

Abstract
Pyoverdines are high affinity siderophores produced by a broad range of pseudomonads to enhance growth under iron deficiency. They are especially relevant for pathogenic and mutualistic strains that inhabit iron-limited environments. Pyoverdines are generated from non-ribosomally synthesized highly modified peptides. They all contain an aromatic chromophore that is formed in the periplasm by intramolecular cyclization steps. Although the cytoplasmic peptide synthesis and side-chain modifications are well characterized, the periplasmic maturation steps are far from understood. Out of five periplasmic enzymes, PvdM, PvdN, PvdO, PvdP, and PvdQ, functions have been attributed only to PvdP and PvdQ. The other three enzymes are also regarded as essential for siderophore biosynthesis. The structure of PvdN has been solved recently, but no function could be assigned. Here we present the first in-frame deletion of the PvdN-encoding gene. Unexpectedly, PvdN turned out to be required for a specific modification of pyoverdine, whereas the overall amount of fluorescent pyoverdines was not altered by the mutation. The mutant strain grew normally under iron-limiting conditions. Mass spectrometry identified the PvdN-dependent modification as a transformation of the N-terminal glutamic acid to a succinamide. We postulate a pathway for this transformation catalyzed by the enzyme PvdN, which is most likely functional in the case of all pyoverdines.
Keywords: decarboxylase, enzyme, iron, pyridoxal phosphate, siderophore, periplasmic tailoring, pyoverdines, siderophore maturation, periplasmic tailoring, Pseudomonas fluorescens
Introduction
Under aerobic conditions in the neutral pH range, iron can form insoluble FeIII oxide hydrates, limiting the amount of readily available iron. Therefore, many organisms produce siderophores that bind and thereby solubilize iron in their surroundings. A special group of these siderophores are the pyoverdines, yellow-green pigments that were first described in 1892 by Gessard (1). Turfitt (2, 3) used the production of pyoverdines for taxonomic classification of “fluorescent pseudomonads,” which include many important pathogenic as well as beneficial pseudomonads. Today it is known that pyoverdines are non-ribosomally synthesized, highly modified peptides whose biosynthesis and regulation involve more than 20 proteins (4). The cytoplasmic biosynthesis reactions are well established, but recently, the periplasmic maturation has gained interest. Of the five periplasmic enzymes, PvdM, PvdN, PvdO, PvdP, and PvdQ, which are found in all known pyoverdine-producing species, functions have been assigned so far only to PvdQ and PvdP, which are involved in a precursor deacylation step and the chromophore cyclization, respectively (5–8). PvdQ has been identified as a potential novel drug target (5). Based on interposon mutagenesis studies, PvdM, PvdN, and PvdO are all considered to be essential for the formation of functional pyoverdines (6, 9–11).
PvdN is translocated via the Tat2 system (11). As heterologously produced PvdN contains as prosthetic group a pyridoxal phosphate cofactor (PLP (12)), the Tat system may transport PvdN together with a bound PLP or a derivative thereof. However, no specific function in pyoverdine biogenesis could be attributed to PvdN.
Here we demonstrate that the enzyme PvdN specifically introduces a side-chain modification into the produced pyoverdine. The side-chain modification did not influence growth under iron-limiting conditions at different pH settings. Fluorescent pseudomonads usually produce more than one isoform of pyoverdine, which is exploited by a method known as siderotyping (13). This method utilizes the different pI values of siderophores, which include modified pyoverdine isoforms. Most of the modifications influencing the pI can be attributed to variations of a side chain at the 3-amino group of the chromophore (4, 14, 15). PvdN is responsible for such a modification. In contrast to results from earlier interposon mutagenesis studies that could not exclude polar effects (9, 10), a scar-less in-frame deletion of pvdN selectively abolished this modification without affecting the formation of functional pyoverdine. A mechanism for the catalyzed reaction of PvdN is proposed that attributes a key function to the bound cofactor. Lys-261 in the cofactor-binding pocket was essential for activity and translocation into the periplasm, suggesting that the Tat system has to translocate PvdN in an active, cofactor-containing conformation.
Results
Pseudomonas fluorescens A506 Produces a Typical Pyoverdine
Based on genetic analyses, it was recently proposed that P. fluorescens strain A506 produces a typical pyoverdine, which is expected to bind FeIII via six ligands provided by two hydroxamates of the peptide moiety and two oxo groups of the catechol fluorophore (Fig. 1A) (4). While the cytoplasmically generated peptide moiety varies in sequence, length, and cyclic/linear structure among the many known pyoverdines, the enzymes involved in the periplasmic maturation of this siderophore are highly conserved in all pyoverdine-producing species and strains, indicating that their activities are required for all known pyoverdines (Fig. 1B). We chose to study these aspects in the fully genome sequenced P. fluorescens strain A506, for which we have established versatile genetic tools such as genomic in-frame knock-outs and complementation vectors, as well as phenotypic assays such as pyoverdine formation analyses. For an initial chemical characterization of the pyoverdine produced by P. fluorescens A506 (referred to as PVDA506 throughout), we carried out the Csáky assay for the detection of hydroxamates (16), the Arnow reaction for the identification of the catechol functionality (17), and a ferric perchlorate assay to detect binding of iron at low pH values (18). Furthermore, we evaluated the expected influence of iron chelation by PVDA506 on spectral properties (19).
FIGURE 1.

Pyoverdine maturation by periplasmic enzymes and their genetic relatedness. A, postulated structure of PVDA506 (24), showing the fluorophore and the position of the modified N-terminal residue (R1), which are the regions where periplasmic maturation occurs. The three shown R1 side chains represent the three structures that are relevant in this study. B, conserved clustered organization of the pvdMNO operon and the pvdP gene, which encode the periplasmic pyoverdine maturation enzymes. PvdQ, which has also functions in other pathways, is not encoded in this cluster.
The fluorescence emission spectrum changed upon chelation of iron by PVDA506 (Fig. 2, A and B). Specifically, the maximum at ∼500 nm was quenched and the intensity of the major emission peak at ∼440 nm was reduced. The electronic absorption spectrum (Fig. 2C) showed a shift of a 405 nm maximum to 400 nm and the formation of a shoulder at 460 nm. PVDA506 reacted positively in the Csáky assay (16) that hydrolyzes the δ-N-formyl-δ-N-hydroxyornithines and thereby forms hydroxylamine that is detectable by a sensitive colorimetric assay (Fig. 2D), indicative for the presence of hydroxamates. This was expected for the δ-N-formyl-δ-N-hydroxyornithines of the siderophore. The Arnow assay (17) is based on the formation of a colored compound with a nitrite-molybdate reagent. As characteristic for pyoverdines (20), PVDA506 did not react positively in this assay despite the presence of a catechol group. As expected, PVDA506 did not show binding of iron in the ferric perchlorate assay (18), as the protonatable catechol is required for efficient binding (PVDA506 is not a Tris(hydroxamate) type siderophore that would react positively).
FIGURE 2.

Initial characterization of the pyoverdine PVDA506. A and B, fluorescence in the range of 350–800 nm of PVDA506 in culture supernatant from P. fluorescens A506, excited at 200–340 nm in the absence (A) or presence (B) of 1 mm FeCl3. AU, arbitrary units. C, electronic absorption spectra of the samples from A and B. D, electronic absorption spectrum of the Csáky assay (16), indicating the presence of hydroxamates. Fresh medium (CAA) was treated in the same manner as the culture supernatant (PVDA506).
The Gene Encoding PvdN Is Not Essential for Pyoverdine Formation
In previous mutagenesis studies, it was already noted that the observed essential phenotype of a pvdN interposon mutation could have been caused by polar effects within the pvdMNO operon (10). To examine the role of PvdN in PVDA506 biosynthesis, we therefore first needed to establish a scar- and marker-less in-frame deletion method for P. fluorescens A506. This was achieved by combining two methods (Fig. 3A) (21, 22). First, a deletion plasmid derived from the suicide plasmid pK18mobsacB (23) was constructed, containing ∼1 kbp of the left and right flanking regions of the target gene. Then, instead of conjugative transfer of the plasmid, we employed electroporation (22), which worked efficiently for P. fluorescens A506. Single crossover integrands were selected on the appropriate antibiotic. Double crossover mutants were selected by successive counter-selection on sucrose-containing medium, and scar- and marker-less deletions were identified and confirmed by PCR and sequencing. We included a deletion of monocistronic pvdP as control, as PvdP has been biochemically confirmed to be essential for the fluorophore formation (8).
FIGURE 3.

A scar- and marker-less in-frame deletion of pvdN in P. fluorescens A506 does not abolish formation of fluorescent pyoverdines. A, scheme for the scar- and marker-less in-frame gene deletion in P. fluorescens A506 (see “Results” for details). LF, left fragment; RF, right fragment; left fragment and right fragment are derived from the upstream or downstream sequences of the region to be deleted. B–E, droplet assays of 1) P. fluorescens A506 ΔpvdP, 2) P. fluorescens A506, 3) P. fluorescens A506 ΔpvdN, 4) P. fluorescens A506 ΔpvdN + pME6010-pvdN-strep, and 5) P. fluorescens A506 ΔpvdN + pME6010-pvdNK261A-strep. Strains were incubated either in the absence (B and C) or in the presence of EDDHA (D and E). The plates were imaged (B and D), and fluorescence was detected on a UV table (C and E).
The resulting ΔpvdN and ΔpvdP deletion strains were tested for growth and pyoverdine production on casamino acid (CAA) medium plates with or without the iron-depleting chelator ethylenediamine di(o-hydroxy)phenylacetic acid (EDDHA) (Fig. 3, B–E). As expected, the ΔpvdP control strain could not produce fluorescent pyoverdine and did not grow on EDDHA-containing medium. However, unlike what was expected from the earlier interposon mutagenesis studies that could not exclude polar effects in the operon (6, 10, 11), the scar-less ΔpvdN mutant strain was not impaired in pyoverdine formation, nor did the deletion affect growth on iron-depleted medium. In fact, the ΔpvdN strain showed the same phenotype as the WT strain on both media, producing approximately equivalent amounts of pyoverdine. Complementation vectors did not further increase the amount of pyoverdine released into the medium. We therefore further investigated the role of PvdN in pyoverdine formation.
PvdN Is Responsible for a Specific Modification of Pyoverdine
To initially assess the impact of the ΔpvdN mutation on pyoverdine formation, we utilized isoelectric focusing (IEF) gels in combination with a chrome azurol S (CAS) overlay assay (Fig. 4A) (13), which detects iron chelators that liberate iron from chrome azurol S, resulting in color reduction. The results revealed that PvdN is involved in the formation of a pyoverdine derivative that can be separated based on its specific pI. As it is known that multiple pyoverdine isoforms with varied side chains attached to the 3-amino group of the chromophore are generally produced in parallel (15), we could clearly attribute such a variation to the enzyme activity of PvdN.
FIGURE 4.

PvdN is responsible for a pyoverdine modification. A, isoelectric focusing indicates that wild type P. fluorescens A506 produces two pyoverdines, whereas the ΔpvdN mutant strain can only form one pyoverdine species (left gel). Besides the wild type and mutant strains, the IEF gel also shows the pyoverdines of strains with complementation vectors that contain either a wild type pvdN gene or a gene encoding the protein with a K261A mutation. Note the functional complementation by the wild type pvdN gene, whereas the exchange of the active site lysine completely abolishes the activity. The CAS overlay assay of the same IEF gel demonstrates the iron binding capacity of both pyoverdines (right gel). B, analysis of pyoverdine content in wild type P. fluorescens A506 and its ΔpvdN mutant. Left and right, reverse phase chromatography elutions (left) and corresponding mass spectra (right) of the succinamide and α-ketoglutarate forms of the pyoverdine (numbers correspond to structures in Fig. 1A). The elution profiles monitor the respective molecular masses during reverse phase chromatography, indicated beside the y-axes, showing that no succinamide is formed by the ΔpvdN mutant.
It was now important to exactly determine the nature of the chemical modification introduced by PvdN. We approached this aspect by UPLC mass spectrometry. Two components were identified in the wild type strain (Fig. 4B). Confirming the postulated structures (24), high resolution mass spectrometry revealed that one component had the mass of 1160.53 Da, which is the exact mass of the succinamide form of the pyoverdine, and the other had the mass of 1189.51 Da, which corresponds to the α-ketoglutarate. The glutamic acid that is initially formed in the periplasm is therefore virtually completely transformed into the two mentioned variants. In the ΔpvdN mutant, only the α-ketoglutarate form of pyoverdine was formed, indicating that the transformation to the succinamide was completely abolished.
Having demonstrated the nature of the chemical modification that depends on PvdN, we carried out complementation analyses to exclude that the missing modification in the ΔpvdN mutant was caused by additional unknown genetic changes. When expressed constitutively from a stable low copy vector, the recombinant pvdN gene resulted in full complementation of the phenotype, demonstrating that the phenotype was indeed exclusively caused by the lack of PvdN and no other genomic mutation (Fig. 4A). There were no new intermediates accumulating in the ΔpvdN mutant, indicating that PvdN most likely transforms the unmodified original glutamic acid residue directly to succinamide at the 3-amino position of the chromophore.
The Modified Pyoverdine Is Not Required for Iron-limited Growth under Specific pH Conditions
PvdN is conserved in all pyoverdine-producing bacteria, and the catalyzed modification must therefore somehow contribute to pyoverdine function. As the major function of pyoverdines is the acquisition of iron under iron limitation, and as we had already observed that the ΔpvdN mutant was able to grow under such conditions at neutral to slightly basic pH (Fig. 3, B–E), we thought about a potential function of the modification at distinct environmental pH conditions. The modification clearly increases the pI of the siderophore (Fig. 4A), and therefore could contribute to iron uptake under pH conditions near the pI of the non-modified pyoverdine and thereby help to ensure the iron supply in a larger pH range. However, this hypothesis turned out to be wrong. The PvdN-dependent modification was not required for growth under iron-limiting conditions under any pH condition tested, ranging from pH 6 to 9 (Fig. 5). Below pH 6, P. fluorescens A506 did not grow at all, irrespective of the iron content.
FIGURE 5.
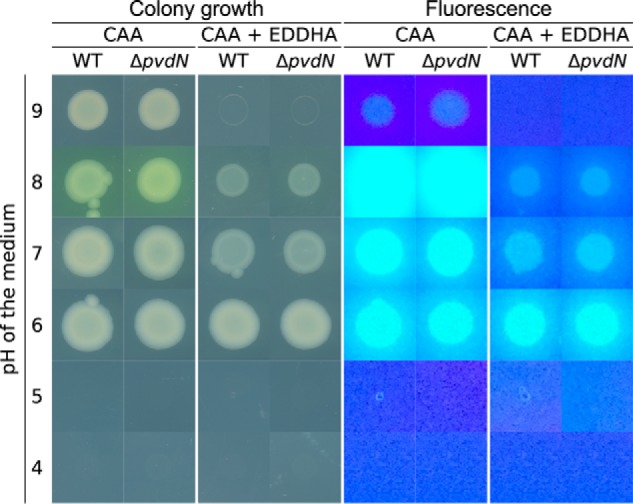
The ΔpvdN mutation does not affect iron acquisition under iron-limiting conditions at pH values that allow growth of strain A506. P. fluorescens (WT and ΔpvdN) was grown under iron-limiting (CAA) and iron starvation (CAA + EDDHA) conditions for 36 h at the indicated pH values, colonies were imaged (left), and the pyoverdine fluorescence was detected (right). Note that there is no detectable difference in growth and pyoverdine formation between the two strains.
PvdN Requires Cytoplasmic Cofactor Assembly for Folding and Tat Transport
As Tat-dependently translocated proteins can be translocated together with bound cofactors, we addressed this question with PvdN, which is believed to contain a PLP cofactor. In related PLP-containing enzymes, a lysine residue is conserved that plays a crucial role in cofactor binding (25). The corresponding residue in PvdN from P. fluorescens A506 is Lys-261. In the published Pseudomonas aeruginosa PvdN crystal structure, PLP had already formed an external aldimine with some substrate and therefore was non-covalently positioned next to the corresponding lysine residue (12). When we produced P. fluorescens A506 PvdN heterologously without its signal peptide in Escherichia coli, wild type PvdN could be obtained with a bound PLP cofactor, whereas a PvdN K261A variant was highly unstable and could not be analyzed, indicating that cofactor binding is likely to be highly important for folding (Fig. 6A). Transport was analyzed with the full-length proteins in P. fluorescens A506. The enzyme was only detectable in membrane and periplasmic fractions (Fig. 6B). While the wild type PvdN was clearly translocated into the periplasm, the PvdN K261A variant was not translocated anymore, and unprocessed full-length protein was detected exclusively in the membrane fraction instead, indicating that the absence of PLP causes structural characteristics that are no longer compatible with functional Tat transport.
FIGURE 6.
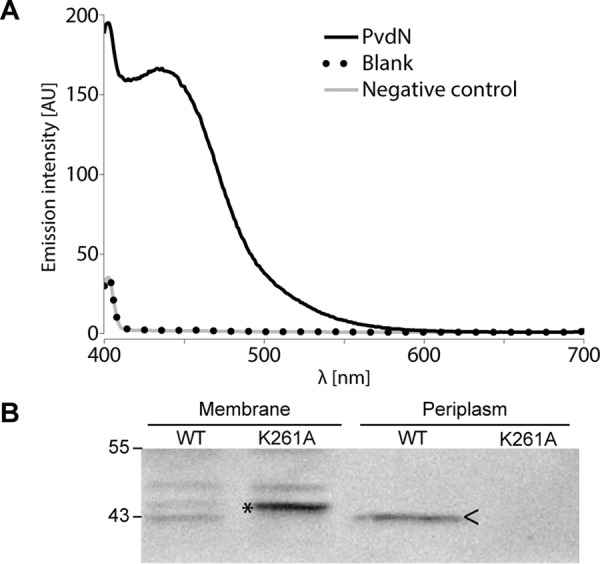
PLP binding is required for translocation of PvdN across the cytoplasmic membrane. A, UV-visible absorption-based PLP detection in PvdN as purified after heterologous production in E. coli ER2566/pEX-mat-pvdN-strep and subsequent transimination with ammonium hydroxide to produce a stable, well detectable PLP oxime (42). AU, arbitrary units. B, Strep-Tactin-HRP conjugate-mediated detection of C-terminally Strep-tagged PvdN or PvdN K261A in membrane and periplasmic fractions of P. fluorescens A506 containing either pME6010-pvdN-strep or pME6010-pvdNK261A-strep by SDS-PAGE/Western blotting. Unprocessed PvdN K261A full-length protein accumulates in membranes and shows abolished translocation, whereas the wild type protein is translocated into the periplasm. >, mature PvdN in the periplasmic fraction; *, full-length PvdN in the membrane fraction. Positions of molecular mass markers (in kDa) are indicated on the left.
Discussion
The periplasmic maturation of pyoverdines has been a mystery for many years. While significant progress has been made with PvdQ and PvdP (5, 8, 26), the function of any of the proteins encoded by the pvdMNO operon has remained unclear. As interposon mutagenesis suggested essential roles of these proteins for pyoverdine production, it was seemingly not possible to address the individual functions of these enzymes (6, 10, 11). Interestingly, we realized that an in-frame deletion of the pvdN gene caused a much milder phenotype than the interposon mutation, suggesting that polar effects might disturb the interpretation of the former approaches, which was already considered by the authors of the original deletion study (10). We were surprised to see that the scar-less ΔpvdN mutation did not reduce the formation and release of fluorescent pyoverdine (Fig. 3, B–E). The only phenotype of the pvdN deletion was the absence of one specifically modified pyoverdine, and this phenotype could be complemented by the single pvdN gene, proving that PvdN is indeed responsible for the modification (Fig. 4). In a more recent study on PvdP, it was already suggested that PvdN, besides PvdM and PvdO, can only be involved in side-chain modifications, as PvdP promotes all steps of chromophore formation (8). Our study now shows that this suggestion was indeed correct. Moreover, the modification that we can now attribute to PvdN is well known and has been observed in numerous structural studies on pyoverdines (reviewed in Ref. 4). Although α-ketoglutarate, succinamide, and succinate chains have been described as side chains in pyoverdines, the enzymes that catalyze their biogenesis starting from glutamic acid, have never been identified, and this study unravels the first of these conversions.
An open question remains the physiological purpose of this highly conserved modification of pyoverdines. Our experiments indicate that it is not the iron acquisition at any specific pH that requires the PvdN-dependent modification (Fig. 5). The amount of released pyoverdine is unaltered, and the affinity to iron seems to be unaffected as well, because the mutant grows equally well on EDDHA-containing iron-limited minimal medium as the wild type strain. The function of the modification might relate to specific environmental conditions or niches that cannot be mimicked in pure cultures and therefore escape our analyses. One could, for example, imagine that certain interactions with abiotic or biotic surfaces are mediated or enhanced by the modifications that somehow contribute to iron supply. Another potential function may relate to signaling pathways involved in quorum sensing or host interactions. Whatever the reason for the modification, its strong conservation among all pyoverdine-producing pseudomonads implies that it cannot be something dispensable under natural conditions.
A highly interesting side aspect of this study was the analysis of the transport of PvdN to the periplasmic space, as the data for the first time suggest a Tat-mediated cotransport of a PLP cofactor with a periplasmic enzyme. PvdN is known to be a Tat-dependently translocated protein (11), and the crystal structure of P. aeruginosa PvdN demonstrated an incorporated PLP cofactor (12). In Pseudomonas taetrolens, it has been demonstrated that PLP could be incorporated into a Sec-dependently translocated periplasmic amino acid racemase after translocation (27), and no PLP insertion into Tat-dependently translocated enzymes has been described so far. Why is PvdN transported by the Tat system if the cofactor could in principle also be incorporated after transport? PvdN requires the PLP-binding lysine residue for functional translocation into the periplasm (Fig. 6B), and our data indicate that PvdN is trapped in the membrane if this cofactor binding is affected in PvdN K261A. The speculation may be allowed that cofactor binding is required for correct folding. This is indeed strongly supported by the published crystal structure (12). PvdN forms a dimer, and the deeply buried cofactor that is located close to the subunit interface likely contributes to folding and stable interaction of the to-be-transported protein (12). The observation that the unprocessed form of PvdN K261A is detected in the membrane fraction may relate to the exposure of hydrophobic core regions, which are known to abolish Tat substrate translocation (28). If PvdN is translocated as a folded dimer, it contains two signal peptides that can mediate its translocation. Such homo-oligomeric Tat substrates are well known, and it is not yet understood how all signal peptides are removed after transport (29, 30).
Mechanistically, it is highly interesting to recognize that PvdN is able to catalyze the production of a succinamide chain at the N terminus of pyoverdines. The substrate of PvdN is likely to be the glutamic acid form of pyoverdine, which is produced by PvdQ after deacylation of the acylated ferribactin (5, 26) immediately after its translocation into the periplasm by PvdE (6). The ΔpvdN mutant still produces α-ketoglutaric acid, which therefore cannot be the product of a PvdN-catalyzed transamination, and because α-ketoglutaric acid cannot be transformed to succinamide by a PLP cofactor, it neither can be the substrate for PvdN (Fig. 7A). PvdN thus apparently catalyzes the oxidative decarboxylation of the glutamic acid to succinamide, a reaction for which the following mechanism can be proposed. As PvdN solely contains a PLP cofactor and no redox-active cofactors that could be involved (see Ref. 12), the situation is analogous to the CcbF-catalyzed PLP-dependent decarboxylation-coupled oxidative deamination (31). Unlike CcbF, an oxo group of the peroxo intermediate at the αC atom is retained, which can be readily explained by standard PLP chemistry, including proton abstraction at the αC atom and electron shuffling (Fig. 7B) (25). This would permit the direct formation of the amide of the decarboxylated amino acid, which would be to our knowledge the first description of such a reaction. The known crystal structure of PvdN strongly supports this hypothetical mechanism, as the deeply buried PLP-containing active site is connected to the surface via two channels. One is large enough to allow entering of the side chain, and the other, more narrow tunnel could permit the passage of CO2 and O2 (Fig. 7C). Future analyses will hopefully clarify the validity of the proposed mechanism, which might also be used in other biosynthetic pathways.
FIGURE 7.

Role and putative mechanism of PvdN catalysis in the biosynthesis of pyoverdines. A, branched modification pathway of the side chains at the N3-position of the pyoverdine fluorophore. B, likely mechanism of PvdN-catalyzed PLP-dependent oxidative decarboxylation under retention of the amine nitrogen. See “Results” for details. C, as expected for the proposed model, two channels connect the PLP-containing active site cavity with the surface. PLP atoms are colored to visualize the channels, which both lead to the reactive side of the cofactor. For this figure, PvdN of P. fluorescens A506 was homology-modeled using the available PvdN structure (PDB5i90; ∼62% sequence identity; global model quality estimation (GMQE) 0.79 (44–46)) as template. The image was generated using the VMD software (47).
Experimental Procedures
Strains and Growth Conditions
P. fluorescens A506 was used for physiological studies, E. coli DH5α λ pir+ was used for cloning, and E. coli ER2566 was used for expressions. P. fluorescens A506 was grown aerobically at 30 °C, and E. coli was grown at 37 °C in LB medium (1% (w/v) Tryptone, 1% (w/v) NaCl, 0.5% (w/v) yeast extract) in the presence of the appropriate antibiotics (100 μg/ml ampicillin, 50 μg/ml kanamycin, 20 μg/ml tetracycline). For subcellular fractionations, the strain was grown in King's B medium (2% (w/v) BD Bacto Proteose Peptone No. 3 (BD Biosciences), 1% (v/v) glycerol, 0.15% (w/v) K2HPO4, 0.15% (w/v) MgSO4).
For the production of pyoverdine, P. fluorescens A506 was grown aerobically in Erlenmeyer flasks with baffles in CAA medium (32) at 30 °C (5 g/liter casamino acids, 5 mm K2HPO4, 1 mm MgSO4 and, optionally, 1.5% (w/v) agar noble for plates). For assessment of the pyoverdine phenotype, 0.5 g/liter EDDHA were added to the medium. Growth was assessed after ∼36 h at 30 °C. Images of the plates were acquired with the Epson Perfection V850 Pro or the Epson Perfection V700 photo scanner (Epson, Meerbusch, Germany).
To assess the impact of pH on iron acquisition, we used CAA and CAA EDDHA agar supplemented with 100 mm of either of the following buffers: Bicine, pH 9.0, HEPES, pH 8.0, HEPES, pH 7.0, MES, pH 6.0, sodium acetate, pH 5.0 or pH 4.0. All buffers were prepared as 1 m stock solutions from the free acid/base, and the pH was adjusted as needed with HCl or NaOH. The growth was assessed after incubation for ∼36 h at 30 °C, and images were acquired as described before. In the above mentioned droplet plate assays, we used LB overnight cultures, which were washed twice with liquid CAA medium and then adjusted to an OD600 of 1.0, from which 10 μl were spotted on a plate.
Genetic Methods and Plasmids
For construction of the scar- and marker-less deletions in P. fluorescens A506, the plasmid pK18mobsacB (23) was utilized, according to a published protocol (21) with slight modifications. In particular, we used 3′- and 5′-flanking regions of ∼1 kbp in length for plasmid integration, and we employed overlap extension PCR (oePCR (33)) to connect both fragments. Furthermore, we avoided the use of conjugation and instead applied a published quick transformation procedure for pseudomonads (22). Additionally, we included an overnight culture step in 5 ml of LB medium after transformant selection on kanamycin plates, which were then diluted 1:1000 before plating 50 μl on 10% (w/v) sucrose counter-selection plates. All incubation steps were carried out at 30 °C. The deletions were confirmed by colony PCR and sequencing of the PCR product.
For complementation of the deletion strain, pvdN was pre-cloned via PCR into the vector pEXH5 (34), adding a Strep-tag with the reverse primer. From there, the gene was cloned into the plasmid pME6010 (35) including its ribosomal binding site from pEXH5 and adding an artificial terminator (International Genetically Engineered Machine (iGEM) part: BBa_B1006). For the construction of a K261A single-point mutation in pvdN, we employed oePCR (33) using the pEXH5-pvdN-strep plasmid as template, subsequently cloning the resulting pvdNK261A-strep fragment into the plasmid pEXH5 and from there into pME6010.
The sequence encoding the mature part of PvdN (starting at position 31, as predicted with the SignalP 4.1 Server (36)) was cloned into the plasmid pEXH5, resulting in pEX-mat-pvdN-strep for overproduction in E. coli ER2566. All primers are listed in Table 1. All plasmids were confirmed by restriction analyses and sequencing.
TABLE 1.
Primers used in this study
| Name | Sequence | Restriction site | Purpose |
|---|---|---|---|
| PfA506-PvdN-F1-MR | CTGTTCGGATCCCGAGCATGAGGTGCGCTATAAGATCATCTCC | BamHI | Forward primer for pvdN left flanking region |
| PfA506-PvdN-F2-MR | CTAACCACCACGAAGTAAGTCCATGACCCGGTTATGACCCCTTTTCCCTTTGAACGAGAC | Forward primer for pvdN right flanking region | |
| PfA506-PvdN-R1-MR | GGTCATGGACTTACTTCGTGGTGGTTAG | Reverse primer for pvdN left flanking region | |
| PfA506-PvdN-R2-MR | GCATCTAAGCTTTGGCCAAACAAGGCTGAACCCTGTAGGAG | HindIII | Reverse primer for pvdN right flanking region |
| PfA506-PvdN-DF-MR | AACGGCGATAACGCTCCGCACAAAC | pvdN genomic deletion control primer | |
| PfA506-PvdN-DR-MR | ACGACGTGCTCAAGACCGAGATTTC | pvdN genomic deletion control primer | |
| PfA506-PvdN-F-MR | TTCGACCATATGACCGACCGCCGTACATTTCTCAAGCAGG | NdeI | Forward primer for cloning PvdN-coding region into pEXH5 |
| PfA506-PvdN-strep-R-MR | CCGATTAAGCTTTTACTTTTCGAACTGCGGGTGGCTCCATAACCGCTGGCTCAGCAGGGTCATGAAG | HindIII | Reverse primer for cloning PvdN-coding region into pEXH5 |
| PfA506-PvdP-DF-MR | GTTGCAGTTCCTTGTCGCTGAGGTTG | pvdP genomic deletion control primer | |
| PfA506-PvdP-DR-MR | CCGGCCAGCATTTCTGGCTATTC | pvdP genomic deletion control primer | |
| PfA506-PvdP-F1-MR | TGCTCTAAGCTTTTGGTCGACATCTGCGACACGTCGATGATCAC | HindIII | Forward primer for pvdP left flanking region |
| PfA506-PvdP-F2-MR | ACGCTTTAAAACGTGTTGCAGGCGCTCTAGG | Forward primer for pvdP right flanking region | |
| PfA506-PvdP-R1-MR | GCGCCTGCAACACGTTTTAAAGCGTTGTCATTGGCTACCTTAGGAAACGGC | Reverse primer for pvdP left flanking region | |
| PfA506-PvdP-R2-MR | TTAAACCCCGGGAGGTACTGGGTAGCCTGTCCAGGCGTATCGACGAG | XmaI | Reverse primer for pvdP right flanking region |
| PfA506-PvdN_A261-F-MR | CTTCTTCATCGCTGGCACCCACGCGTGGATGTTCGGCCCGCGGGGCACCGGCC | Primer for K261A exchange by oePCR | |
| PfA506-PvdN_A261-R-MR | CGCGTGGGTGCCAGCGATGAAGAAGTCGCAGTG | Primer for K261A exchange by oePCR | |
| PfA506-matPvdN-F-MR | ATACGGCATATGGCCGCCACGAACGACCCATGGACGGGATTGAAACAAC | NdeI | Forward primer for cloning mature PvdN-coding region into pEXH5 |
| pEXH5-RBS-F-MR | GGCGCGGGATCCGTTTAACTTTAAGAAGGAGATATAC | EcoRI | Forward primer for subcloning from pEXH5 into pME6010 |
| pEXPT7-strep-term-R-MR | CCCCTTGAATTCAAAAAAAACCCCGCCCTGTCAGGGGCGGGGTTTTTTTTTTTACTTTTCGAACTGCGGGTGGCTCC | BamHI | Reverse primer for subcloning from pEXH5 into pME6010 |
Biochemical Methods
SDS-PAGE and subsequent Western blotting or Colloidal Coomassie staining were carried out by standard procedures (37–40). The Western blots were developed employing Strep-Tactin-HRP conjugate according to the manufacturer's instructions (IBA, Göttingen, Germany). Images of the Western blots were acquired utilizing the MF-ChemiBIS 4.2 imaging system (DNR Bio-Imaging Systems, Jerusalem, Israel).
For the overproduction of mature PvdN, the main culture was inoculated with 1% of its volume of an overnight pre-culture. The main culture was grown at 37 °C until it reached an optical density at 600 nm of ∼0.6 and was then induced with 1 mm isopropyl β-d-1-thiogalactopyranoside (from a 1 m stock solution) for 3 h. The cells (from a 1-liter culture) were pelleted at 5,000 × g for 30 min at 4 °C, resuspended in 25 ml of lysis buffer (100 mm Tris/HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA with freshly added 1 mm PMSF, 25 μg/ml DNase, and 1 mm DTT), and lysed with the French press (Thermo Fisher Scientific, Dreieich, Germany) at 20,000 p.s.i. cell pressure. The protein was then purified over 1.5-ml gravity flow Strep-Tactin® Sepharose columns according to the manufacturer's instructions (IBA). Subcellular fractionation was carried out according to established protocols (41) with slight modifications: 100-ml cultures were grown to OD600 of 1 and fractionated with 1-ml fractions.
Binding of PLP to PvdN was examined as described previously (42), with slight modifications. Briefly, the protein was purified as described above, but the cells where lysed in a buffer containing 1 mm PLP. After purification, the buffer was exchanged against 0.1 m potassium phosphate buffer, pH 7.2, employing 5-ml HiTrapTM desalting columns according to the manufacturer's instructions (GE Healthcare, Solingen, Germany). The protein concentration was evaluated with Roti®-Nanoquant according to the instruction manual (Carl Roth, Karlsruhe, Germany) using the SpectraMax M3 spectrophotometer (Molecular Devices, Biberach an der Riss, Germany). 0.5 mg of protein in 1.5 ml were treated with 5 mm hydroxylamine at 4 °C for ∼72 h, followed by filtration through a Vivaspin® 6 concentrator with a cutoff of 3 kDa (Sartorius, Göttingen, Germany). Subsequently, the fluorescence of the PLP oxime was detected at 446 nm with excitation at 353 nm employing the Jasco FP-6500 spectrofluorometer (Jasco, Gross-Umstadt, Germany).
Extraction of pyoverdine was performed with Amberlite XAD-4 resin as described previously (43) with minor modifications. Briefly, the culture was centrifuged at 20,000 × g at 4 °C for 30 min and subsequently sterile-filtered through 0.2-μm filters. The pH was adjusted to pH 6.0, and 20 g/liter XAD-4 were added. The mixture was incubated for 3 h at 4 °C under constant stirring. Thereafter, the XAD-4 resin was filtered and resuspended in half of the original volume of pure (>18 megaohms) water. The mixture was stirred for 1 h at 4 °C. Successively, the XAD-4 resin was filtered and resuspended in a fifth of the original volume of 15% (v/v) methanol and incubated at 4 °C under constant stirring for 15 min. Consequently, the XAD-4 resin was filtered again, resuspended in 15% of the original volume of 50% (v/v) methanol, and incubated at 4 °C under constant stirring for 1 h. Thereafter, the XAD-4 resin was removed by filtration, and the filtrate was reduced to dryness in vacuo, never heating the liquid above 25 °C. The dried extract was stored at 4 °C and then dissolved in pure water for MS or IEF analysis. The dissolved substance was stored at −20 °C.
LC-MS analysis was performed using a Q-Tof Premier mass spectrometer (Waters, Eschborn, Germany) equipped with a LockSprayTM unit, an electrospray ionization ion source (3-kV capillary voltage, 30-V sampling cone voltage, 250 °C nitrogen gas at a flow of 650 liters/h), and an ACQUITY UPLC (Waters). Separation was performed on a Waters ACQUITY UPLC HSS T3 column (1.8 μm, 2.1 × 100 mm) using the following linear gradient of solvent A (double-distilled water with 0.1% (v/v) formic acid) and solvent B (acetonitrile with 0.1% (v/v) formic acid) at a flow of 0.4 ml/min: 10% B (0 min), 90% B (10 min), 90% B (13.00 min), 10% B (13.10 min), 10% B (15 min).
To characterize the produced pyoverdines, we carried out the Csáky assay (16), the Arnow assay (17), and the ferric perchlorate assay as described previously (18). Furthermore, we recorded absorption and emission spectra with the Jasco V-650 spectrophotometer and the Jasco FP-6500 spectrofluorometer (Jasco), respectively, with and without the addition of 1 mm FeCl3. To qualitatively estimate the produced amount of pyoverdine, 3 ml of the culture were centrifuged at 16,000 × g for 2 min. 2 ml of the supernatant were adjusted to pH 8.0 by adding 50 mm HEPES, pH 8.0, from a 1 m stock solution. Thereafter, absorption spectra were recorded with the Jasco V-650 spectrophotometer (Jasco). All previously mentioned assays were performed on culture supernatant without further workup. To further characterize the pyoverdines, the XAD-4-extracted samples were subjected to IEF electrophoresis with subsequent chrome azurol S overlay detection as described previously (13) with minor modifications. Briefly, the sample amount was normalized to an absorption at 400 nm of ∼0.1, utilizing the NanoDrop 2000 spectrophotometer (Thermo Scientific). For IEF electrophoresis, we employed vertical precast SERVAGelTM IEF 3-10 gels with 12 wells (SERVA Electrophoresis, Heidelberg, Germany) together with an SE260 Mighty Small II Deluxe Mini vertical electrophoresis unit (Hoefer, Holliston, MA), an EPS 601 electrophoresis power supply (GE Healthcare), and a MultiTemp III cooling system (GE Healthcare). For IEF analysis, we used 35 μl of sample premixed with the appropriate loading buffer. The IEF gels were run with the cooling set to 4 °C with voltage settings according to the manufacturer's instructions, except that the electrophoresis time at 200 V was extended to 3 h. The gels were imaged immediately after running with the MF-ChemiBIS 4.2 imaging system (DNR Bio-Imaging Systems) with top UV lamps turned on. The CAS overlay solution was always prepared fresh from stock solutions, and 30 ml were poured into a 10 × 10-cm Petri dish (Sarstedt, Nümbrecht, Germany). When the CAS overlay solution had solidified, the IEF gels were wetted with pure water and placed on top of the cast gel. The incubation time was adjusted until bands were visible in the CAS overlay solution (∼5 min). The CAS assay was imaged with the same lamp settings as the IEF gels.
Author Contributions
M. T. R. performed the experiments, contributed to the preparation of the figures, and analyzed the data together with T. B. G. D. performed the MS analyses. T. B. conceived and coordinated the study, and T. B. and M. T. R. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Joyce E. Loper for kindly providing the strain P. fluorescens A506, Jens Boch for the kind gift of the plasmid pK18mobsacB, Guido V. Bloemberg for kindly providing the plasmid pME6010, and Andreas Kirschning for fruitful discussions. We thank Sybille Traupe for technical support.
This work was supported by the German Research Foundation (DFG) as a project of the GRK1798 “Signaling at the Plant-Soil Interface.” The authors declare that they have no conflicts of interest with the contents of this article.
- Tat
- twin-arginine translocation
- oePCR
- overlap extension polymerase chain reaction
- EDDHA
- ethylenediamine di(o-hydroxy)phenylacetic acid
- PLP
- pyridoxal phosphate
- IEF
- isoelectric focusing
- CAS
- chrome azurol S
- CAA
- casaminoacid
- UPLC
- ultra-performance liquid chromatography
- Bicine
- N,N-bis(2-hydroxyethyl)glycine.
References
- 1. Gessard M. C. (1892) Sur la fonction fluorescigène des microbes. Ann. Inst. Pasteur 4, 88–102 [Google Scholar]
- 2. Turfitt G. E. (1937) Bacteriological and biochemical relationships in the pyocyaneus-fluorescens group: Investigations on the green fluorescent pigment. Biochem. J. 31, 212–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Turfitt G. E. (1936) Bacteriological and biochemical relationships in the pyocyaneus-fluorescens group: The chromogenic function in relation to classification. Biochem. J. 30, 1323–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cézard C., Farvacques N., and Sonnet P. (2015) Chemistry and biology of pyoverdines, Pseudomonas primary siderophores. Curr. Med. Chem. 22, 165–186 [DOI] [PubMed] [Google Scholar]
- 5. Drake E. J., and Gulick A. M. (2011) Structural characterization and high-throughput screening of inhibitors of PvdQ, an NTN hydrolase involved in pyoverdine synthesis. ACS Chem. Biol. 6, 1277–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yeterian E., Martin L. W., Guillon L., Journet L., Lamont I. L., and Schalk I. J. (2010) Synthesis of the siderophore pyoverdine in Pseudomonas aeruginosa involves a periplasmic maturation. Amino Acids 38, 1447–1459 [DOI] [PubMed] [Google Scholar]
- 7. Hannauer M., Schäfer M., Hoegy F., Gizzi P., Wehrung P., Mislin G. L. A., Budzikiewicz H., and Schalk I. J. (2012) Biosynthesis of the pyoverdine siderophore of Pseudomonas aeruginosa involves precursors with a myristic or a myristoleic acid chain. FEBS Lett. 586, 96–101 [DOI] [PubMed] [Google Scholar]
- 8. Nadal-Jimenez P., Koch G., Reis C. R., Muntendam R., Raj H., Jeronimus-Stratingh C. M., Cool R. H., and Quax W. J. (2014) PvdP is a tyrosinase that drives maturation of the pyoverdine chromophore in Pseudomonas aeruginosa. J. Bacteriol. 196, 2681–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ochsner U. A., Wilderman P. J., Vasil A. I., and Vasil M. L. (2002) GeneChip® expression analysis of the iron starvation response in Pseudomonas aeruginosa: identification of novel pyoverdine biosynthesis genes. Mol. Microbiol. 45, 1277–1287 [DOI] [PubMed] [Google Scholar]
- 10. Lamont I. L., and Martin L. W. (2003) Identification and characterization of novel pyoverdine synthesis genes in Pseudomonas aeruginosa. Microbiology 149, 833–842 [DOI] [PubMed] [Google Scholar]
- 11. Voulhoux R., Filloux A., and Schalk I. J. (2006) Pyoverdine-mediated iron uptake in Pseudomonas aeruginosa: the Tat system is required for PvdN but not for FpvA transport. J. Bacteriol. 188, 3317–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Drake E. J., and Gulick A. M. (2016) 1.2 Å resolution crystal structure of the periplasmic aminotransferase PvdN from Pseudomonas aeruginosa. Acta Crystallogr. F Struct. Biol. Commun. 72, 403–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koedam N., Wittouck E., Gaballa A., Gillis A., Höfte M., and Cornelis P. (1994) Detection and differentiation of microbial siderophores by isoelectric focusing and chrome azurol S overlay. Biometals 7, 287–291 [DOI] [PubMed] [Google Scholar]
- 14. Meyer J. M. (2000) Pyoverdines: pigments, siderophores and potential taxonomic markers of fluorescent Pseudomonas species. Arch. Microbiol. 174, 135–142 [DOI] [PubMed] [Google Scholar]
- 15. Schäfer H., Taraz K., and Budzikiewicz H. (1991) [On the genesis of the dicarboxylic acids bound amidically to the chromophor of the pyoverdins] Z. Naturforsch. C 46, 398–406 [Google Scholar]
- 16. Csáky T. Z., Hassel O., Rosenberg T., Lång S., Turunen E., and Tuhkanen A. (1948) On the estimation of bound hydroxylamine in biological materials. Acta Chem. Scand. 2, 450–454 [Google Scholar]
- 17. Arnow L. E. (1937) Colorimetric determination of the components of 3,4-dihydroxyphenylalanine-tyrosine mixtures. J. Biol. Chem. 118, 531–537 [Google Scholar]
- 18. Payne S. M. (1994) Detection, isolation, and characterization of siderophores. Methods Enzymol. 235, 329–344 [DOI] [PubMed] [Google Scholar]
- 19. Meyer J. M., and Abdallah M. A. (1978) The fluorescent pigment of Pseudomonas fluorescens: biosynthesis, purification and physicochemical properties. J. Gen. Microbiol. 107, 319–328 [Google Scholar]
- 20. Cox C. D., and Adams P. (1985) Siderophore activity of pyoverdin for Pseudomonas aeruginosa. Infect. Immun. 48, 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kvitko B. H., and Collmer A. (2011) Construction of Pseudomonas syringae pv. tomato DC3000 mutant and polymutant strains. Methods Mol. Biol. 712, 109–128 [DOI] [PubMed] [Google Scholar]
- 22. Choi K.-H., Kumar A., and Schweizer H. P. (2006) A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397 [DOI] [PubMed] [Google Scholar]
- 23. Schäfer A., Tauch A., Jäger W., Kalinowski J., Thierbach G., and Pühler A. (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145, 69–73 [DOI] [PubMed] [Google Scholar]
- 24. Hartney S. L., Mazurier S., Girard M. K., Mehnaz S., Davis E. W. 2nd, Gross H., Lemanceau P., and Loper J. E. (2013) Ferric-pyoverdine recognition by Fpv outer membrane proteins of Pseudomonas protegens Pf-5. J. Bacteriol. 195, 765–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jansonius J. N. (1998) Structure, evolution and action of vitamin B6-dependent enzymes. Curr. Opin. Struct. Biol. 8, 759–769 [DOI] [PubMed] [Google Scholar]
- 26. Nadal Jimenez P., Koch G., Papaioannou E., Wahjudi M., Krzeslak J., Coenye T., Cool R. H., and Quax W. J. (2010) Role of PvdQ in Pseudomonas aeruginosa virulence under iron-limiting conditions. Microbiology 156, 49–59 [DOI] [PubMed] [Google Scholar]
- 27. Matsui D., Oikawa T., Arakawa N., Osumi S., Lausberg F., Stäbler N., Freudl R., and Eggeling L. (2009) A periplasmic, pyridoxal-5′-phosphate-dependent amino acid racemase in Pseudomonas taetrolens. Appl. Microbiol. Biotechnol. 83, 1045–1054 [DOI] [PubMed] [Google Scholar]
- 28. Richter S., Lindenstrauss U., Lücke C., Bayliss R., and Brüser T. (2007) Functional Tat transport of unstructured, small, hydrophilic proteins. J. Biol. Chem. 282, 33257–33264 [DOI] [PubMed] [Google Scholar]
- 29. Nurizzo D., Halbig D., Sprenger G. A., and Baker E. N. (2001) Crystal structures of the precursor form of glucose-fructose oxidoreductase from Zymomonas mobilis and its complexes with bound ligands. Biochemistry 40, 13857–13867 [DOI] [PubMed] [Google Scholar]
- 30. Ma X., and Cline K. (2010) Multiple precursor proteins bind individual Tat receptor complexes and are collectively transported. EMBO J. 29, 1477–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang M., Zhao Q., Zhang Q., and Liu W. (2016) Differences in PLP-dependent cysteinyl processing lead to diverse S-functionalization of lincosamide antibiotics. J. Am. Chem. Soc. 138, 6348–6351 [DOI] [PubMed] [Google Scholar]
- 32. Ochsner U. A., Snyder A., Vasil A. I., and Vasil M. L. (2002) Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 99, 8312–8317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Higuchi R., Krummel B., and Saiki R. (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16, 7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brüser T., Deutzmann R., and Dahl C. (1998) Evidence against the double-arginine motif as the only determinant for protein translocation by a novel Sec-independent pathway in Escherichia coli. FEMS Microbiol. Lett. 164, 329–336 [DOI] [PubMed] [Google Scholar]
- 35. Heeb S., Itoh Y., Nishijyo T., Schnider U., Keel C., Wade J., Walsh U., O'Gara F., and Haas D. (2000) Small, stable shuttle vectors based on the minimal pVS1 replicon for use in gram-negative, plant-associated bacteria. Mol. Plant Microbe Interact. 13, 232–237 [DOI] [PubMed] [Google Scholar]
- 36. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 37. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 38. Burnette W. (1981) “Western Blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112, 195–203 [DOI] [PubMed] [Google Scholar]
- 39. Towbin H., Staehelin T., and Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Neuhoff V., Arold N., Taube D., and Ehrhardt W. (1988) Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9, 255–262 [DOI] [PubMed] [Google Scholar]
- 41. Ize B., Viarre V., and Voulhoux R. (2014) Cell fractionation. Methods Mol. Biol. 1149, 185–191 [DOI] [PubMed] [Google Scholar]
- 42. Ojha S., Wu J., LoBrutto R., and Banerjee R. (2002) Effects of heme ligand mutations including a pathogenic variant, H65R, on the properties of human cystathionine β-synthase. Biochemistry 41, 4649–4654 [DOI] [PubMed] [Google Scholar]
- 43. Meyer J. M., Stintzi A., De Vos D., Cornelis P., Tappe R., Taraz K., and Budzikiewicz H. (1997) Use of siderophores to type pseudomonads: the three Pseudomonas aeruginosa pyoverdine systems. Microbiology 143, 35–43 [DOI] [PubMed] [Google Scholar]
- 44. Benkert P., Biasini M., and Schwede T. (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arnold K., Bordoli L., Kopp J., and Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 47. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]