by CO2 overdose and exsanguination.
Tissue preparation
The left lung was fixed in situ in the distended state by infusion of 0.8% agarose into the trachea and then placed in 10% PFA before paraffin embedding for histology. The right lung was frozen in liquid nitrogen and stored at -80ºC. Peripheral blood from mice was collected in EDTA-coated tubes (Sarstedt) and run on a Wooley ABC (mouse) analyser.
Quantification of leukocyte recruitment to the lung
A polyclonal rabbit anti-human MPO antibody was used at 1/1000 dilution to stain fixed mouse lung sections. Images (n=10; 400x magnification) were taken of the lung parenchyma and ImageJ software was used to count the number of leukocytes within each field of view (FOV). Fixed mouse lung sections were stained with haematoxylin and eosin (H&E). Images (n=20; 400x magnification) were taken of the alveolar-associated vessels on H&E-stained mouse lung sections. ImageJ software was used to count the number of leukocytes within the alveolar-associated vessels. All images were taken and analysed with the operator blinded as to the experimental conditions.
Determination of mouse lung neutrophil elastase (NE) activity
Individual frozen mouse lung lobes were placed into 2 ml tubes containing a stainless steel bead (Qiagen) and 500 µl of PBS (pH 6.0) supplemented with hexadecyltrimethylammonium bromide (HTAB, 0.5%) and EDTA (5 mM). Mouse lungs were homogenised using a TissueLyser II (Qiagen) for 60 s, four times. Mouse lung homogenates were centrifuged for 10 min at 10,000g at 4ºC and the supernatant collected. Neutrophil elastase activity in mouse lung homogenates was quantified using a Enzchek® elastase activity assay kit, which measures the ability of neutrophil elastase to cleave non fluorescent DQ-elastin substrate into fluorescent fragments. DQ-elastin substrate (reconstituted in dH2O to 1mg/ml) was diluted 1:20 in 1x reaction buffer and 100 µl added to the required wells of a 96 well ELISA plate. Neat sample (100 µl) was added to DQ-elastin wells in duplicate. Reaction buffer (100 µl) was added for negative controls. Porcine pancreatic elastase (100 µl; PPE, 0.2 U/ml) supplied with the assay kit was added for a positive control. Samples were incubated in the dark at room temperature for 30 min and measured at 485/535 nm by a fluorescence multiwell plate reader (Victor 3 multilabel plate reader, Perkin Elmer). Background fluorescence in negative control wells was averaged and subtracted from each sample reading.
Statistical analysis
Unpaired Student T-tests were used for comparisons between two groups. Comparisons between three or more groups was performed by Ordinary one-way ANOVA with Tukey’s multiple comparisons. Normality of data distribution was assessed using a D'Agostino & Pearson omnibus normality test. All data are reported as mean ± SEM.
Results
BMP9 increases TLR4 in blood outgrowth endothelial cells (BOECs) through ALK1 and Smad1/5 signalling
First, we investigated the potential role of BMP9 as a regulator of TLR4 expression in blood outgrowth endothelial cells (BOECs), an EC population derived from peripheral blood, which have been shown previously to behave like mature vascular ECs in terms of gene and protein expression and function (37–40). BMP9 increased TLR4 mRNA in BOECs, an effect not seen with LPS, a known negative regulator of TLR4 (Figure 1A) (41). To identify the BMP9 receptor mediating the induction of TLR4, siRNA knockdowns of ALK1 and ALK2 were performed. ALK1 knockdown reduced the BMP9-induced increase of TLR4, whereas ALK2 knockdown had no effect (Figure 1B). Knockdown of ALK1 and ALK2 in combination did not result in a further reduction in TLR4 induction (Figure 1B). ALK1 and ALK2 siRNA knockdown efficiency was confirmed in BOECs by qPCR, which showed >90% reduction of the target gene (Figure S1A,B). Next, the involvement of Smad1/5 (canonical downstream mediators of BMP signalling) was investigated by transfecting BOECs with siSmad1 and siSmad5. Knockdown of Smad1/5 in combination inhibited BMP9-induced TLR4 upregulation (Figure 1C). Knockdown with siSmad1 and siSmad5 produced >90% reduction in gene expression of both targets (Figure S1C,D). Taken together, these data indicate that BMP9 upregulates TLR4 expression through ALK1 and Smad1/5.
Figure 1.
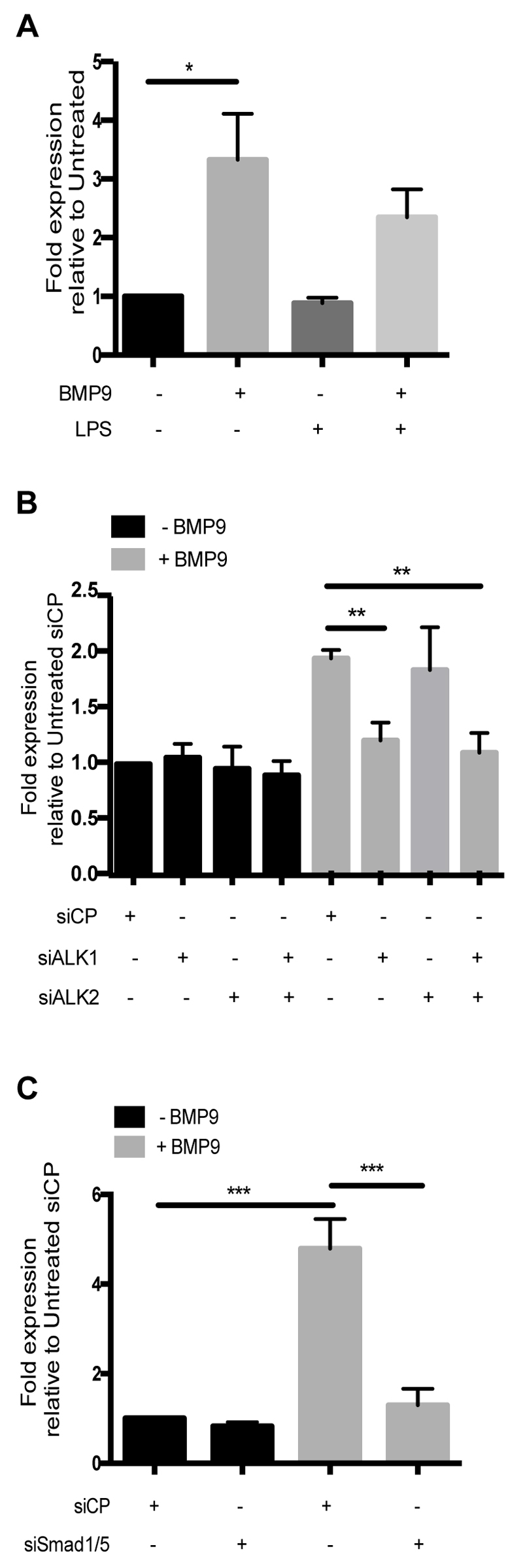
BMP9 increases TLR4 expression in blood outgrowth endothelial cells (BOECs) via ALK1 and Smad1/5 signalling. BOECs were treated with BMP9 (16 h; 5 ng/ml) prior to the addition of LPS (4 h; 100 ng/ml). (A) BMP9 increased TLR4 mRNA levels. (B) Knockdown of ALK1 inhibited the BMP9-induced up-regulation of TLR4 mRNA, whilst ALK2 knockdown did not. (C). Knockdown of Smad1/5 inhibited the BMP9-induced upregulation of TLR4 mRNA. TLR4 mRNA levels were quantified using qPCR and were expressed as fold expression relative to untreated BOECs transfected with siRNA Control Pool (siCP). Error bars correspond to ± SEM; n=3-4.
BMP9 causes a synergistic increase in leukocyte recruitment to endothelial cells
Next, we investigated the effects of BMP9 and LPS on leukocyte recruitment to endothelial cells using an in vitro flow adhesion assay, a system that allows for the real-time visualisation of endothelial:leukocyte interactions under conditions of flow. BMP9 alone did not induce neutrophil recruitment to BOECs (Figure 2A). Whilst LPS induced neutrophil recruitment, BMP9 pre-treatment caused a major and synergistic increase in the total number of neutrophils recruited to LPS-stimulated BOECs (Figure 2A). BMP9 pre-treatment of LPS-stimulated BOECs, however did not affect the percentage of rolling, arrested or transmigrated neutrophils, suggesting that BMP9 enhanced all aspects of the recruitment cascade (Figure 2B). The effect of BMP9 on leukocyte recruitment was examined in another endothelial cell type, human pulmonary arterial endothelial cells (HPAECs); these experiments again showed no effect of BMP9 alone, but a similar synergistic increase in leukocyte recruitment when LPS-stimulated HPAECs were pre-treated with BMP9 (Figure 2C). Again no change in the percentage of rolling arrested or transmigrated neutrophils was observed (Figure 2D).
Figure 2.

BMP9 increases neutrophil recruitment to LPS-stimulated endothelial cells. Neutrophils were perfused over confluent endothelial monolayers treated with BMP9 (16 h; 5 ng/ml) prior to the addition of LPS (4 h; 100 ng/ml). Total neutrophil recruitment to (A) BOECs and (C) HPAECs. Neutrophil behaviours of rolling, arrest and transmigration were expressed as a percentage of total recruitment to (B) BOECs and (D) HPAECs. Data was collected from 3 experiments, and error bars correspond to ± SEM. *p<0.05 **p<0.01 *** p<0.001 ****p<0.0001 compared with untreated cells and #p<0.05 ##p<0.01 ###0.001 ####p.0001 compared with LPS-stimulated cells. BOECs were transfected with siALK1 and siALK2 and treated with BMP9 (16 hours; 5 ng/ml) prior to the addition of LPS (4 hours; 100 ng/ml). (E) Representative images (100x) showing surface arrested neutrophils (SA) as bright phase and transmigrated (TM) neutrophils as dark phase. (F) Total recruitment to siALK1 and siALK2 transfected BOECs. Data was collected from 3-4 experiments, and error bars correspond to ± SEM, **p<0.01.
Using siRNA, we next showed that ALK1 knockdown impaired neutrophil recruitment to BOECs treated with BMP9 and LPS, whilst ALK2 knockdown had no effect (Figure 2E,F). A combined knockdown of ALK1 and ALK2 had no further effect on neutrophil recruitment to BOECs treated with BMP9 and LPS (Figure 2 G). Together, these data demonstrate that BMP9 pre-treatment enhances LPS-induced neutrophil recruitment in two different EC populations, BOECs and HPAECs. Further, this process can be inhibited completely with siRNA knockdown of ALK1.
BMP9 causes a synergistic increase in the surface expression of adhesion molecules expressed on LPS-stimulated endothelial cells
Next, we used flow cytometry to explore which, if any, surface expressed endothelial selectins and adhesion molecules might be regulated by BMP9. As anticipated, LPS upregulated E-selectin and VCAM-1 in BOECs and HPAECs. BMP9 pre-treatment caused a major and synergistic increase in E-selectin and VCAM-1 in both BOECs and HPAECs treated with LPS, compared to levels expressed in ECs treated with LPS alone (Figure 3 A,B,D,E). Treatment with BMP9 alone induced VCAM-1 expression in BOECs (Figure 3B), but not in HPAECs (Figure 3E). Both BOECs and HPAECs expressed ICAM-1 during basal conditions and treatment with BMP9 alone reduced ICAM-1 expression in BOECs and HPAECs. LPS increased ICAM-1 levels, but pre-treatment with BMP9 before LPS-stimulation did not change ICAM-1 expression in BOECs or HPAECs (Figure 3C,F). P-selectin was not detected on either HPAECs or BOECs with any of the treatments used in this study (data not shown). Taken together, these results show that BMP9 mediates a synergistic increase in surface expression of both E-selectin and VCAM-1 in both BOECs and HPAECs. Conversely, BMP9 reduced ICAM-1 expression, indicating that ICAM-1 gene regulation is through a different mechanism to E-selectin and VCAM-1.
Figure 3.

BMP9 increases surface expression of E-selectin and VCAM-1, but not ICAM-1 on LPS-stimulated ECs. ECs were treated with BMP9 (16 h; 5 ng/ml) prior to the addition of LPS (4 h; 100 ng/ml). Flow cytometry was performed to assess surface expression of E-selectin (FITC-conjugated anti-human E-selectin), VCAM-1 (PE-Cy5-conjugated anti-human VCAM-1) and ICAM-1 (APC-conjugated anti-human ICAM-1) on BOECs (A-C) and HPAECs (D-F). Forward scatter and side scatter gating was applied to the endothelial populations. Histograms are representative of 3-4 experiments. Graphs show median fluorescence intensity (MFI) expressed as fold change relative to untreated ECs. Error bars correspond to ± SEM, n=3-4. *p<0.05 **p<0.01 *** p<0.001 ****p<0.0001 compared with untreated cells and #p<0.05 ##p<0.01 ###0.001 ####p.0001 compared with LPS-stimulated cells.
BMP9-mediated expression of E-selectin and VCAM-1 is concentration dependent
BMP9 is reported to circulate at concentrations ranging from 200-400 pg/ml (42). Thus, we treated BOECs with BMP9 concentrations ranging from 0-5 ng/ml in the presence or absence of LPS and assessed expression of E-selectin, VCAM-1 and ICAM-1 (Figure 4). BMP9 at a concentration of 5 ng/ml induced higher levels of E-selectin in the presence of LPS, compared with BOECs treated with LPS alone (Figure 4A). VCAM-1 surface expression was significantly elevated in LPS-stimulated BOECs pre-treated with 1 or 5 ng/ml BMP9. VCAM-1 induction was also observed in BOECs treated with BMP9 alone at concentrations of 1 or 5 ng/ml BMP9, but at levels almost 2 fold lower than that seen from BOECs treated with BMP9 and LPS in combination (Figure 4B). ICAM-1 inhibition was observed at concentrations of 1 and 5 ng/ml BMP9, with or without LPS (Figure 4C). Collectively, these data reveal that BMP9 upregulates E-selectin and VCAM-1 expression in ECs at concentrations that are approximately 2-fold higher than circulating levels, and maximal expression requires co-stimulation with LPS.
Figure 4.

BMP9-mediated upregulation of E-selectin and VCAM-1 is concentration dependent. Blood outgrowth endothelial cells (BOECs) were treated with 5 different concentrations of BMP9 (ranging from 0-5 ng/ml) for 16 h prior to the addition of LPS (4 h; 100 ng/ml). Flow cytometry was performed to assess surface expression of (A) E-selectin (FITC-conjugated anti-human E-selectin), (B) VCAM-1 (PE-Cy5-conjugated anti-human VCAM-1) and (C) ICAM-1 (APC-conjugated anti-human ICAM-1). Forward scatter and side scatter gating was applied to the BOEC population. Histograms are representative of 3 experiments. BMP9 dose response curves show median fluorescence intensity (MFI) as fold change relative to untreated BOECs. Error bars correspond to ± SEM, n=3, *p<0.05.
BMP9-induced endothelial expression of E-selectin and VCAM-1 is mediated through different BMP/TGFβ type I receptors
Next, we assessed the regulation of E-selectin, VCAM-1 and ICAM-1 in BOECs with siRNAs targeting ALK1 and ALK2. The synergistic increase of E-selectin expression observed in BOECs treated with both BMP9 and LPS was inhibited by ALK2 knockdown (Figure 5A). ALK1 knockdown had no effect on E-selectin expression with any of the treatments (Figure 5A). Conversely, ALK1 knockdown impaired VCAM-1 expression in BOECs treated with BMP9 alone, and in BOECs treated with BMP9 and LPS (Figure 5B). ALK2 knockdown had no effect on VCAM-1 expression with any of the treatment conditions (Figure 5B). Knockdown of both ALK1 and ALK2 in combination, did not result in any further reduction in either E-selectin or VCAM-1 expression with any of the treatments (Figure 5A,B). Combined knockdown of ALK1 and ALK2 significantly increased BMP9-induced ICAM-1 expression (Figure 5C). Together, these data indicate that BMP9 upregulates endothelial surface expression of E-selectin through ALK2 and VCAM-1 through ALK1.
Figure 5.

Knockdown of ALK1 and ALK2 inhibits the BMP9-mediated induction of E-selectin and VCAM-1, but not ICAM-1 in blood outgrowth endothelial cells (BOECs). siRNA-transfected BOECs were treated with BMP9 (16 h; 5 ng/ml) prior to the addition of LPS (4 hours; 100 ng/ml). Flow cytometry was performed to assess surface expression of (A) E-selectin (FITC-conjugated anti-human E-selectin) and (B) VCAM-1 (PE-Cy5-conjugated anti-human VCAM-1). (C) ICAM-1 (APC-conjugated anti-ICAM-1). Forward scatter and side scatter gating was applied to the BOEC population. Histograms are representative of 3-4 independent experiments. Graphs show median fluorescence intensity (MFI) expressed as fold change relative to BOECs that were transfected with siRNA control pool (siCP) and cultured under the same treatment conditions. Error bars correspond to ± SEM, n=3-4, *p<0.05 **p<0.01 ****p<0.0001.
Smad1/5 signals downstream of ALK1 and ALK2 to induce BMP9-mediated E-selectin and VCAM-1 expression
Next, we employed knockdown of Smad1 and Smad5 in combination (Smad1/5) to determine the contributions of Smads to BMP9-induced upregulation of E-selectin and VCAM-1 in LPS-stimulated BOECs. Smad1/5 knockdown inhibited BMP9-induced upregulation of E-selectin and VCAM-1 in LPS-stimulated BOECs (Figure 6A,B). Smad1/5 knockdown also inhibited VCAM-1 expression in BOECs treated with BMP9 alone (Figure 6B), and whilst there was a trend towards increased ICAM-1 expression after Smad1/5 knockdown, this did not reach significance (Figure 6C). Together, these results show that Smad1/5 signal downstream of ALK1 and ALK2 to induce BMP9-mediated E-selectin and VCAM-1 expression.
Figure 6.

Knockdown of Smad1/5 inhibits the BMP9-mediated upregulation of E-selectin and VCAM-1, but not ICAM-1 in LPS-stimulated blood outgrowth endothelial cells (BOECs). siRNA-transfected BOECs were treated with BMP9 (16 h; 5 ng/ml) prior to the addition of LPS (4 h; 100 ng/ml). Flow cytometry was performed to assess surface expression of (A) E-selectin (FITC-conjugated anti-human E-selectin), (B) VCAM-1 (PE-Cy5-conjugated anti-human VCAM-1). (C) ICAM-1 (APC-conjugated anti-ICAM-1). Forward scatter and side scatter gating was applied to the BOEC population. Histograms are representative of 4 independent experiments. Graphs show median fluorescence intensity (MFI) expressed as fold change relative to BOECs that were cultured under the same treatment conditions and transfected with siRNA control pool (siCP). Error bars correspond to ± SEM, n=4, *p<0.05.
BMP6 causes a synergistic increase in E-selectin, VCAM-1 and ICAM-1 expression in LPS-stimulated BOECs
BMP6 is a known ligand for ALK2, but not ALK1, and has previously been associated with endothelial inflammation and osteogenesis (43). Thus, we investigated whether BMP6 might also induce surface expression of E-selectin, VCAM-1 and ICAM-1 in BOECs. BOECs treated with BMP6 and LPS in combination displayed higher levels of E-selectin and VCAM-1 compared to BOECs treated with LPS alone (Figure 7A,B). In contrast to BMP9, BMP6 pre-treatment increased ICAM-1 expression above levels seen from BOECs treated with LPS alone (Figure 7C).
Figure 7.

BMP6 increases surface expression of E-selectin, VCAM-1 and ICAM-1 on LPS-stimulated blood outgrowth endothelial cells (BOECs). BOECs were treated with BMP6 (16 h; 50 ng/ml) prior to the addition of LPS (4 h; 100 ng/ml). Flow cytometry was performed to assess surface expression of (A) E-selectin (FITC-conjugated anti-human E-selectin), (B) VCAM-1 (PE-Cy5-conjugated anti-human VCAM-1) and (C) ICAM-1 (APC-conjugated anti-human ICAM-1). Forward scatter and side scatter gating was applied to BOECs. Histograms are representative of 3 replicates. Graphs show median fluorescence intensity (MFI) expressed as fold change relative to untreated BOECs. Error bars correspond to ± SEM, n=3.
BMP9 increases leukocyte recruitment to the lung during acute endotoxemia in mice
To determine the potential relevance of our findings to an in vivo setting we next explored the effect of BMP9 on leukocyte recruitment, using an acute endotoxemia model in mice. LPS challenge produced the anticipated influx of leukocytes within the pulmonary vasculature, with the majority staining positive for MPO, (an enzyme that is predominantly stored in granules within cells of myeloid origin, and is extensively used as a marker of granulocytes) (Figure 8A,B,C,D). The MPO-positive leukocytes predominantly remained within the pulmonary vessels, as seen in previous studies using a similar endotoxemia model.(32) Lung neutrophil elastase activity was increased in mice treated with LPS, indicating the increased presence of neutrophils within the lung (Figure 8E). Mice that received a single dose of BMP9 24 hours before systemic challenge with LPS (4 hours), showed a modest, but significant increase in leukocyte sequestration within the pulmonary vessels, an increase in MPO-positive staining and an increase in neutrophil elastase activity within the lung compared to mice that only received LPS (Figure 8A,B,C,D,E). BMP9 alone did not influence the recruitment of leukocytes to the lung and showed minimal MPO staining and very little neutrophil elastase activity in the lung (Figure 8A,B,C,D,E). Circulating leukocyte counts were lower in mice that received LPS, in the presence of BMP9 (Figure 8F). These data indicate that while BMP9 alone does not lead to leukocyte sequestration into the lung, exogenous delivery of BMP9 in an acute endotoxemia model, significantly increases leukocyte recruitment in the lung.
Figure 8.

BMP9 enhances leukocyte recruitment to the lung in an acute endotoxemia mouse. Systemic endotoxemia was induced by administration of LPS (3 mg/kg; IP inj) for 4 h in mice. Some mice were also treated with BMP9 (50 ng; IP inj) 24 h prior to LPS administration. Representative images of formalin-fixed mouse lung sections stained with (A) myeloperoxidase (MPO) and (B) haematoxylin and eosin (H&E), 400x magnification. (C) MPO-positive cells within the parenchyma were counted and quantified (10 images/slide). (D) The number of leukocytes within the vessels associated with alveolar ducts were counted (20 images/slide). (E) Mouse lungs were homogenised and neutrophil elastase activity was quantified and normalised to total protein concentration of the lung lysate. (F) Mouse blood was collected into EDTA-coated tubes and the total number of circulating leukocytes was assessed. Error bars correspond to ± SEM, n=8-16, *p<0.05.
Discussion
Here we report for the first time that BMP9 and LPS synergise to enhance leukocyte recruitment in an in vitro flow adhesion assay and in an acute endotoxemia mouse model. BMP9 alone increased TLR4 expression in BOECs, but did not induce leukocyte recruitment either in vitro or in vivo, suggesting that at high concentrations of BMP9 serves to prime the endothelium to mount a more intensive response when faced with an inflammatory stimuli, such as endotoxin (LPS). While previous studies have shown that the inhibitory Smad, Smad6, can negatively regulate TLR4 expression (44, 45), how BMP9/ALK1/Smad1/5 signalling regulates TLR4 expression has not been elucidated.
Excessive neutrophil recruitment to the lung is associated with changes in pulmonary vascular permeability and aberrant levels of inflammatory cytokines (46), which can lead to acute lung injury (47, 48). However, neutrophil recruitment to the lung is not always pathological (49) and is an essential part of the innate immune response, which is responsible for the clearance of pathological bacteria (50–52). For example, artificial TLR4 stimulation has been shown to enhance clearance of Pseudomonas aeruginosa from the lungs of mice through augmentation of neutrophil recruitment (53). It remains unclear whether the BMP9-enhanced neutrophil recruitment to the lung observed in the present study represents a healthy physiological or potentially pathological host response and further investigation is needed to elucidate this.
This is not the first study to implicate BMP9 signalling in leukocyte recruitment. Endoglin is a BMP co-receptor and essential to BMP9 signalling (2, 3, 54). Endoglin heterozygous mice display impaired leukocyte recruitment to the lungs and peritoneum in response to LPS and carrageenan. Moreover, endoglin overexpression increased leukocyte recruitment in vitro, through the activation of β1-intergrins on the surface of the leukocytes. Moreover, BOECs isolated from HHT patients (deficient in endoglin), showed impaired leukocyte recruitment in vitro (24). Endoglin has also been shown to be important for endothelial:mural cell adhesion (55). Whilst the previous endoglin studies strongly support the findings from our study, we report for the first time the direct impact of BMP9 on endothelial-mediated leukocyte recruitment.
Using a similar in vitro flow adhesion assay to the one described in our current study, Burton and colleagues demonstrated enhanced leukocyte recruitment to TGFβ-stimulated HPAECs transfected with siRNA targeting BMPRII, a BMP type II receptor that binds to BMP9 in the presence of ALK1 (56). Burton and colleagues also showed that loss of BMPRII from HPAECs increased expression of IL-8 and its ligand CXCR1/2 after stimulation with TGFβ (56) and pharmacological inhibition of CXCR1/2 reversed pulmonary hypertension in BMPRII deficient mice (56, 57). These BMPRII studies further implicate BMP9 signalling in leukocyte recruitment.
Using siRNA knockdown, we investigated which BMP/TGFβ type I receptor was responsible for the BMP9-induced neutrophil recruitment in LPS-stimulated endothelial cells. Our data revealed that ALK1 was essential for BMP9-induced TLR4 and VCAM-1 expression, and the enhanced neutrophil recruitment seen in the presence of LPS. Our data is somewhat surprising as VCAM-1 does not adhere to the β2-intregrins LFA-1 or MAC-1, which are the predominant integrins known to mediate neutrophil adhesion (through binding to ICAM-1 on endothelial cells) (58). VCAM-1/β1-integrin interaction mediates monocyte (59) and lymphocyte (60, 61) adhesion to the vascular endothelium, but there is very little evidence for VCAM-1/β1-integrin adhesion with neutrophils. β1-integrins are expressed on the surface of human neutrophils, albeit at a lower density than β2-integrins (62–64), but the reasons why VCAM-1/β1-integrins are not involved in neutrophil recruitment remain unclear. One theory is that VCAM-1/β1-integrin adhesion is more complex than adhesion through ICAM-1/β2-integrins, and β1-integrins require an additional activation stage before association with VCAM-1 (65, 66). However, when given the appropriate activation, VCAM-1/β1-integrins can mediate neutrophil adhesion (63, 65).
As mentioned previously, endoglin can activate β1-integrins on lymphocytes and monocytes (24) and BMP9 upregulates endoglin expression in endothelial cells (25). To explain our surprising findings, we hypothesise that BMP9 is upregulating endoglin expression in the endothelial cells, which is then activating β1-integrins on the neutrophils, thus allowing for VCAM-1/β1-adhesion.
ALK2 has previously been linked to inflammatory and osteogenic processes, through BMP6 signalling (43). We showed that BMP6 was able to induce the expression of E-selectin, VCAM-1 and ICAM-1 in the presence of LPS, but to a lesser extent than that seen with BMP9. We showed that ALK2 was responsible for BMP9-induced E-selectin expression in LPS-stimulated BOECs. ALK2 knockdown did not impact on neutrophil recruitment. Whilst LPS induced an upregulation of ICAM-1 expression, BMP9 did not enhance ICAM-1 expression further in either BOECs or HPAECs, and treatment with BMP9 alone reduced ICAM-1 expression, suggesting that ICAM-1 regulation is controlled through different mechanisms than E-selectin and VCAM-1. The broader relevance of our findings was supported as two endothelial subtypes BOECs and HPAECs responded to BMP9 and LPS in a near identical manner in terms of neutrophil recruitment and surface expression of the selectins and adhesion molecules.
BMP9 has been characterised as an ‘endothelial circulating quiescence factor’ because of its anti-angiogenic properties in adult vascular endothelial cells (1). BMP9 inhibits LPS-induced endothelial leak, and TNFα-induced apoptosis in vitro, and reverses pulmonary hypertension in rats and mice (22). Taken together, these observations suggest that BMP9 while inhibiting extravasation of fluid enhances the recruitment and transmigration of neutrophils. Further investigation is required to establish whether the increased leukocyte recruitment represents a healthy response to promote bacterial clearance or contributes to a pathological response.
In summary, our data demonstrate that BMP9 increases LPS-induced leukocyte adhesion to endothelial cells; while BMP9 in isolation does not promote leukocyte recruitment it primes the endothelium to enhance leukocyte recruitment when subsequently challenged with an inflammatory stimulus such as LPS. This occurs in an ALK1/2-dependent manner, and was observed under both in vitro and in vivo conditions. These findings inform our understanding of BMP9’s role in vascular homeostasis and have major implications for the use of BMP9 as a potential therapeutic agent.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by funding from the British Heart Foundation, Papworth Hosptial R&D Department and the NIHR Cambridge C. C.G.M holds a Wellcome Trust PhD Fellowship in Metabolic and Cardiovascular Disease. K.H. was a British Lung Foundation Graduate Student and K.L. holds a Wellcome Trust Clinical Training Fellowship.
Glossary
Abbreviations
- ALK
activin receptor-like kinase
- BMP
bone morphogenetic protein
- TGFβ
transforming growth factor beta
- siRNA
small interfering RNA
Footnotes
Authorship: S.L.A. designed and performed the research, analysed the data and wrote the paper. C.M. performed the research. A.C. performed the research. K.H. performed the research. K.L. performed the research. P.D.U. wrote the paper. C.M.Y. performed the research and analysed the data. G.B.N. designed the research, analysed the data and wrote the paper. E.R.C. designed the research, analysed the data and wrote the paper. N.W.M. designed the research, analysed the data and wrote the paper.
Conflict of interest disclosure: NWM is a founder and director of Morphogen-IX.
References
- 1.David L, Mallet C, Keramidas M, Lamande N, Gasc JM, Dupuis-Girod S, Plauchu H, Feige JJ, Bailly S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ Res. 2008;102:914–922. doi: 10.1161/CIRCRESAHA.107.165530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109:1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- 3.Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, Lowik CW, ten Dijke P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J Cell Sci. 2007;120:964–972. doi: 10.1242/jcs.002949. [DOI] [PubMed] [Google Scholar]
- 4.Upton PD, Davies RJ, Trembath RC, Morrell NW. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J Biol Chem. 2009;284:15794–15804. doi: 10.1074/jbc.M109.002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 6.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 7.Wooderchak-Donahue WL, McDonald J, O'Fallon B, Upton PD, Li W, Roman BL, Young S, Plant P, Fulop GT, Langa C, Morrell NW, et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. American journal of human genetics. 2013;93:530–537. doi: 10.1016/j.ajhg.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. American journal of human genetics. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 10.van den Driesche S, Mummery CL, Westermann CJ. Hereditary hemorrhagic telangiectasia: an update on transforming growth factor beta signaling in vasculogenesis and angiogenesis. Cardiovasc Res. 2003;58:20–31. doi: 10.1016/s0008-6363(02)00852-0. [DOI] [PubMed] [Google Scholar]
- 11.Townson SA, Martinez-Hackert E, Greppi C, Lowden P, Sako D, Liu J, Ucran JA, Liharska K, Underwood KW, Seehra J, Kumar R, et al. Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J Biol Chem. 2012;287:27313–27325. doi: 10.1074/jbc.M112.377960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei Z, Salmon RM, Upton PD, Morrell NW, Li W. Regulation of bone morphogenetic protein 9 (BMP9) by redox-dependent proteolysis. J Biol Chem. 2014;289:31150–31159. doi: 10.1074/jbc.M114.579771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebisawa T, Tada K, Kitajima I, Tojo K, Sampath TK, Kawabata M, Miyazono K, Imamura T. Characterization of bone morphogenetic protein-6 signaling pathways in osteoblast differentiation. J Cell Sci. 1999;112(Pt 20):3519–3527. doi: 10.1242/jcs.112.20.3519. [DOI] [PubMed] [Google Scholar]
- 14.Hoodless PA, Haerry T, Abdollah S, Stapleton M, O'Connor MB, Attisano L, Wrana JL. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell. 1996;85:489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- 15.Liu F, Hata A, Baker JC, Doody J, Carcamo J, Harland RM, Massague J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature. 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature. 1997;389:85–89. doi: 10.1038/38008. [DOI] [PubMed] [Google Scholar]
- 17.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoodless PA, Tsukazaki T, Nishimatsu S, Attisano L, Wrana JL, Thomsen GH. Dominant-negative Smad2 mutants inhibit activin/Vg1 signaling and disrupt axis formation in Xenopus. Developmental biology. 1999;207:364–379. doi: 10.1006/dbio.1998.9168. [DOI] [PubMed] [Google Scholar]
- 19.Verrecchia F, Vindevoghel L, Lechleider RJ, Uitto J, Roberts AB, Mauviel A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene. 2001;20:3332–3340. doi: 10.1038/sj.onc.1204448. [DOI] [PubMed] [Google Scholar]
- 20.Ricard N, Ciais D, Levet S, Subileau M, Mallet C, Zimmers TA, Lee SJ, Bidart M, Feige JJ, Bailly S. BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood. 2012;119:6162–6171. doi: 10.1182/blood-2012-01-407593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levet S, Ciais D, Merdzhanova G, Mallet C, Zimmers TA, Lee SJ, Navarro FP, Texier I, Feige JJ, Bailly S, Vittet D. Bone morphogenetic protein 9 (BMP9) controls lymphatic vessel maturation and valve formation. Blood. 2013;122:598–607. doi: 10.1182/blood-2012-12-472142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long L, Ormiston ML, Yang X, Southwood M, Graf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med. 2015;21:777–785. doi: 10.1038/nm.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res. 2007;101:234–247. doi: 10.1161/CIRCRESAHA.107.151860b. [DOI] [PubMed] [Google Scholar]
- 24.Rossi E, Sanz-Rodriguez F, Eleno N, Duwell A, Blanco FJ, Langa C, Botella LM, Cabanas C, Lopez-Novoa JM, Bernabeu C. Endothelial endoglin is involved in inflammation: role in leukocyte adhesion and transmigration. Blood. 2013;121:403–415. doi: 10.1182/blood-2012-06-435347. [DOI] [PubMed] [Google Scholar]
- 25.Young K, Conley B, Romero D, Tweedie E, O'Neill C, Pinz I, Brogan L, Lindner V, Liaw L, Vary CP. BMP9 regulates endoglin-dependent chemokine responses in endothelial cells. Blood. 2012;120:4263–4273. doi: 10.1182/blood-2012-07-440784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akira S, Takeda K. Toll-like receptor signalling. Nature reviews. Immunology. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 27.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nature immunology. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 28.Craig A, Mai J, Cai S, Jeyaseelan S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infection and immunity. 2009;77:568–575. doi: 10.1128/IAI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akashi S, Saitoh S, Wakabayashi Y, Kikuchi T, Takamura N, Nagai Y, Kusumoto Y, Fukase K, Kusumoto S, Adachi Y, Kosugi A, et al. Lipopolysaccharide interaction with cell surface Toll-like receptor 4-MD-2: higher affinity than that with MD-2 or CD14. J Exp Med. 2003;198:1035–1042. doi: 10.1084/jem.20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeyaseelan S, Chu HW, Young SK, Freeman MW, Worthen GS. Distinct roles of pattern recognition receptors CD14 and Toll-like receptor 4 in acute lung injury. Infection and immunity. 2005;73:1754–1763. doi: 10.1128/IAI.73.3.1754-1763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 32.Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, Kubes P. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest. 2003;111:1011–1020. doi: 10.1172/JCI16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andonegui G, Zhou H, Bullard D, Kelly MM, Mullaly SC, McDonald B, Long EM, Robbins SM, Kubes P. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest. 2009;119:1921–1930. doi: 10.1172/JCI36411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LS, Marchesan D, Yang J, Suntharalingam J, Soon E, Exley A, Stewart S, et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009;180:780–787. doi: 10.1164/rccm.200810-1662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cooke BM, Usami S, Perry I, Nash GB. A simplified method for culture of endothelial cells and analysis of adhesion of blood cells under conditions of flow. Microvasc Res. 1993;45:33–45. doi: 10.1006/mvre.1993.1004. [DOI] [PubMed] [Google Scholar]
- 36.Butler LM, Jeffery HC, Wheat RL, Rae PC, Townsend K, Alkharsah KR, Schulz TF, Nash GB, Blackbourn DJ. Kaposi's sarcoma-associated herpesvirus infection of endothelial cells inhibits neutrophil recruitment through an interleukin-6-dependent mechanism: a new paradigm for viral immune evasion. Journal of virology. 2011;85:7321–7332. doi: 10.1128/JVI.00021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105:71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medina RJ, O'Neill CL, O'Doherty TM, Wilson SE, Stitt AW. Endothelial progenitors as tools to study vascular disease. Stem Cells Int. 2012;2012:346735. doi: 10.1155/2012/346735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Medina RJ, O'Neill CL, Sweeney M, Guduric-Fuchs J, Gardiner TA, Simpson DA, Stitt AW. Molecular analysis of endothelial progenitor cell (EPC) subtypes reveals two distinct cell populations with different identities. BMC Med Genomics. 2010;3:18. doi: 10.1186/1755-8794-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toshner M, Dunmore BJ, McKinney EF, Southwood M, Caruso P, Upton PD, Waters JP, Ormiston ML, Skepper JN, Nash G, Rana AA, et al. Transcript analysis reveals a specific HOX signature associated with positional identity of human endothelial cells. PLoS One. 2014;9:e91334. doi: 10.1371/journal.pone.0091334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato S, Nomura F, Kawai T, Takeuchi O, Muhlradt PF, Takeda K, Akira S. Synergy and cross-tolerance between toll-like receptor (TLR) 2- and TLR4-mediated signaling pathways. J Immunol. 2000;165:7096–7101. doi: 10.4049/jimmunol.165.12.7096. [DOI] [PubMed] [Google Scholar]
- 42.Kienast Y, Jucknischke U, Scheiblich S, Thier M, de Wouters M, Haas A, Lehmann C, Brand V, Bernicke D, Honold K, Lorenz S. Rapid Activation of Bone Morphogenic Protein 9 by Receptor-mediated Displacement of Pro-domains. J Biol Chem. 2016;291:3395–3410. doi: 10.1074/jbc.M115.680009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yung LM, Sanchez-Duffhues G, Ten Dijke P, Yu PB. Bone morphogenetic protein 6 and oxidized low-density lipoprotein synergistically recruit osteogenic differentiation in endothelial cells. Cardiovasc Res. 2015;108:278–287. doi: 10.1093/cvr/cvv221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee YS, Park JS, Jung SM, Kim SD, Kim JH, Lee JY, Jung KC, Mamura M, Lee S, Kim SJ, Bae YS, et al. Inhibition of lethal inflammatory responses through the targeting of membrane-associated Toll-like receptor 4 signaling complexes with a Smad6-derived peptide. EMBO Mol Med. 2015;7:577–592. doi: 10.15252/emmm.201404653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee YS, Park JS, Kim JH, Jung SM, Lee JY, Kim SJ, Park SH. Smad6-specific recruitment of Smurf E3 ligases mediates TGF-beta1-induced degradation of MyD88 in TLR4 signalling. Nature communications. 2011;2:460. doi: 10.1038/ncomms1469. [DOI] [PubMed] [Google Scholar]
- 46.DiStasi MR, Ley K. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends in immunology. 2009;30:547–556. doi: 10.1016/j.it.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 49.Summers C, Singh NR, White JF, Mackenzie IM, Johnston A, Solanki C, Balan KK, Peters AM, Chilvers ER. Pulmonary retention of primed neutrophils: a novel protective host response, which is impaired in the acute respiratory distress syndrome. Thorax. 2014;69:623–629. doi: 10.1136/thoraxjnl-2013-204742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garvy BA, Harmsen AG. The importance of neutrophils in resistance to pneumococcal pneumonia in adult and neonatal mice. Inflammation. 1996;20:499–512. doi: 10.1007/BF01487042. [DOI] [PubMed] [Google Scholar]
- 51.Jeyaseelan S, Young SK, Yamamoto M, Arndt PG, Akira S, Kolls JK, Worthen GS. Toll/IL-1R domain-containing adaptor protein (TIRAP) is a critical mediator of antibacterial defense in the lung against Klebsiella pneumoniae but not Pseudomonas aeruginosa. J Immunol. 2006;177:538–547. doi: 10.4049/jimmunol.177.1.538. [DOI] [PubMed] [Google Scholar]
- 52.Tateda K, Moore TA, Deng JC, Newstead MW, Zeng X, Matsukawa A, Swanson MS, Yamaguchi K, Standiford TJ. Early recruitment of neutrophils determines subsequent T1/T2 host responses in a murine model of Legionella pneumophila pneumonia. J Immunol. 2001;166:3355–3361. doi: 10.4049/jimmunol.166.5.3355. [DOI] [PubMed] [Google Scholar]
- 53.Nakamura S, Iwanaga N, Seki M, Fukudome K, Oshima K, Miyazaki T, Izumikawa K, Yanagihara K, Miyazaki Y, Mukae H, Kohno S. Toll-like Receptor 4 Agonistic Antibody Promotes Host Defense against Chronic Pseudomonas aeruginosa Lung Infection in Mice. Infection and immunity. 2016 doi: 10.1128/IAI.01384-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alt A, Miguel-Romero L, Donderis J, Aristorena M, Blanco FJ, Round A, Rubio V, Bernabeu C, Marina A. Structural and functional insights into endoglin ligand recognition and binding. PLoS One. 2012;7:e29948. doi: 10.1371/journal.pone.0029948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rossi E, Smadja DM, Boscolo E, Langa C, Arevalo MA, Pericacho M, Gamella-Pozuelo L, Kauskot A, Botella LM, Gaussem P, Bischoff J, et al. Endoglin regulates mural cell adhesion in the circulatory system. Cellular and molecular life sciences: CMLS. 2016;73:1715–1739. doi: 10.1007/s00018-015-2099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burton VJ, Ciuclan LI, Holmes AM, Rodman DM, Walker C, Budd DC. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood. 2011;117:333–341. doi: 10.1182/blood-2010-05-285973. [DOI] [PubMed] [Google Scholar]
- 57.Burton VJ, Holmes AM, Ciuclan LI, Robinson A, Roger JS, Jarai G, Pearce AC, Budd DC. Attenuation of leukocyte recruitment via CXCR1/2 inhibition stops the progression of PAH in mice with genetic ablation of endothelial BMPR-II. Blood. 2011;118:4750–4758. doi: 10.1182/blood-2011-05-347393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 59.Luscinskas FW, Kansas GS, Ding H, Pizcueta P, Schleiffenbaum BE, Tedder TF, Gimbrone MA., Jr Monocyte rolling, arrest and spreading on IL-4-activated vascular endothelium under flow is mediated via sequential action of L-selectin, beta 1-integrins, and beta 2-integrins. J Cell Biol. 1994;125:1417–1427. doi: 10.1083/jcb.125.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alon R, Kassner PD, Carr MW, Finger EB, Hemler ME, Springer TA. The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J Cell Biol. 1995;128:1243–1253. doi: 10.1083/jcb.128.6.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berlin C, Bargatze RF, Campbell JJ, von Andrian UH, Szabo MC, Hasslen SR, Nelson RD, Berg EL, Erlandsen SL, Butcher EC. alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- 62.Bikoue A, George F, Poncelet P, Mutin M, Janossy G, Sampol J. Quantitative analysis of leukocyte membrane antigen expression: normal adult values. Cytometry. 1996;26:137–147. doi: 10.1002/(SICI)1097-0320(19960615)26:2<137::AID-CYTO7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 63.Reinhardt PH, Elliott JF, Kubes P. Neutrophils can adhere via alpha4beta1-integrin under flow conditions. Blood. 1997;89:3837–3846. [PubMed] [Google Scholar]
- 64.van den Berg JM, Mul FP, Schippers E, Weening JJ, Roos D, Kuijpers TW. Beta1 integrin activation on human neutrophils promotes beta2 integrin-mediated adhesion to fibronectin. European journal of immunology. 2001;31:276–284. doi: 10.1002/1521-4141(200101)31:1<276::AID-IMMU276>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 65.Lomakina EB, Waugh RE. Adhesion between human neutrophils and immobilized endothelial ligand vascular cell adhesion molecule 1: divalent ion effects. Biophysical journal. 2009;96:276–284. doi: 10.1016/j.bpj.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waugh RE, Lomakina EB. Active site formation, not bond kinetics, limits adhesion rate between human neutrophils and immobilized vascular cell adhesion molecule 1. Biophysical journal. 2009;96:268–275. doi: 10.1016/j.bpj.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.