

Abstract
β-Site APP-cleaving enzyme 1 (BACE1) cleaves amyloid β-protein precursor (APP) at the bond between Met671 and Asp672 (β-site) to generate the carboxyl-terminal fragment (CTFβ/C99). BACE1 also cleaves APP at another bond between Thr681 and Gln682 (β′-site), yielding CTFβ′/C89. Cleavage of CTFβ/C99 by γ-secretase generates Aβ(1-XX), whereas cleavage of CTFβ′/C89 generates Aβ(11-XX). Thus, β′-site cleavage by BACE1 is amyloidolytic rather than amyloidogenic. β′ cleavage of mouse APP is more common than the corresponding cleavage of human APP. We found that the H684R substitution within human Aβ, which replaces the histidine in the human protein with the arginine found at the corresponding position in mouse, facilitated β′ cleavage irrespective of the species origin of BACE1, thereby significantly increasing the level of Aβ(11-XX) and decreasing the level of Aβ(1-XX). Thus, amino acid substitutions within the Aβ sequence influenced the selectivity of alternative β- or β′-site cleavage of APP by BACE1. In familial Alzheimer's disease (FAD), the APP gene harbors pathogenic variations such as the Swedish (K670N/M671L), Leuven (E682K), and A673V mutations, all of which decrease Aβ(11–40) generation, whereas the protective Icelandic mutation (A673T) increases generation of Aβ(11–40). Thus, A673T promotes β′ cleavage of APP and protects subjects against AD. In addition, CTFβ/C99 was cleaved by excess BACE1 activity to generate CTFβ′/C89, followed by Aβ(11–40), even if APP harbored pathogenic mutations. The resultant Aβ(11–40) was more metabolically labile in vivo than Aβ(1–40). Our analysis suggests that some FAD mutations in APP are amyloidogenic and/or amyloidolytic via selection of alternative BACE1 cleavage sites.
Keywords: Alzheimer disease, amyloid precursor protein (APP), amyloid-β (Aβ), β-secretase 1 (BACE1), secretase
Introduction
β-Site APP-cleaving enzyme 1 (BACE1),3 a type I transmembrane aspartic protease, was identified as the β-secretase that cleaves amyloid β-protein precursor (APP) to generate neurotoxic amyloid β (Aβ) (1–4). BACE1 cleaves APP at the peptide bond between Met671 and Asp672 (β-site; sequence numbering refers to the APP770 isoform) (5, 6). This primary cleavage of APP generates the secreted form of the amino-terminal large fragment (sAPPβ) and the membrane-associated carboxyl-terminal fragment (CTFβ/C99) (reviewed in Ref. 7). Because CTFβ/C99 includes the complete amino acid sequence of the Aβ region, and subsequent cleavage of CTFβ/C99 by γ-secretase generates the Aβ(1-XX) peptides (Aβ1 indicates the position of Asp672), the β-site cleavage is referred to as amyloidogenic processing of APP (reviewed in Ref. 8). Alternatively, APP can also be cleaved by α-secretase (mainly ADAM10/17), at the peptide bond between Lys687 and Leu688, generating sAPPα and CTFα/C83 including the Aβ(17-XX) region; accordingly, this cleavage is referred to as amyloidolytic processing of APP (9–11). BACE1 also cleaves APP at another peptide bond between Tyr681 and Gln682 (β′-site), resulting in generation of sAPPβ′ and CTFβ′/C89 (12, 13). Cleavage of CTFβ′/C89 by γ-secretase yields Aβ(11-XX), which lacks the first 10 amino acids of the Aβ domain. This β′-site cleavage of APP is also thought to be amyloidolytic, because the structural analysis suggests that Aβ(11–40) and Aβ(11–42) are less toxic than Aβ(1–40) and Aβ(1–42) (14), and the Aβ(11–42) showed reduced neurotoxicity compared with Aβ(1–42) and Aβ(3–42) in a study with transgenic fly (15), although the neurotoxicity of Aβ(11-XX) in human brain is controversial (16).
Aβ is the major protein component of senile plaques observed in the brain of Alzheimer's disease (AD) subjects, and soluble Aβ oligomer(s) are thought to impair synaptic functions prior to Aβ deposition in the brain (17–19). Therefore, to understand AD pathogenesis and develop effective AD therapies, it is important to elucidate the molecular mechanisms of Aβ generation and degradation. Regulation of BACE1 activity represents a promising therapeutic option for decreasing Aβ production (reviewed in Ref. 20).
In familial AD (FAD), several amino acid mutations have been reported in the APP gene, especially in or around the Aβ sequence. Some of these mutations are pathogenic, e.g. the Swedish (K670N/M671L), Leuven (E682K), and A673V mutations increase Aβ generation by promoting APP β-site cleavage by BACE1 (21–23). On the other hand, at least one protective mutation exists: the Icelandic mutation, A673T, decreases Aβ generation (24, 25). Although the effect of the Swedish mutation, which is located outside the amino terminus of the Aβ sequence, is obvious, the molecular mechanisms by which amino acid alterations within Aβ affect the level of Aβ in the brain remain controversial. Because BACE1 cleaves APP either at the amyloidogenic β-site or the amyloidolytic β′-site, we investigated the effect of alternative BACE1 cleavage sites in APP on generation and degradation of Aβ.
Results
Alternative Cleavage of APP by BACE1 Depends on the Amino Acid Sequence within the Aβ Region, but Not on Species-specific Differences in BACE1
APP, a membrane protein, is highly conserved among animals. The cytoplasmic region, with which many conserved cytoplasmic proteins interact to regulate the metabolism and/or functions of APP, is completely conserved in human and mouse, and even in the electric ray (reviewed in Ref. 26; 27, 28). However, the amino acid sequence of the Aβ region differs slightly between mouse and human (Fig. 1A). In mouse APP (mAPP), Arg at position 676 in human APP (hAPP) is Gly (R676G), Tyr681 is Phe (Y681F), and His684 is Arg (H684R). These three amino acid substitutions in mouse Aβ are located near the β- and/or β′-sites of BACE1 cleavage. Rodent primary cultured neurons generate more Aβ(11-XX) than Aβ(1-XX) from endogenous APP into conditioned media, which were identified immunoprecipitation MS and radiosequencing analyses (29). When human APP was exogenously expressed in rodent primary cultured neurons, human Aβ(1–40) was predominant (30). Furthermore, analysis of Aβ42 species in human cerebrospinal fluid (CSF) with a surface-enhanced laser desorption/ionization technology revealed Aβ(1–42) as a major form, whereas Aβ(11–42) was less in quantity (31). This was thought to be a consequence of the species differences in the specificity of BACE1 (30). However, the molecular mechanism by which the β- or β′-site is selected by BACE1 remains controversial.
FIGURE 1.

Amino acid sequence of the human and mouse Aβ region and secretase cleavage sites, and generation of various forms of Aβ in cells overexpressing human or mouse APP. A, amino acid sequence of the human and mouse Aβ regions. Cleavage sites of α-secretase (α-site), β-secretase (BACE1; β- and β′-sites), and γ-secretase (γ-sites) to generate Aβ(XX-40) and Aβ(XX-42) are indicated by arrows. Numbering is for the APP-(770) isoform. Amino acid substitutions observed in FAD mutations are underlined. Mouse Aβ differs from human Aβ at three positions, indicated by a gray background. Aβ(1–40) and Aβ(11–40) are indicated by lines. B, representative MS spectra of Aβ species secreted from human and mouse cells expressing either hAPP or mAPP. Aβ forms generated from human and mouse APP expressed in mouse N2a and human SHSY5Y neuroblastoma cells were analyzed by MALDI-MS/MS analysis. Aβ(1-XX) is the product of cleavage at the β-site, whereas Aβ(11-XX) is the product of cleavage at the β′-site. C, levels of Aβ(11-XX) production from each APP in cells. Aβ(11–40)/Aβ(1–40) ratios from human and mouse APP in N2a and SHSY5Y cells were compared (left), along with the Aβ(11–34)/Aβ(1–34) ratio from hAPP and mAPP in N2a cells (right). Data are shown as mean ± S.E. Statistical significance was examined by Student's t test (n = 3), and p values are indicated (*, p < 0.05; ****, p < 0.0001).
To explore this issue, we first compared the generation of Aβ from mAPP and hAPP (Fig. 1B). To this end, we transiently expressed mAPP-(695) and hAPP-(695) in mouse N2a and human SHSY5Y neuroblastoma cells, and then analyzed the secreted Aβ forms by MALDI-TOF/MS following immunoprecipitation with pan-Aβ antibody 4G8, which recognizes an epitope within the Aβ(17–24) region. We identified several Aβ species in which the amino-terminal amino acid was Asp (Aβ(1-XX), a product of β-site cleavage) or Glu (Aβ(11-XX), a product of β′-site cleavage). For convenience, we focused our investigation of alternative APP cleavage by BACE1 on Aβ(1–40) and Aβ(11–40). In both human and mouse cells, much more Aβ(1–40) was generated from hAPP than Aβ(11–40). By contrast, Aβ(11–40) was the predominant form generated from mAPP in both types of cells (Fig. 1C, left). The preferential cleavage of mAPP at the β′-site was also observed for Aβ(11–34) and Aβ(1–34) in mouse cells (Fig. 1C, right), but these were not quantified in human cells because of less amounts. Non-transfected cells did not yield detectable Aβ signals in these MS spectra. These results indicate that mAPP is subject to amyloidolytic rather than amyloidogenic cleavage by both human and mouse BACE1. In other words, the difference between hAPP and mAPP is not due to species-specific differences in BACE1. Instead, the alternative cleavage of APP by BACE1, i.e. the selection of either the β- or β′-site, depends on the amino acid sequence within the Aβ region.
This was further confirmed by a in vitro β-secretase assay with substrate peptides of human and mouse amino acid sequences (Fig. 2). Human and mouse APP-(662–691) peptides (Fig. 2A) were incubated with a recombinant human BACE1, and the generated peptides cleaved at the β′-site (APP-(662–681)) and β-site (APP-(672–691)) were analyzed and quantified. In vitro assay, the β-site cleavage was predominant in both human and mouse substrates (Fig. 2B, left). However, the ratio of β′-cleavage to β-cleavage (β′/β) was significantly high in mouse substrate compared with human (Fig. 2B, right). The result supports the conclusion that the mouse APP sequence is preferentially cleaved at the β′-site by BACE1 in the cell (Fig. 1), although the magnitude of preferential β′-site cleavage is slightly different in an in vitro study from a cell study, and we cannot exclude an affect of second cleavage of the first cleaved product.
FIGURE 2.

Cleavage of peptides including the human and mouse Aβ sequence by human BACE1 in vitro. A, amino acid sequences of substrate human peptide and peptides cleaved at β- and β′-site by BACE1. When substrate APP-(662–691) is cleaved at β-site or β′-site, the generated APP-(672–691) and APP-(662–681) are shown, respectively. B, representative MS spectra of products cleaved at β- and β′-sites of human and mouse substrates as indicated in panel A. Spectra of human (upper) and mouse (lower) substrates were shown (β, peptide cleaved at β-site; β′, peptide cleaved at β′-site). Levels of the peptide cleaved at the β′-site are indicated as the ratio to that of the peptide cleaved at β-site, a β′/β ratio, and compared between human and mouse substrate. Data are shown as mean ± S.E. Statistical significance was examined by Student's t test (n = 4), and p values are indicated (**, p < 0.01).
To confirm this conclusion, we next investigated whether amino acid substitutions within the Aβ sequence would influence alternative selection of BACE1 cleavage sites. We focused on three amino acid positions within mouse Aβ, which differs from human Aβ by R676G, Y681F, and H684R (Fig. 1A). hAPP harboring the single mutation R676G, Y681F, or H684R, or the triple mutation R676G/Y681F/H684R (3mut, i.e. fully recapitulating the mouse sequence) were expressed in N2a cells along with innate hAPP and mAPP, and the secreted forms of Aβ in the medium were analyzed (Fig. 3A). The Aβ(11–40)/Aβ(1–40) ratio was compared with the ratio of Aβ secreted from cells expressing innate hAPP and mAPP (Fig. 3B). Among the single-substitution mutants, hAPP H684R had a significantly elevated Aβ(11–40)/Aβ(1–40) ratio; however, the ratio was lower than those of hAPP-3mut and innate mAPP. hAPP R676G showed a tendency an elevated Aβ(11–40)/Aβ(1–40) ratio, but did not have a significantly higher Aβ(11–40)/Aβ(1–40) ratio. The result indicates that the amino acid at position 684 plays an important role in the alternative selection of BACE1 cleavage sites of hAPP, although we cannot rule out a slight contribution of amino acid at position 676. Changes in amino acids within the Aβ region can increase the balance between amyloidogenic and amyloidolytic cleavage, in other words, the amino acid sequence within the Aβ region can determine the alternative of cleavage sites by BACE1.
FIGURE 3.

Alternative cleavage of human and mouse APP by β-secretase. A, representative MS spectra of Aβ with amino-terminal β- and β′-cleavage sites generated from hAPP (with or without amino acid substitutions found in the mouse protein) and mAPP. Spectra of Aβ species from hAPP (a, WT), hAPP R676G (b), hAPP Y681F (c), hAPP H684R (d), hAPP R676G/Y681F/H684R (e, 3mut), and innate mAPP (f, WT) expressed in N2a cells were analyzed by IP-MS. B, levels of Aβ(11-XX) production from each APP are indicated as the Aβ(11–40)/Aβ(1–40) ratio. Data are shown as mean ± S.E. Statistical significance to WT was examined by Dunnett's multiple comparisons test (n = 5), and p values are indicated (***, p < 0.001).
The Protective A673T Mutation, but Not the A673V Mutation, within the Aβ Sequence Promotes β′-Site Cleavage of hAPP by BACE1
FAD-associated mutations of the APP gene include some variations within the Aβ sequence. Substitution of Val for Ala673 (A673V) is pathogenic, resulting in elevated production of Aβ, whereas substitution of Thr at the same position (A673T, the Icelandic mutation) is protective (23, 25). However, the molecular mechanism underlying altered Aβ generation remains controversial, especially regarding the protective A673T mutation.
To investigate the role of the A673T mutation, we examined Aβ generation in N2a cells expressing hAPP harboring A673T or A673V along with the wild-type hAPP (WT) and hAPP harboring the Swedish mutation (K670N/M671L), which is predominantly cleaved at the β site (Fig. 4). Levels of APP were identical in cells expressing each variant except for the Swedish mutation, which is susceptible to β-site cleavage (Fig. 4A). The CTFβ/C99 level of hAPP A673T was very similar to that of hAPP (WT); however, as reported (25), generation of Aβ(1–40) decreased significantly, and the level of Aβ(1–42) tended to decrease (Fig. 4B). The Swedish mutation, used as a positive pathogenic control, markedly promoted formation of both CTFβ/C99 and Aβ(1–40) and Aβ(1–42) compared with hAPP (WT), consistent with earlier work (32–34). As expected from a previous report (35), the A673V mutation was pathogenic, with elevated CTFβ/C99 and Aβ(1–40) generation, although the slight increase in Aβ(1–42) generation was not significant.
FIGURE 4.

Alternative cleavage of human APP harboring FAD mutations by β-secretase, and the resultant generation of Aβ. A, levels of APP and APP CTFs in cells. N2a cells expressing hAPP (WT), Swedish hAPP K670N/M671L, hAPP A673V, or Icelandic hAPP A673T were lysed, and cell lysates (10 μg of protein) were analyzed by Western blotting with anti-APP antibody and anti-α-tubulin antibody (to confirm equal protein loading). Mature (N- and O-glycosylated form) and immature (N-glycosylated form) hAPP (upper), CTFβ/C99, CTFβ′/C89, and CTFα/C83 (middle), and α-tubulin (lower) are indicated. Numbers indicate molecular size markers (in kDa). The levels of APP and APP CTFβ/C99 are indicated as ratios relative to the levels of hAPP (WT), which was assigned a reference value 1.0. Statistical analysis was performed using Dunnett's multiple comparison test. p values are provided for comparison with the hAPP wild-type (WT) (mean ± S.E., n = 6; *, p < 0.05). B, levels of Aβ(1–40) and Aβ(1–42) secreted from cells. Aβ levels in culture media of cells expressing hAPP variants, as indicated in panel A, were assayed by sELISA. Statistical analysis was performed using Dunnett's multiple comparison test. p values are provided for the comparison with the hAPP wild-type (WT) (mean ± S.E., n = 6; **, p < 0.01; ***, p < 0.001). C, representative MS spectra of Aβ with amino-terminal β- and β′-cleavage sites generated from hAPP with or without FAD mutation. Aβ(1-XX) is the product of cleavage at the β-site, whereas Aβ(11-XX) is the product of cleavage at the β′-site. D, relative levels of Aβ(11–40) production and the Aβ(11–40)/Aβ(1–40) ratio. Levels of Aβ(11–40) production from each APP are indicated as ratios relative to the level of hAPP (WT), which was assigned a reference value of 1.0 (left). The Aβ(11–40)/Aβ(1–40) ratio is also indicated as a relative ratio (right). Statistical significance was determined by Dunnett's multiple comparisons test (n = 6), and p values are indicated (**, p < 0.01; ***, p < 0.001). Data are shown as mean ± S.E.
Next, we analyzed the alternative cleavage of hAPP harboring A673T and A673V by BACE1 (Fig. 4C). Interestingly, the β′-site cleavage product Aβ(11–40) was present at significantly higher levels in cells expressing hAPP A673T: Aβ(11–40) production was 2.5-fold higher, and the Aβ(11–40)/Aβ(1–40) ratio was 3-fold higher, than in cells expressing hAPP (WT) (Fig. 4D). By contrast, cells expressing hAPP A673V significantly decreased Aβ(11–40) production, as did those expressing the Swedish mutation; in both cases, the Aβ(11–40)/Aβ(1–40) ratio was significantly lower than that of hAPP (WT). This result clearly shows that the protective effect of hAPP A673T is due to elevated β′-site cleavage of hAPP, resulting in higher levels of Aβ(11-XX), rather than to the metabolic lability of Aβ(1-XX) containing the A673T mutation. A673V is pathogenic, but the detailed mechanism underlying elevated Aβ generation was unclear (23). In this analysis, we clarified that the pathogenic effect of A673V is mediated by a decrease in β′-site cleavage of hAPP along with an increase in β-site cleavage, resulting in significantly higher levels of Aβ(1–40) and a slight increase in the level of Aβ(1–42).
Elevated BACE1 Activity Cleaves CTFβ/C99, the Precursor of Aβ(1-XX), to Generate Aβ(11-XX)
BACE1 activity generates the amyloidogenic fragment CTFβ/C99 from hAPP. This β-site cleavage of hAPP is facilitated by pathogenic mutations that attenuate β′ cleavage of hAPP. Because BACE1 can use CTFβ/C99 as well as APP as substrates (12), we investigated whether elevated BACE1 activity can cleave CTFβ/C99 at the β′-site subsequent to the primary cleavage. BACE1 overexpression was previously reported to decrease brain Aβ levels in vivo (36), but the mechanism remains controversial.
For these experiments, we expressed hAPP and CTFβ/C99 in N2a cells with or without BACE1. Protein levels of hAPP, CTFβ/C99, and BACE1 were confirmed by Western blotting (Fig. 5A). hAPP can exist in both mature (N- and O-glycosylated) and immature (N-glycosylated) forms (reviewed in Ref. 26). Overexpression of BACE1 increases formation of CTFβ/C99 and decreases the proportion of mature hAPP (Fig. 5B). Because only mature APP is a bona fide substrate of APP secretases (37), almost all mature hAPP is subject to cleavage upon overexpression of BACE1, along with less cleavage by endogenous APP α-secretase. Overexpression of BACE1 decreases the level of mature APP by disturbing axonal transport in the brain in vivo (36); however, based on our observations in undifferentiated N2a cells (Fig. 5A), it is reasonable to speculate that the decrease in the level of mature hAPP is due to proteolysis by overexpressed BACE1 along with elevated generation of CTFβ/C99 and CTFβ′/C89.
FIGURE 5.

Elevated BACE1 activity cleaves CTFβ/C99 at the β′-site to generate Aβ(11-XX). A, protein levels of APP, APP CTFs, and BACE1 in cells. N2a cells were expressed with hAPP or hAPP CTFβ/C99 in the presence (+) or absence (−) of BACE1-FLAG expression. Cell lysates (10 μg of protein) were analyzed by Western blotting with anti-APP, anti-FLAG, and anti-actin antibodies. hAPP (first row), CTFβ/C99, CTFβ′/C89, and CTFα/C83 (second row), BACE1 (third row), and actin (fourth row) are indicated. Numbers indicate molecular size markers (in kDa). B, the levels of APP and APP CTFs derived from hAPP are indicated as ratios relative to the levels of hAPP and CTFs without (−) BACE1-FLAG expression, which was assigned a reference value 1.0. C, the levels of APP CTFs derived from hC99 are indicated as ratios relative to the levels of CTFs without (−) BACE1-FLAG expression, which was assigned a reference value 1.0. Statistical analysis was performed using Student's t test. p values are provided for the comparison with the value in the absence of BACE1 expression (mean ± S.E., n = 6; *, p < 0.05; ***, p < 0.001; ****, p < 0.0001). D, representative MS spectra of Aβ with amino-terminal β- and β′-cleavage sites generated from hAPP and CTFβ/C99 (left, sets of spectra). Culture media of cells expressing hAPP (left) and CTFβ/hC99 (right) in the presence (lower) and absence (upper) of BACE1 expression were analyzed by IP-MS. Aβ(1-XX) is the product of cleavage at the β-site, whereas Aβ(11-XX) is the product of cleavage at the β′-site. Relative levels of Aβ(11–40)/Aβ(1–40) ratio from hAPP and CTFβ/hC99 (right figure). The Aβ(11–40)/Aβ(1–40) ratio in cells expressed either hAPP or CTFβ/hC99 were indicated as a relative ratio and compared with (+) or without (−) overexpression of BACE1. Statistical significance was determined by Student's t test (n = 3), and p values are indicated (***, p < 0.001). Data are shown as mean ± S.E.
As expected based on a previous report (12), overexpression of BACE1 significantly increased the level of C89/CTFβ′ (Fig. 5, A and B). We speculated that CTFβ/C99 might be further cleaved at the β′-site due to the elevated activity of overexpressed BACE1. We tested this possibility in cells expressing C99/CTFβ with BACE1. Cells overexpressing BACE1 contained lower levels of CTFβ/C99 but dramatically higher levels of CTFβ′/C89 (Fig. 5C), indicating that CTFβ/C99 was cleaved again at the β′-site by elevated BACE1 activity, as reported (12). To confirm this conclusion, we analyzed Aβ species secreted from cells that did or did not overexpress BACE1 (Fig. 5D). In medium from cells expressing hAPP without BACE1 overexpression, Aβ(1–40) was again the major form, accompanied by a small amount of Aβ(11–40). By contrast, in cells overexpressing BACE1, Aβ(1–40) generation was remarkably reduced and a great deal of Aβ(11–40) was detected, consistent with the results of Western blot analysis (Fig. 5D). In the medium of cells expressing CTFβ/C99, overexpression of BACE1 remarkably decreased Aβ(1–40) generation, but increased the generation of Aβ(11–40) (Fig. 5D). The elevated Aβ(11–40)/Aβ(1–40) ratio in cells with BACE1 overexpression was statistically significant in cells expressed by either hAPP or CTFβ/C99 (Fig. 5D, right). These results clearly showed that CTFβ/C99, the product of hAPP primarily cleaved at the β-site by BACE1, can be further cleaved at the β′-site by excess BACE1 activity. These data suggest that excess BACE1 activity functions amyloidolytic instead of amyloidogenic, at least under our experimental conditions, rather than by decreasing the levels of its substrates (mature APP and CTFβ/C99) by altering the intracellular APP transport system in in vivo neurons (36).
Next, we performed similar studies on hAPP harboring pathogenic mutations (Fig. 6). Again, N2a cells expressed hAPP in the presence or absence of BACE1 overexpression. Expression of BACE1 decreased APP, irrespective of the presence of a FAD mutation (Fig. 6, A and B), as shown in Fig. 5. Cells expressing hAPP with FAD mutations expressed significantly (Swedish; K670N/M671L) or slightly without a statistically significance (Leuven; E682K) higher level of CTFβ/C99 compared with those expressing hAPP (WT) (Fig. 6C, left). Moreover, overexpression of BACE1 remarkably increased the level of CTFβ′/C89 in cells expressing hAPP, irrespective of the presence of a FAD mutation (Fig. 6C, middle). We also examined Aβ species in the medium (Fig. 6D). When BACE1 was overexpressed, the ratio of Aβ(1–40) to Aβ(11–40) significantly decreased, whereas the Aβ(11–40)/Aβ(1–40) ratio increased significantly, in cells expressing hAPP (WT) and hAPP (Swedish) (Fig. 6D, right). In cells expressing hAPP (E682K), the level of Aβ(1–40) was greatly decreased by BACE1 overexpression, as in the case of hAPP (WT) and hAPP (Swedish); unexpectedly, however, no increase in Aβ(11–40) was detected (Fig. 6D). Position 11 of the Aβ sequence is Glu682, the site of the Leuven mutation. CTFβ′/C89 with substitution of Lys for Glu at Aβ position 11 (amino terminus of Aβ(11-XX)) may be preferentially cleaved by α-secretase to generate CTFα/C83. In other words, Aβ(11–40) harboring the Leuven E682K mutation is more likely to be cleaved by α-secretase; therefore, no increase in the level of Aβ(11–40) was detected in the medium of cells expressing hAPP E682K, in contrast to cells expressing hAPP (WT) and hAPP (Swedish), which have Glu at position 682 (Fig. 6D). The preferential cleavage of CTFβ′/C89 (including E682K) by α-secretase was reflected by the significantly higher level of CTFα/C83 in cells overexpressing BACE1 (Fig. 6C, right). Overexpression of BACE1 decreased the level of CTFα/C83, significantly in hAPP (Swedish) and with a trend of decrease in hAPP (WT) (Fig. 6C, right), because mature APP was predominantly cleaved by overexpressed BACE1 but not by α-secretase (Figs. 5A and 6A). Subsequently, the resultant CTFβ′/C89 was further cleaved by γ-secretase to yield Aβ(11-XX). By contrast, the level of CTFα/C83 derived from hAPP (E682K) was equivalent to that in cells overexpressing BACE1 (Fig. 6C, right). Based on these observations, it is reasonable to explain that the higher level of CTFα/C83 derived from hAPP (E682K) in BACE1-overexpressing cells was due to α-site cleavage of CTFβ′/C89.
FIGURE 6.

Elevated BACE1 activity cleaves hAPP at the β′-site to decrease the level of Aβ(1-XX), even if hAPP harbors FAD mutations. A, protein levels of APP, APP CTFs, and BACE1 in cells. N2a cells expressed hAPP with or without FAD mutation in the presence (+) or absence (−) of BACE1-FLAG expression. Cell lysates (10 μg of protein) were analyzed by Western blotting with anti-APP, anti-FLAG, and anti-actin antibodies. hAPP (first row), CTFβ/C99, CTFβ′/C89, and CTFα/C83 (second row), BACE1 (third row), and actin (fourth row) are indicated. Numbers indicate molecular size markers (in kDa). B, the levels of APP are indicated as ratios relative to the levels of hAPP (WT) without BACE1 expression, which was assigned a reference value 1.0. Statistical analysis was performed by Student's t test. p values are provided for the comparison with the APP without (−) BACE1 expression (mean ± S.E., n = 6; **, p < 0.001; ****, p < 0.0001). C, the levels of APP CTFs are indicated as ratios relative to the levels of hAPP without BACE1 expression, which was assigned a reference value 1.0. Statistical analysis was performed by Student's t test. p values are provided for comparison with the CTF without (−) BACE1 expression (mean ± S.E., n = 6; *, p < 0.05; **, p < 0.01; ***, p < 0.001). In another case, statistical analysis was performed using Dunnett's multiple comparison test. p values are provided for comparison with the hAPP wild-type (WT) (mean ± S.E., n = 6; ##, p < 0.001; ###, p < 0.001). D, representative MS spectra of Aβ with amino-terminal β- and β′-cleavage sites generated from innate hAPP (WT) or hAPP harboring the Swedish (middle) or E682K FAD mutation. Culture media of cells expressing wild-type hAPP (upper), hAPP Swedish (middle), and hAPP E682K (lower) in the presence (right) and absence (left) of BACE1 expression were analyzed by IP-MS. Aβ(1-XX) is the product of cleavage at the β site, whereas Aβ(11-XX) is the product of cleavage at the β′-site. Changes of the Aβ(11–40)/Aβ(1–40) ratio from hAPP harboring FAD-associated mutations. The Aβ(11–40)/Aβ(1–40) ratios were indicated as a relative ratio and compared with (+) or without (−) overexpression of BACE1. Statistical significance was determined by Student's t test (n = 3), and p values are indicated (***, p < 0.001). Data are shown as mean ± S.E.
BACE1 can cleave the peptide bond between hAPP Leu705 (position 34 in Aβ) and Met706 (position 35 in Aβ) (38). Accordingly, as shown in Figs. 5D and 6D, cells overexpressing BACE1 contained higher levels of Aβ(1–34) and Aβ(11–34) fragments. Overexpression of BACE1 promoted cleavage at this position to increase production of Aβ(1–34) and Aβ(11–34). Again, Aβ(11–34) was not detectable in medium of cells expressing hAPP E682K (Fig. 6D). We speculate that, like Aβ(11–40), Aβ(11–34) harboring E682K was cleaved preferentially by α-secretase. Therefore, the excess activity of BACE1 can function amyloidolytic, at least in cell studies, even in APP harboring pathogenic variations such as the Swedish and Leuven mutations.
In Mouse, Aβ(11–40) Is Less Abundant Than Aβ(1–40) in Vivo
We showed above that mAPP is predominantly cleaved at the β′-site in cells, resulting in preferential generation of Aβ(11-XX) rather than Aβ(1-XX) as observed typically in Aβ(11–40) and Aβ(1–40) species (Fig. 1). To extend this finding, we asked whether Aβ(11–40) is also predominantly generated from endogenous APP in mouse neurons in vivo (Fig. 7). Using IP-MS, we analyzed the Aβ forms present in the medium of mouse primary mixed cultured neurons (cerebrum cortex plus hippocampus) and in the adult mouse CSF (mixture of 3 individuals 6 months old), as well as those in the medium of N2a cells overexpressing mAPP. As shown in Fig. 1, N2a cells again generated more Aβ(11–40) than Aβ(1–40). Mouse neurons also expressed Aβ(11–40) as the major form, as reported previously (29). Unexpectedly, mouse CSF included Aβ(1–40) as the major form, and the level of Aβ(11–40) was remarkably lower (Fig. 7A). The Aβ(11–40)/Aβ(1–40) ratio was clearly lower in CSF when compared with those generated N2a cells and primary cultured neurons. Although the analysis is a duplicate study without statistical analysis, and we cannot rule out a possibility that the Aβ species generated from adult neurons may differ from those secreted from embryonic cultured neurons, these data suggest that mouse neurons predominantly generate Aβ(11-XX) rather than Aβ(1-XX), but that Aβ(11-XX) is metabolically labile in vivo and subject to degradation or faster clearance than Aβ(1-XX).
FIGURE 7.
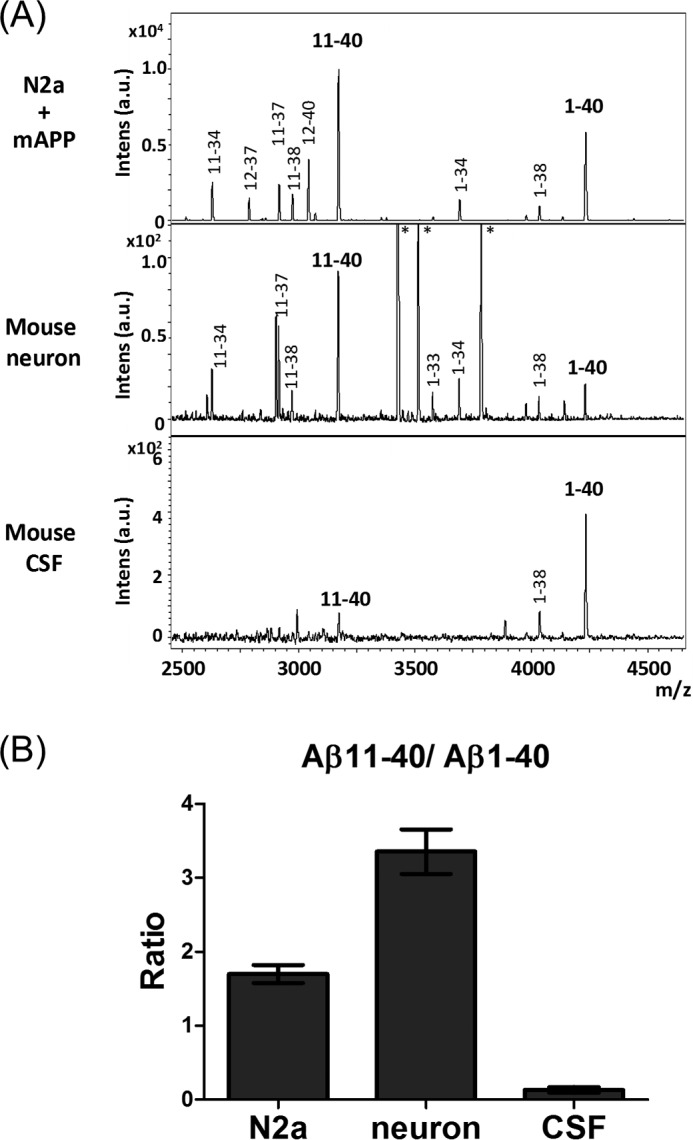
Comparison of alternative products of mAPP cleaved at the β- and β′-site in cells and in vivo. A, representative MS spectra of Aβ with amino-terminal β- and β′-cleavage sites secreted into the culture media from N2a cells expressing mAPP (upper), culture media from mouse primary neurons (middle), and mouse CSF (lower). Samples were analyzed by IP-MS. Aβ(1-XX) is the product of cleavage at the β site, whereas Aβ(11-XX) is the product of cleavage at the β′-site. Asterisk (*) indicates a nonspecific product. B, relative levels of Aβ(11–40)/Aβ(1–40) ratio in cultured medium of N2a cell, mouse primary neurons, and adult mouse CSF (n = 2).
To explore this possibility that Aβ(11–40) is less stable than Aβ(1–40) in vivo, Aβ(1–40) and Aβ(11–40) were incubated in vitro with neprilysin, which is a major extracellular Aβ-degrading enzyme (39), and lability of Aβ peptides was analyzed (Fig. 8). In the presence of neprilysin, the level of Aβ(11–40) lowered quickly and significantly (by 40% at 1 h and 20% at 2 h), whereas the levels of Aβ(1–40) remained over 50% during incubation time. Although the amounts of both peptides tended to decrease in the absence of enzyme, the results were standardized with values of reaction in the absence of neprilysin. Although it may be difficult to exclude other possibilities of the lower level of Aβ(11–40) in CSF (Fig. 7), this in vitro study supports that Aβ(11–40) is less stable rather than Aβ(1–40), at least in degradation by neprilysin. In other words, the Aβ species generated by β′-site cleavage are subject to more rapid clearance than those generated by β-site cleavage.
FIGURE 8.
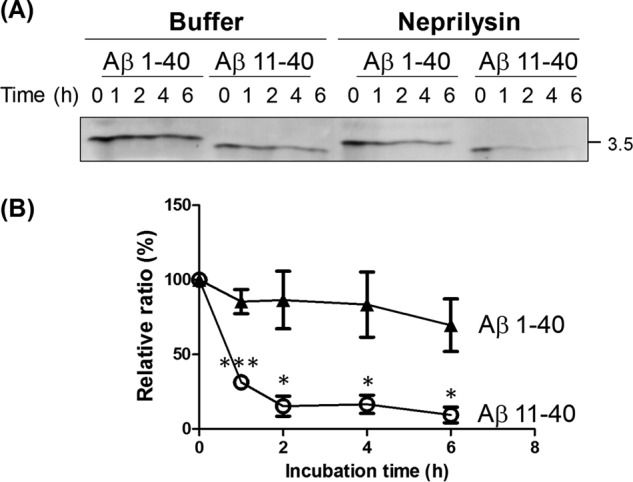
Degradation ratios of Aβ(1–40) and Aβ(11–40) by neprilysin. A, changes of Aβ(1–40) and Aβ(11–40) amounts by incubations with neprilysin. Synthetic human Aβ(1–40) and Aβ(11–40) peptides were incubated with (Neprilysin) or without (Buffer) a recombinant human neprilysin for indicated time (h). The reactions were subject to Western blot analysis with anti-pan Aβ antibody 4G8. The number indicates a molecular size marker (in kDa). B, the levels of Aβ(1–40) (closed triangle) and Aβ(11–40) (open circle) in the presence of neprilysin were standardized by the levels of peptides in the absence of neprilysin, and indicated as ratios to the levels shown in 0 h, which was assigned a reference value of 100 (%). Statistical significance was determined by Student's t test (n = 3), and p values are indicated (*, p < 0.05; ***, p < 0.001). Data are shown as mean ± S.E.
Discussion
It is widely accepted that β-secretase BACE1 is the primary APP-cleaving enzyme responsible for generation of the Aβ(1-XX) species, and that the combined cleavage of APP by BACE1 and the γ-secretase complex generates multiple types of Aβ species with distinct carboxyl termini, e.g. Aβ(1–40) and Aβ(1–42) (reviewed in Refs. 8 and 40). Previous work showed that BACE1 also cleaves APP at the β′-site, but the biological significance of this reaction remained unclear because the β′-cleaved products of hAPP, CTFβ′/C89, and Aβ(11-XX) are scarce relative to CTFβ/C99 and Aβ(1-XX) (12, 41). Along with previously reported data, our results confirm that mAPP is preferentially cleaved at the β′-site within the Aβ sequence and predominantly generates Aβ(11-XX); by contrast, hAPP mostly generates Aβ(1-XX) in vivo and in vitro (29–31).
Over the course of this study, we made several discoveries regarding the role of BACE1 in APP metabolism. First, we showed that alternative selection of cleavage sites by BACE1 in both human and mouse is determined by the amino acid at position 684 (His in human, Arg in mouse). This suggests that some FAD-associated pathogenic and/or protective mutations within the Aβ region of the APP gene influence the selection of cleavage sites by BACE1. Second, we showed that the pathogenic mutation A673V decreased β′-site cleavage of hAPP, whereas the protective Icelandic mutation A673T increased β′-site cleavage. This observation strongly indicates that amino acid substitutions at position 673 influenced the alternative selectivity of the cleavage site by BACE1. In other words, A673V induces the pathogenic β-site cleavage of APP by BACE1, whereas A673T raises the protective β′-site cleavage of APP. This is the primary cause of FAD harboring APP A673V, and this is the primary effect to protect AD in subjects harboring APP A673T. Third, we demonstrated that elevation of BACE1 activity due to overexpression of the enzyme results in secondary cleavage of CTFβ/C99 at the β′-site, dramatically increasing Aβ(11-XX) production. This is true, at least in the cell study, even for APP harboring the Swedish mutation, which predominantly produces Aβ(1-XX), although a pilot study in a BACE-overexpressing transgenic mouse suggested that reduction in Aβ deposition is mediated by another mechanism (36). Finally, we showed that Aβ(11-XX) may be more metabolically labile than Aβ(1-XX) in vivo and in vitro.
Thus, contrary to the popular view, our findings may suggest that activation of BACE1 in AD subjects (including FAD patients), using procedures other than overexpression of BACE1, represents a promising target for AD therapies aimed at decreasing the level of Aβ. Development of inhibitors of BACE1 and/or γ-secretase activities has been pursued since these enzymes were identified (reviewed in Ref. 42). However, we believe that more attention should have been devoted to γ-secretase inhibitors. It is reasonable to predict that attenuation of γ-secretase activity by an inhibitor would decrease generation of neurotoxic Aβ generation. In contrast to this early idea, recent progress in understanding the molecular mechanism by which γ-secretase cleaves APP suggested to us that attenuating γ-secretase activity would result in reduced production of Aβ(1–38) and elevated production of Aβ(1–42), which is a precursor of Aβ(1–38) and is more neurotoxic than Aβ(1–38) and Aβ(1–40) (43) (reviewed in Ref. 44).
Our results suggest that β-secretase inhibitors may face some of the same problems as γ-secretase inhibitors. Suppression of APP cleavage using a BACE1 inhibitor is a potential therapeutic strategy for decreasing Aβ generation. However, many other substrates of BACE1 have been reported to date, suggesting that BACE1 inhibition could have considerable side effects. Nevertheless, suppression of BACE1 activity specifically in the brain may be a practical means for treating AD patients (reviewed in Ref. 42). Our results show that cleavage of APP by excess BACE1 activity can degrade Aβ by cleaving APP at the β′-site. Moreover, if CTFβ/C99 is abundant in FAD subjects, sufficient levels of BACE1 activity could cleave CTFβ/C99 again at the β′-site to generate Aβ(11-XX), which is more metabolically labile. Consequently, the total amount of Aβ in the brain would decrease, as demonstrated in BACE1 transgenic mice (36). Because Aβ(1–34) is degraded by neprilysin, an Aβ-degrading enzyme, faster than Aβ(1–40) (45), Aβ(11-XX) may also be degraded more easily by such enzymes than Aβ(1-XX) (46). At least we showed that Aβ(11–40) was degraded quickly rather than Aβ(1–40) in a study in vitro. Therefore, to treat AD patients (e.g. FAD subjects harboring the Swedish mutation), activation of BACE1 might be a more effective therapy than administration of a BACE1 inhibitor. Moreover, in contrast to development of γ-secretase modulators that enhance the peptidase-like activity of γ-secretase to reduce Aβ(1–42) generation while promoting formation of Aβ(1–38), it may be difficult to develop a drug that modifies the selectivity of BACE1 because selection of a cleavage site (i.e. the β- or β′-site) depends on the sequence of the substrate Aβ domain. However, we cannot rule out other possibilities that the intracellular environment of BACE1 can influence in the selectivity of cleavage site of APP by BACE1.
In general, it is possible that both BACE1 and γ-secretase activities may decrease with age, although at least one report showed that the BACE1 level is elevated in AD (47). Reduction of BACE1 activity may attenuate the amyloidolytic β′-site cleavage of APP relative to amyloidogenic β-site cleavage, resulting in the generation of Aβ(1-XX). Attenuation of γ-secretase activity, especially its carboxypeptidase-like activity, promotes production of Aβ(XX-42) at the expense of Aβ(XX-38). Therefore, the combination of altering and/or weakening the activities of both BACE1 and γ-secretase increases production of the most neurotoxic species, Aβ(1–42), whereas decreasing production of Aβ(11-XX) and Aβ(XX-38) species. Based on our understanding of the mechanisms of APP cleavage by BACE1 and γ-secretase, administration of compounds to regulate BACE1 and γ-secretase activities to AD subjects should proceed with scrupulous caution.
Experimental Procedures
Plasmid Construction, Expression in Cells, and Western Blot Assays
Cloning of human APP-(695) into pcDNA3, yielding pcDNA3-hAPP695, was described previously (48). Various mutant hAPP plasmids harboring amino acid substitutions were generated from pcDNA3-hAPP-(695) by oligonucleotide-based PCR mutagenesis. pcDNA3.1-hC99 was prepared by PCR as described previously. The encoded protein, SPA4CT-DA, consists of the APP signal peptide fused to the APP C-terminal 99 region, separated by a dipeptide linker Leu + Glu (49). pcDNA3.1-hBACE1 was kindly supplied by Dr. Robert W. Doms (50), and recloned into pcDNA3.1 with a 5′ Kozak sequence and a 3′ FLAG tag sequence to generate pcDNA3.1-hBACE1-FLAG. Mouse APP-(695) and BACE1 were cloned from mouse brain mRNA by RT-PCR and inserted into pcDNA3 at the HindIII/XbaI sites to generate pcDNA3-mAPP-(695), or inserted into pcDNA3.1 with a 5′ Kozak sequence and a 3′ FLAG tag sequence at the BamHI/NotI sites to generate pcDNA3.1-mBACE1-FLAG.
Mouse Neuro 2a (N2a) and human SHSY5Y neuroblastoma cell lines were cultured in DMEM (Wako Pure Chemicals, Osaka, Japan) supplemented with 10% (v/v) fetal bovine serum (MP Biomedicals, Santa Ana, CA). Cells (0.3–1.0 × 106) were transfected with plasmids (total 0.4 μg) in Lipofectamine 2000 (Invitrogen/Thermo Fisher Scientific, Carlsbad, CA). After 24 h, the cells were harvested and lysed in a radioimmune precipitation assay (RIPA) buffer (50 mm Tris-HCl, pH 8.0, 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate, 1% Nonidet P-40, and 150 mm NaCl). The proteins from cell lysates (∼10 μg of protein/lane) were separated by electrophoresis on a 9% (w/v) polyacrylamide Tris glycine gel, or on a 17.5% (w/v) Tris-Tricine gel for APP CTFs, transferred onto a nitrocellulose membrane, incubated with the indicated antibodies, and detected by ECL (GE Healthcare Bio-Sciences, Little Chalfont, UK).
Immunoprecipitation MALDI-TOF/MS (IP-MS) and -MS/MS Analysis and Quantitation of Aβ Species
Aβ peptides secreted into the medium (1 ml) and mixed mouse CSF (C57BL/6, 100 μl) were recovered by immunoprecipitation with anti-pan Aβ antibody 4G8, which was raised to Aβ17–24 epitope (BioLegend, San Diego, CA) plus Protein G-Sepharose 4B (GE Healthcare Bio-Sciences) following pre-clearing with the same beads without antibody. The beads were sequentially washed, and the bound proteins were eluted with trifluoroacetic acid/acetonitrile/water (1:20:20) saturated with sinapinic acid as described (51). The dissolved samples were dried on a target plate, and matrix-assisted laser desorption ionization time-of-flight tandem mass spectrometry (MALDI-TOF/MS) analysis was performed using an UltraflexII TOF/TOF (Bruker Daltonics, Bremen, Germany). MS/MS analysis was performed to confirm the amino acid sequence of Aβ(11–40) and Aβ(1–40) (supplemental Fig. S1). The quantitative accuracy of mass spectrometric analysis was confirmed by studies with a mixture of synthetic human Aβ(1–40) and Aβ(11–40) peptides (supplemental Fig. S2) with a procedure described previously (51, 52). Molecular masses were calibrated using a peptide calibration standard (Bruker Daltonics). Secretion of Aβ40 and Aβ42 into the medium was quantitated by sandwich ELISA (sELISA) as described previously, except that biotinylated 6E10 antibody (catalog number 803003; BioLegend), which was raised Aβ(1–16), was used instead of the 2D1 antibody, which was raised to Aβ(1–27) (37).
In Vitro β-Secretase Assay
Synthetic human and mouse substrate peptides, APP-(662–691) (GeneScript, Piscataway, NJ), were synthesized. Substrate (10 μm) in a reaction buffer (100 mm sodium acetate, pH 4.5) including 4 μg of recombinant human BACE1 (catalog number. 931-AS; R&D Systems, Minneapolis, MN) was incubated at 37 °C for 12 h. To the reaction mixture, 0.8 μl of 0.2% trifluoroacetic acid (TFA) (v/v) and 8.6 ng of p3-Alcβ40 (51) as an internal standard peptide were added, subject to desalination with Zip Tip C18 (Millipore/Sigma), and eluted with 2 μl of TA solution (0.1% TFA and 50% acetonitrile) saturated with α-cyano-4-hydroxycinnamic acid. The sample was dried on a target plate, washed with washing buffer (10 mm ammonium phosphate in 0.1% TFA), and subject to MALDI-TOF/MS analysis using Ultraflex II TOF/TOF (Bruker Daltonics). The cleaved peptides, APP-(672–691) and APP-(662–681), were quantified with the amount of internal control peptide p3-Alcβ40.
Antibodies and Mice
Mouse monoclonal anti-pan Aβ 4G8 (catalog number 800702; BioLegend, San Diego, CA), which recognizes the amino acid sequence in Aβ(17–24), anti-α-tubulin DM1A (catalog number SC-32293; Santa Cruz Biotechnology, Santa Cruz, TX), anti-actin C4 (catalog number MAB1501; Chemicon/Millipore, Billerica, MA), and anti-FLAG M2 (catalog number F1804; Sigma) antibodies were purchased from the indicated suppliers. Rabbit polyclonal anti-APP cytoplasmic G369 antibody was kindly supplied by Dr. Sam Gandy (53). HRP-linked sheep anti-mouse IgG (catalog number NA9310) and anti-rabbit IgG (catalog numbers NA-9340) antibodies were purchased from GE Healthcare Bio-Sciences.
Studies with mice were conducted in compliance with the guidelines of the Animal Studies Committee of Hokkaido University. The C57BL/6 mice used in this study were housed in a specific pathogen-free environment with MicroVent units (Allentown Inc., Allentown, NJ) throughout the study period.
Mouse Primary Cultured Neurons
The primary culture of mixed mouse cortical and hippocampal neurons was performed with a modified version of the method described by Bartlet and Banker (54). C57BL/6 wild-type were used as described (55). In brief, the cortex and hippocampus of mice at embryonic day 15.5 were dissected, and neurons were spread in a buffer containing papain and cultured at 5 × 104 cells/cm2 in a medium composed of Neurobasal Medium (Life Technologies) containing 30% Nerve-Cell Culture Medium (DS Pharma Biomedical), 2% B-27 supplement (Thermo Fisher Scientific), Glutamax I (4 mm), 5% heat-inactivated horse serum (Thermo Fisher Scientific), and antibiotics (Thermo Fisher Scientific) on poly-d-lysine-treated plates for 5 days. All animal studies were conducted in compliance with the guidelines of the Animal Studies Committee of Hokkaido University. Mice were housed in a specific pathogen-free environment.
Degradation of Aβ(1–40) and Aβ(11–40) by Neprilysin in Vitro
Aβ(1–40) and Aβ(11–40) (15 ng; GeneScript, Piscataway, NJ) were incubated in a buffer (50 mm Tris-HCl, pH 7.5, including 0.4% bovine serum albumin) with or without 1.5 ng of recombinant Neprilysin (catalog number 1182-ZNC; R&D Systems) at 37 °C for the indicated times. Peptides in the reaction tube were analyzed by Western blotting with anti-pan-Aβ antibody 4G8.
Statistical Analysis
Statistical analyses were performed with GraphPad Prism (GraphPad Software, San Diego, CA). The threshold for significance is indicated with p value.
Author Contributions
A. K., S. H., and T. S. participated in the design of study, and A. K. and S. H. carried out all studies. T. S. conceived the study, and T. S. and S. H. wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
This work was supported in part by Grants-in-aid for Scientific Research 262930110 and 16K14690 (to T. S.) and 15K18854 (to S. H.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the Bilateral Joint Research Project of the Japan Society for the Promotion of Science (to S. H.). The authors declare that they have no conflict of interest with the contents of this article.

This article contains supplemental Figs. S1 and S2.
- BACE1
- β-site APP-cleaving enzyme 1
- AD
- Alzheimer's disease
- FAD
- familial Alzheimer's disease
- APP
- amyloid β-protein precursor
- Aβ
- amyloid β-protein
- sAPP
- soluble large extracellular N-terminal domain of APP truncated at the primary cleavage site
- CSF
- cerebrospinal fluid
- CTF
- C-terminal fragment of APP truncated at the primary cleavage site
- IP-MS
- immunoprecipitation MALDI-TOF/MS
- 3mut
- triple mutation R676G/Y681F/H684R
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Hussain I., Powell D., Howlett D. R., Tew D. G., Meek T. D., Chapman C., Gloger I. S., Murphy K. E., Southan C. D., Ryan D. M., Smith T. S., Simmons D. L., Walsh F. S., Dingwall C., and Christie G. (1999) Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol. Cell Neurosci. 14, 419–427 [DOI] [PubMed] [Google Scholar]
- 2. Sinha S., Anderson J. P., Barbour R., Basi G. S., Caccavello R., Davis D., Doan M., Dovey H. F., Frigon N., Hong J., Jacobson-Croak K., Jewett N., Keim P., Knops J., Lieberburg I., et al. (1999) Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 402, 537–540 [DOI] [PubMed] [Google Scholar]
- 3. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., et al. (1999) Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 4. Yan R., Bienkowski M. J., Shuck M. E., Miao H., Tory M. C., Pauley A. M., Brashier J. R., Stratman N. C., Mathews W. R., Buhl A. E., Carter D. B., Tomasselli A. G., Parodi L. A., Heinrikson R. L., and Gurney M. E. (1999) Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature 402, 533–537 [DOI] [PubMed] [Google Scholar]
- 5. Haass C., Schlossmacher M. G., Hung A. Y., Vigo-Pelfrey C., Mellon A., Ostaszewski B. L., Lieberburg I., Koo E. H., Schenk D., Teplow D. B., and Selkoe D. J. (1992) Amyloid β-peptides is produced by cultured cells during normal metabolism. Nature 359, 322–325 [DOI] [PubMed] [Google Scholar]
- 6. Roher A. E., Lowenson J. D., Clarke S., Wolkow C., Wang R., Cotter R. J., Reardon I. M., Zürcher-Neely H. A., Heinrikson R. L., Ball M. J., and Greenberg B. D. (1993) Structural alteration in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer's disease. J. Biol. Chem. 268, 3072–3083 [PubMed] [Google Scholar]
- 7. Cole S. L., and Vassar R. (2008) The role of amyloid precursor protein processing by BACE1, the β-secretase, in Alzheimer's disease pathophysiology. J. Biol. Chem. 283, 29621–29625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thinakaran G., and Koo E. H. (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buxbaum J. D., Liu K. N., Luo Y., Slack J. L., Stocking K. L., Peschon J. J., Johnson R. S., Castner B. J., Cerretti D. P., and Black R. A. (1998) Evidence that tumor necrosis factor α converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 273, 27765–27767 [DOI] [PubMed] [Google Scholar]
- 10. Lammich S., Kojro E., Postina R., Gilbert S., Pfeiffer R., Jasionowski M., Haass C., and Fahrenholz F. (1999) Constitutive and regulated α-secretase cleavage of Alzhemer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allinson T. M., Parkin E. T., Turner A. J., and Hooper N. M. (2003) ADAMs family members as amyloid precursor protein α-secretase. J. Neurosci. Res. 74, 342–352 [DOI] [PubMed] [Google Scholar]
- 12. Liu K., Doms R. W., and Lee V. M. (2002) Glu11 site cleavage and N-terminally truncated Aβ production upon BACE overexpression. Biochemistry 41, 3128–3136 [DOI] [PubMed] [Google Scholar]
- 13. Huse J. T., Liu K., Pijak D. S., Carlin D., Lee V. M., and Doms R. W. (2002) β-Secretase processing in the trans-Golgi network preferentially generated truncated amyloid specific that accumulate in Alzheimer's disease brain. J. Biol. Chem. 277, 16278–16284 [DOI] [PubMed] [Google Scholar]
- 14. Meral D., and Urbanc B. (2013) Discrete molecular dynamics study of oligomer formation by N-terminally truncated amyloid β-protein. J. Mol. Biol. 425, 2260–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jonson M., Pokrzywa M., Starkenberg A., Hammarstrom P., and Thor S. (2015) Systematic Aβ analysis in Drosophila reveals high toxicity for the 1–42, 3–42 and 11–42 peptides, and emphasizes N- and C-terminal residues. PLoS ONE 10, e0133272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu K., Solano I., Mann D., Lemere C., Mercken M., Trojanowski J. Q., and Lee V. M. (2006) Characterization of Aβ11–40/42 peptide deposition in Alzheimer's disease and young Down's syndrome brains: implication of N-terminally truncated Aβ species in the pathogenesis of Alzheimer's disease. Acta Neuropathol. 112, 163–174 [DOI] [PubMed] [Google Scholar]
- 17. Lue L. F., Kuo Y. M., Roher A. E., Brachova L., Shen Y., Sue L., Beach T., Kurth J. H., Rydel R. E., and Rogers J. (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., and Masters C. L. (1999) Soluble pool of Aβ amyloid as determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 19. Lesné S. E., Sherman M. A., Grant M., Kuskowski M., Schneider J. A., Bennett D. A., and Ashe K. H. (2013) Brain amyloid-β oligomers in aging and Alzheimer's disease. Brain 136, 1383–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan R., Fan Q., Zhou J., and Vassar R. (2016) Inhibiting BACE1 to reverse synaptic dysfunction in Alzheimer's disease. Neurosci. Biobehav. Rev. 65, 326–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., and Lannfelt L. (1992) A pathogenic mutation for probable Alzheimer's disease in the APP gene at N-terminus of β-amyloid. Nat. Genet. 1, 345–347 [DOI] [PubMed] [Google Scholar]
- 22. Zhou L., Brouwers N., Benilova I., Vandersteen A., Mercken M., Van Laere K., Van Damme P., Demedts D., Van Leuven F., Sleegers K., Broersen K., Van Broeckhoven C., Vandenberghe R., and De Strooper B. (2011) Amyloid precursor protein mutation E682K at the alternative β-secretase cleavage β′-site increase Aβ generation. EMBO Mol. Med. 3, 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Di Fede G., Catania M., Morbin M., Rossi G., Suardi S., Mazzoleni G., Merlin M., Giovagnoli A. R., Prioni S., Erbetta A., Falcone C., Gobbi M., Colombo L., Bastone A., Beeg M., et al. (2009) A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323, 1473–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peacock M. L., Warren J. T. Jr, Roses A. D., and Fink J. K. (1993) Novel polymorphism in the A4 region of the amyloid precursor protein gene in a patient without Alzheimer's disease. Neurology 43, 1254–1256 [DOI] [PubMed] [Google Scholar]
- 25. Jonsson T., Atwal J. K., Steinberg S., Snaedal J., Jonsson P. V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., et al. (2012) A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature 488, 96–99 [DOI] [PubMed] [Google Scholar]
- 26. Suzuki T., and Nakaya T. (2008) Regulation of APP by phosphorylation and protein interactions. J. Biol. Chem. 283, 29633–29637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iijima K., Lee D.-S., Okutsu J., Tomita S., Hirashima N., Kirino Y., and Suzuki T. (1998) cDNA isolation of Alzheimer's amyloid precursor protein from cholinergic nerve terminals of the electric organ of the electric ray. Biochem. J. 330, 29–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Motodate R., Saito Y., Hata S., and Suzuki T. (2016) Expression and localization of X11 family proteins in neurons. Brain Res. 1646, 227–234 [DOI] [PubMed] [Google Scholar]
- 29. Gouras G. K., Xu H., Jovanovic J. N., Buxbaum J. D., Wang R., Greengard P., Relkin N. R., and Gandy S. (1998) Generation and regulation of β-amyloid peptide variants by neurons. J. Neurochem. 71, 1920–1925 [DOI] [PubMed] [Google Scholar]
- 30. Cai H., Wang Y., McCaethy D., Wen H., Borchelt D. R., Price D. L., and Wong P. C. (2001) BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat. Neurosci. 4, 233–234 [DOI] [PubMed] [Google Scholar]
- 31. Vanderstichele H., De Meyer G., Andreasen N., Kostanjevecki V., Wallin A., Olsson A., Blennow K., and Vanmechelen E. (2005) Amino-truncated β-amyloid42 peptide in cerebrospinal fluid and prediction of progression of mild cognitive impairment. Clin. Chem. 51, 1650–1660 [DOI] [PubMed] [Google Scholar]
- 32. Citron M., Oltersdorf T., Haass C., McConlogue L., Hung A. Y., Seubert P., Vigo-Pelfrey C., Lieberburg I., and Selkoe D. J. (1992) Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production. Nature 360, 672–674 [DOI] [PubMed] [Google Scholar]
- 33. Felsenstein K. M., Hunihan L. W., and Roberts S. B. (1994) Altered cleavage and secretion of a recombinant β-APP bearing the Swedish familial Alzheimer's disease mutation. Nat. Genet. 6, 251–255 [DOI] [PubMed] [Google Scholar]
- 34. Johnston J. A., Cowburn R. F., Norgren S., Wiehager B., Venizelos N., Winblad B., Vigo-Pelfrey C., Schenk D., Lannfelt L., and O'Neill C. (1994) Increased β-amyloid release and levels of amyloid precursor protein (APP) in fibroblast cell lines from family members with the Swedish Alzheimer's disease APP670/671 mutation. FEBS Lett. 354, 274–278 [DOI] [PubMed] [Google Scholar]
- 35. Messa M., Colombo L., del Favero E., Cantù L., Stoilova T., Cagnotto A., Rossi A., Morbin M., Di Fede G., Tagliavini F., and Salmona M. (2014) The peculiar role of the A2V mutation in amyloid-β (Aβ) 1–42 molecular assembly. J. Biol. Chem. 289, 24143–24152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee E. B., Zhang B., Liu K., Greenbaum E. A., Doms R. W., Trojanowski J. Q., and Lee V. M. (2005) BACE overexpression alters the subcellular processing of APP and inhibits Aβ deposition in vivo. J. Cell Biol. 168, 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tomita S., Kirino Y., and Suzuki T. (1998) Cleavage of Alzheimer's amyloid precursor protein by secretases occurs after O-glycosylation of APP in the protein secretory pathway. J. Biol. Chem. 273, 6277–6284 [DOI] [PubMed] [Google Scholar]
- 38. Fluhrer R., Multhaup G., Schlicksupp A., Okochi M., Takeda M., Lammich S., Willem M., Westmeyer G., Bode W., Walter J., and Haass C. (2003) Identification of a β-secretase activity, which truncates amyloid β-peptide after its presenilin-dependent generation. J. Biol. Chem. 278, 5531–5538 [DOI] [PubMed] [Google Scholar]
- 39. Iwata N., Tsubuki S., Takaki Y., Watanabe K., Sekiguchi M., Hosoki E., Kawashima-Morishima M., Lee H. J., Hama E., Sekine-Aizawa Y., and Saido T. C. (2000) Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150 [DOI] [PubMed] [Google Scholar]
- 40. Steiner H., Fluhrer R., and Haass C. (2008) Intramembrane proteolysis by γ-secretase. J. Biol. Chem. 283, 29627–29631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moore B. D., Chakrabarty P., Levites Y., Kukar T. L., Baine A. M., Moroni T., Ladd T. B., Das P., Dickson D. W., and Golde T. E. (2012) Overlapping profiles of Aβ peptides in the Alzheimer's disease and pathological aging brains. Alzheimers Res. Ther. 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan R., and Vassar R. (2014) Targeting the β secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 13, 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takami M., Nagashima Y., Sano Y., Ishihara S., Morishima-Kawashima M., Funamoto S., and Ihara Y. (2009) γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 29, 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takami M., and Funamoto S. (2012) γ-Secretase-dependent proteolysis of transmembrane domain of amyloid precursor protein: successive tri- and tetrapeptide release in amyloid β-protein production. Int. J. Alzheimers Dis. 10.1055/2012/591392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Caillava C., Ranaldi S., Lauritzen I., Bauer C., Fareh J., Abraham J.-D., and Checler F. (2014) Study on Aβ34 biology and detection in transgenic mice brains. Neurobiol. Aging. 35, 1570–1581 [DOI] [PubMed] [Google Scholar]
- 46. Iwata N., Tsubuki S., Takaki Y., Shirotani K., Lu B., Gerard N. P., Gerard C., Hama E., Lee H. J., and Saido T. C. (2001) Metabolic regulation of brain Aβ by neprilysin. Science 292, 1550–1552 [DOI] [PubMed] [Google Scholar]
- 47. Fukumoto H., Cheung B. S., Hyman B. T., and Irizarry M. C. (2002) β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 59, 1381–1389 [DOI] [PubMed] [Google Scholar]
- 48. Tomita S., Kirino Y., and Suzuki T. (1998) A basic amino acid in the cytoplasmic domain of Alzheimer's β-amylid precursor protein (APP) is essential for cleavage of APP at the α-site. J. Biol. Chem. 273, 19304–19310 [DOI] [PubMed] [Google Scholar]
- 49. Lichtenthaler S. F., Multhaup G., Masters C. L., and Beyreuther K. (1999) A novel substrate for analyzing Alzheimer's disease γ-secretase. FEBS Lett. 453, 288–292 [DOI] [PubMed] [Google Scholar]
- 50. Huse J. T., Pijak D. S., Leslie G. J., Lee V. M., and Doms R. W. (2000) Maturation and endosomal targeting of β-site amyloid precursor protein-cleaving enzyme. J. Biol. Chem. 275, 33729–33737 [DOI] [PubMed] [Google Scholar]
- 51. Hata S., Fujishige S., Araki Y., Kato N., Araseki M., Nishimura M., Hartmann D., Saftig P., Fahrenholz F., Taniguchi M., Urakami K., Akatsu H., Martins R. N., Yamamoto K., Maeda M., Yamamoto T., Nakaya T., Gandy S., and Suzuki T. (2009) Alcadein cleavages by APP α- and γ-secretases generate small peptides p3-Alcs indicating Alzheimer disease-related γ-secretase dysfunction. J. Biol. Chem. 284, 36024–36033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Piao Y., Kimura A., Urano S., Saito Y., Taru H., Yamamoto T., Hata S., and Suzuki T. (2013) Mechanism of intramembrane cleavage of Alcadeins by γ-secretase. PLoS ONE 8, e62431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Oishi M., Nairn A. C., Czernik A. J., Lim G. S., Isohara T., Gandy S. E, Greengard P., and Suzuki T. (1997) The cytoplasmic domain of the Alzheimer's β-amyloid precursor protein is phosphorylated at Thr-654, Ser-655 and Thr-668 in adult rat brain and cultured cells. Mol. Med. 3, 111–123 [PMC free article] [PubMed] [Google Scholar]
- 54. Bartlett W. P., and Banker G. A. (1984) An electron microscopic study of the development of axpns and dendrites by hippocampal neurons in culture: I. cells which develop without intercellular contacts. J. Neurosci. 4, 1944–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chiba K., Araseki M., Nozawa K., Furukori K., Araki Y., Matsushima T., Nakaya T., Hata S., Saito Y., Uchida S., Okada Y., Nairn A. C., Davis R. J., Yamamoto T., Kinjo M., Taru H., and Suzuki T. (2014) Quantitative analysis of APP axonal transport in neurons: role of JIP1 in APP anterograde transport. Mol. Biol. Cell 25, 3569–3580 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.