

Abstract
δ, a small protein found in most Gram-positive bacteria was, for a long time, thought to be a subunit of RNA polymerase (RNAP) and was shown to be involved in recycling of RNAP at the end of each round of transcription. However, how δ participates in both up-regulation and down-regulation of genes in vivo remains unclear. We have recently shown, in addition to the recycling of RNAP, δ functions as a transcriptional activator by binding to an A-rich sequence located immediately upstream of the −35 element, consequently facilitating the open complex formation. The result had explained the mechanism of up-regulation of the genes by δ. Here, we show that Bacillus subtilis δ could also function as a transcriptional repressor. Our results demonstrate that δ binds to an A-rich sequence located near the −35 element of the spo0B promoter, the gene involved in the regulatory cascade of bacterial sporulation and inhibits the open complex formation due to steric clash with σ region 4.2. We observed a significant increase in the mRNA level of the spo0B gene in a δ-knock-out strain of B. subtilis compared with the wild-type. Thus, the results report a novel function of δ, and suggest the mechanism of down-regulation of genes in vivo by the protein.
Keywords: Bacillus, bacterial transcription, gene regulation, repressor protein, sporulation
Introduction
δ factor is found in most of the Gram-positive bacteria, including Bacillus subtilis. Because δ factor is often found to be associated with the purified RNA polymerase (RNAP)3 core enzyme from these bacteria, it was initially thought that δ factor is a subunit of the polymerase. Assuming δ to be a subunit of RNAP, most of the earlier studies demonstrated that δ functions by modulating RNAP in transcription initiation and recycling of the polymerase (1–7). An additional mechanism of function was depicted for δ in which the protein δ mediates a change in the requirement of iNTP by RNAP to stabilize the open complex formation at certain promoters (8). However, the fact that δ does not exhibit any affinity to RNAP holo (RNAP core + σ), but functions with RNAP holo remains ambiguous (9–11). Our recent report (11) suggested that δ functions as a transcriptional regulator, as well as, is involved in the recycling of polymerase from any paused ternary complex. In certain promoters, the protein acts as a transcriptional activator by binding at the A-rich sequence immediately upstream of the −35 element of the promoter and enhancing the rate of the open complex formation. This finding also explains the observations why δ factor does not bind to RNAP holo but still functions with the holoenzyme.
Despite the extensive study on the mechanism of function of δ, the in vivo role of the protein still remains unclear. Removal of the rpoE gene (encoding δ) from the bacterial genome does not result in any distinctive phenotype or major change in growth rate suggesting that the protein is non-essential for the bacteria (10, 12). On the other hand, disruption of the rpoE gene, although, did not significantly change the sporulation efficiency, could rescue the sporulation defect in certain mutant bacteria (10, 13). This result indicated that δ factor might be indirectly involved in sporulation. In addition, few reports suggested that the protein was required for virulence in certain bacteria (14–16). This finding was further supported by the transcriptome analysis of Staphylococcus aureus strains with and without δ, which showed that the protein was involved in down-regulation of the genes that encode virulence factors as well as in up-regulation of certain genes (17, 18). Our recent observation that δ functions as a transcriptional activator could explain the mechanism of up-regulation of genes (11). However, how the same protein could function in down-regulation of genes remains unknown.
Based on the fact that δ binds to A-rich sequence, we predicted that δ may function as a transcriptional repressor for a promoter on which the binding sites of δ and RNAP overlap. We found that spo0B of B. subtilis, the gene involved in sporulation of the bacteria, contains a promoter with a putative δ binding site at the −35 element (19, 20). Using in vitro biochemical assays, we show that δ represses transcription from the spo0B promoter by inhibiting the open complex formation. The mRNA level of the spo0B gene is significantly elevated in a δ knock-out strain. Thus, transcriptional repression by δ suggests the mechanism of down-regulation of genes by δ.
Results
δ Represses the spo0B Promoter
The spo0B promoter of B. subtilis contains an A-rich sequence at the −35 element of its promoter (19). Thus, the promoter does not contain a consensus −35 element, but contains an extended −10-like element (Fig. 1A). Because δ exhibits affinity to the A-rich sequence, we examined the effect of δ on this promoter. We prepared a linear double-stranded (ds)DNA fragment (−105/+57) containing the spo0B promoter and performed in vitro transcription assays on this DNA fragment in the absence and presence of increasing concentrations of δ. Two sets of in vitro transcription assays were performed. First, we incubated the promoter fragment with δ to allow binding of the protein to DNA prior to addition of RNAP (Fig. 1B). The result showed that δ inhibits transcription from the spo0B promoter with an approximate IC50 of 53 ± 10 nm. In the second assay, RNAP was first incubated with the promoter fragment to form an open complex and then added with δ before transcription initiation. To our surprise, we also observed a significant extent of inhibition by δ in the in vitro transcription assay (Fig. 1C). Thus, δ inhibits transcription from the spo0B promoter regardless of the order of addition of the protein on the DNA-RNAP complex. The result further suggests that the open complex formation at this promoter is not stable enough and could be easily destabilized by δ.
FIGURE 1.

δ represses the spo0B promoter. A, nucleotide sequence of spo0B promoter DNA (−52/+1): the rectangular box represents the −10 element, the putative δ binding site is underlined. B, in vitro transcription assay: δ was incubated with promoter DNA in transcription buffer at 25 °C for 15 min prior to open complex formation. Open complex was formed by addition of 400 nm RNAP holo at 37 °C for 20 min. Transcription reaction was initiated with addition of NTP (final concentrations: 250 μm ATP, GTP, and UTP, and 25 μm [α-32P]CTP (0.2 μCi)). Run-off transcript size is 57 nt. Each experiment was repeated three times and the mean fold-decrease in the amount of transcript at each concentration of δ with respect to the amount in the absence of δ were plotted as a bar graph (shown below of each panel). C, same as in B, but δ was incubated for 10 min once the open complex was formed. D, in vivo recombinant reporter assay; three-plasmid expression system in E. coli. The bars represent relative mCherry fluorescence of E. coli cells containing the pFPVmCherry-spo0B and plasmids encoding (i) δ (pAcYcDuet-rpoE), (ii) BsRNAP core (pNG219), (iii) BsRNAP holo (pNG219 + pAcYcDuet-rpoD), and (iv) BsRNAP holo + δ (pNG219 pAcYcDuet-rpoD-rpoE). DNA fragments (−105/+12) of spo0B were inserted upstream of the mCherry gene. Each set of assay was repeated three times, and the mean values of relative mCherry fluorescence of the cells were plotted. Fluorescence of the cells containing BsRNAP holo were normalized to 1.
To further test the interaction of δ with the spo0B promoter in vivo, we employed a recombinant reporter assay as used previously (11). The assay involved a three plasmid expression system in Escherichia coli: one plasmid for expression of RNAP core (pNG219 (21)), one for expression of σA and δ (pYcACDuet-rpoD-rpoE), and the third for expression of mCherry, which was controlled by the spo0B promoter. All three plasmids were transformed in E. coli B834 (DE3) and the cells were grown at 16 °C for 16 h after isopropyl 1-thio-β-d-galactopyranoside induction. To rule out the possible interference by E. coli (Ec) RNAP on the mCherry expression, we performed control assays by omitting BsRNAP expressing plasmid, pNG219. The assays were carried out with and without δ (Fig. 1D). The levels of mCherry expression by BsRNAP holo in the presence of δ was normalized to 1. mCherry fluorescence from the control assay with E. coli harboring only pFPVmCherry-spo0B was very small and was considered as the background fluorescence. On the other hand, expression of the BsRNAP core or δ in the control assays increased mCherry expression marginally compared with the background. In contrast, expression of BsRNAP holo resulted in at least a 4-fold increase in the mCherry expression level from the spo0B promoter. The presence of δ decreased mCherry expression from the spo0B promoter to a level comparable with the controls. Thus, the result indicated a repression of the spo0B promoter by δ. Although, the levels of expression of mCherry, and the extent of repression of the spo0B promoter by δ in our reporter assay in E. coli could be different if being performed in B. subtilis, our results unequivocally confirm the in vivo interaction of δ with the spo0B promoter.
A-rich Sequence Is Required for δ Function
To test whether the A-rich sequence around the −35 element of the spo0B promoter is responsible for the δ-mediated inhibition of transcription, we, first, generated a spo0B promoter derivative (spo0B mut1) by mutating the AA nucleotide at −34, −35 positions of the template strand by T and C, respectively (Fig. 2A). When a in vitro transcription assay was performed with this promoter derivative, we observed a significant enhancement of transcript yield in the presence of δ, instead of a loss of inhibition of transcription (Fig. 2B). To find out the possible explanation of this result, we noticed that the spo0B promoter fragment contains an additional A-rich sequence upstream of the −35 element (sequence from −44 to −48). Previously, we had shown that δ activates transcription from a promoter when it binds to the A-rich DNA immediately upstream of the −35 element (11). Therefore, we reasoned that this sequence element might be responsible for the δ-mediated increase in the transcript yield on the spo0B mutant derivative. This prediction was subsequently verified by generating another spo0B promoter derivative (spo0B mut2) in which the −45, −46, and −47 nucleotides of the non-template strand of the spo0B mut1 was replaced by three C nucleotides (Fig. 2A). The in vitro transcription assay with this promoter derivative showed no or little change in the transcript yield by δ (Fig. 2C). Similarly, when the spo0B mut2 promoter was used in the in vivo reporter assay, no significant change of the mCherry expression was observed in the presence of δ (Fig. 2D).
FIGURE 2.

A-rich sequence required for δ function. A, nucleotide sequence of the mutant spo0B promoter DNA fragments (−52/+1). The rectangular box represents the −10 promoter element. The putative δ binding site is underlined. The mutated bases are highlighted. B, in vitro transcription assay: 400 nm RNAP holo, 100 nm spo0B mut1 promoter DNA were used in the absence and presence of δ. Run-off transcript size is 57 nt. Each experiment was repeated three times and the mean of the relative amount of transcript at each concentration of δ with respect to the amount in the absence of δ were plotted as a bar graph (shown below of each panel). C, same as in B, but for the spo0B mut2 promoter DNA. D, in vivo recombinant reporter assay: same as in Fig. 1D but with the pFPVmCherry-spo0B mut2. E and F, binding of δ to A-rich DNA fragments: fluorescence anisotropy assay, and 20 nm TMR-labeled δ, was added with different spo0B promoter derivatives. Fluorescence anisotropy of the labeled δ was monitored at excitation 540 nm and emission 580 nm. Each data set represents mean of three replicates. The Kd values of δ to A-rich DNA template was estimated using the sigmoidal function.
Because the results of both in vitro transcription assay and in vivo reporter assay indicated that the A-rich sequence is required for δ-mediated transcription inhibition, we tested the ability of δ to bind the A-rich sequence by fluorescence anisotropy assay. We used the same spo0B DNA fragment (−105/+57) used for the transcription assay. For control, we used a DNA fragment of the spo0B promoter in which the putative A-rich binding sites for δ were altered by site-directed mutagenesis (spo0B mut2, sequence shown in Fig. 2A). When fluorescence anisotropy assays were performed with tetramethylrhodamine (TMR)-labeled δ, we observed that the protein bound to the spo0B DNA fragments with a dissociation constant of 37 ± 3 nm (Fig. 2E), but not to the spo0B mut2 that did not contain any putative site for δ (Fig. 2F). Thus, the result confirms that the A-rich sequence around the −35 element is required for binding of δ.
δ Inhibits RNAP-Promoter Complex Formation
Because δ binds to the A-rich sequence near the −35 element of the spo0B promoter, a site that overlaps with the RNAP binding region, it is likely that δ inhibits RNAP-promoter complex formation. This was confirmed by EMSA of the RNAP-DNA complex formed in the presence of δ. For detection of the RNAP-DNA complex, we labeled the spo0B promoter DNA fragment (−105/+30) with γ-32P at the 5′ end of the template strand. The upper band in the EMSA that corresponds to the RNAP-promoter complex decreased with the increasing amount of δ (Fig. 3A). Thus, the result clearly indicates that δ prevents the binding of RNAP to the spo0B promoter DNA, hence, subsequently prevents the RNAP-DNA complex formation on this promoter. When the A-rich sequence at the −35 region was mutated to abolish the δ binding to the spo0B promoter (spo0B mut1), the protein is unable to prevent RNAP-DNA complex formation (Fig. 3B).
FIGURE 3.

δ inhibits RNAP-promoter complex formation. A, EMSA: 200 nm RNAP holo samples were added to 25 nm 32P-labeled spo0B promoter DNA following incubation with δ for 25 °C for 15 min in transcription buffer and further incubated at 37 °C for 30 min. The products were challenged by 400 nm unlabeled DNA before resolving on 5% PAGE in 0.5× TBE buffer. The gels were scanned by phosphorimaging. B, same as A, but with spo0B mut1 promoter DNA.
Steric Clash with σ Region 4.2 Is Responsible for δ-mediated Transcriptional Inhibition
The spo0B promoter contains an extended −10 element. The −35 region includes a stretch of T nucleotides instead of the consensus sequence. In a normal promoter, σ region 2.4 interacts with the −10 element and σ region 4.2 interacts with the −35 element to form an RNAP-promoter complex (22). In contrast, for the promoter that contains an extended −10 element, the interaction between σ region 4.2 and −35 element is not required for the RNAP-promoter complex formation. In the extended −10 promoter, the σ region 4.2, although not clearly known, is possibly located near the −35 element. Because the spo0B promoter contains a −35 element that includes a binding site for δ, we speculated that the binding of δ might involve a steric clash with σ region 4.2 that occludes the binding of RNAP to the promoter. To test this hypothesis, we prepared a σA derivative that lacks region 4.2 (σAΔ4.2) (Fig. 4A) and assessed the effect of δ in transcription by the RNAP derivative containing σAΔ4.2. The result showed that removal of σ region 4.2 did not affect the ability of the σ derivative to initiate transcription from the spo0B promoter (Fig. 4B, lane 1), however, δ-mediated inhibition of transcription at the promoter was abrogated. Only a small extent of inhibition was observed at very high concentrations of δ (Fig. 4B). When the assay was performed with the spo0B mut2, an increase in the transcription yield was observed due to the effect of δ bound at the A-rich sequence upstream of the −35 element (Fig. 4C). The result suggests that upon removal of σ region 4.2, the RNAP derivative is able to form an RNAP-DNA complex even when the −35 element is occupied by δ. This was verified by EMSA with the RNAP derivative, which showed that δ-mediated inhibition of RNAP-DNA complex formation on the spo0B promoter was forfeited (Fig. 4D). The simplest interpretation of the result is that removal of σ region 4.2 creates enough space to accommodate δ without disturbing the RNAP-promoter complex. Thus, both RNAP and δ simultaneously bind to the promoter without affecting the open complex formation, and subsequent transcription initiation.
FIGURE 4.

Steric clash with σ region 4.2 is responsible for δ-mediated transcriptional inhibition. A, SDS-PAGE of σA and σAΔ4.2. M represents molecular mass marker in kDa. B, in vitro transcription assay: 400 nm RNAP holo (prepared by incubating 400 nm RNAP core and 3 μm σAΔ4.2), 100 nm spo0B promoter DNA were used in the absence and presence of δ. Run-off transcript size is 57 nucleotides. Each experiment was repeated three times and the mean of relative amount of transcript at each concentration of δ with respect to the amount in the absence of δ were plotted as a bar graph (shown below of each panel). C, same as in B, but for the spo0B mut1 promoter DNA. Run-off transcript size is 57 nucleotides. D, EMSA: as in Fig. 3A but with RNAP-σAΔ4.2 holo.
δ Down-regulates the spo0B Gene in Vivo
Our in vitro results clearly indicate that δ inhibits transcription from the spo0B promoter. To verify the in vivo effect of δ on the expression of the spo0B gene, we monitored the mRNA expression level of the same gene in wild-type and δ knock-out strains. As a control, the mRNA levels of the rpoA (the gene for α subunit of RNAP), a housekeeping gene, was monitored for both the strains. Real time PCR data of the cDNA generated from the mRNA pool showed that the level of expression of spo0B is ∼1.7-fold less in the wild-type strain than in δ knock-out strain (Fig. 5). The result suggests that δ down-regulates the spo0B gene in vivo.
FIGURE 5.
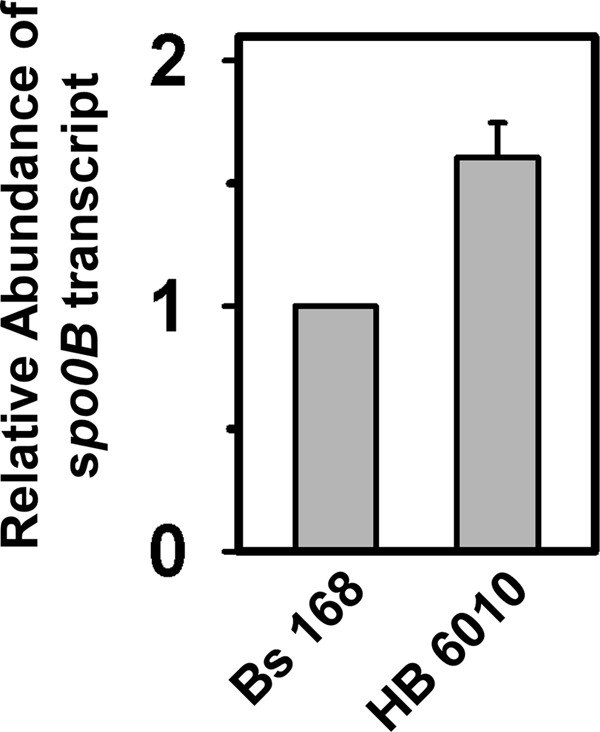
δ down-regulates the spo0B gene in vivo: qRT-PCR analysis. qRT-PCR analysis of spo0B mRNA in Bs168 and HB 6010 (ΔrpoE) cells. The relative abundance of gene transcripts was determined using duplicate samples and from two independent experiments.
Discussion
Previously we showed that δ acts as a transcriptional activator of certain genes. At these promoters, δ binds to DNA immediately upstream of the −35 element at an A-rich sequence and enhances the rate of open complex formation (11). In this study, we show that δ also functions as a transcriptional repressor on the spo0B promoter. In this promoter, δ binds to the A-rich sequence located at the −35 element and inhibits the open complex formation. Thus, the protein could act both as a transcriptional activator and repressor depending on the location of its binding site. This finding is consistent with the previous observation that δ is involved in both up-regulation and down-regulation of several genes in bacteria (17, 18).
spo0B, the promoter used in this study, does not contain any −35 element, instead contains a binding site for δ at the −35 element and has an extended −10-like promoter sequence (19). In the RNAP-DNA complex at the extended −10 promoter, σ region 4.2 does not interact with the −35 element. When this σ region 4.2 is omitted from the initiation factor, both the RNAP derivative and δ could simultaneously occupy the promoter region. As a result δ fails to prevent the binding of the RNAP derivative to the promoter and thereby, is unable to inhibit transcription. On the other hand, the presence of σ region 4.2, although is not required for the promoter recognition, induces a steric clash with δ preventing the binding of RNAP at the spo0B promoter. Our studies with the mutant spo0B promoter derivatives further confirm the role of δ binding at the −35 element in inhibiting the RNAP-promoter interaction and transcription. It is important to note that δ is able to inhibit transcription from this promoter even when added after the formation of the open complex. This observation suggests that the RNAP-DNA interaction at this promoter is not very stable. Similar to the spo0B promoter, we found δ-mediated repression of transcription both in vitro and in vivo on four other promoters, e.g. xynA (23), purT (24), cydA (25), and glpD (26) (supplemental Fig. S1). Notably, all of these four promoters contain an extended −10-like promoter element and putative δ binding site(s) around the ill-defined −35 element. However, when we tried to examine the effect of δ on sinIR (27) which contains an A-rich sequence within the promoter region, the protein was unable to inhibit transcription (supplemental Fig. S2). The sinIR promoter contains a −35 element that partially resembles the consensus sequence. Thus, the interactions between the −35 element and σ region 4.2 could occur at this promoter. Although further investigation is required, it is likely that the RNAP-DNA interaction on the sinIR promoter is too strong to be destabilized by δ. We conclude that not all the promoters that contain an A-rich binding site for δ within the promoter region would be inhibited by δ.
Our observation that δ inhibits transcription from the spo0B promoter in vitro is consistent with the in vivo result. The level of mRNA of the spo0B gene was reduced by 2-fold in the wild-type strain than in δ-knock-out strain suggesting a down-regulation of the spo0B gene by δ. Although the spo0B gene is involved in controlling several genes that are responsible for sporulation, and the gene is down-regulated by δ, there is no significant effect of δ on the sporulation efficiency (see previous observations in Refs. 10 and 13).4 Interestingly, few other genes involved in sporulation, e.g. spo0A, spo0E, and spo0F (20, 28), were transcriptionally activated by δ.4 Therefore, we speculate that the effect of repression of spo0B is revoked by activation of the other genes involved in sporulation and could be one of the possible reasons why the effect of δ in sporulation is not apparent. However, when δ was overexpressed in the δ-knock-out strain, the sporulation efficiency was increased by several fold4 compared with the wild-type strain. Thus, at a normal level δ does not have any effect on sporulation, however, at an elevated level δ could function by a different mechanism to increase the sporulation efficiency. Further studies are required to unravel the precise role of δ in sporulation.
Overall, in this study, we report a novel function of δ of B. subtilis as a transcriptional repressor of the spo0B gene. The mechanism of transcription repression has been demonstrated using biochemical and biophysical assays in which the protein binds to the spo0B promoter at a site that overlaps with the −35 element of the promoter and induces a steric clash with σ region 4.2 to inhibit the RNAP-promoter complex formation and transcription from this promoter. We further demonstrate that δ down-regulates the spo0B gene in vivo. Therefore, our result indicates the possible mechanism of down-regulation of the genes in vivo by δ.
Experimental Procedures
Purification of Bs RNAP Core, δ, σA, and σAΔ4.2
For purification of B. subtilis (Bs) RNAP core, E. coli B834 (DE3) cells were transformed with respective plasmids pNG540 and pNG545 (21) and grown in Luria-Bertani (LB) medium supplemented with 0.1% dextrose and antibiotics (100 μg ml−1 ampicillin and 35 μg ml−1 of chloramphenicol). For purification of δ, pAcYcDuet-rpoE was transformed into E. coli BL21 (DE3) cells and grown in 2× YT medium (16 g of tryptone, 10 g of yeast extract, and 5 g of NaCl/liter) supplemented with 35 μg ml−1 of chloramphenicol. For the expression of σA, E. coli C43 (DE3) cells were transformed with pET28a-rpoD and grown in 2× YT medium supplemented with 50 μg ml−1 of kanamycin. Proteins were purified as described by Prajapati et al. (11). For preparation of σAΔ4.2, we first generated mutant plasmid pET28a-rpoDΔ4.2 by introducing a stop codon at amino acid position 330 of rpoD by site-directed mutagenesis using oligo primers (Table 1). The σA derivative was purified following the same method as used for σA.
TABLE 1.
Template strand sequence is shown. The extended −10 element is underlined and the +1 nucleotide is shown in bold
| Oligo primers | Sequence |
|---|---|
| spo0B (−105) F | AAGAATTCGTTCTGCCTGGCTGCAAATC |
| spo0B (−105 KpnI) F | AAGGTACCGTTCTGCCTGGCTGCAAATC |
| spo0B (+49) R | AAGGATCCCGCACTCCCAATCATTTAATTTC |
| spo0B (+30) R | TTTCTTATTTAGGAGTCTGTATAAGTGTG |
| spo0B (+12) R | AAGGATCCGTATAAGTGTGTATTATGAATTATATCAGAGAAG |
| spoB mut1 F | GGGAAAAACAATTGTTGATCTAACAAAGCC |
| spoB mut1 R | GGCTTTGTTAGATCAACAATTGTTTTTCCC |
| spoB mut2 F | GGGACCCACAATTGTTGATCTAACAAAGCC |
| spoB mut2 R | GGCTTTGTTAGATCAACAATTGTGGGTCCC |
| spo0B RT F | GGCCATTCCCGGCATGATTGGATGAATAAG |
| spo0B RT R | GGATCGGCAAAGGCGCCGTGAAAATCAAGG |
| rpoA F | CTTCATATCGCGACTCTTGGTGAGAATGCG |
| rpoA R | GATCCAATTCTTCAATTGTCATTTC |
| rpoE F | AAGATATCATGGGGAGTGTCCGACCATGGGTATCAAAC |
| rpoE R | AAGGTACCCTATATTTAATTTCCTCTTCTTCATCATC |
| rpoEL51C F | GTGAAAAAAGAAGAGTGTGGAGACCGC |
| rpoEL51C R | GCGGTCTCCACACTCTTCTTTTTTCAC |
| rpoD F | GGATCCATGGCTGATAAACCCACG |
| rpoD R | GAATTCAAGCTTTTATTCAAGGAAATCTTTCAAACGTTTACTTC |
| rpoDΔ4.2 F | GACGGCCGTACATGAACATTAGAAG |
| rpoDΔ4.2 R | CTTCTAATGTTCATGTACGGCCGTC |
| DNA | |
| spo0B DNA template | AAGAATTCGTTCTGCCTGGCTGCAAATCAACCAAAGCCAGAAAAAAGGAACATGATATTTCTGGGAAAAACAATTGTTTTTCTAACAAAGCCTTCTCTGTTATAATTCATAATACACACTTATACAGACTCCTAAATAAGAAATTAAATGATTGGGAGTGCGGGATCCTT |
Preparation of DNA Fragments
The spo0B promoter DNA fragment from −105 to +49 was amplified by PCR from genomic DNA (isolated from Bs168) using oligo primers (Table 1) and was cloned in pUC19 using EcoRI-BamHI. spo0B promoter derivatives, spo0B mut1 (mutation at positions −34, −35) and spo0B mut2 (mutation at positions −34, −35, −45, −46, −47) were generated by PCR using primers and site-directed mutagenesis kits (Stratagene Inc.). spo0B promoter DNA fragment and its mutant derivatives were prepared by PCR on respective plasmids using primers (Table 1).
In Vitro Transcription Assay
400 nm RNAP core was mixed with 1.6 μm σA or 3.0 μm σAΔ4.2 in 1× transcription buffer (18 mm Tris-Cl, pH 8.0, 10 mm NaCl, 8 mm β-mercaptoethanol, 10 mm MgCl2) and incubated on ice for 30 min followed by 10 min at 25 °C to form the holoenzyme. 100 nm promoter DNA fragments were added to RNAP holo and incubated at 37 °C for 20 min to form the open complex. δ was incubated either with DNA at 25 °C for 15 min prior to the open complex formation or incubated after the open complex was formed (indicated in the figures). Transcription was initiated with NTP (final concentrations: 250 μm ATP, GTP, and UTP, and 25 μm [α-32P]CTP (0.2 μCi)) at 37 °C for 30 min. The reactions were terminated by the addition of 2.5 μl of FLB dye (80% formamide, 10 mm EDTA, 0.01% bromphenol blue, 0.01% xylene cyanol), resolved in 8 or 12% urea-PAGE (30) and scanned by storage phosphor scanner (Typhoon trio+, GE Healthcare).
Fluorescence Anisotropy Assays
The single-cysteine derivative of δ (Cys at residue 51) was generated, purified, and labeled with TMR-6-maleimide as described in Prajapati et al. (11). The labeling efficiency is 98%, and activity of the labeled protein was confirmed by in vitro transcription assay.
20 nm TMR-labeled δ in 60 μl of transcription buffer was titrated with increasing concentrations of DNA at 37 °C and fluorescence intensity and anisotropy values were measured (λex = 540 nm, λem = 580 nm) using a PTI Fluorescence Master QM400 system fitted with automatic polarizers. Normalized fluorescence anisotropy increments (ΔA/A0, where A and A0 are the anisotropy value of δ bound to DNA and free protein, respectively, and ΔA = A − A0) were plotted against titer concentration of DNA using the Sigmaplot software (Systat software Inc.). The dissociation constants (Kd) of the bindings for DNA were determined by fitting the data to single parameter sigmoidal functions.
EMSA
The reverse primer for spo0B DNA fragment was labeled using [γ-32P]ATP and T4 polynucleotide kinase (New England Biolabs) following the manufacturer's protocol. The promoter DNA fragment was amplified by PCR using the above 32P-labeled primer and an unlabeled forward. The amplified DNA was purified using 2% agarose gel. RNAP holoenzyme was formed by incubating BsRNAP core and σA or its derivative as indicated in the transcription assay. Two sets of EMSA were performed. In the first set, 200 nm RNAP holo samples were incubated with 25 nm 32P-labeled DNA in 10 μl of transcription buffer at 37 °C for 20 min and then δ was added and further incubated at 37 °C for 10 min, challenged by 400 nm unlabeled DNA before resolving on 5% PAGE in 0.5× TBE buffer. In the second set, δ was mixed with DNA in 1× transcription buffer at 25 °C for 15 min before addition of RNAP holo. The gels were scanned by phosphorimaging (Typhoon trio+, GE Healthcare). EMSA with promoter DNA derivatives spo0B mut1 (containing mutation at −34, −35) and using σA or σAΔ4.2 were performed as above after labeling the DNA fragment with [γ-32P]ATP.
In Vivo Recombinant Assay
A recombinant in vivo reporter assay using a three-plasmid expression system in E. coli was employed essentially as in Ref. 29. Plasmid pNG 219 (a kind gift from Dr. Peter J. Lewis, The University of Newcastle (21)) containing the genes rpoA, rpoB, and rpoC, respectively, of B. subtilis was used for BsRNAP core expression. The plasmids pAcYcDuet-rpoD and pAcYcDuet-rpoD-rpoE were used for expression of σA and both σA and δ, respectively. The spo0B promoter fragment (−105 to +12) was amplified by PCR from genomic DNA (isolated from Bs168) using oligo primers (Table 1) and cloned pFPVmCherry using KpnI-BamHI.
E. coli B834 (DE3) was transformed with: (i) pFPVmCherry-spo0B alone; (ii) pFPVmCherry-spo0B + pAcYcDuet-rpoE (for expression of δ); (iii) pFPVmCherry-spo0B + pNG219 (for expression of BsRNAP core); (iv) pFPVmCherry-spo0B + pNG219 + pAcYcDuet-rpoD (for expression of BsRNAP holo); and (v) pFPVmCherry-spo0B+ pNG219+pAcYcDuet-rpoD-rpoE (for expression of BsRNAP holo + δ). The cells were grown in 50 ml of LB medium supplement with appropriate antibiotics (100 μg ml−1 of ampicillin, 35 μg ml−1 of chloramphenicol, whenever required) at 37 °C up to 0.5 OD, added with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and were grown further for 16 h at 16 °C. Cells from each set were diluted to obtain equal OD and their fluorescence intensities were measured at 610 nm with excitation at 592 nm. An identical assay was performed with the pFPVmCherry-spo0Bmut2 promoter. The spo0B promoter in pFPVmCherry was mutated by site-directed mutagenesis using primers (Table 1) to generate the pFPVmCherry-spo0Bmut2.
Real-time PCR Assay
The B. subtilis strains Bs168 and HB6010 (a kind gift from Prof. John D. Helmann, Cornell University (10)), were grown separately at 37 °C in 1× LB medium until OD 1.5 (600 nm), 5-ml cultures were harvested by centrifugation. The total RNA was isolated using RNA extracting kit (Pure LinkTM RNA Mini Kit, Life Technologies). On column DNase I treatment was performed to avoid any DNA contaminant. The RNA concentration was estimated by measuring absorbance at 260 nm using a UV spectrophotometer (PerkinElmer Life Sciences). 5 μg of total RNA from each sample was used for cDNA synthesis using Super Reverse Transcriptase MuL V Kit (BioBharti LifeScience Pvt. Ltd.). qPCR analysis was performed with KAPA SYBR® FAST qPCR kit (KAPA BIOSYSTEMS) as per the manufacturer's protocol. The partial sequence of rpoA was used as an internal control. The PCR amplification used 35 cycles of 94 °C for 15 s, 55 °C for 30 s, 72 °C for 30 s on 7500 fast Real-Time PCR system (Applied Biosystem). The difference in cycle number (ΔCt) for a specific fluorescence threshold was calculated between the cDNA of spo0B and rpoA. The relative abundances of spo0B RNA from the two strains were estimated using the formula EΔCt, where E is the primer efficiency and was estimated as ∼2. Each set of real-time PCR was repeated 3 times with independent RNA samples and the relative amount of spo0B RNA were determined from the average of the data. The amount of RNA from the wild-type strain was normalized to 1.
Author Contributions
J. M. and R. S. conceived and designed the experiments; R. K P. performed the experiments; R. K. P. and J. M. analyzed the data; and J. M. wrote the paper.
Supplementary Material
Acknowledgment
We thank A. B. Datta (Bose Institute) for critically reading the manuscript and comments.
This work was supported in part by Department of Biotechnology, India Research Grants BT/PR 5345/MED/29/648/2012 and BT/PR 5270/BRB/10/1066/2012. The authors declare no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1 and S2.
R. Prajapati and J. Mukhopadhyay, unpublished data.
- RNAP
- RNA polymerase
- TMR
- tetramethylrhodamine
- qPCR
- quantitative PCR.
References
- 1. Tjian R., Losick R., Pero J., and Hinnebush A. (1977) Purification and comparative properties of the δ and σ subunits of RNA polymerase from Bacillus subtilis. Eur. J. Biochem. 74, 149–154 [DOI] [PubMed] [Google Scholar]
- 2. Williamson V. M., and Doi R. H. (1979) σ Factor is not released during transcription in Bacillus subtilis. Mol. Gen. Genet. 174, 47–52 [DOI] [PubMed] [Google Scholar]
- 3. Dickel C. D., Burtis K. C., and Doi R. H. (1980) δ Factor increases promoter selectivity of Bacillus subtilis vegetative cell RNA polymerase. Biochem. Biophys. Res. Commun. 95, 1789–1795 [DOI] [PubMed] [Google Scholar]
- 4. Achberger E. C., and Whiteley H. R. (1981) The role of the δ peptide of the Bacillus subtilis RNA polymerase in promoter selection. J. Biol. Chem. 256, 7424–7432 [PubMed] [Google Scholar]
- 5. Dobinson K. F., and Spiegelman G. B. (1987) Effect of the delta subunit of Bacillus subtilis RNA polymerase on initiation of RNA synthesis at two bacteriophage φ29 promoters. Biochemistry 26, 8206–8213 [DOI] [PubMed] [Google Scholar]
- 6. Juang Y. L., and Helmann J. D. (1994) The δ subunit of Bacillus subtilis RNA polymerase: an allosteric effector of the initiation and core-recycling phases of transcription. J. Mol. Biol. 239, 1–14 [DOI] [PubMed] [Google Scholar]
- 7. López de Saro F. J., Woody A. Y., and Helmann J. D. (1995) Structural analysis of the Bacillus subtilis δ factor: a protein polyanion which displaces RNA from RNA polymerase. J. Mol. Biol. 252, 189–202 [DOI] [PubMed] [Google Scholar]
- 8. Rabatinová A., S˘anderová H., Jirát Matĕjc˘kova J., Korelusová J., Sojka L., Barvík I., Papous˘ková V., Sklenár V., Z˘ídek L., and Krásný L. (2013) The δ subunit of RNA polymerase is required for rapid changes in gene expression and competitive fitness of the cell. J. Bacteriol. 195, 2603–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hyde E. I., Hilton M. D., and Whiteley H. R. (1986) Interactions of Bacillus subtilis RNA polymerase with subunits determining the specificity of initiation: σ and δ peptides can bind simultaneously to core. J. Biol. Chem. 261, 16565–16570 [PubMed] [Google Scholar]
- 10. López de Saro F. J., Yoshikawa N., and Helmann J. D. (1999) Expression, abundance, and RNA polymerase binding properties of the δ factor of Bacillus subtilis. J. Biol. Chem. 274, 15953–15958 [DOI] [PubMed] [Google Scholar]
- 11. Prajapati R. K., Sengupta S., Rudra P., and Mukhopadhyay J. (2016) Bacillus subtilis δ factor functions as a transcriptional regulator by facilitating the open complex formation. J. Biol. Chem. 291, 1064–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lampe M., Binnie C., Schmidt R., and Losick R. (1988) Cloned gene encoding the δ subunit of Bacillus subtilis RNA polymerase. Gene 67, 13–19 [DOI] [PubMed] [Google Scholar]
- 13. Gao H., and Aronson A. I. (2004) The δ subunit of RNA polymerase functions in sporulation. Curr. Microbiol. 48, 401–404 [DOI] [PubMed] [Google Scholar]
- 14. Seepersaud R., Needham R. H., Kim C. S., and Jones A. L. (2006) Abundance of the δ subunit of RNA polymerase is linked to the virulence of Streptococcus agalactiae. J. Bacteriol. 188, 2096–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xue X., Tomasch J., Sztajer H., and Wagner-Döbler I. (2010) The δ subunit of RNA polymerase, RpoE, is a global modulator of Streptococcus mutans environmental adaptation. J. Bacteriol. 192, 5081–5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xue X., Sztajer H., Buddruhs N., Petersen J., Rohde M., Talay S. R., and Wagner-Döbler I. (2011) Lack of the δ subunit of RNA polymerase increases virulence related traits of Streptococcus mutans. PloS One 6, e20075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xue X., Li J., Wang W., Sztajer H., and Wagner-Döbler I. (2012) The global impact of the delta subunit RpoE of the RNA polymerase on the proteome of Streptococcus mutans. Microbiology 158, 191–206 [DOI] [PubMed] [Google Scholar]
- 18. Weiss A., Ibarra J. A., Paoletti J., Carroll R. K., and Shaw L. N. (2014) The δ subunit of RNA polymerase guides promoter selectivity and virulence in Staphylococcus aureus. Infect. Immun. 82, 1424–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bouvier J., Stragier P., Bonamy C., and Szulmajster J. (1984) Nucleotide sequence of the spo0B gene of Bacillus subtilis and regulation of its expression. Proc. Natl. Acad. Sci. U.S.A. 81, 7012–7016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sonenshein A. L. (2000) Control of sporulation initiation in Bacillus subtilis. Curr. Opin. Microbiol. 3, 561–566 [DOI] [PubMed] [Google Scholar]
- 21. Yang X., and Lewis P. J. (2008) Overproduction and purification of recombinant Bacillus subtilis RNA polymerase. Protein Exp. Purif. 59, 86–93 [DOI] [PubMed] [Google Scholar]
- 22. Campbell E. A., Muzzin O., Chlenov M., Sun J. L., Olson C. A., Weinman O., Trester-Zedlitz M. L., and Darst S. A. (2002) Structure of the bacterial RNA polymerase promoter specificity σ subunit. Mol. Cell 9, 527–539 [DOI] [PubMed] [Google Scholar]
- 23. Garcia-Vallvé S., Palau J., and Romeu A. (1999) Horizontal gene transfer in glycosyl hydrolases inferred from codon usage in Escherichia coli and Bacillus subtilis. Mol. Biol. Evol. 16, 1125–1134 [DOI] [PubMed] [Google Scholar]
- 24. Saxild H. H., Jacobsen J. H., and Nygaard P. (1995) Functional analysis of the Bacillus subtilis purT gene encoding formate-dependent glycinamide ribonucleotide transformylase. Microbiology 141, 2211–2218 [DOI] [PubMed] [Google Scholar]
- 25. Winstedt L., Yoshida K., Fujita Y., and von Wachenfeldt C. (1998) Cytochrome bd biosynthesis in Bacillus subtilis: characterization of the cydABCD operon. J. Bacteriol. 180, 6571–6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holmberg C., and Rutberg B. (1991) Expression of the gene encoding glycerol-3-phosphate dehydrogenase (glpD) in Bacillus subtilis is controlled by antitermination. Mol. Microbiol. 5, 2891–2900 [DOI] [PubMed] [Google Scholar]
- 27. Shafikhani S. H., Mandic-Mulec I., Strauch M. A., Smith I., and Leighton T. (2002) Postexponential regulation of sin operon expression in Bacillus subtilis. J. Bacteriol. 184, 564–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grossman A. D. (1995) Genetic networks controlling the initiation of sporulation and the development of genetic competence in Bacillus subtilis. Annu. Rev. Genet. 29, 477–508 [DOI] [PubMed] [Google Scholar]
- 29. Banerjee R., Rudra P., Saha A., and Mukhopadhyay J. (2015) Recombinant reporter assay using transcriptional machinery of Mycobacterium tuberculosis. J. Bacteriol. 197, 646–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sambrook J., and Russel D. W. (2001) Molecular cloning: a laboratory manual, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.