

Abstract
A proportion of homologous recombination (HR) events in mammalian cells resolve by “long tract” gene conversion, reflecting copying of several kilobases from the donor sister chromatid prior to termination. Cells lacking the major hereditary breast/ovarian cancer predisposition genes, BRCA1 or BRCA2, or certain other HR-defective cells, reveal a bias in favor of long tract gene conversion, suggesting that this aberrant HR outcome might be connected with genomic instability. If termination of gene conversion occurs in regions lacking homology with the second end of the break, the normal mechanism of HR termination by annealing (i.e., homologous pairing) is not available and termination must occur by as yet poorly defined non-canonical mechanisms. Here we use a previously described HR reporter to analyze mechanisms of non-canonical termination of long tract gene conversion in mammalian cells. We find that non-canonical HR termination can occur in the absence of the classical non-homologous end joining gene XRCC4. We observe obligatory use of microhomology (MH)-mediated end joining and/or nucleotide addition during rejoining with the second end of the break. Notably, non-canonical HR termination is associated with complex breakpoints. We identify roles for homology-mediated template switching and, potentially, MH-mediated template switching/microhomology-mediated break-induced replication, in the formation of complex breakpoints at sites of non-canonical HR termination. This work identifies non-canonical HR termination as a potential contributor to genomic instability and to the formation of complex breakpoints in cancer.
Author Summary
Complex breakpoints are a recognized feature of cancer genome rearrangements, but the mechanisms that lead to their formation are undefined. Although homologous recombination (HR) is considered a potentially error-free pathway, cells lacking critical HR genes, such as the major hereditary breast/ovarian cancer predisposition genes, BRCA1 or BRCA2, frequently engage error-prone homologous recombination mechanisms in which HR termination does not occur in a timely fashion. We show here that aberrant termination of HR in mammalian cells involves the use of error-prone alternative end joining mechanisms and can lead to the formation of complex breakpoints by means of template switching mechanisms. This suggests that defective termination of homologous recombination underlies some of the complex breakpoints observed in cancer cells.
Introduction
Double strand breaks (DSBs) are dangerous lesions, the misrepair of which can contribute to genomic instability and cancer predisposition, premature aging and immunological deficiency in mammals [1–3]. A major trigger to chromosome breakage occurs during attempted replication across a damaged DNA template [4–8]. Such replication-associated DSBs may be repaired by sister chromatid recombination (SCR)—a potentially error-free pathway of homologous recombination (HR) in which the broken chromosome uses the neighboring sister chromatid as a template for repair [9–12]. Germ line mutation of HR genes contributes to hereditary breast/ovarian cancer susceptibility, Fanconi anemia and other cancer-prone or developmental disorders [1, 13–15]. Other recognized DSB repair pathways include classical non-homologous end joining (C-NHEJ), alternative end-joining (A-EJ, i.e., end-joining in the absence of one or more C-NHEJ genes) and single strand annealing (SSA) [2]. A-EJ is characterized by the dominant use of microhomology (MH)-mediated end joining (MMEJ)—rejoining events in which the two DNA ends share short stretches of homology at the breakpoint [16, 17].
Cancer genomes commonly reveal complex patterns of chromosomal rearrangement. This complexity may take the form of multiple breakpoints at the site of a chromosome rearrangement with insertion of short stretches of DNA sequence derived from ectopic loci [18–20]. The breakpoints of cancer rearrangements frequently reveal MH, but homeologous breakpoints (i.e., breakpoints with extensive but imperfect homology) and breakpoints with untemplated nucleotide addition (N-addition) are also observed [18]. Such complex rearrangements could entail rejoining of simultaneously arising chromosome breaks, break-induced copying from ectopic templates, or both [21].
A major pathway of HR repair in somatic cells is “Synthesis-dependent strand annealing” (SDSA) [22]. SDSA entails DNA end resection, loading of the Rad51 recombinase onto single stranded (ss)DNA and Rad51-mediated homologous invasion of the donor DNA molecule, such as the neighboring sister chromatid, by one of the two DNA ends. Extension of the invading/nascent strand by repair synthesis is followed by its release (“displacement”) and termination of SDSA normally occurs by annealing (i.e., homologous pairing) of the displaced nascent strand with complementary ssDNA sequences on the resected second end of the DSB. The majority of HR events triggered by a DSB resolve by “short tract” gene conversion (STGC), which typically entails repair synthesis of <100 base pairs from the donor [23–25]. A proportion of HR events resolve as “long tract” gene conversions (LTGC), in which several kilobases (up to ~10 kb) of the neighboring, undamaged sister chromatid are copied into the break site of the damaged chromosome [26, 27]. LTGC and crossing over can produce similar rearrangements in the context of an HR reporter. Where studied, these outcomes have proven to be mediated by LTGC and not by crossing over [26, 28–30]. Genetic inactivation of the major hereditary breast/ovarian cancer predisposition HR genes BRCA1 or BRCA2, or of other HR genes such as the Rad51 paralogs Rad51C, XRCC2 or XRCC3 biases HR in favor of LTGC [28–34]. Thus, understanding the mechanisms underlying LTGC in mammalian cells may yield insight into mechanisms of genomic instability in HR-defective hereditary breast/ovarian cancer-predisposition syndromes.
Very long gene conversions in Saccharomyces cerevisiae are mediated by break-induced replication (BIR), which can copy >100 kilobases from the donor molecule [35–37]. The BIR copying mechanism in S. cerevisiae is conservative, rather than the semi-conservative mechanism of a conventional replication fork [38, 39]. BIR in S. cerevisiae is dependent on the Pif1 helicase and entails a migrating bubble mechanism [39, 40]. Gene conversions in S. cerevisiae that ultimately resolve as BIR may reveal homologous template switches during the early stages of the process, suggesting that the initial steps of BIR can be mediated by less robust copying mechanisms [41]. Further, spontaneous somatic gene conversions in S. cerevisiae reveal a bimodal distribution of tract lengths, with median peaks at 6 kb and >50 kb [42]. Taken together, these studies suggest that classical BIR and LTGC, although topologically similar processes, retain some mechanistic differences.
If the site of HR termination lacks homology with the second (non-invading) end of the DSB, the classical SDSA mechanism of termination by annealing with the resected second end of the DSB is not available. Under these circumstances, HR termination may be mediated by end joining mechanisms [26, 27, 43, 44]. Breakpoints of non-canonical HR termination often reveal MH, suggesting a role for A-EJ in this process [43, 44]. However, the genetic regulation of non-canonical HR termination in mammalian cells is currently undefined. In Drosophila melanogaster, non-homologous termination of HR repair of a transposase-induced break is independent of the C-NHEJ gene LIG4 and is mediated by the error-prone DNA polymerase PolΘ, encoded by the POLQ gene [45, 46]. Here, we use a previously described mammalian reporter of LTGC between sister chromatids [27] to analyze mechanisms of non-canonical LTGC termination in XRCC4 conditional and isogenic XRCC4 null mouse embryonic stem (ES) cells [47, 48]. Our work reveals that non-canonical termination of HR in mammalian cells is independent of XRCC4 and can lead to the formation of complex breakpoints, mediated by template switching. This suggests that non-canonical termination of HR may contribute to the formation of complex breakpoints in the cancer genome.
Results
Non-canonical termination of mammalian HR does not require XRCC4
We previously described a HR reporter that enables positive selection of both short tract (STGC) and long tract gene conversions (LTGC) between sister chromatids in response to a site-specific DSB induced by the rare-cutting homing endonuclease I-SceI (Fig 1) [27]. Briefly, we positioned two artificial exons of the gene encoding blasticidin S deaminase (here termed “BsdR”) in a non-productive orientation between the two GFP copies of an HR reporter. Parental cells, or products of STGC, remain blasticidin sensitive (BsdR–; Fig 1A). In contrast, LTGC duplicates the BsdR cassette, thereby allowing expression of wild type (wt) BsdR by splicing (Fig 1A). LTGC is experimentally defined here as a gene conversion of >1.03kb—sufficient to duplicate exon B of the blasticidin cassette.
Fig 1. Method for identifying non-canonical HR termination products in mammalian cells.

(A) Schematic of the HR reporter. Duplication of a blasticidin resistance cassette during LTGC allows expression of wt BsdR by splicing. Thus, I-SceI-induced STGCs are GFP+ and Bsd sensitive (BsdR–), while I-SceI-induced LTGCs are GFP+ and Bsd resistant (BsdR+). Most LTGCs resolve as “GFP triplication” events, but a small fraction of LTGCs resolve by non-canonical mechanisms. Non-canonical LTGC termination products can be distinguished by the structure of the LTGC product, as shown. (B) Characterization of XRCC4fl/fl and XRCC4Δ/Δ Cre-treated HR reporter clones. Upper panel: Southern blotting, as described in Materials and Methods. Lower panel: western blotting for XRCC4 or for ß-tubulin loading control.
The most abundant I-SceI-induced HR product is STGC, in which the broken copy of GFP is converted to wild type GFP, leaving the reporter structure otherwise unchanged (Fig 1A). In wild type cells, approximately 5% of all I-SceI-induced GFP+ products resolve by LTGC [28, 29, 47, 49]. LTGC frequently results in triplication of the GFP copies within the repaired sister chromatid (Fig 1A). However, a small proportion of I-SceI-induced LTGCs are terminated in regions lacking homology with the second end of the DSB [26, 27, 29]. These LTGCs must be terminated by non-canonical mechanisms (Fig 1A).
To study the contribution of C-NHEJ to non-canonical HR termination, we introduced the above-noted “long tract” HR/SCR reporter into mouse embryonic stem (ES) cells carrying biallelic conditional (“floxed”) alleles of XRCC4 (XRCC4fl/fl ES cells) [48, 50]. We identified individual clones in which a single, intact copy of the reporter had been integrated into the ROSA26 locus, as described previously and in Materials and Methods [49]. We transduced two distinct XRCC4fl/fl HR/SCR reporter ES cell clones with adenovirus encoding the Cre recombinase and screened Cre-treated cells for derivative clones that either had or had not undergone biallelic Cre-mediated deletion of XRCC4. Southern and western blotting identified XRCC4Δ/Δ and XRCC4fl/fl derivatives of these cells (examples in Fig 1B). We transfected XRCC4fl/fl and, in parallel, XRCC4Δ/Δ HR/SCR reporter ES cells with I-SceI (with appropriate controls as described in Materials and Methods), and scored HR products as the frequency of I-SceI-induced GFP+ and BsdR+ events (LTGCs). The ratio LTGC:Total HR (BsdR+ GFP+: Total GFP+) is a measure of the probability that a given HR event will resolve as LTGC. This value was ~3% in each cell type, suggesting that XRCC4 does not directly influence the probability of engaging LTGC during I-SceI-induced HR.
We amplified I-SceI-induced BsdR+ colonies from two XRCC4fl/fl HR/SCR reporter clones (n = 163) and two isogenic XRCC4Δ/Δ HR/SCR reporter clones (n = 211), prepared genomic DNA (gDNA), and analyzed the underlying structure of the LTGC product by Southern blotting, as described in Materials and Methods—results summarized in Table 1. We noted examples of non-canonical LTGC termination in both XRCC4fl/fl (6/163; 3.7%) and XRCC4Δ/Δ (5/211; 2.4%) HR/SCR reporter cells (difference not significant by Fisher’s exact test). This establishes that non-canonical HR termination can occur in the absence of the C-NHEJ gene XRCC4. A proportion of LTGCs produced aberrant Southern blot patterns, either in the form of off-size bands or additional GFP-hybridizing bands, which defied easy interpretation. 14/211 (6.6%) of all LTGCs examined in XRCC4Δ/Δ HR/SCR reporter cells were aberrant; the equivalent proportion in XRCC4fl/fl HR/SCR reporter cells was 2/163 (1.2%); (P = 0.0102 by Fisher’s exact test). The higher proportions of aberrant LTGCs noted in XRCC4Δ/Δ HR/SCR reporter cells is consistent with the known role of XRCC4 in suppressing chromosomal rearrangements [51, 52]. Analysis of one of these aberrant LTGCs in XRCC4Δ/Δ HR/SCR reporter cells is presented below.
Table 1. I-SceI-induced LTGC products in XRCC4fl/fl and XRCC4Δ/Δ cells.
| Genotype | XRCC4fl/fl | XRCC4Δ/Δ |
|---|---|---|
| GFP triplication | 155 | 192 |
| LTGC non-canonical termination | 6 | 5 |
| Aberrant | 2 | 14 |
Table 1 summarizes Southern blot analysis of I-SceI-induced blasticidin-resistant clones in XRCC4fl/fl (n = 163) and XRCC4Δ/Δ (n = 211) SCR reporter cells. Fisher’s exact test XRCC4fl/fl vs. XRCC4Δ/Δ for GFP triplication vs. non-canonically terminated LTGC: not significant. Fisher’s exact test XRCC4fl/fl vs. XRCC4Δ/Δ for GFP triplication vs. aberrant LTGCs (excludes non-canonically terminated LTGC products): P = 0.0102.
Microhomology-mediated end joining mediates non-canonical LTGC termination
The unrearranged parental reporter and the major “GFP triplication” LTGC product produce predictable patterns of hybridization following gDNA digestion with a panel of restriction endonucleases (Fig 2). We made the assumption that non-canonical termination of LTGC normally entails rejoining with the second end of the DSB and used the specific pattern of Southern blot hybridizations to deduce the likely site of non-canonical LTGC termination in XRCC4fl/fl or XRCC4Δ/Δ LTGC clones. Two such examples are shown in Fig 3. We were able to clone the breakpoints of six XRCC4fl/fl and three XRCC4Δ/Δ non-canonical LTGC termination products (see Materials and Methods). The cloned breakpoints did indeed reflect rejoining to the second end of the DSB, which had undergone varying degrees of resection (Fig 4). Each breakpoint revealed use of MMEJ or untemplated nucleotide addition (N-addition) at the breakpoint. It has been suggested that N-addition breakpoints of the type observed here might also be products of MMEJ-type rejoining [45]. There were no blunt-ended non-homologous breakpoints in this limited sample and no breakpoints were suggestive of dual homologous invasions by both ends of the original I-SceI-induced DSB. Thus, non-canonical termination of HR can occur in the absence of the C-NHEJ gene XRCC4 and entails use of MMEJ/N-addition rejoining mechanisms, implicating A-EJ as a contributing mechanism.
Fig 2. Restriction mapping of parental reporter and of LTGC “GFP triplication” products.

(A) Expected GFP-hybridizing gDNA restriction fragment sizes for HR reporter at the ROSA26 locus. Upper panel: parental reporter; lower panel: “GFP triplication” outcome of LTGC. GFP copies within the reporter are shown. Filled ovals: artificial BsdR exons A and B. Restriction enzyme sites shown are SpeI (Sp), EcoRI (E), BamHI (B), HindIII (H) and SacI (Sa). Note that each of these restriction endonucleases, which cut target sites between the two GFP copies within the parental reporter, generate an additional 3.2kb GFP-hybridizing band in the context of the “GFP triplication” outcome. (B) Genomic DNA from parental and “GFP triplication” LTGC clones, as shown, was digested with the restriction enzymes shown (code as described above) and analyzed by Southern blotting (GFP probe). The 3.2kb amplification product characteristic of the “GFP triplication” LTGC outcome is marked with an arrowhead.
Fig 3. Restriction mapping of products of non-canonical LTGC termination.

Genomic DNA from two clones in which LTGC was terminated by non-canonical mechanisms was digested with the restriction enzymes shown and analyzed by Southern blotting (GFP probe). Restriction enzymes used were SacI (Sa), HindIII (H), BamHI (B), EcoRI (E) and SpeI (Sp). Cartoons on right show restriction fragment sizes observed for HR reporter at the ROSA26 locus. The presence or absence of the 3.2kb amplification product in each restriction digest helps to localize the site of LTGC termination within the reporter. (A) XRCC4fl/fl clone in which termination of LTGC occurred between HindIII and EcoRI sites within the HR reporter. EcoRI and SpeI digests lack the 3.2kb amplification product. (B) XRCC4Δ/Δ clone in which termination of LTGC occurred between SacI and HindIII sites within the HR reporter. HindIII, EcoRI and SpeI digests lack the 3.2kb amplification product. In this clone, the right hand arms of the SpeI and HindIII digests are much smaller (SpeI) or larger (HindIII) than would be predicted. This is explained by the deletion of ~3.5kb from the second end of the DSB, as revealed by sequencing (see Fig 6B).
Fig 4. Breakpoints of non-canonical LTGC termination in five XRCC4fl/fl and two XRCC4Δ/Δ clones.

Cartoon shows approximate positions of breakpoints. Black numbers mark site of LTGC termination; paired blue numbers mark extent of second end resection for the same clone (not to scale). Numbers correlate with the numbered clones in lower panel, showing length of gene conversion tract (black) and extent of second end resection (blue) in each clone, with genotype as indicated. Red nucleotides: N-insertions at the breakpoint. Dual black/blue nucleotide sequences at the breakpoint represent microhomology.
Analysis of an aberrant LTGC product of XRCC4Δ/Δ HR/SCR reporter cells
We used a similar restriction mapping approach to analyze one aberrant LTGC product identified in XRCC4Δ/Δ HR/SCR reporter cells. As discussed above, aberrant LTGC products characteristically reveal off-size or additional GFP-hybridizing bands by Southern blotting. One such aberrant clone is shown in Fig 5. Southern analysis appeared to show two groups of GFP-hybridizing bands with distinct intensities. Importantly, these groups were not separated by recloning of the cells, indicating that all the GFP fragments visualized by Southern blotting reside within one nucleus. We interpret the Southern blot pattern as a case of non-canonical LTGC termination (blue arrow-heads Fig 5) in which LTGC termination occurred between the SacI and HindIII sites within the reporter. However, all restriction fragments involving enzymes beyond the SacI site (i.e., HindIII, EcoRI and SpeI) reveal off-size GFP-hybridizing bands (Fig 5B). These fragments do not match restriction fragment patterns of ROSA26 sequence up to 50 kb beyond the second end of the DSB. This suggests that LTGC termination in this case entailed incorporation of ectopic chromosomal sequences. We interpret the fainter GFP-hybridizing bands in this Southern blot (orange arrow-heads) as possible products of the second end of the break (Fig 5C). If so, the rearrangement underlying this aberrant LTGC product could entail a gross chromosomal rearrangement (GCR) initiated by non-canonical LTGC termination. Alternatively, the ectopic sequences (grey bars) depicted in Fig 5A and 5B might be part of one single insertion of several kilobases between the site of LTGC termination and the second end of the break. In this regard, the solitary ~9 kb SpeI fragment in Fig 5A, which appears to have a higher intensity than all other bands, could potentially span this insertion, while retaining GFP sequences from both sides of the termination breakpoint. However, our attempts to amplify such a putative insertion product between the two ends of the break have not yet been successful. The notion that non-canonical LTGC termination might lead to GCR is consistent with the expected greater availability of free DNA ends in XRCC4Δ/Δ cells, where efficient C-NHEJ mechanisms are compromised. This clone is an example of non-canonical LTGC termination that presents with an aberrant LTGC pattern by Southern blotting. However, until this and other aberrant LTGC products are mapped and sequenced, it would not be valid to conclude that all aberrant LTGC outcomes arise from non-canonical LTGC termination.
Fig 5. Restriction mapping of an aberrant LTGC rearrangement in XRCC4Δ/Δ HR/SCR reporter cells.

(A) Restriction analysis of aberrant LTGC product in XRCC4Δ/Δ HR/SCR reporter cells. Genomic DNA was digested with the restriction enzymes shown and analyzed by Southern blotting (GFP probe). Restriction enzymes used were SacI (Sa), HindIII (H), BamHI (B), EcoRI (E) and SpeI (Sp). Patterns suggest that LTGC was terminated by non-canonical mechanisms. Blue arrow-heads: deduced products of non-canonical LTGC termination. Blue star: off-size restriction fragments of HindIII, EcoRI and SpeI digests are inconsistent with rejoining with the second end of the original I-SceI-induced DSB (compare with Fig 2). Orange arrow-heads: fainter bands may represent the half-copy of GFP retained by the second end of the original I-SceI-induced DSB. Note that the upper band of SpeI-restricted gDNA has a greater intensity than other bands, suggesting presence of two distinct co-migrating GFP-hybridizing fragments, or a single fragment containing >1 copy of GFP. (B) Deduced rearrangement of the non-canonically-terminated LTGC. Blue star: off-size restriction fragments of HindIII, EcoRI and SpeI digests. Note that each of these off-size fragments spans the predicted breakpoint of LTGC termination. This suggests that this LTGC event terminated by rejoining to ectopic chromosomal sequences (grey bar in the figure). (C) Deduced rearrangement of the second end of the DSB. Note that GFP-hybridizing fragments of SpeI, BamHI and HindIII restriction digest are off-size, potentially consistent with rejoining of the second end of the DSB with ectopic chromosomal sequences (grey bar).
Complex breakpoints associated with non-canonical termination of LTGC
In one XRCC4fl/fl clone in which LTGC had been terminated by non-canonical mechanisms, sequencing revealed two distinct breakpoints: one homologous and one N-addition breakpoint. The homologous breakpoint reflected incorporation of sequences from the episomal I-SceI expression vector within the repaired sister chromatid (Fig 6A). The vector sequence had been incorporated at a site of perfect and extensive homology between the chromosomally integrated HR/SCR reporter and the episomal plasmid, based upon shared rabbit β-globin intron sequences [27, 53]. Following LTGC using the sister chromatid as template, a template switching mechanism allowed the displaced nascent strand to invade homologous sequences on the episomal plasmid. After further nascent strand synthesis of ≥342 bp (the exact point of homologous invasion of the episomal plasmid is not definable), the newly extended nascent strand was displaced from the plasmid template and was joined to the second end of the I-SceI-induced chromosomal break, with insertion of one nucleotide at this second (non-homologous) breakpoint (Fig 6A). Thus, non-canonical termination of LTGC can entail homologous template switching—a phenomenon known to be associated with LTGC and BIR in S. cerevisiae [41, 54].
Fig 6. Template switching and complex breakpoints during non-canonical LTGC termination.
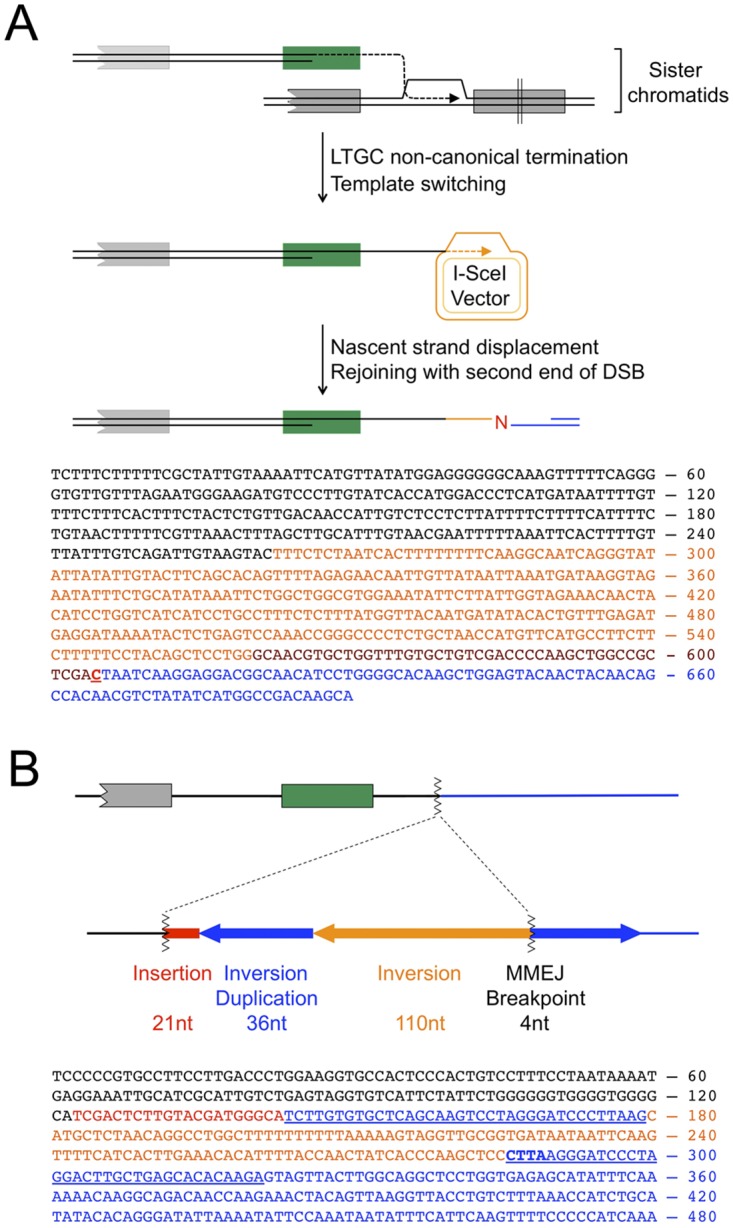
(A) Homologous template switching during non-canonical LTGC termination in a XRCC4fl/fl clone. Upper panel: Cartoon depicts the HR-mediated template switch between the displaced nascent strand product of LTGC (black) and identical rabbit β-globin intron sequences within the episomal I-SceI expression vector. Lower panel: Sequence of the homologous template switch complex breakpoint. Orange: intron sequences copied from the I-SceI expression vector. Brown: additional sequence copied from the episomal I-SceI expression vector. Red: single N-addition at second breakpoint. Blue: second end of the DSB, resected 9 bp prior to end joining with the twice-displaced nascent strand. (B) Microhomology-mediated complex breakpoint formation during non-canonical LTGC termination in a XRCC4Δ/Δ clone. Southern blotting analysis of this clone is presented in Fig 3B. Upper panel: Cartoon shows map of the complex breakpoint, which involved rearrangement of the second (non-invading) DNA end. Red: 21nt insertion. Blue arrows: duplicated 36bp sequence from second end of DSB (located 3579-3614bp from the I-SceI site). Orange arrow: Inverted 110bp sequence adjacent to duplicated sequence (located 3469-3578bp from the I-SceI site). The MMEJ breakpoint within the second DNA end is located 3579-3582bp downstream of the I-SceI site. Blue sequences (including correctly oriented blue arrow) to the right of MH breakpoint are unrearranged ROSA26 locus. With the exception of the inverted 110 bp sequence, a segment of the second DNA end ~3.5kb adjacent to the I-SceI site was deleted during the rearrangement. Lower panel: Sequence of the MH-mediated complex breakpoint. Black: LTGC product (gene conversion tract length was 1249bp). Red: 21nt insertion. First blue underlined: inverted 36bp repeat. Orange: 110bp inversion. Second blue underlined: correctly oriented 36bp repeat, contiguous with unrearranged ROSA26 sequence. Bold underlined blue: 4bp MH breakpoint. Hypothetical model of this complex breakpoint is presented in Fig 7.
A second complex breakpoint of non-canonical LTGC termination was present in one XRCC4Δ/Δ clone. Sequencing of the breakpoint revealed an inversion/duplication rearrangement of the second end of the DSB (Fig 6B; Southern blot analysis of this clone is shown in Fig 3B), involving at least two breakpoints in close proximity to one another. The first breakpoint entailed a 21bp insertion at the site of non-canonical LTGC termination, showing 16bp identity with several heterologous loci in the mouse genome (if templated, this 21bp insertion could represent two independent breakpoints). The second was a 4bp MH breakpoint generated during ligation to the second end of the DSB, with an accompanying complex deletion/inversion/duplication rearrangement of the second end of the DSB. Although the mechanisms underlying this complex rearrangement are a matter of speculation, the rearrangement suggests that the nascent strand, having been displaced from the donor sister chromatid during LTGC termination, underwent further rounds of MH-mediated template switches and short nascent strand extension—a process termed “microhomology-mediated BIR” (MMBIR) [55]. Fig 7 depicts how this MMBIR rearrangement could have arisen through a fork stalling and template switching (FoSTeS) mechanism [56]. Notably, the 146 bp inversion fragment (Fig 6B) is of a size consistent with FoSTeS-type copying from a lagging strand donor.
Fig 7. MMBIR model of complex breakpoint shown in Fig 5B.

Strand separation occurs within the DNA of the second end of the break ~3.5 kb from the I-SceI site. One possible source depicted here is a stalled replication fork. The pale orange and blue arrows flanking the stalled fork represent the exposed ssDNA sequences that template the inversion (orange) and inversion-duplication (blue) sequences identified within the LTGC breakpoint (A) The displaced nascent strand product of LTGC (black) acquires a ≥21bp insertion (red; whether templated or untemplated is unknown). (B) Microhomology-mediated base-pairing between the 3’ end of the displaced nascent strand and ssDNA of the stalled replication fork. (C) The lagging strand template enables retrograde nascent strand extension (“MMBIR”), generating the inversion sequences as shown. (D) Displacement of the nascent strand. (E) Four base pair MH-mediated (Fig 6B) annealing of the 3’ end of the displaced nascent strand with the 5’ end of the duplicated region on the leading strand. Black arrowheads: sites of endonucleolytic cleavage that would enable completion of rearrangement by MMEJ-mediated rejoining. Alternatively, more extensive MMBIR copying could complete the rearrangement.
Discussion
We used the positive selective power of a HR/SCR reporter to capture rare LTGCs in which HR had been terminated by non-canonical mechanisms in XRCC4fl/fl and XRCC4Δ/Δ mouse ES cells. Rejoining with the second end of the chromosomal break entails use of XRCC4-independent MMEJ (i.e. A-EJ), in agreement with previous studies in D. melanogaster [45, 46]. A notable finding of the current study is that non-canonical HR termination in mammalian cells may entail homologous template switching or MH-mediated template switching (i.e., MMBIR) prior to rejoining with the second DNA end, leading to the formation of complex breakpoints at the site of HR termination. Long gene conversions during gap repair in D. melanogaster have been proposed to entail cycles of invasion and displacement of the nascent strand, with an implied potential for template switching [57]. Both homologous template switches and MMBIR have been described in S. cerevisiae during LTGC/BIR, suggesting that these error-prone mechanisms of HR termination are evolutionarily conserved [41, 54, 58]. Our findings provide direct evidence of homologous template switching during mammalian HR, highlighting the extreme reactivity of the displaced nascent strand and its potential significance as an instigator of genomic instability. Given the likely importance of template switching mechanisms in the formation of complex breakpoints in cancer cells, our findings suggest that aberrant HR termination may underlie some of the complex breakpoints observed in cancer genomes [18–21].
A striking feature of the breakpoints associated with non-canonical LTGC termination is the frequent use of MMEJ/insertional rejoining mechanisms. The channeling of repair into an MMEJ mechanism is likely best explained by the DNA structures that are presented for rejoining. Both the displaced nascent strand and the resected second end of the break possess extended 3’ ssDNA tails. These are poor substrates for Ku binding and, hence, for C-NHEJ-mediated rejoining, leading to a preference for A-EJ [59]. Completion of non-canonical LTGC by MMEJ-mediated rejoining to the second end of the DSB may suppress more deleterious outcomes, such as template switching, BIR and chromosome translocation, at sites of non-canonical HR termination. Direct testing of this hypothesis must await the development of more readily quantifiable systems for studying non-canonical HR termination in mammalian cells. However, this idea is strongly corroborated by work on the A-EJ mediator PolΘ, which suppresses genomic instability in mammalian cells and prevents large deletions at sites of replication arrest or at transposase-induced gaps in model organisms [46, 60–64]. Conversely, unrestrained LTGC in BRCA mutant and other HR-defective cells might channel HR towards these deleterious outcomes as a mechanism of genomic instability in tumorigenesis [28–30].
In the cell lines studied here, non-canonical LTGC termination accounts for ~3% of all LTGCs in XRCC4fl/fl cells, corresponding to ~0.1% of all measured GFP+ I-SceI-induced HR events. These low frequencies may nonetheless be highly significant for genomic instability and cancer predisposition, since cancer initiation and progression result from stochastic events on a “per cell” basis. The significance of non-canonical termination of LTGC may be greater than is suggested by the above calculations, since the repetitive structure of the HR reporter used here presents two opportunities for HR termination by annealing: during STGC and in the termination of LTGC by “GFP triplication” (Fig 1). In contrast, when gene conversion occurs within non-repetitive sequences, STGC alone provides an opportunity for HR to be terminated by annealing. In this more natural setting, presumably all LTGCs must resolve either by non-canonical termination mechanisms or by BIR. In this regard, it is relevant that mammalian cells lacking the major hereditary breast/ovarian cancer predisposition genes BRCA1 or BRCA2 or other cancer predisposition HR genes reveal a bias towards LTGC [28, 31–34]. This bias is even more marked at stalled replication forks, where >80% of HR events may resolve as LTGCs in BRCA/HR-defective cells [30]. In this setting, the arrival of a converging replication fork and the activity of stalled fork endonucleases may be additional determinants of genomic instability [65]. The work described here identifies mechanisms by which dysregulated LTGC may contribute to genomic instability in BRCA/HR-defective cells and in general tumorigenesis.
Materials and Methods
Plasmids—The sister chromatid recombination reporter was previously characterized. Expression plasmids for I-SceI and GFP were described previously [27, 49]. New constructs described here were generated by standard cloning procedures.
Cell Lines and Cell Culture—XRCC4fl/fl mouse embryonic stem (ES) cells were obtained from Catherine Yan and Frederick Alt and have been described previously [48]. ES cells were maintained in ES medium on either irradiated MEF feeder cells or gelatinized plates. To generate SCR reporter stable lines, 20μg of KpnI-linearized SCR reporter plasmid was electroporated into 2x107 XRCC4fl/fl ES cells and cells were seeded into 60mm dishes with neomycin resistant feeder mouse embryonic fibroblasts and 400μg/mL G418 (Sigma-Aldrich) was added to the medium 1 day after electroporation. Beginning 1 week after continuous selection, G418-resistant colonies were isolated and screened by Southern blotting for single-copy SCR reporter integration. To generate isogenic XRCC4fl/fl, XRCC4fl/Δ and XRCC4Δ/Δ SCR cell lines, adeno-Cre infection was performed as described previously [49], followed by screening of derivative cell lines by Southern blotting.
Recombination Assays—1.6x105 trypsinized ES cells were transfected with 0.5μg plasmid DNA using Lipofectamine 2000 (Invitrogen) in a 24-well plate. Transfection efficiency was measured by parallel transfection of wtGFP expression vector (at 1:10 dilution in empty vector). GFP+ frequencies were measured 72 hr post-treatment by flow cytometry using an FC500 (Beckman Coulter) as described previously [27]. To assay LTGC events, cells were counted and replated at 1-3x105 cells per gelatinized 100mm dish in triplicate into media containing 5μg/mL blasticidin (Invitrogen). Approximately 2 weeks later, blasticidin resistant colonies were stained and counted or expanded for molecular analysis. Plating efficiency was determined by plating 3-5x102 cells per gelatinized 100mm dish in triplicate into media lacking selection. HR measurements were corrected for background levels of HR events, transfection efficiency and plating efficiency, as described previously [49].
Southern Blotting—Genomic DNA was extracted from 5-20x106 cells using the ArchivePure Cell/Tissue Kit (5 PRIME). GFP and XRCC4 Southern blots were carried out as previously described [27, 47, 50, 66].
Western Blotting—Cell lysates were prepared using RIPA buffer (50 mM Tris-HCl [pH 8.0], 1.0% NP-40, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS) containing protease inhibitors (Roche). Protein concentration was estimated using Bradford’s Reagent (Sigma-Aldrich). Cellular proteins were resolved by SDS-PAGE on NuPAGE Novex Bis-Tris Gels (Invitrogen), transferred to nitrocellulose membrane (Bio-Rad semi-dry transfer system, 40 mA overnight). The membrane was blocked with 5% nonfat milk in 0.05% PBST (0.05% Tween 20, in PBS) and incubated with rabbit polyclonal anti-XRCC4 1:200 (Sigma-Aldrich) or mouse monoclonal anti-β-tubulin 1:200 (Abcam) at room temperature for 3 hrs. Membranes were washed in 0.05% PBST, incubated with peroxidase-conjugated Protein A (GE Healthcare) or goat anti-mouse antibody (Jackson ImmunoResearch) and developed using high-sensitivity ECL (PerkinElmer).
PCR and Sequencing—Breakpoints were amplified using AccuPrime Taq DNA Polymerase High Fidelity (Invitrogen) according to manufacturers instructions. The PCR products were excised from the gel and purified using the QIAquick Gel Extraction Kit (QIAGEN) and subsequently cloned into the pGEM-T Easy vector (Promega). Sequencing was performed at the Dana-Farber/Harvard Cancer Center DNA Resource Core.
Acknowledgments
We thank Drs. James Haber, Fred Alt, Mitch McVey, Lorraine Symington, Anna Malkova and members of the Scully lab for helpful comments and critiques.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by NIH grants R01CA095175 and R01GM073894 (to RS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. Epub 2010/10/23. S1097-2765(10)00747-1 [pii] 10.1016/j.molcel.2010.09.019 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423(2):157–68. ; 10.1042/BJ20090942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152(3):417–29. Epub 2013/02/05. 10.1016/j.cell.2013.01.007 S0092-8674(13)00009-3 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404(6773):37–41. 10.1038/35003501 [DOI] [PubMed] [Google Scholar]
- 5.Lambert S, Watson A, Sheedy DM, Martin B, Carr AM. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005;121(5):689–702. 10.1016/j.cell.2005.03.022 [DOI] [PubMed] [Google Scholar]
- 6.Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326(5960):1698–701. Epub 2009/12/08. 10.1126/science.1182372 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan KL, Hickson ID. New insights into the formation and resolution of ultra-fine anaphase bridges. Seminars in cell & developmental biology. 2011;22(8):906–12. Epub 2011/07/26. 10.1016/j.semcdb.2011.07.001 S1084-9521(11)00087-5 [pii]. . [DOI] [PubMed] [Google Scholar]
- 8.Duxin JP, Walter JC. What is the DNA repair defect underlying Fanconi anemia? Current opinion in cell biology. 2015;37:49–60. 10.1016/j.ceb.2015.09.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11(3):196–207. Epub 2010/02/24. 10.1038/nrm2851 nrm2851 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagaraju G, Scully R. Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair (Amst). 2007;6(7):1018–31. 10.1016/j.dnarep.2007.02.020 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr AM, Lambert S. Replication Stress-Induced Genome Instability: The Dark Side of Replication Maintenance by Homologous Recombination. J Mol Biol. 2013. Epub 2013/05/07. S0022-2836(13)00271-4 [pii] 10.1016/j.jmb.2013.04.023 . [DOI] [PubMed] [Google Scholar]
- 12.Lambert S, Mizuno K, Blaisonneau J, Martineau S, Chanet R, Freon K, et al. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell. 2010;39(3):346–59. Epub 2010/08/14. 10.1016/j.molcel.2010.07.015 . [DOI] [PubMed] [Google Scholar]
- 13.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408(6811):429–32. 10.1038/35044000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11(2):103–5. 10.1016/j.ccr.2007.01.010 [DOI] [PubMed] [Google Scholar]
- 15.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108(2):171–82. . [DOI] [PubMed] [Google Scholar]
- 16.McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends in genetics: TIG. 2008;24(11):529–38. 10.1016/j.tig.2008.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, et al. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A. 2013;110(19):7720–5. Epub 2013/04/24. 10.1073/pnas.1213431110 1213431110 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bignell GR, Santarius T, Pole JC, Butler AP, Perry J, Pleasance E, et al. Architectures of somatic genomic rearrangement in human cancer amplicons at sequence-level resolution. Genome research. 2007;17(9):1296–303. 10.1101/gr.6522707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462(7276):1005–10. Epub 2009/12/25. nature08645 [pii] 10.1038/nature08645 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, Layer RM, et al. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome research. 2013;23(5):762–76. 10.1101/gr.143677.112 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willis NA, Rass E, Scully R. Deciphering the Code of the Cancer Genome: Mechanisms of Chromosome Rearrangement. Trends Cancer. 2015;1(4):217–30. 10.1016/j.trecan.2015.10.007 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiology and molecular biology reviews: MMBR. 1999;63(2):349–404. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol Cell Biol. 1998;18(1):93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sweetser DB, Hough H, Whelden JF, Arbuckle M, Nickoloff JA. Fine-resolution mapping of spontaneous and double-strand break-induced gene conversion tracts in Saccharomyces cerevisiae reveals reversible mitotic conversion polarity. Mol Cell Biol. 1994;14(6):3863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taghian DG, Nickoloff JA. Chromosomal double-strand breaks induce gene conversion at high frequency in mammalian cells. Mol Cell Biol. 1997;17(11):6386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. Embo J. 2000;19(13):3398–407. 10.1093/emboj/19.13.3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puget N, Knowlton M, Scully R. Molecular analysis of sister chromatid recombination in mammalian cells. DNA Repair (Amst). 2005;4(2):149–61. 10.1016/j.dnarep.2004.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagaraju G, Odate S, Xie A, Scully R. Differential regulation of short- and long-tract gene conversion between sister chromatids by Rad51C. Mol Cell Biol. 2006;26(21):8075–86. 10.1128/MCB.01235-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandramouly G, Kwok A, Huang B, Willis NA, Xie A, Scully R. BRCA1 and CtIP suppress long-tract gene conversion between sister chromatids. Nat Commun. 2013;4:2404 Epub 2013/09/03. 10.1038/ncomms3404 ncomms3404 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, et al. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature. 2014;510(7506):556–9. 10.1038/nature13295 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brenneman MA, Wagener BM, Miller CA, Allen C, Nickoloff JA. XRCC3 controls the fidelity of homologous recombination: roles for XRCC3 in late stages of recombination. Molecular Cell. 2002;10(2):387–95. [DOI] [PubMed] [Google Scholar]
- 32.Tauchi H, Kobayashi J, Morishima K, van Gent DC, Shiraishi T, Verkaik NS, et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature. 2002;420(6911):93–8. 10.1038/nature01125 [DOI] [PubMed] [Google Scholar]
- 33.Saleh-Gohari N, Helleday T. Strand invasion involving short tract gene conversion is specifically suppressed in BRCA2-deficient hamster cells. Oncogene. 2004;23(56):9136–41. Epub 2004/10/14. 1208178 [pii] 10.1038/sj.onc.1208178 . [DOI] [PubMed] [Google Scholar]
- 34.Nagaraju G, Hartlerode A, Kwok A, Chandramouly G, Scully R. XRCC2 and XRCC3 regulate the balance between short- and long-tract gene conversion between sister chromatids. Mol Cell Biol. 2009. 10.1128/MCB.01406-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anand RP, Lovett ST, Haber JE. Break-induced DNA replication. Cold Spring Harb Perspect Biol. 2013;5(12). Epub 2013/07/25. 10.1101/cshperspect.a010397 a010397 [pii] cshperspect.a010397 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malkova A, Haber JE. Mutations arising during repair of chromosome breaks. Annual review of genetics. 2012;46:455–73. Epub 2012/11/14. 10.1146/annurev-genet-110711-155547 . [DOI] [PubMed] [Google Scholar]
- 37.Llorente B, Smith CE, Symington LS. Break-induced replication: what is it and what is it for? Cell Cycle. 2008;7(7):859–64. 10.4161/cc.7.7.5613 [DOI] [PubMed] [Google Scholar]
- 38.Donnianni RA, Symington LS. Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A. 2013;110(33):13475–80. Epub 2013/07/31. 10.1073/pnas.1309800110 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, et al. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502(7471):389–92. Epub 2013/09/13. 10.1038/nature12584 nature12584 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, et al. Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013;502(7471):393–6. Epub 2013/09/13. 10.1038/nature12585 nature12585 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447(7140):102–5. 10.1038/nature05723 [DOI] [PubMed] [Google Scholar]
- 42.Yim E, O'Connell KE, St Charles J, Petes TD. High-resolution mapping of two types of spontaneous mitotic gene conversion events in Saccharomyces cerevisiae. Genetics. 2014;198(1):181–92. 10.1534/genetics.114.167395 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson C, Jasin M. Coupled homologous and nonhomologous repair of a double-strand break preserves genomic integrity in mammalian cells. Molecular & Cellular Biology. 2000;20(23):9068–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, et al. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014;343(6166):88–91. 10.1126/science.1243211 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu AM, McVey M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. 2010;38(17):5706–17. 10.1093/nar/gkq379 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan SH, Yu AM, McVey M. Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS genetics. 2010;6(7):e1001005 10.1371/journal.pgen.1001005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie A, Hartlerode A, Stucki M, Odate S, Puget N, Kwok A, et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol Cell. 2007;28(6):1045–57. 10.1016/j.molcel.2007.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449(7161):478–82. 10.1038/nature06020 [DOI] [PubMed] [Google Scholar]
- 49.Xie A, Puget N, Shim I, Odate S, Jarzyna I, Bassing CH, et al. Control of sister chromatid recombination by histone H2AX. Mol Cell. 2004;16(6):1017–25. 10.1016/j.molcel.2004.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95(7):891–902. . [DOI] [PubMed] [Google Scholar]
- 51.Ferguson DO, Sekiguchi JM, Chang S, Frank KM, Gao Y, DePinho RA, et al. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci U S A. 2000;97(12):6630–3. 10.1073/pnas.110152897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nature structural & molecular biology. 2010;17(4):410–6. 10.1038/nsmb.1773 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88(2):265–75. [DOI] [PubMed] [Google Scholar]
- 54.Anand RP, Tsaponina O, Greenwell PW, Lee CS, Du W, Petes TD, et al. Chromosome rearrangements via template switching between diverged repeated sequences. Genes Dev. 2014;28(21):2394–406. 10.1101/gad.250258.114 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS genetics. 2009;5(1):e1000327 Epub 2009/01/31. 10.1371/journal.pgen.1000327 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131(7):1235–47. 10.1016/j.cell.2007.11.037 . [DOI] [PubMed] [Google Scholar]
- 57.McVey M, Adams M, Staeva-Vieira E, Sekelsky JJ. Evidence for multiple cycles of strand invasion during repair of double-strand gaps in Drosophila. Genetics. 2004;167(2):699–705. 10.1534/genetics.103.025411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sakofsky CJ, Ayyar S, Deem AK, Chung WH, Ira G, Malkova A. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol Cell. 2015;60(6):860–72. 10.1016/j.molcel.2015.10.041 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foster SS, Balestrini A, Petrini JH. Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol Cell Biol. 2011;31(21):4379–89. Epub 2011/08/31. 10.1128/MCB.05854-11 MCB.05854-11 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shima N, Munroe RJ, Schimenti JC. The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol Cell Biol. 2004;24(23):10381–9. 10.1128/MCB.24.23.10381-10389.2004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roerink SF, van Schendel R, Tijsterman M. Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome research. 2014;24(6):954–62. 10.1101/gr.170431.113 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yousefzadeh MJ, Wood RD. DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair (Amst). 2013;12(1):1–9. 10.1016/j.dnarep.2012.10.004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yousefzadeh MJ, Wyatt DW, Takata K, Mu Y, Hensley SC, Tomida J, et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS genetics. 2014;10(10):e1004654 10.1371/journal.pgen.1004654 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wyatt DW, Feng W, Conlin MP, Yousefzadeh MJ, Roberts SA, Mieczkowski P, et al. Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol Cell. 2016. 10.1016/j.molcel.2016.06.020 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, et al. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science. 2015;349(6249):742–7. 10.1126/science.aaa8391 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nature structural & molecular biology. 2009;16(8):814–8. 10.1038/nsmb.1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.