

Abstract
Activation of the blood vessel endothelium is a critical step during inflammation. Endothelial cells stimulated by pro-inflammatory cytokines play an essential part in the adhesion and extravasation of circulating leukocytes into inflamed tissues. The endothelial egfl7 gene (VE-statin) represses endothelial cell activation in tumors, and prior observations suggested that it could also participate in the regulation of endothelial cell activation during inflammation. We show here that Egfl7 expression is strongly repressed in mouse lung endothelial cells during LPS- and TNFα-induced inflammation in vivo. LPS have a limited effect on Egfl7 expression by endothelial cells in vitro, whereas the pro-inflammatory cytokine TNFα strongly represses Egfl7 expression in endothelial cells. TNFα regulates the egfl7 gene promoter through regions located between −7585 and −5550 bp ahead of the main transcription start site and via an NF-κB-dependent mechanism. Conversely, Egfl7 regulates the response of endothelial cells to TNFα by restraining the induced expression of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin, resulting in a decreased adhesion of leukocytes onto endothelial cells stimulated by TNFα. Egfl7 regulates the expression of these adhesion molecules through the NF-κB and MEK/Erk pathways, in particular by preventing the proteasome-mediated degradation of IkBα both in non-activated endothelial cells and during activation. Egfl7 is thus an endogenous and constitutive repressor of blood vessel endothelial cell activation in normal and inflammatory conditions and participates in a loop of regulation of activation of these cells by pro-inflammatory cytokines.
Keywords: adhesion, endothelium, inflammation, leukocyte, NF-kappa B (NF-KB), vascular biology
Introduction
Endothelial cells line the luminal side of blood vessels in direct contact with the circulation. Under normal conditions, the endothelium forms a non-adhesive and non-thrombogenic surface on which blood cells slide with minimal interactions with the vascular wall. During inflammation, endothelial cells become activated in response to pro-inflammatory cytokines, which promote a strong increase in the expression levels of leukocyte adhesion molecules such as E-selectin, intercellular adhesion molecule-1 (ICAM-1),4 and vascular cell adhesion molecule-1 (VCAM-1). These adhesion molecules are expressed at the endothelial cell surface and participate in the rolling, arrest, firm adhesion, and extravasation of immune cells from the circulation through the endothelium and toward the tissues (1). Endothelial cell activation in response to inflammation and its subsequent capture of leukocytes is thus a vital response, and its regulation is critical. Excessive or aberrant local activation of endothelial cells leads to inflammatory disorders such as atherosclerosis, chronic inflammation, multiple sclerosis, and rheumatoid arthritis. Activation of the endothelium is transitory, and endothelial cells resume a basal, non-activated condition when pro-inflammatory cytokines levels recess. Thus, most of the time, the endothelium is non-activated, and there are now several lines of evidence suggesting that this condition depends on the active expression of endogenous genes, which, when repressed, spontaneously trigger endothelial cell activation. Epidermal growth factor-like domain 7 (egfl7 or VE-statin) is mainly expressed by endothelial cells during embryonic development and in the adult. Egfl7 codes for a secreted protein that represses smooth muscle cell migration, regulates elastogenesis (2, 3), and is essential to blood vessel lumen formation during development (4–6). We have previously shown that the ectopic expression of Egfl7 by cancer cells reduces the expression of leukocyte adhesion molecules in tumor blood vessels and favors tumor escape from immunity (7) and that high expression levels of Egfl7 correlate with low endothelial cell activation in peritumoral vessels of human breast cancer (8). Egfl7 was also shown to inhibit ICAM-1 expression in response to injuries such as hypoxia/reoxygenation (9) and calcineurin inhibition (10) in human coronary endothelial cells. These observations were made in situations where the endothelium was severely altered (cancer) or chemically injured and suggested that Egfl7 could possibly regulate the endothelial activation during inflammation, but the exact roles of Egfl7 in this process have not been studied. Furthermore, there is currently no report on the regulation of Egfl7 expression during endothelial cell activation in response to pro-inflammatory stimuli.
Here, we show that Egfl7 participates in the regulation of endothelial cell activation during inflammation. Egfl7 expression is transitorily reduced under LPS- and TNFα-induced inflammatory conditions in vivo and in endothelial cells treated with pro-inflammatory cytokines in vitro. TNFα represses egfl7 gene transcription in endothelial cells via the NF-κB pathway. Conversely, Egfl7 represses the TNFα-induced activation of endothelial cells and adhesion of leukocytes, notably by limiting the expression of ICAM-1, VCAM-1, and E-selectin through the repression of the NF-κB and the MEK/Erk pathways. Egfl7 participates in the stabilization of IκBα and inhibits its degradation by the proteasome.
Results
Egfl7 Is Repressed in Endothelial Cells in Inflammatory Conditions in Vivo and in Vitro
Egfl7 is mainly expressed by blood vessel endothelial cells during development and in the adult (2, 6, 11). Comparing in situ hybridization of Egfl7 and immunostaining of CD31 in parallel slides of normal mouse lungs, Egfl7 expression was observed mostly in CD31+ endothelial cells (Fig. 1A). When lungs were dissociated and cells purified by immunoaffinity against cell surface CD31, expression of Egfl7 was detected mostly in the CD31+ enriched fraction (Fig. 1B), confirming that in normal lungs, CD31+ endothelial cells represent the main cell type that expresses Egfl7. To check whether Egfl7 expression was regulated during inflammation, LPS were instilled in mouse lungs as to produce an acute and transitory inflammatory condition. The efficacy of this LPS treatment in inducing lung endothelium activation was confirmed by the observed 3.5- and 2-fold up-regulation of ICAM-1 and VCAM-1 RNA expression levels after 10 h of LPS treatment, respectively (Fig. 1C). The expression levels of E-selectin were up-regulated 250-fold after 10h of stimulation by LPS. All levels were back to almost basal values after 24 h. During this LPS treatment, Egfl7 transcript levels decreased 75% after 10 h when compared with PBS controls and resumed values close to controls after 24 h (Fig. 1C), thus showing a simultaneous and inverse regulation of expression of Egfl7 when compared with leukocyte adhesion molecules. To check whether the down-regulation of Egfl7 could be due to a direct effect of LPS on endothelial cells, primary human umbilical vein endothelial cells (HUVEC) cultured in vitro were treated with increasing amounts of LPS and expression of Egfl7 assessed. The most active dose of LPS (0.1 μg/ml for 4 h) induced a 20% decrease in Egfl7 transcript levels (Fig. 1D), and a time course treatment of HUVEC with that same dose showed a maximal 20% reduction in expression of Egfl7 after 4 and 8 h (Fig. 1D). This indicated that in lung tissues inflamed using LPS, the much stronger repression of Egfl7 observed was probably not due to a direct effect of the LPS on endothelial cells. Because an LPS treatment in vivo induces the release of TNFα and pro-inflammatory interleukins in tissues (12, 13), we then checked whether TNFα could regulate the expression of Egfl7 in vivo. Instillation of TNFα in mice induced a strong increase in ICAM-1, VCAM-1, and E-selectin expression levels after 10 h, confirming activation of the lung endothelium (Fig. 1E). Under these conditions, TNFα induced a 50% decrease in Egfl7 expression after 10 h and 69% after 24 h when compared with PBS controls at the same time points (Fig. 1E).
FIGURE 1.
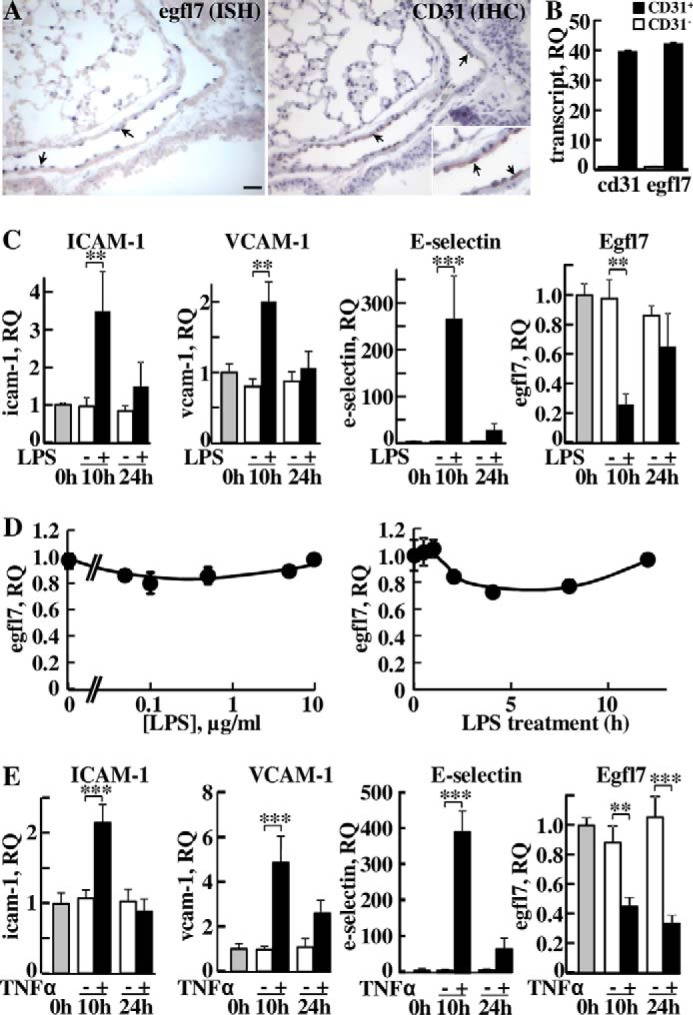
Egfl7 is repressed in endothelial cells under inflammatory conditions in vivo. A, left panel, in situ hybridization detection of Egfl7 transcripts in endothelial cell nuclei of adult mouse lungs (blue staining, arrows). Right panel and inset, CD31 immunostaining (brown, arrows) and hematoxylin counterstaining of a parallel section of the same area. Bar, 25 μm. B, expression levels of CD31 and Egfl7 transcripts in CD31− cells (white bars) and CD31+ cells (black bars) isolated from mouse lungs using immunoaffinity and measured by duplex RT-qPCR using a mouse CD31-FAM or a mouse Egfl7-FAM TaqMan probe mixed with a mouse β-actin-VIC probe (see “Experimental Procedures”). The results are plotted as quantities relative to CD31− controls values set to 1. RQ, relative quantities. C, LPS (5 mg/kg, +) or LPS-free PBS (−) was instilled in mice nostrils, and animals were sacrificed at the onset of treatment (0 h) or after 10 or 24 h; the lungs were dissected and processed for total RNA isolation. Expression levels of ICAM-1, VCAM-1, E-selectin, and Egfl7 were measured by duplex RT-qPCR using the indicated FAM-labeled TaqMan probe for the mouse transcript of interest and a mouse β-actin-VIC-labeled TaqMan probe and expressed as 2−ΔΔCT quantities relative to t = 0 h values set to 1. *, p < 0.05; **, p < 0.01; ***, p < 0,001. The results are representative of three experiments performed in triplicate. RQ, relative quantities. D, left panel, HUVEC were treated with increasing doses of LPS for 4 h and Egfl7 transcript levels assessed by duplex RT-qPCR. Right panel, expression levels of Egfl7 transcripts in primary HUVEC cells treated with 0.1 μg/ml of LPS for the indicated length of time and assessed by duplex RT-qPCR. E, TNFα (0.25 mg/kg, +) or LPS-free PBS (−) was instilled in mice nostrils and lungs processed as in C for the analysis of expression levels of ICAM-1, VCAM-1, E-selectin, and Egfl7 expressed as relative to t = 0 values set to 1. RQ, relative quantities. **, p < 0.01; ***, p < 0,001.
In vitro, a time course treatment of endothelial cells with TNFα showed a 60% decrease of the levels of Egfl7 transcripts after 6 h of stimulation; these levels were back to control values after 24 h (Fig. 2A). Treating endothelial cells with the other pro-inflammatory cytokine IL1β resulted in a similar but prolonged response while treating with IL6 had no significant effects on Egfl7 expression when compared with controls (Fig. 2B). Accordingly, upon TNFα treatment, Egfl7 protein levels in endothelial cells decreased at 8 h and thereafter when compared with non-stimulated cells (Fig. 2C), and this decrease in expression was also detected by immunofluorescence of Egfl7 in endothelial cells treated with TNFα (Fig. 2D). Because TNFα and IL1β are active angiogenic factors in vivo (14, 15), we tested whether Egfl7 could be regulated by other angiogenic factors such as FGF-2 and VEGF-A165, but neither factor induced significant variations in the expression levels of Egfl7, at any of the concentration tested (Fig. 2E).
FIGURE 2.
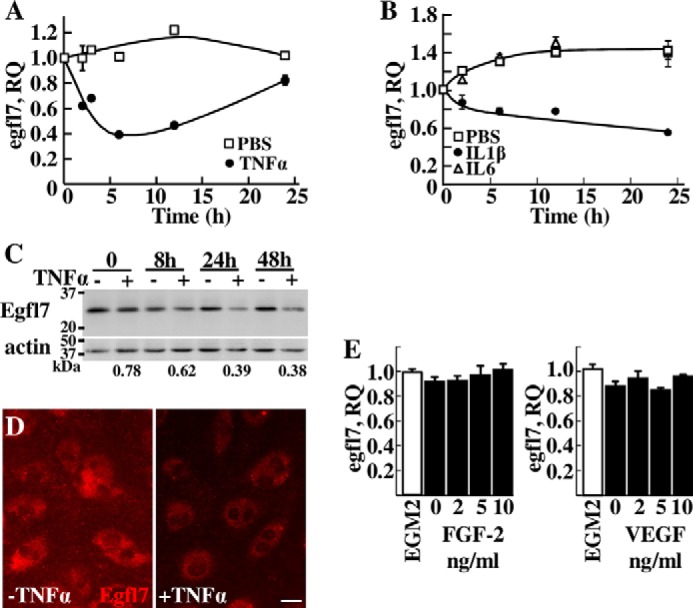
Expression of Egfl7 in endothelial cells is repressed by pro-inflammatory cytokines. A and B, expression levels of Egfl7 measured by duplex RT-qPCR in HUVEC treated with PBS (□) or with 10 ng/ml TNFα (●) (A) for the indicated length of time or with PBS (□), 10 ng/ml IL1β (●), or 10 ng/ml IL6 (▵) (B). The results are representative of three experiments performed in triplicate. C, Western blotting analysis of endogenous Egfl7 or of actin in HUVEC treated with TNFα as in A for the indicated length of time. The numbers below indicate the TNFα treated/non-treated ratio of Egfl7 protein levels normalized to actin levels taken at the same time points and assessed by densitometry. The results are representative of two experiments. D, immunostaining of Egfl7 in confluent HUVEC monolayers treated for 4 h with or without TNFα. Bar, 25 μm. E, expression levels of Egfl7 measured by duplex RT-qPCR in HUVEC treated for 24 h with complete medium (EGM-2) or in basal medium supplemented with the indicated amounts of FGF-2 or VEGF-A165. The results are representative of two experiments performed in triplicate.
TNFα Represses the Transcription of the egfl7 Gene
Egfl7 repression by TNFα was not due to a shorter half-life of its mRNA because the kinetics of decay of the Egfl7 transcripts were similar in HUVEC treated or not with TNFα prior to blocking transcription (Fig. 3A). On the other hand, the decrease in Egfl7 transcript levels induced by TNFα depended on active transcription because it was abolished by treating the cells with actinomycin D before TNFα stimulation (Fig. 3B). Similar results were obtained when treating endothelial cells with IL1β (not shown).
FIGURE 3.
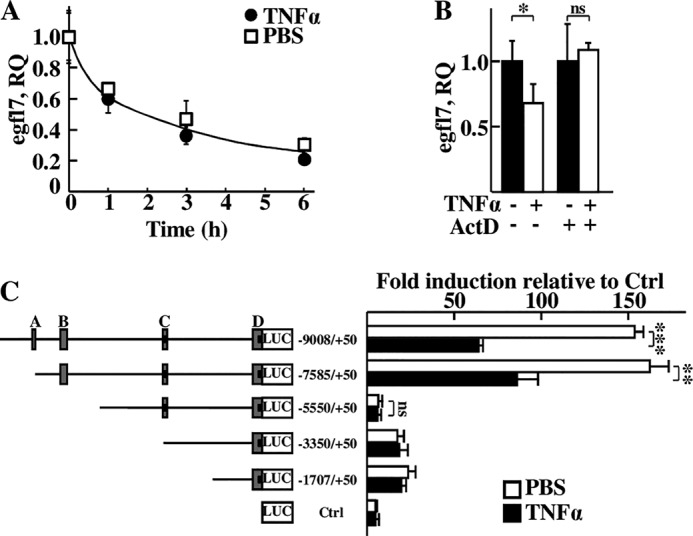
Egfl7 expression in endothelial cells is regulated by TNFα at the transcriptional level. A, Egfl7 transcript levels measured by duplex RT-qPCR in confluent HUVEC treated with 10 ng/ml TNFα (●) or with PBS (□) for 1 h and then with 10 μg/ml actinomycin D (ActD, t = 0) and assessed during the next 6 h. B, Egfl7 transcripts levels measured by duplex RT-qPCR in confluent HUVEC treated with DMSO or 10 μg/ml actinomycin D for 1 h before stimulation with or without 10 ng/ml TNFα for 6 h. *, p < 0.05; **, ns, non-significant. C, luciferase activities measured in HUVEC transfected with pGL3basic (Ctrl) or with the indicated reporter constructs containing fragments of the human egfl7 gene promoter and with the pCMV-β-Gal normalizing vector. The cells were then treated with 10 ng/ml TNFα (black bars) or with PBS (white bars) and lysed 18 h later. The letters correspond to conserved promoter regions (16). The numbers indicate the base position relative to the exon 1b transcription initiation site (2). Activities were normalized with β-galactosidase values, folds of induction were calculated using pGL3basic values as reference; the results are representative of three experiments performed in triplicate. **, p < 0.01; ***, p < 0.001; ns, non-significant.
These observations suggested that the egfl7 gene promoter was regulated when treating endothelial cells with TNFα. To address this point, several successive deletion reporter vectors based on the location of conserved regions between the mouse and human egfl7 gene promoters (16) were cloned and transfected in HUVEC, and cells were then stimulated or not with TNFα. The −9008/+50 and −7585/+50 promoter regions were highly active in endothelial cells (Fig. 3C), and treating with TNFα reduced the activity of these fragments by 58 and 46%, respectively. Deletion of the fragment encompassing region B resulted in a loss of sensitivity of the constructs to TNFα: the −5550/+50Luc reporter showed an almost complete lack of sensitivity to TNFα (6% difference versus non-treated) together with a large decrease in global reporter activity when compared with the longer constructs. Further deletions showed no significant differences of activity upon treatment with TNFα either, as seen in shorter mutants (Fig. 3C), which nevertheless maintained a detectable luciferase activity, like in the mouse egfl7 promoter (16).
TNFα and IL1, which repress Egfl7, are known potent inducers of the nuclear factor-κB (NF-κB) pathway in HUVEC (17, 18). On the other hand, IL6, which does not the activate the NF-κB pathway in endothelial cells in vitro, including in HUVEC (17, 19, 20), has no effects on Egfl7 expression. We thus suspected that the NF-κB pathway could be involved in the regulation of egfl7 gene expression by TNFα in these cells. Indeed, treating HUVEC with the NF-κB inhibitor BAY117085 prevented the repression of egfl7 induced by TNFα (Fig. 4A), and transfecting cells with the constitutively active NF-κB super-repressor Iκ-Bα S32/36A (21) relieved the repression of the −7585/+50Luc construct induced by TNFα (Fig. 4B).
FIGURE 4.
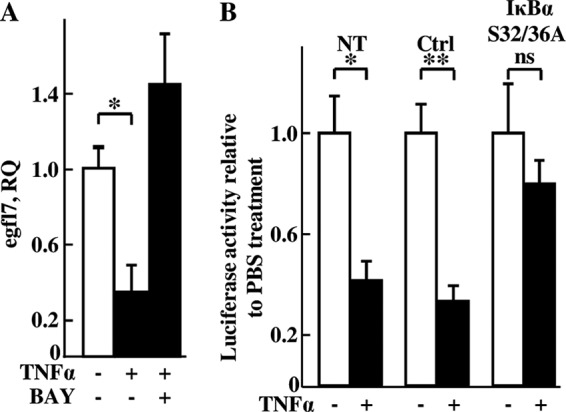
Egfl7 repression by TNFα is mediated via the NF-κB pathway. A, confluent HUVEC were cultured in the presence or not of 5 ng/ml TNFα and of 10 μm of the NF-κB inhibitor BAY117085 (BAY) for 6 h, after which RNA were isolated and Egfl7 transcripts were quantified by duplex RT-qPCR. *, p < 0.05. B, HUVEC were transfected with pGL3basic (Ctrl) or with the −7585/+50Luc construct in the absence (NT) or presence of pRC-CMV (Ctrl) or of pRC-CMV-IκBα S32/36A vector and with pCMV-β-Gal. 24 h later, the cells were treated with 10 ng/ml TNFα (+, black bars) or with PBS (−, white bars) and lysed after 18 h, and luciferase and β-galactosidase activities were measured in cell extracts. The results are representative of two experiments performed in triplicate. *, p < 0.05; **, p < 0.01; ns, non-significant.
TNFα-induced Endothelial Cell Activation Is Repressed by Endogenous Egfl7
Altogether, the results above show that Egfl7 expression is down-regulated under inflammatory conditions in vivo and in vitro by a direct regulation of its gene in endothelial cells. We had previously observed that repressing Egfl7 could activate endothelial cells in the absence of inflammatory cytokines (7). In such conditions, repressing Egfl7 increased the expression levels of ICAM-1, VCAM-1, and E-selectin transcripts and the number of T-lymphocytes spontaneously adhering onto endothelial cells. This added to the present observations suggested that the proper activation of endothelial cells during inflammation may require the simultaneous repression of Egfl7 in these cells.
To address this point, we set up RNA interference conditions using two different siRNA which specifically down-regulated Egfl7 expression without affecting that of miR126-3p and miR126-5p (Fig. 5A). This point was particularly important because miR126-3p and miR126-5p are embedded within the egfl7 gene seventh intron, and both are known to affect endothelial cell activation (22, 23). The effects of siEgfl7 in HUVEC could be rescued by overexpressing Egfl7 by the means of an expression plasmid. This plasmid induced an ∼6-fold overexpression of Egfl7 over the endogenous Egfl7 transcript and abolished the effects of siEgfl7 (Fig. 5B). Importantly, overexpression of Egfl7 in these conditions did not affect the expression levels of miR126-3p nor those of miR126-5p (Fig. 5B).
FIGURE 5.
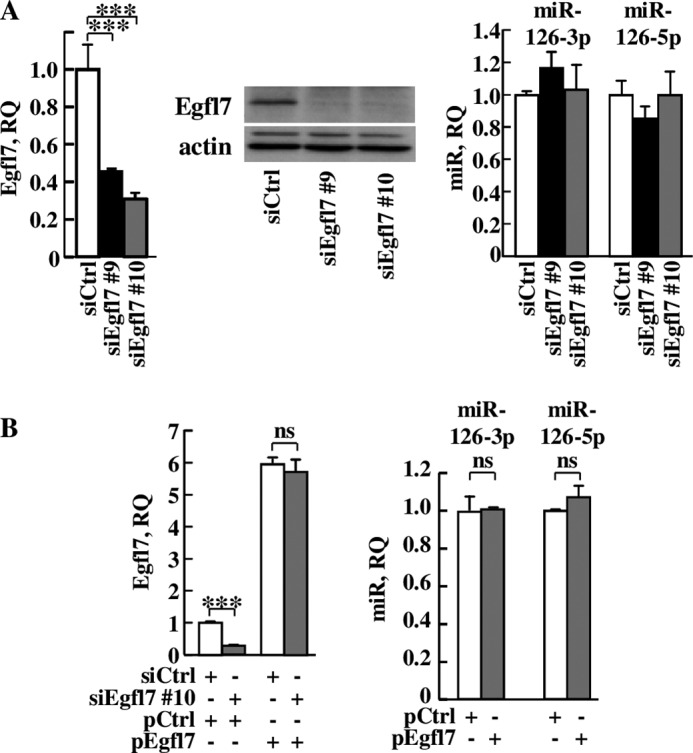
Specific targeting and rescue of egfl7 do not affect expression of the miR-126 locus. A, HUVEC were transfected with siCtrl (white) or with two different siRNA targeting Egfl7: siEgfl7 #9 (black) or siEgfl7 #10 (gray) and cultured for 4 days. Left panel, total RNA were prepared and analyzed by duplex RT-qPCR for Egfl7 expression. ***, p < 0.001. Middle panel, protein extracts were prepared and analyzed for the presence of Egfl7 or actin by 12% SDS-PAGE and Western blotting. Right panel, HUVEC were treated as in A, and analysis of miR126-3p and miR126-5p expression was performed by RT-qPCR, using the U6 snRNA as normalizer. B, HUVEC were transfected with siCtrl or siEgfl7#10 as in A and then with pcDNA3 (pCtrl) or a pcDNA3-human Egfl7 expression plasmid (pEgfl7). Left panel, RT-qPCR analysis of Egfl7 expression on triplicate experimental samples. Right panel, RT-qPCR analysis of expression of miR126-3p and of miR126-5p in pCtrl- or in pEgfl7-transfected HUVEC on triplicate experimental samples. ***, p < 0.001; ns, non-significant.
Repression of Egfl7 in endothelial cells by RNA interference followed by TNFα stimulation resulted in a strongly exacerbated activation of the cells: expression levels of ICAM-1 doubled on average at each time point in TNFα-stimulated conditions when compared with control, reaching the highest values after 6 h (Fig. 6A). VCAM-1 expression levels increased 50% over control after 6 h of stimulation when Egfl7 was repressed (Fig. 6A). After 2 h, the levels of expression of E-selectin were not significantly higher than controls when Egfl7 was repressed. However, the peak of expression of E-selectin lasted longer, reaching its maximal values after 6 h in the siEgfl7 condition when expression levels had already dropped by half in the siCtrl condition. To confirm the specific role of Egfl7 in these observations, expression of Egfl7 was rescued using pEgfl7 in HUVEC treated with TNFα for 6 h. Of note, the Egfl7 rescue was as effective in PBS-treated HUVEC as in TNFα-treated HUVEC, thus allowing comparison of the conditions (Fig. 6B). In the pEgfl7-rescued conditions, the expression levels of ICAM-1, VCAM-1, and E-selectin dropped back almost to siCtrl values in all cases (Fig. 6B), therefore confirming that the effects obtained with siEgfl7 were indeed due to the repression of Egfl7 itself. In good correlation with these results, overexpression of Egfl7 using pEgfl7 also cancelled the stimulating effect of TNFα on the adhesion of leukocytes to the endothelial cells monolayer (Fig. 6C), altogether showing that Egfl7 is actually an efficient endogenous repressor of endothelial activation that can counteract the endothelial cell biological response to inflammatory cytokines.
FIGURE 6.

Egfl7 represses the activation of endothelial cells by TNFα. A, expression levels of ICAM-1 (left panel), VCAM-1 (middle panel), and E-selectin (right panel) measured using duplex RT-qPCR in HUVEC transfected with siCtrl (□) or siEgfl7 (●) and stimulated 3 days later with TNFα. The results are representative of two experiments performed in triplicate. At time 0, the levels of Egfl7, ICAM-1, VCAM-1, and E-selectin in the siEgfl7 condition relative to siCtrl condition were 0.31 ± 0.01, 3.18 ± 0.23, 1.00 ± 0.10, and 2.14 ± 0.29, respectively. B, left panel, HUVEC were transfected with pCtrl or pEgfl7 and treated or not for 6 h with TNFα and expression levels of Egfl7 assessed by RT-qPCR on triplicate experimental samples. The numbers indicate the pEgfl7/pCtrl ratio of Egfl7 expression. ***, p < 0.001. Right three panels, HUVEC were transfected with siCtrl or siEgfl7, rescued with pCtrl or pEgfl7, and stimulated or not 2 days later with TNFα for 6 h. Expression levels of ICAM-1, VCAM-1, and E-selectin were assessed by duplex RT-qPCR on triplicate experimental samples. *, p < 0.05; ***, p < 0.001. C, left panel, adhesion of fluorescently labeled Jurkat T-lymphocytes plated onto monolayers of HUVEC transfected 2 days earlier with pCtrl or pEgfl7 and stimulated or not with TNFα. The results are representative of two experiments performed in triplicate. Right panel, mean numbers of adherent fluorescent cells counted over 10 microscopic fields in each condition. *, p < 0.05; ns, non-significant
Egfl7 Regulates the Expression of Leukocyte Adhesion Molecules through the NF-κB and MEK/Erk Pathways
Pro-inflammatory cytokines promote the expression of adhesion molecules by endothelial cells mainly through the activation of the NF-κB pathway (18). Treating HUVEC with TNFα in our conditions increased the expression levels of ICAM-1, VCAM-1, and E-selectin and this effect was abolished in the presence of the inhibitor of NF-κB activation BAY117085 (24) (not shown). Because Egfl7 was previously shown to repress NF-κB nuclear translocation in response to hypoxia/reoxygenation and NF-κB DNA binding in response to calcineurin treatment (9, 10), we then assessed whether Egfl7 would also repress endothelial activation through this signaling pathway. Treating endothelial cells with the NF-κB inhibitor BAY117085 suppressed the increase in T-cell adhesion onto endothelial cells transfected with siEgfl7 (Fig. 7A). Treatment with the inhibitor also cancelled the stimulating effects of siEgfl7 on VCAM-1 and E-selectin expression but not on ICAM-1 expression (Fig. 7B).
FIGURE 7.

Egfl7 regulates the activation of endothelial cells through the NF-κB and MEK/Erk pathways. A, left panel, microscopic fields of fluorescent Jurkat T-lymphocytes adhering onto HUVEC transfected 3 days earlier with siCtrl or siEgfl7. DMSO or BAY117085 (BAY, 10 μm) was added 20 h before plating T-lymphocytes. Right panel, mean numbers of adherent Jurkat T-lymphocytes counted in 15 non-overlapping microscope fields. *, p < 0.05. B, expression levels of ICAM-1, VCAM-1, and E-selectin transcripts measured using duplex RT-qPCR in HUVEC transfected with siCtrl or siEgfl7 and treated with DMSO or BAY117085 (10 μm). The results are representative of three experiments performed in triplicate. C, left panel, microscopic fields of fluorescent T-lymphocytes adhering onto HUVEC transfected 3 days earlier with siCtrl or siEgfl7. DMSO or U0126 (10 μm) was added 20 h before plating T-lymphocytes. Right panel, mean numbers of adherent Jurkat T-lymphocytes counted in 15 non-overlapping microscope fields. The results are representative of two experiments performed in triplicate. *, p < 0.05. D, expression levels of ICAM-1, VCAM-1, and E-selectin measured using duplex RT-qPCR in HUVEC transfected with siCtrl or siEgfl7 and treated with DMSO or 10 μm of the MEK1/2 inhibitor U0126 for 16 h. The results are representative of two experiments performed in triplicate. *, p < 0.05.
The MAPK/Erk pathway is also known to regulate endothelial cell activation (25–27). Treating HUVEC with the MEK/Erk inhibitor U0126 increased T-cell adhesion onto endothelial cells and cancelled the promoting effects of siEgfl7 on leukocyte adhesion (Fig. 7C). This treatment cancelled the effects of the siEgfl7 on expression of ICAM-1, VCAM-1, and E-selectin (Fig. 7D), thus showing that in endothelial cells, Egfl7 constitutively represses ICAM-1, VCAM-1, and E-selectin through the MEK/Erk pathway.
Egfl7 Represses the Degradation of IkBα in Endothelial Cells
To more precisely identify the mechanisms by which Egfl7 regulates the NF-κB pathway during inflammation, we then performed a detailed analysis of activation of this pathway during TNFα stimulation of endothelial cells. Briefly, in non-stimulated cells, dimers of NF-κB/Rel transcription factors are complexed with the inhibitory IκB protein as inactive factors in the cytosol. Upon TNFα stimulation, phosphorylation of the IκB kinase (IKK) complex, which contains the IKKα and IKKβ catalytic subunits, activates this IKK complex, which in turn phosphorylates IκBα on Ser32 and Ser36. This signal promotes the ubiquitin-mediated proteasome-dependent degradation of IκBα. NF-κB dimers are thus released and translocate into the nucleus. During this process, NF-κB p65 phosphorylation at Ser536 promotes nuclear localization and transcriptional activity (28). When Egfl7 was down-regulated using siEgfl7 and endothelial cells treated with TNFα, the phosphorylation of NF-κB p65 on Ser536, taken as an indicator of NF-κB activation, was detected earlier (5min) and lasted longer (up to 2 h) than in cells transfected with siCtrl (Fig. 8A). Quantification of the signals showed that the phosphorylation levels of NFκB p65 on Ser536 were higher at almost every time point in cells transfected with siEgfl7 than with siCtrl, including without stimulation (time 0; Fig. 8B). This confirmed that Egfl7 repressed the NF-κB pathway both in normal conditions and during activation of endothelial cells. The hyperactivation of the NF-κB pathway mediated by Egfl7 repression was not due to a difference in activation of the IKK complex because total and phosphorylation levels of IKKα and IKKβ were not noticeably different between cells treated with siCtrl and siEgfl7 (Fig. 9). Regarding IκBα phosphorylation, the kinetics under TNFα treatment were slightly different as phosphorylation was detected sooner (5 min) in siEgfl7 condition than siCtrl (Fig. 10A, P-IκBα (Ser32)). The most remarkable effect observed was a large reduction in total IκBα protein levels in cells where Egfl7 expression had been repressed, both without treatment (time 0) and during early TNFα treatment (Fig. 10A, Total IκBα). The repression of Egfl7 also cancelled the neo-synthesis of IκBα observed after 2–4 h of treatment with TNFα in the siCtrl condition. The effects of Egfl7 on IκBα protein levels were not due to variations in the expression levels of the IκBα transcripts because they were not significantly different between cells expressing Egfl7 or not (not shown). Upon its phosphorylation, the stability of IκBα is dependent on its ubiquitination and subsequent degradation by the proteasome (28), and the previous observations suggested that Egfl7 could affect the stability of IκBα in endothelial cells. Indeed, when HUVEC were treated with the proteasome inhibitor MG132 after transfection with siEgfl7 but prior to stimulation with TNFα, the levels of IκBα were partially restored (Fig. 10B). They were also higher during stimulation by TNFα, thus showing that in endothelial cells, Egfl7 constitutively represses the activation of the NF-κB pathway by preventing the degradation of IκBα by the proteasome.
FIGURE 8.
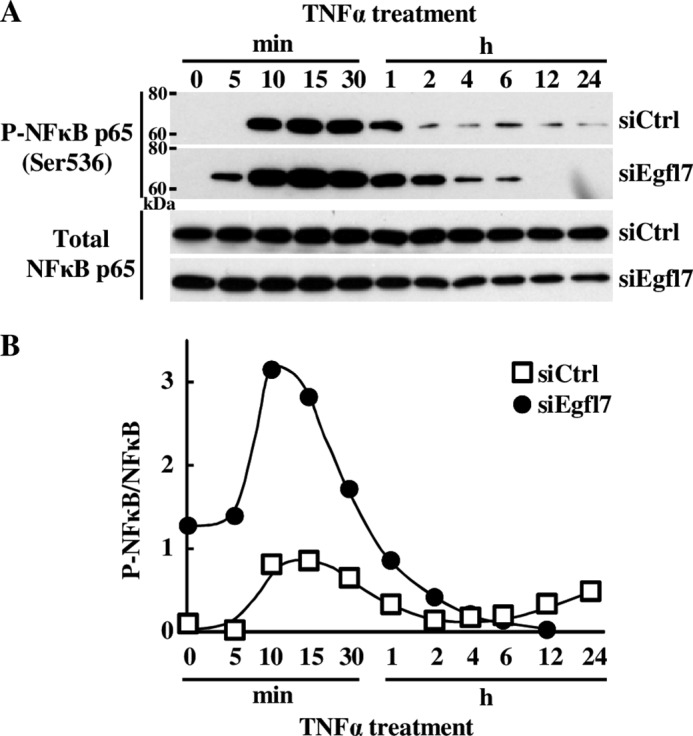
Egfl7 prevents the activation of the NF-κB pathway in endothelial cells. A, HUVEC were transfected with siCtrl or siEgfl7, treated 4 days later with 10 ng/ml TNFα, and lysed at the indicated time points. The cell extracts were prepared, and proteins (10–20 μg) were analyzed by Western blotting using antibodies against the indicated proteins (left). The results are representative of three experiments. B, membranes were exposed to a Las3000 system, and intensities were quantified using the Multigauge V3.0 software.
FIGURE 9.
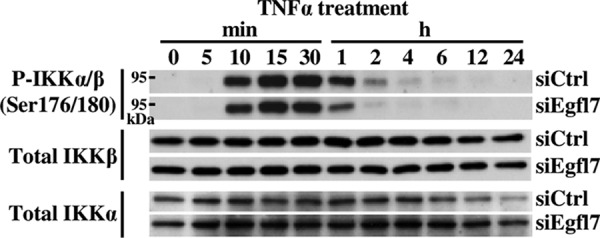
Egfl7 does not affect the IKK complex. HUVEC were transfected with siCtrl or siEgfl7, treated 4 days later with 10 ng/ml TNFα, and lysed at the indicated time points. Cell extracts were prepared, and proteins (10–20 μg) were analyzed by Western blotting using antibodies against the indicated proteins (left). The results are representative of three experiments.
FIGURE 10.
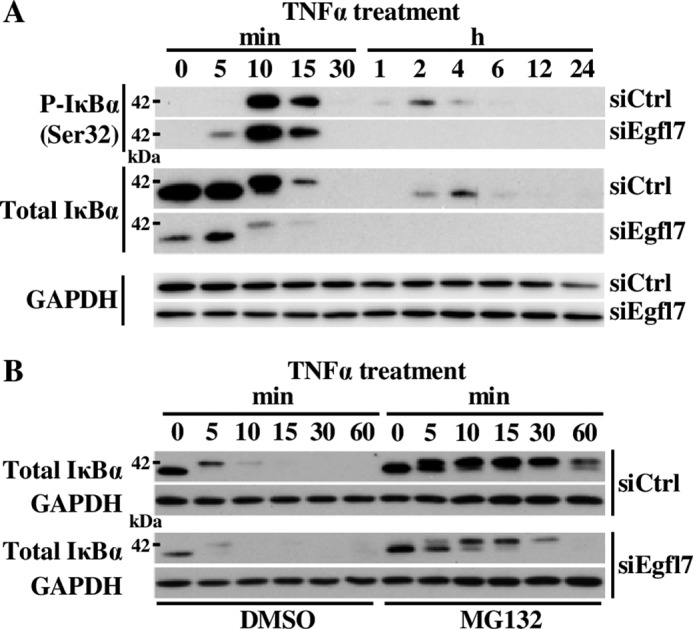
Egfl7 regulates the degradation of IκBα by the proteasome. A, HUVEC were transfected with siCtrl or siEgfl7, treated 4 days later with 10 ng/ml TNFα, and lysed at the indicated time points. The cell extracts were prepared, and proteins (10–20 μg) were analyzed by Western blotting using antibodies against the indicated proteins (left). The results are representative of three experiments. B, HUVEC were transfected with siCtrl or siEgfl7 and treated 4 days later with the proteasome inhibitor MG132 for 2 h prior to adding 10 ng/ml TNFα for the indicated time. The cells were lysed, and proteins (5 μg) were analyzed by Western blotting against total IκBα or GAPDH. The results are representative of two experiments.
Discussion
We show here that Egfl7 participates in a loop of regulation of endothelial cell activation during inflammation: in normal conditions, Egfl7 expressed by endothelial cells maintains high levels of IκBα, thus constitutively repressing the NF-κB pathway and activation of the cells. During inflammation and following stimulation by pro-inflammatory cytokines, Egfl7 is repressed, and endothelial cell activation in response to TNFα is favored (Fig. 11). Whether the repression of Egfl7 during LPS- or TNFα-induced activation is a prerequisite to a proper cell response in vivo is, however, not clear yet. Timewise, in lungs of LPS- or TNFα-treated animals, Egfl7 transcripts are repressed at the same time as leukocyte adhesion molecules are induced, with optimal variations at 6 h of treatment (Fig. 1, C and E). Similarly, in HUVEC treated with TNFα, Egfl7 transcripts are down-regulated following the same timing as the increase in leukocyte adhesion molecules (Figs. 2A and 6A). These observations suggest that the down-regulation of Egfl7, which occurs at the earlier steps of endothelial cell activation rather than during the resolution of vascular activation (29), is needed for proper activation of the cells. The functional effects of Egfl7 also suggest that its repression is needed for proper cell activation during inflammation, because Egfl7 represses the effects of TNFα on leukocyte adhesion (Fig. 6C). These observations suggest that the down-regulation of Egfl7 during the earlier steps of vascular activation is needed for a more effective activation of the cells in response to inflammation.
FIGURE 11.

Egfl7 participates in a loop of regulation of endothelial activation during inflammation. In normal, unstimulated conditions (left panel), Egfl7 constitutively expressed by endothelial cells represses IκBα phosphorylation and degradation, thus promoting the accumulation of inactive NF-κB dimers complexed to IκBα. Egfl7 contributes to low expression levels of leukocyte adhesion molecules and low cell activation. Upon TNFα stimulation (right panel), activation of the NF-κB pathway leads to the repression of Egfl7, which participates in the destabilization of IκBα and triggers endothelial cell activation.
The molecular mechanisms that maintain the endothelium in a non-activated state in normal conditions are not well described. There is a very limited number of known endogenous repressors of endothelial cell activation such as Egfl7, and most of them have been identified as regulators of cytokine stimulation. Repression of the sphingosine-1 P2 receptor in HUVEC endothelial cells alters their activation by TNFα (30). The cannabinoid-2 receptors expressed by coronary artery endothelial cells also inhibit their activation after treatment with TNFα (31). The G-protein-coupled receptor-30 expressed by endothelial cells represses ICAM-1 and VCAM-1 in response to TNFα (32). A NO donor prevents the activation of endothelial cells by TNFα, whereas a NO synthase inhibitor activates these cells (33). Among these, the Erg factor is of particular interest in regard to the role of Egfl7 in this process. Erg is a member of the ETS family of transcription factors, which is mainly expressed by endothelial cells (34, 35). Erg maintains endothelial cells in a non-activated state by constitutively repressing NF-κB activity and ICAM-1 expression after TNFα stimulation (36, 37). Erg also reduces the expression of inflammatory cytokines such as interleukin-8 (38), and its down-regulation leads to spontaneous neutrophil attachment to non-stimulated endothelial cells. Furthermore and like Egfl7, Erg expression is down-regulated in endothelial cells stimulated by TNFα (38). Thus, most of the functions described for Egfl7 here are shared with the Erg transcription factor. This, added to the fact that Erg was shown to directly regulate the egfl7 gene promoter in endothelial cells (16), strongly suggests that the reported effects of Erg on endothelial activation are mediated, at least in part, by its target gene egfl7.
The factors that regulate the expression of Egfl7 in endothelial cells are not known. Egfl7 expression has been described in several physiological and pathological conditions in vivo. It is almost exclusively expressed by endothelial cells during embryonic development (2, 6, 11, 39). Its expression is reduced in adult blood vessels (6, 40) and is up-regulated during active angiogenesis or after arterial injury such as ballooning or a chemical insult in rat (40). Egfl7 is also up-regulated in neonatal rat brains in response to hypoxia (41) and repressed in lungs after hyperoxic exposure (42). We found here that the expression of Egfl7 is down-regulated by TNFα and IL1β in vivo and in endothelial cells, and this is the first instance that describes the regulation of the gene in endothelial cells stimulated by specific exogenous factors. In addition to FGF-2 and VEGF165, we have tested other growth and angiogenic factors such as HGF, TGFβ, or PDGF, none of which significantly regulated Egfl7 expression in endothelial cells (not shown). Aside from egfl7 and the erg gene mentioned above, several other genes expressed by endothelial cells are down-regulated upon TNFα treatment, such as the endothelial variant HoxA9EC (43) or the del-1 gene, which product antagonizes integrin-mediated firm adhesion of leukocytes to the endothelium (44, 45). Microarray analyses performed on endothelial cells treated or not with TNFα identified a number of other such genes (46, 47), but most of them now require experimental confirmation and functional studies.
We addressed here the role of Egfl7 as an endogenously produced molecule in endothelial cells and showed that it represses cell activation in the context of inflammation. Previously, Badiwala et al. (10) reported that treating human coronary artery endothelial cells stimulated by cyclosporin A or tacrolimus with exogenously added recombinant Egfl7 repressed neutrophil adhesion to these cells, decreased NF-κB DNA-binding, and lowered ICAM-1 expression. We had also reported earlier that an Egfl7-producing tumor cell-conditioned medium prevented the adhesion of T-cells to the endothelial cells (7). Although the stimuli and conditions used in these different studies were quite different, they showed similar activities for Egfl7 whether it was produced directly by endothelial cells or whether it was added to their medium. Because Egfl7 bears a signal peptide and is a secreted molecule (2), these observations suggest that Egfl7 produced and secreted by endothelial cells may act as an autocrine factor regulating their own activation and response to inflammatory cytokines. Although there is no reported specific receptor for Egfl7, the protein was shown to inhibit the Notch pathway and to down-regulate the levels of expression of Notch target genes when overexpressed in endothelial cells in vivo (48). Egfl7 was also found to activate Notch, depending on the environment and the experimental conditions (49, 50). Whether Egfl7 produced by endothelial cells regulates their activation through Notch is not known, but it is interesting to note that the Notch pathway has been itself linked to the regulation of activation of endothelial cells: the knockdown of Notch-4 in arterial endothelial cells enhances the expression of VCAM-1 in the absence or presence of a pro-inflammatory stimulus (51). Furthermore, Notch-1 induces the expression of VCAM-1 by endothelial cells in the absence of inflammatory cytokines and potentiates the IL1β-dependent VCAM-1 up-regulation by interacting with the NF-kB pathway (52).
VCAM-1 and E-selectin repression by Egfl7 is mediated through the NF-κB and the MEK/Erk pathways, whereas ICAM-1 repression seems solely dependent on the MEK/Erk pathway. Although ICAM-1, VCAM-1, and E-selectin are concomitantly overexpressed upon activation of endothelial cells by pro-inflammatory cytokines such as TNFα, it was already known that their regulation of expression were actually not strictly similar. For example, histone acetylase inhibitors block TNFα-induced expression of VCAM-1 and leukocyte adhesion but do not affect ICAM-1 and E-selectin expression (53). Shear stress increases ICAM-1 expression but decreases VCAM-1 expression induced by TNFα in HUVEC (54). Treatment of HUVEC with the CD40 ligand increases expression of VCAM-1 but does not affect expression of ICAM-1, whereas co-stimulation with IL4 and CD40 ligand enhances the expression of P-selectin and VCAM-1 but inhibits that of ICAM-1 and E-selectin. The involvement of the MEK/Erk pathway in the regulation of ICAM-1 and VCAM-1 expression in endothelial cells remains quite controversial (26). In correlation with our observations, it is interesting to note that overexpression of Erk1 or Erk2 in HUVEC repressed ICAM-1 without any change in phosphorylation of IκBα, suggesting that MEK/Erk is indeed able to regulate ICAM-1 expression independently of NF-κB in endothelial cells (26).
In conclusion, under normal conditions, Egfl7 constitutively expressed by endothelial cells participates in the control of their activation. By down-regulating Egfl7, pro-inflammatory cytokines induce a more potent and efficient activation of the endothelium. It is very likely that this loop of control is deregulated in immune and inflammatory disorders and that it participates in favoring tumor escape from immunity by down-regulating endothelial cell activation and immune infiltration of tumors.
Experimental Procedures
Materials
The pRC-CMV and pRC-CMV-IκBα S32/36A vectors were a kind gift from Dr. Robert Weil (Institut Pasteur, Paris, France). Human recombinant TNFα, human recombinant IL1β, human recombinant IL6, and the NF-κB inhibitor BAY117085 were from R&D Systems (Lille, France).
Cells
Jurkat immortalized T-lymphocytes cells were obtained from the ATCC (LGC Standards, Molsheim, France) and not further characterized. They were cultured in RPMI, 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin. Primary HUVEC were obtained from Lonza (Levallois, France), cultured in EGM-2 medium in 25-cm2 ventilated flasks, and subcultured as recommended by the manufacturer. They were used between passages 2 and 7. The cells were maintained in humidified 95% air, 5% CO2 atmosphere incubators at 37 °C. The cells were checked for mycoplasma contamination on a monthly basis and discarded if found positive.
Cloning
egfl7 gene luciferase reporter vectors were prepared by cloning the −9008/+50, −7585/+50, −5550/+50, −3350/+50, and −1707/+50 regions of the human egfl7 gene relative to the transcription start of exon 1b (2) isolated from a BAC vector in front of the luciferase gene in the pGL3basic reporter vector. All vectors were sequenced to avoid spurious mutations.
Transfections
For RNA interference assays, HUVEC were plated in 4- or 9.6-cm2 well plates (25,000 cells/cm2) and transfected the next day with 10 nm siRNA (Dharmacon, GE Healthcare Europe, Velizy-Villacoublay, France) in Primefect siRNA reagent (Lonza) mixed with EGM-2. The cells were cultured for the indicated time with a medium change every other day. For transactivation assays, HUVEC were plated in 9.6-cm2 culture wells (15,000/cm2) and transfected the next day with 63 fmol of the indicated vectors and 42 fmol of pCMV-β-gal normalizing vector in EGM-2 containing 10 μl/well Superfect (Qiagen). After 3 h, the transfection medium was changed for EGM-2, and cells were further cultured for 24 h, at which time they were stimulated or not with TNFα for 18 h, rinsed in PBS, and lysed in reporter lysis buffer (Promega). Luciferase and β-galactosidase activities were measured in cell extracts using a Lumat luminometer (Berthold, Thoiry, France). For expression assays, HUVEC were transfected with 11.2 fmol/15,000 cells of pcDNA3 plasmid (pCtrl) or with a pcDNA3 plasmid coding for a full-length, HA-tagged, human Egfl7 cDNA (pEgfl7) and used 2 days later. For rescue experiments, HUVEC were transfected with siCtrl or siEgfl7 as in RNA interference assays, cultured for 24 h, and then transfected with pCtrl or pEgfl7 as in expression assays; the cells were used 2 days later.
Isolation of CD31+ Cells
CD31+ cells were purified from mouse lungs using anti-CD31-coated magnetic Dynabeads (Life Technologies) exactly as described in (23).
RT-qPCR
Cells and lungs were homogenized in Trizol (Life Technologies), total RNA were extracted and reverse transcribed using high capacity cDNA reverse transcription (Life Technologies). All qPCR were performed in duplex PCR mixing cDNA with both the TaqMan FAM-labeled probe of the tested gene (Life Technologies) and a β-actin- or a β2-microglobulin-VIC-labeled probe and processed for qPCR in a StepOne machine. CT values were calculated at the upper linear range of the logarithm-2 amplification curve using the StepOne v2.3 software. The data are expressed as 2−ΔΔCT (55), where ΔCT = CT of transcript of interest − CT of reference (β-actin or β2-microglobulin) measured in the same tube, and ΔΔCT = mean ΔCT experimental samples − mean ΔCT control samples of the same experiment. The relative quantity is 2−ΔΔCT and transforms the logarithmic-2 data into decimal values.
Immunoblots
The anti-human Egfl7 antibody (AF3638, lot WNK0109011) was from R&D Systems. The antibodies anti-IKKα (3G12) 11930 lot 5, IKKβ (D30C6) 8943 lot 3, phospho-IKKα/β (Ser176/180) (16A6) 2697 lot 16, NF-κB p65 (D14E12) 8242 lot 4, phospho-NF-κB p65 (Ser536) (93H1) 3033 lot 14, Iκ-Bα (L35A5) 4814 lot 17, and phospho-Iκ-Bα (Ser32) (14D4) 2859 lot 14 were from Cell Signaling (Ozyme, St. Quentin en Yvelines, France). Antibodies against GAPDH (6C5) (sc-32233 lot C1315) and actin (sc-1615 lot G0513) were from Santa Cruz Biotechnology (Clinisciences, Nanterre, France). Antibodies were used at 1/1000 dilution except for actin and GAPDH (1/10000). Proteins were extracted in radioimmune precipitation assay buffer containing protease and phosphatase inhibitors (Complete, Phostop, Roche Diagnostics, Meylan, France), analyzed by 10% or 12% SDS-PAGE, blotted onto Immobilon-P (Millipore, Molsheim, France), processed with antibody incubation, and revealed using ECL-Prime (GE Healthcare Europe) and chemiluminescence detected by film exposure or using a Luminescent image system (LAS3000, Fujifilm) and quantified using Multigauge v3.0 software.
Immunofluorescence
The cells cultured on 14-mm Ø glass coverslips were fixed in PBS, 4% paraformaldehyde for 20 min at room temperature and washed three times with PBS. Fixed cells were incubated in PBS, 2% BSA, 0.1 m glycine at room temperature for 5 min and washed with PBS. The cells were incubated in PBS, 2% BSA at 37 °C for 30 min and then with antibodies against human Egfl7 (1/250) in PBS, 1% BSA for 1 h at 37 °C, and washed three times with PBS. The cells were incubated with Alexa 594 donkey anti-goat antibody (1/250; Jackson ImmunoResearch Laboratories, Interchim, Montluçon, France) in PBS, 1% BSA for 30 min at room temperature, stained with 1 μg/ml DAPI in PBS for 5 min, and washed three times in PBS. The coverslips were finally mounted on glass slides in Mowiol reagent and analyzed under AxioImagerZ1 apotome (Zeiss).
Induced Lung Inflammation
BALB/c mice (Charles River, L'Arbresle, France) were anesthetized with 2% aerosolized isoflurane. LPS from Escherichia coli serotype 0111:B4 (Sigma-Aldrich) or recombinant human TNFα were given by intranasal instillation at the dose of 5 and 0.25 mg/kg, respectively, in 40 μl of LPS-free PBS. Control mice were instilled with 40 μl of LPS-free PBS. 10 and 24 h after instillation, mouse lungs were excised for mRNA extraction or fixed and embedded in paraffin for immunostaining.
In Situ Hybridization and Immunohistochemistry
A murine antisense egfl7 probe was synthesized from a pcDNA3-egfl7 vector linearized by EcoRI digestion and using the Sp6 RNA polymerase in the presence of 350 μm digoxigenin-11-UTP (Roche) and ZG1 tissue array slides (SuperBiochips, Clinisciences) were processed for in situ hybridization as described (2). For CD31 immunostaining, tissue slides were treated with 250 ng/ml trypsin (Life Technologies) and then in TNB blocking solution (TSA Biotin System; Perkin Elmer) and incubated for 1 h in rat monoclonal anti-CD31 antibody (1/100, BD Pharmingen) in TNB. The slides were washed three times for 5 min in 0.1 m Tris-HCl, pH 7.5, 0.15 m NaCl, 0.05% Tween 20 and then incubated with a biotinylated anti-rat IgG (1/250; Vector Labs, Clinisciences) and revealed using the DAB kit (Vector Labs). The slides were counterstained using hematoxylin and mounted in Vectamount (Vector Labs).
Adhesion Assays
Human Jurkat T-lymphocytes (1 × 106 cells) were incubated with DiI (2 μm; Molecular Probes, Life Technologies) for 10 min at 37 °C and allowed to adhere (105 cells/cm2) onto monolayers of confluent HUVEC for 20 min at 37 °C. The cells were rinsed three times in PBS and fixed in PBS, 4% paraformaldehyde for 20 min at room temperature. Fluorescent cells were photographed under a UV microscope, and the numbers of adherent cells were estimated by counting cells over 10–15 microscope fields.
Ethics
Mice were housed according to European legislation. Protocols were approved by the local ethics committee. S. P., C. D., V. M., and F. S. have level 1 animal experimentation diplomas.
Author Contributions
S. P. and B. C. performed most of the experiments; A. L. B., C. D., V. M., and F. S. performed experiments and analyzed data; C. H., G. V., and R. D. provided technical assistance; V. M. and F. S. wrote the paper; and F. S. designed the project. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Corinne Abbadie, Véronique Baud, David Tulasne, and Robert Weil for reagents and discussions.
This work was supported by grants from the Fondation ARC pour la Recherche sur le Cancer, Ligue contre le Cancer and the Institut National du Cancer. The authors declare that they have no conflicts of interest with the contents of this article.
- ICAM-1
- intercellular adhesion molecule-1
- VCAM-1
- vascular cell adhesion molecule-1
- HUVEC
- human umbilical vein endothelial cell(s)
- siCtrl
- control siRNA
- siEgfl7
- siRNA targeting Egfl7
- IKK
- IκB kinase
- qPCR
- quantitative PCR.
References
- 1. Muller W. A. (2009) Mechanisms of transendothelial migration of leukocytes. Circ. Res. 105, 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soncin F., Mattot V., Lionneton F., Spruyt N., Lepretre F., Begue A., and Stehelin D. (2003) VE-statin, an endothelial repressor of smooth muscle cell migration. EMBO J. 22, 5700–5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lelièvre E., Hinek A., Lupu F., Buquet C., Soncin F., and Mattot V. (2008) VE-statin/egfl7 regulates vascular elastogenesis by interacting with lysyl oxidases. EMBO J. 27, 1658–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Charpentier M. S., Christine K. S., Amin N. M., Dorr K. M., Kushner E. J., Bautch V. L., Taylor J. M., and Conlon F. L. (2013) CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev. Cell 25, 132–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charpentier M. S., Tandon P., Trincot C. E., Koutleva E. K., and Conlon F. L. (2015) A distinct mechanism of vascular lumen formation in Xenopus requires EGFL7. PLoS One 10, e0116086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parker L. H., Schmidt M., Jin S. W., Gray A. M., Beis D., Pham T., Frantz G., Palmieri S., Hillan K., Stainier D. Y., De Sauvage F. J., and Ye W. (2004) The endothelial-cell-derived secreted factor Egfl7 regulates vascular tube formation. Nature 428, 754–758 [DOI] [PubMed] [Google Scholar]
- 7. Delfortrie S., Pinte S., Mattot V., Samson C., Villain G., Caetano B., Lauridant-Philippin G., Baranzelli M.-C., Bonneterre J., Trottein F., Faveeuw C., and Soncin F. (2011) Egfl7 promotes tumor escape from immunity by repressing endothelial cell activation. Cancer Res. 71, 7176–7186 [DOI] [PubMed] [Google Scholar]
- 8. Pannier D., Philippin-Lauridant G., Baranzelli M.-C., Bertin D., Bogart E., Delprat V., Villain G., Mattot V., Bonneterre J., and Soncin F. (2016) High expression levels of egfl7 correlate with low endothelial cell activation in peritumoral vessels of human breast cancer. Oncol. Lett. 12, 1422–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Badiwala M. V., Tumiati L. C., Joseph J. M., Sheshgiri R., Ross H. J., Delgado D. H., and Rao V. (2010) Epidermal growth factor-like domain 7 suppresses intercellular adhesion molecule 1 expression in response to hypoxia/reoxygenation injury in human coronary artery endothelial cells. Circulation 122, S156–61 [DOI] [PubMed] [Google Scholar]
- 10. Badiwala M. V., Guha D., Tumiati L., Joseph J., Ghashghai A., Ross H. J., Delgado D. H., and Rao V. (2011) Epidermal growth factor-like domain 7 is a novel inhibitor of neutrophil adhesion to coronary artery endothelial cells injured by calcineurin inhibition. Circulation 124, S197–S203 [DOI] [PubMed] [Google Scholar]
- 11. Fitch M. J., Campagnolo L., Kuhnert F., and Stuhlmann H. (2004) Egfl7, a novel epidermal growth factor-domain gene expressed in endothelial cells. Dev Dyn. 230, 316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Togbe D., Schnyder-Candrian S., Schnyder B., Doz E., Noulin N., Janot L., Secher T., Gasse P., Lima C., Coelho F. R., Vasseur V., Erard F., Ryffel B., Couillin I., and Moser R. (2007) Toll-like receptor and tumour necrosis factor dependent endotoxin-induced acute lung injury. Int. J. Exp. Pathol. 88, 387–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vandenbroucke E., Mehta D., Minshall R., and Malik A. B. (2008) Regulation of endothelial junctional permeability. Ann. N.Y. Acad. Sci. 1123, 134–145 [DOI] [PubMed] [Google Scholar]
- 14. Fràter-Schröder M., Risau W., Hallmann R., Gautschi P., and Böhlen P. (1987) Tumor necrosis factor type α, a potent inhibitor of endothelial cell growth in vitro, is angiogenic in vivo. Proc. Natl. Acad. Sci. U.S.A. 84, 5277–5281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. BenEzra D., Hemo I., and Maftzir G. (1990) In vivo angiogenic activity of interleukins. Arch. Ophthalmol. 108, 573–576 [DOI] [PubMed] [Google Scholar]
- 16. Le Bras A., Samson C., Trentini M., Caetano B., Lelievre E., Mattot V., Beermann F., and Soncin F. (2010) VE-statin/egfl7 expression in endothelial cells is regulated by a distal enhancer and a proximal promoter under the direct control of Erg and GATA-2. PLoS One 5, e12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Montgomery K. F., Osborn L., Hession C., Tizard R., Goff D., Vassallo C., Tarr P. I., Bomsztyk K., Lobb R., and Harlan J. M. (1991) Activation of endothelial-leukocyte adhesion molecule 1 (ELAM-1) gene transcription. Proc. Natl. Acad. Sci. U.S.A. 88, 6523–6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rao R. M., Yang L., Garcia-Cardena G., and Luscinskas F. W. (2007) Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ. Res. 101, 234–247 [DOI] [PubMed] [Google Scholar]
- 19. Romano M., Sironi M., Toniatti C., Polentarutti N., Fruscella P., Ghezzi P., Faggioni R., Luini W., van Hinsbergh V., Sozzani S., Bussolino F., Poli V., Ciliberto G., and Mantovani A. (1997) Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 6, 315–325 [DOI] [PubMed] [Google Scholar]
- 20. Barnes T. C., Anderson M. E., and Moots R. J. (2011) The many faces of interleukin-6: the role of IL-6 in inflammation, vasculopathy, and fibrosis in systemic sclerosis. Int. J. Rheumatol. 2011, 721608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Traenckner E. B., Pahl H. L., Henkel T., Schmidt K. N., Wilk S., and Baeuerle P. A. (1995) Phosphorylation of human IκB-α on serines 32 and 36 controls IκB-α proteolysis and NF-κB activation in response to diverse stimuli. EMBO J. 14, 2876–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang S., Aurora A. B., Johnson B. A., Qi X., McAnally J., Hill J. A., Richardson J. A., Bassel-Duby R., and Olson E. N. (2008) The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 15, 261–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poissonnier L., Villain G., Soncin F., and Mattot V. (2014) miR126-5p repression of ALCAM and SetD5 in endothelial cells regulates leucocyte adhesion and transmigration. Cardiovasc. Res. 102, 436–447 [DOI] [PubMed] [Google Scholar]
- 24. Pierce J. W., Schoenleber R., Jesmok G., Best J., Moore S. A., Collins T., and Gerritsen M. E. (1997) Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 272, 21096–21103 [DOI] [PubMed] [Google Scholar]
- 25. Kuldo J. M., Westra J., Asgeirsdóttir S. A., Kok R. J., Oosterhuis K., Rots M. G., Schouten J. P., Limburg P. C., and Molema G. (2005) Differential effects of NF-κB and p38 MAPK inhibitors and combinations thereof on TNF-α- and IL-1β-induced proinflammatory status of endothelial cells in vitro. Am. J. Physiol. Cell Physiol. 289, C1229–1239 [DOI] [PubMed] [Google Scholar]
- 26. Maeng Y.-S., Min J.-K., Kim J. H., Yamagishi A., Mochizuki N., Kwon J.-Y., Park Y.-W., Kim Y.-M., and Kwon Y.-G. (2006) ERK is an anti-inflammatory signal that suppresses expression of NF-κB-dependent inflammatory genes by inhibiting IKK activity in endothelial cells. Cell Signal. 18, 994–1005 [DOI] [PubMed] [Google Scholar]
- 27. Zhou Z., Connell M. C., and MacEwan D. J. (2007) TNFR1-induced NF-κB, but not ERK, p38MAPK or JNK activation, mediates TNF-induced ICAM-1 and VCAM-1 expression on endothelial cells. Cell Signal. 19, 1238–1248 [DOI] [PubMed] [Google Scholar]
- 28. Kanarek N., and Ben-Neriah Y. (2012) Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol. Rev. 246, 77–94 [DOI] [PubMed] [Google Scholar]
- 29. Winsauer G., and de Martin R. (2007) Resolution of inflammation: intracellular feedback loops in the endothelium. Thromb. Haemost. 97, 364–369 [PubMed] [Google Scholar]
- 30. Zhang W., An J., Jawadi H., Siow D. L., Lee J.-F., Zhao J., Gartung A., Maddipati K. R., Honn K. V., Wattenberg B. W., and Lee M.-J. (2013) Sphingosine-1-phosphate receptor-2 mediated NFκB activation contributes to tumor necrosis factor-α induced VCAM-1 and ICAM-1 expression in endothelial cells. Prostaglandins Other Lipid Mediat. 106, 62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rajesh M., Mukhopadhyay P., Bátkai S., Haskó G., Liaudet L., Huffman J. W., Csiszar A., Ungvari Z., Mackie K., Chatterjee S., and Pacher P. (2007) CB2-receptor stimulation attenuates TNF-α-induced human endothelial cell activation, transendothelial migration of monocytes, and monocyte-endothelial adhesion. Am. J. Physiol. Heart Circ. Physiol. 293, H2210–H2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chakrabarti S., and Davidge S. T. (2012) G-protein coupled receptor 30 (GPR30): a novel regulator of endothelial inflammation. PLoS One 7, e52357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khan B. V., Harrison D. G., Olbrych M. T., Alexander R. W., and Medford R. M. (1996) Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 93, 9114–9119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dhordain P., Dewitte F., Desbiens X., Stehelin D., and Duterque-Coquillaud M. (1995) Mesodermal expression of the chicken erg gene associated with precartilaginous condensation and cartilage differentiation. Mech. Dev. 50, 17–28 [DOI] [PubMed] [Google Scholar]
- 35. Hollenhorst P. C., Jones D. A., and Graves B. J. (2004) Expression profiles frame the promoter specificity dilemma of the ETS family of transcription factors. Nucleic Acids Res. 32, 5693–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dryden N. H., Sperone A., Martin-Almedina S., Hannah R. L., Birdsey G. M., Khan S. T., Layhadi J. A., Mason J. C., Haskard D. O., Göttgens B., and Randi A. M. (2012) The transcription factor Erg controls endothelial cell quiescence by repressing activity of nuclear factor (NF)-κB p65. J. Biol. Chem. 287, 12331–12342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sperone A., Dryden N. H., Birdsey G. M., Madden L., Johns M., Evans P. C., Mason J. C., Haskard D. O., Boyle J. J., Paleolog E. M., and Randi A. M. (2011) The transcription factor Erg inhibits vascular inflammation by repressing NF-κB activation and proinflammatory gene expression in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 31, 142–150 [DOI] [PubMed] [Google Scholar]
- 38. Yuan L., Nikolova-Krstevski V., Zhan Y., Kondo M., Bhasin M., Varghese L., Yano K., Carman C. V., Aird W. C., and Oettgen P. (2009) Antiinflammatory effects of the ETS factor ERG in endothelial cells are mediated through transcriptional repression of the interleukin-8 gene. Circ. Res. 104, 1049–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Poissonnier L., Villain G., Soncin F., and Mattot V. (2014) Egfl7 is differentially expressed in arteries and veins during retinal vascular development. PLoS One 9, e90455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Campagnolo L., Leahy A., Chitnis S., Koschnick S., Fitch M. J., Fallon J. T., Loskutoff D., Taubman M. B., and Stuhlmann H. (2005) EGFL7 is a chemoattractant for endothelial cells and is up-regulated in angiogenesis and arterial injury. Am. J. Pathol. 167, 275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gustavsson M., Mallard C., Vannucci S. J., Wilson M. A., Johnston M. V., and Hagberg H. (2007) Vascular response to hypoxic preconditioning in the immature brain. J. Cereb. Blood Flow Metab. 27, 928–938 [DOI] [PubMed] [Google Scholar]
- 42. Xu D., Perez R. E., Ekekezie I. I., Navarro A., and Truog W. E. (2008) Epidermal growth factor-like domain 7 protects endothelial cells from hyperoxia-induced cell death. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L17–L23 [DOI] [PubMed] [Google Scholar]
- 43. Patel C. V., Sharangpani R., Bandyopadhyay S., and DiCorleto P. E. (1999) Endothelial cells express a novel, tumor necrosis factor-α-regulated variant of HOXA9. J. Biol. Chem. 274, 1415–1422 [DOI] [PubMed] [Google Scholar]
- 44. Hidai C., Zupancic T., Penta K., Mikhail A., Kawana M., Quertermous E. E., Aoka Y., Fukagawa M., Matsui Y., Platika D., Auerbach R., Hogan B. L., Snodgrass R., and Quertermous T. (1998) Cloning and characterization of developmental endothelial locus-1: an embryonic endothelial cell protein that binds the αvβ3 integrin receptor. Genes Dev. 12, 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Choi E. Y., Chavakis E., Czabanka M. A., Langer H. F., Fraemohs L., Economopoulou M., Kundu R. K., Orlandi A., Zheng Y. Y., Prieto D. A., Ballantyne C. M., Constant S. L., Aird W. C., Papayannopoulou T., Gahmberg C. G., Udey M. C., Vajkoczy P., Quertermous T., Dimmeler S., Weber C., and Chavakis T. (2008) Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science 322, 1101–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Viemann D., Goebeler M., Schmid S., Klimmek K., Sorg C., Ludwig S., and Roth J. (2004) Transcriptional profiling of IKK2/NF-κB- and p38 MAP kinase-dependent gene expression in TNF-α-stimulated primary human endothelial cells. Blood 103, 3365–3373 [DOI] [PubMed] [Google Scholar]
- 47. Viemann D., Goebeler M., Schmid S., Nordhues U., Klimmek K., Sorg C., and Roth J. (2006) TNF induces distinct gene expression programs in microvascular and macrovascular human endothelial cells. J. Leukoc. Biol. 80, 174–185 [DOI] [PubMed] [Google Scholar]
- 48. Nichol D., Shawber C., Fitch M. J., Bambino K., Sharma A., Kitajewski J., and Stuhlmann H. (2010) Impaired angiogenesis and altered Notch signaling in mice overexpressing endothelial Egfl7. Blood 116, 6133–6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nichol D., and Stuhlmann H. (2012) EGFL7: a unique angiogenic signaling factor in vascular development and disease. Blood 119, 1345–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmidt M. H., Bicker F., Nikolic I., Meister J., Babuke T., Picuric S., Müller-Esterl W., Plate K. H., and Dikic I. (2009) Epidermal growth factor-like domain 7 (EGFL7) modulates Notch signalling and affects neural stem cell renewal. Nat. Cell Biol. 11, 873–880 [DOI] [PubMed] [Google Scholar]
- 51. Quillard T., Coupel S., Coulon F., Fitau J., Chatelais M., Cuturi M. C., Chiffoleau E., and Charreau B. (2008) Impaired Notch4 activity elicits endothelial cell activation and apoptosis: implication for transplant arteriosclerosis. Arterioscler. Thromb. Vasc. Biol. 28, 2258–2265 [DOI] [PubMed] [Google Scholar]
- 52. Verginelli F., Adesso L., Limon I., Alisi A., Gueguen M., Panera N., Giorda E., Raimondi L., Ciarapica R., Campese A. F., Screpanti I., Stifani S., Kitajewski J., Miele L., Rota R., and Locatelli F. (2015) Activation of an endothelial Notch1-Jagged1 circuit induces VCAM1 expression, an effect amplified by interleukin-1β. Oncotarget 6, 43216–43229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Inoue K., Kobayashi M., Yano K., Miura M., Izumi A., Mataki C., Doi T., Hamakubo T., Reid P. C., Hume D. A., Yoshida M., Aird W. C., Kodama T., and Minami T. (2006) Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler. Thromb. Vasc. Biol. 26, 2652–2659 [DOI] [PubMed] [Google Scholar]
- 54. Chiu J.-J., Lee P.-L., Chen C.-N., Lee C.-I., Chang S.-F., Chen L.-J., Lien S.-C., Ko Y.-C., Usami S., and Chien S. (2004) Shear stress increases ICAM-1 and decreases VCAM-1 and E-selectin expressions induced by tumor necrosis factor-α in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 24, 73–79 [DOI] [PubMed] [Google Scholar]
- 55. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]