

Abstract
Aldehyde dehydrogenases (ALDHs) catalyze the NAD(P)+-dependent oxidation of aldehydes to carboxylic acids and are important for metabolism and detoxification. Although the ALDH superfamily fold is well established, some ALDHs contain an uncharacterized domain of unknown function (DUF) near the C terminus of the polypeptide chain. Herein, we report the first structure of a protein containing the ALDH superfamily DUF. Proline utilization A from Sinorhizobium meliloti (SmPutA) is a 1233-residue bifunctional enzyme that contains the DUF in addition to proline dehydrogenase and l-glutamate-γ-semialdehyde dehydrogenase catalytic modules. Structures of SmPutA with a proline analog bound to the proline dehydrogenase site and NAD+ bound to the ALDH site were determined in two space groups at 1.7–1.9 Å resolution. The DUF consists of a Rossmann dinucleotide-binding fold fused to a three-stranded β-flap. The Rossmann domain resembles the classic ALDH superfamily NAD+-binding domain, whereas the flap is strikingly similar to the ALDH superfamily dimerization domain. Paradoxically, neither structural element performs its implied function. Electron density maps show that NAD+ does not bind to the DUF Rossmann fold, and small-angle X-ray scattering reveals a novel dimer that has never been seen in the ALDH superfamily. The structure suggests that the DUF is an adapter domain that stabilizes the aldehyde substrate binding loop and seals the substrate-channeling tunnel via tertiary structural interactions that mimic the quaternary structural interactions found in non-DUF PutAs. Kinetic data for SmPutA indicate a substrate-channeling mechanism, in agreement with previous studies of other PutAs.
Keywords: enzyme kinetics, flavoprotein, nicotinamide adenine dinucleotide (NAD), oligomerization, protein domain, small-angle X-ray scattering (SAXS), X-ray crystallography, aldehyde dehydrogenase superfamily, proline catabolism, proline utilization A
Introduction
The aldehyde dehydrogenase (ALDH)2 superfamily is a vast collection of enzymes that catalyze the NAD(P)+-dependent oxidation of aldehydes to carboxylic acids. Humans have 19 functional genes and three pseudogenes. The genes encode ALDHs in families 1–9, 16, and 18. The ALDH fold has been characterized by numerous crystallographic studies, dating back to the first crystal structure of an ALDH in 1997 (1). The ALDH fold consists of a Rossmann NAD+ binding domain, a catalytic domain that provides the nucleophilic Cys residue, and an oligomerization domain. The latter domain mediates dimerization in all ALDHs studied to date by forming a domain-swapped interaction with the catalytic domain. Although this fold is conserved throughout the superfamily, at least two ALDH families (ALDH4A1 and ALDH16A1) contain a domain of unknown function (DUF) near the C terminus of the polypeptide chain (C-terminal domain (CTD)). To characterize the ALDH superfamily DUF, we targeted bifunctional l-glutamate-γ-semialdehyde dehydrogenase (GSALDH) enzymes that contain the CTD for crystal structure determination.
GSALDH (also called ALDH4A1 and Δ1-pyrroline-5-carboxylate dehydrogenase) is the second enzyme of proline catabolism and catalyzes the oxidation of l-glutamate-γ-semialdehyde (GSAL) to glutamate (Fig. 1A). In some bacteria, GSALDH is combined into a bifunctional enzyme with proline dehydrogenase (PRODH), which catalyzes the first step of proline catabolism. PRODH catalyzes the FAD-dependent oxidation of proline to Δ1-pyrroline-5-carboxylate (P5C). Hydrolysis of P5C generates GSAL. The bifunctional PRODH-GSALDH enzymes are known as proline utilization A (PutA) (2, 3). An interesting aspect of PutAs is that the intermediate P5C/GSAL is channeled between the two active sites through a 75 Å tunnel (4, 5).
FIGURE 1.

The reactions of proline catabolism and the domains of PutA and ALDH16A1. A, the reactions catalyzed by PutA. B, domain diagrams of PutAs and ALDH16A1. The small N-terminal domain in type C PutAs is a ribbon-helix-helix DNA-binding domain.
Three types of PutA are distinguished by domain architecture (Fig. 1B). Type A PutAs contain the minimal domain set required for catalysis, which consists of an N-terminal PRODH module and a C-terminal GSALDH module. Type B and C PutAs additionally contain the ALDH superfamily CTD. Type C PutAs also have an N-terminal ribbon-helix-helix DNA-bonding domain, which endows them with autogenous transcriptional repressor functionality (6). The CTD is also present in ALDH16A1 (Fig. 1B), a protein that is widely distributed in mammals but whose function is unknown (7, 8).
We have determined the first crystal structure of a PutA containing the CTD. The structures show that the CTD contains a Rossmann dinucleotide-binding fold fused to a β-flap that resembles the oligomerization domain of other ALDHs. Paradoxically, the CTD Rossmann fold does not bind NAD+, and the flap does not mediate oligomerization. The CTD is proposed to be a structural domain that facilitates substrate channeling and GSALDH activity.
Results
Structure of the PutA Protomer
The structure of the type B PutA from Sinorhizobium meliloti (SmPutA) complexed with NAD+ and the proline analog l-tetrahydrofuroic acid (THFA) was determined in space groups P21 and P3121 at 1.7 and 1.9 Å resolution, respectively (Table 1). As observed in type A PutAs, which lack the CTD (4, 5), SmPutA contains spatially separated PRODH and GSALDH active sites (Fig. 2A). The root mean square deviation (rmsd) of SmPutA to the type A PutA from Bradyrhizobium japonicum (BjPutA, PDB code 3HAZ (4)) is 1.5 Å over 830 residues. As expected, the PRODH active site is located in a (βα)8 barrel. THFA binds at the si face of the FAD, and its interactions with the enzyme are consistent with other PRODHs complexed with this inhibitor (Fig. 2B) (5, 9, 10). Like other ALDHs, the GSALDH module has a Rossmann dinucleotide-binding domain and an α/β catalytic domain (Fig. 2A). The GSALDH active site features a conserved nucleophilic Cys residue (Cys844), which is near the nicotinamide of the bound NAD+ (Fig. 2C). The FAD and Cys844 are separated by a linear distance of 42 Å and connected by a 75 Å tunnel (Fig. 2A). The dimensions of the tunnel are similar to those of type A PutAs.
TABLE 1.
Data collection and refinement statistics
The values for the outer resolution shell of data are given in parentheses.
| Monoclinic | Trigonal | |
|---|---|---|
| Space group | P21 | P3121 |
| Beamline | ALS 4.2.2 | ALS 4.2.2 |
| Unit cell parameters (Å, °) | a = 101.4, b = 102.3, c = 125.9, β = 106.5 | a = 128.8, c = 150.5 |
| Wavelength (Å) | 1.000 | 1.000 |
| Resolution (Å) | 60.3–1.70 (1.73–1.70) | 62.4–1.90 (1.93–1.90) |
| Unique reflections | 265,575 | 113,632 |
| Rmerge(I) | 0.066 (0.460) | 0.113 (1.305) |
| Rmeas(I) | 0.077 (0.578) | 0.120 (1.405) |
| Rpim(I) | 0.041 (0.343) | 0.040 (0.510) |
| Mean I/σ | 13.3 (2.0) | 15.5 (1.5) |
| Completeness (%) | 98.3 (90.3) | 99.9 (97.3) |
| Multiplicity | 3.6 (2.7) | 8.6 (7.1) |
| No. of protein residues | 2420 | 1214 |
| No. of atoms | ||
| Protein | 17,827 | 9004 |
| FAD | 106 | 53 |
| THFA | 16 | 8 |
| NAD+ | 88 | 44 |
| Water | 1286 | 604 |
| Rcryst | 0.190 (0.260) | 0.174 (0.287) |
| Rfreea | 0.222 (0.318) | 0.203 (0.317) |
| rmsd bond lengths (Å) | 0.007 | 0.006 |
| rmsd bond angles (°) | 0.898 | 0.839 |
| Ramachandran plotb | ||
| Favored (%) | 97.67 | 98.34 |
| Outliers (%) | 0.08 | 0.00 |
| Clashscore (percentile)b | 2.04 (99) | 2.30 (99) |
| MolProbity score (percentile)b | 1.04 (100) | 1.01 (100) |
| Average B (Å2) | ||
| Protein | 21.3 | 32.4 |
| FAD | 16.2 | 31.4 |
| THFA | 23.3 | 38.4 |
| NAD+ | 15.4 | 20.2 |
| Water | 24.4 | 31.2 |
| Coordinate error (Å)c | 0.21 | 0.19 |
| PDB code | 5KF6 | 5KF7 |
a 5% test set.
b Generated with MolProbity.
c Maximum likelihood-based coordinate error estimate from PHENIX.
FIGURE 2.
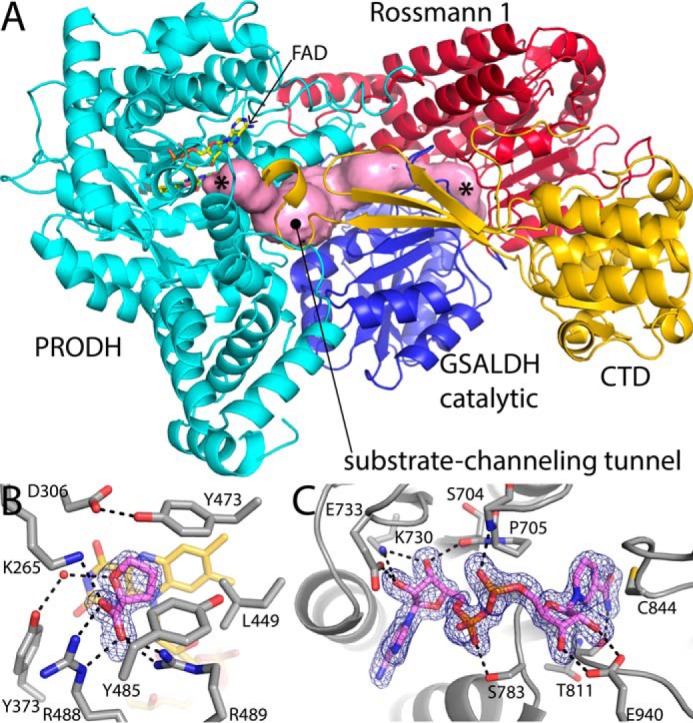
Structure of SmPutA. A, cartoon drawing of the protomer. The PRODH module is colored cyan. The Rossmann NAD+-binding and catalytic domains of the GSALDH module are colored red and blue, respectively. The CTD is colored gold. The pink surface represents the substrate-channeling tunnel. The asterisks indicate the locations of the two active sites in the tunnel, with the PRODH site on the left and the GSALDH site on the right. B, electron density and interactions for the proline analog THFA bound to the SmPutA PRODH active site (space group P21, chain A). The cage represents a simulated annealing σA-weighted Fo − Fc omit map (3.0 σ). C, electron density and interactions for the NAD+ bound to SmPutA (space group P21). The cage represents a simulated annealing σA-weighted Fo − Fc omit map (3.0 σ).
Structure of the CTD
The CTD (residues 1034–1233) consists of an α/β domain with a protruding β-flap (Fig. 3A). The α/β domain packs against the Rossmann fold domain of the GSALDH module, whereas the flap extends toward the PRODH active site (Fig. 2A).
FIGURE 3.
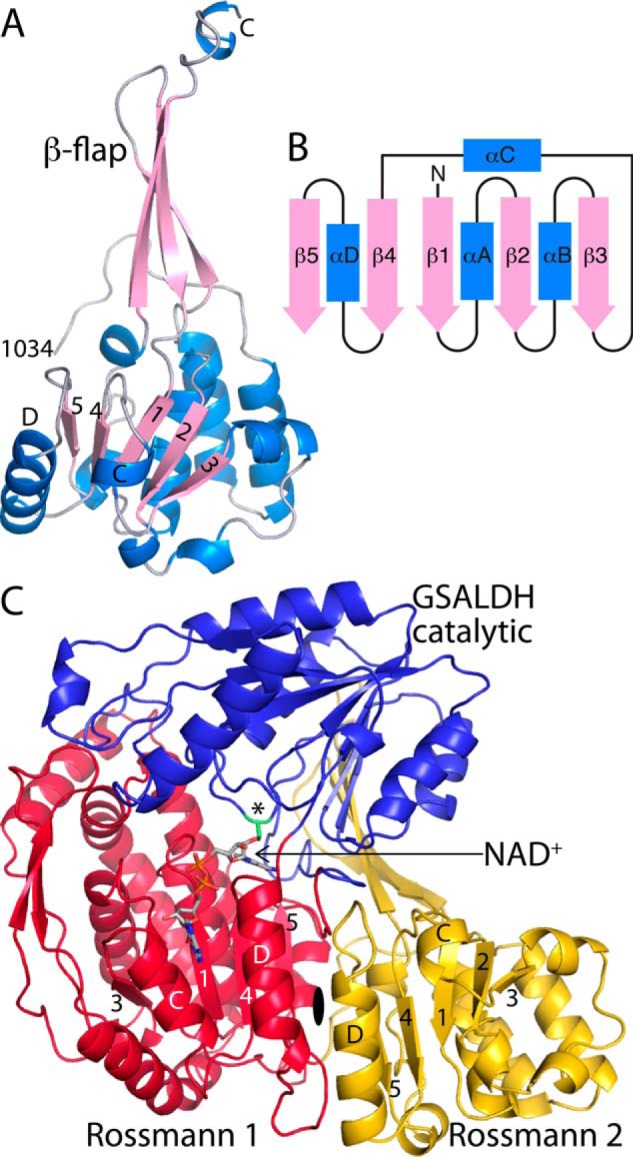
The fold and tertiary structural interactions of the CTD. A, cartoon drawing of the CTD. Selected β-strands and α-helices of the Rossmann domain are indicated. B, topology diagram of the Rossmann dinucleotide-binding fold found in ALDHs. C, tertiary structural interactions between the CTD (gold) and the GSALDH module. The asterisk indicates catalytic Cys844.
The α/β domain of the CTD (residues 1034–1078 and 1098–1211) has the Rossmann dinucleotide-binding fold (Fig. 3A). The classic Rossmann fold, which was first observed in a crystal structure of lactate dehydrogenase, consists of a pair of βαβαβ motifs related by a 2-fold axis parallel to the sheet, which results in a six-stranded parallel β-sheet having strand order 321456, or equivalently, 654123 (11, 12). ALDHs use an abbreviated version of the archetype that lacks the final helix and strand (Fig. 3B). The α/β domain of the CTD clearly exhibits the ALDH variation of the Rossmann fold (Fig. 3A).
SmPutA thus has tandem ALDH superfamily Rossmann domains: one in the GSALDH module (Rossmann 1) and a second one in the CTD (Rossmann 2). The two domains pack against each other and are related by a pseudo 2-fold axis perpendicular to the sheets (Fig. 3C). The two Rossmann folds are structurally similar and superimpose with an rmsd of 2.7 Å over 137 residues (Fig. 4, A and B). A major difference is that Rossmann 1 is not fused to a long β-hairpin as in the CTD (Fig. 4B). Another notable difference is that αB and αC are abbreviated in the CTD (Fig. 4A).
FIGURE 4.

Comparison of the CTD to ALDH superfamily domains. A, the CTD of SmPutA. B, the Rossmann NAD+-binding domain in the GSALDH module of SmPutA. C, the Rossmann NAD+-binding and oligomerization domains of BjPutA (PDB code 3HAZ). D, the Rossmann and oligomerization domains of benzaldehyde dehydrogenase (PDB code 3R64). In all panels, the diagnostic strands and helices of the Rossmann fold are indicated.
The β-flap of the CTD is a bipartite substructure consisting of a β-hairpin (residues 1079–1097) and the C terminus of the chain (residues 1212–1233) (Fig. 3A). The flap resembles the oligomerization domain found in all other ALDH superfamily structures reported to date, including type A PutAs. The similarity of the CTD flap to the oligomerization domain of the type A BjPutA is striking (Fig. 4, A and C).
The CTD contains structural elements that resemble the ALDH superfamily domains that function in binding NAD+ (Rossmann 2) and oligomerization (β-flap). The similarity of the CTD to the ALDH superfamily fold was confirmed by querying the PDB with PDBeFold (13). The top match for the CTD in terms of Q score (a compromise between rmsd and alignment length) is a benzaldehyde dehydrogenase (Fig. 4D). Like the CTD, benzaldehyde dehydrogenase features a β-flap fused to a five-stranded Rossmann fold domain. The CTD and benzaldehyde dehydrogenase superimpose with an rmsd of 2.8 Å over 168 residues, despite having negligible (14%) sequence identity. The NAD+ binding and oligomerization domains of BjPutA, a type A PutA, was also identified in this search (Fig. 4C). BjPutA and the CTD superimpose with an rmsd of 2.4 Å over 161 residues (27% sequence identity).
The Rossmann Fold of the CTD Does Not Bind NAD+ in Crystallo
The presence of tandem Rossmann dinucleotide-binding domains is unexpected for an enzyme that requires only one equivalent of NAD+ per catalytic cycle (Fig. 1A). This observation motivated crystallographic studies of NAD+ binding.
Strong electron density for NAD+ bound to Rossmann 1 is present in both crystal forms, whereas no evidence for NAD+ bound to the CTD was observed (Fig. 2C). NAD+ binds at the C termini of the strands of the Rossmann 1 β-sheet, which is the canonical location (Fig. 3C). The C4 atom of the nicotinamide is 2.9 Å from the S atom of catalytic Cys844 (Fig. 2C). This distance is consistent with the accepted ALDH catalytic mechanism (14) in which a hydride is transferred to C4 from the hemithioacetal intermediate bound to Cys844.
The co-factor makes several interactions with the enzyme, including hydrogen bonds with Lys730, Glu733, Ser783, and Glu940 (Fig. 2C). A hydrogen bond with the backbone carbonyl of Thr811 establishes the orientation of the nicotinamide carboxamide group.
The conformation of NAD+ and its protein environment are indicative of a bona fide co-factor binding site. Furthermore, the conformation of NAD+ is identical to that observed in monofunctional GSALDH (15, 16) and type A PutA (4). These in crystallo results suggest that only Rossmann 1 participates directly in catalysis by binding NAD+ and imply that the Rossmann domain in the CTD has a purely structural role.
SmPutA Forms a Concentration-dependent Dimer in Solution
The observation of an apparent oligomerization flap in the CTD motivated studies of the oligomeric state and quaternary structure of SmPutA using small-angle X-ray scattering (SAXS). Several samples were analyzed at Beamline 12.3.1 at the Advanced Light Source through the SIBYLS mail-in program (17).
The shape of the SAXS curve varies with protein concentration (Fig. 5A). At the lowest concentration, the curve is relatively featureless, but a bump appears in the curve at q = 0.10–0.14 Å−1 as the protein concentration is increased. The prominence of the bump correlates with an increase in the radius of gyration (Rg). The Rg from Guinier analysis ranges from 34.5 Å for the lowest concentration sample to 38.6 Å at the highest concentration (Fig. 5A, inset). For reference, the Rg calculated from an SmPutA monomer is 32.6 Å. Similarly, the real space Rg from calculations of the distance distribution function increases from 34.8 to 39.2 Å with increasing concentration (Fig. 5B). The distance distribution function exhibits a single maximum, the position of which increases with concentration from 39.8 to 49.5 Å (Fig. 5B). The dependence of the SAXS structural parameters on protein concentration is consistent with dynamic self-association.
FIGURE 5.
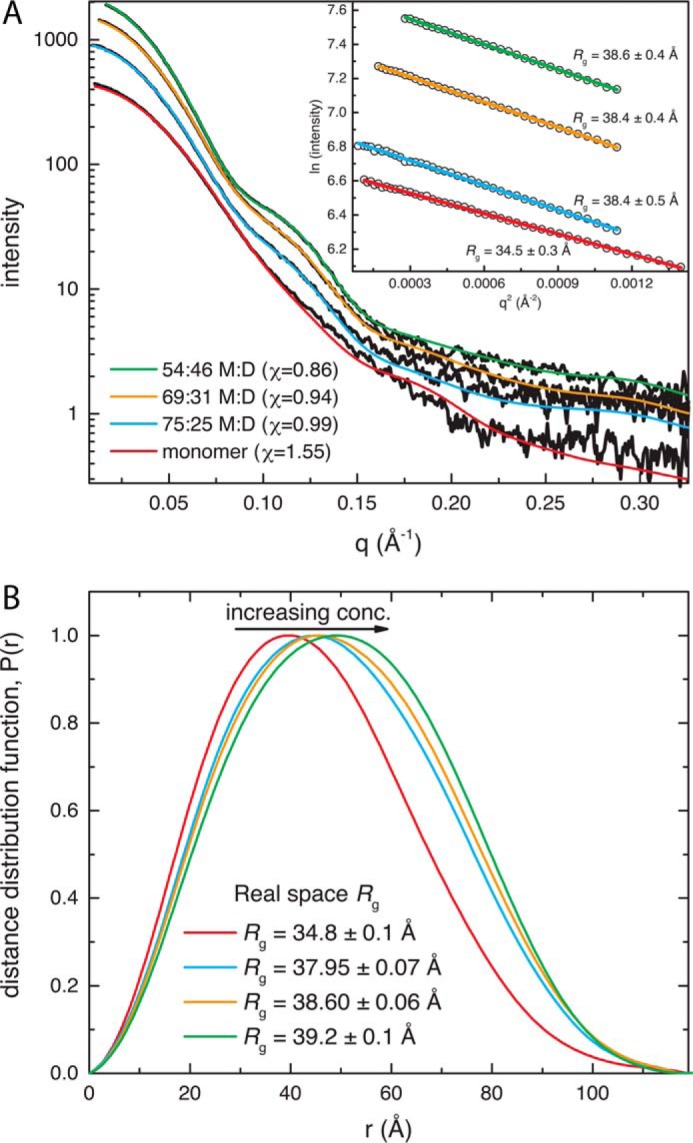
SAXS analysis of SmPutA. A, SAXS curves from samples at different protein concentrations. The black curves represent the experimental data. The smooth curves represent theoretical SAXS curves calculated from atomic models. The inset shows Guinier plots. B, distance distribution functions calculated from the SAXS curves.
The experimental SAXS curves were compared with theoretical ones calculated from atomic models (18, 19) to characterize the structural basis of self-association (Fig. 5A). Oligomer models were obtained by analyzing the protein-protein interfaces in the crystal structures using PDBePISA (20). This analysis identified a potentially stable dimer with Rg of 40.3 Å. The dimer is present in the P21 asymmetric unit. It is also found in the P3121 lattice, where the dimer is generated from the protomer in the asymmetric unit by the 2-fold axis coincident with the shorter diagonal of the a-b plane. The lowest concentration SAXS curve agrees well with the curve calculated from a monomer (goodness of fit parameter, χ = 1.55). Consideration of a monomer-dimer ensemble using MultiFoXS (18, 19) did not improve the fit for the lowest concentration sample. In contrast, the SAXS curves from the three higher concentration samples could not be satisfactorily fit with either the monomer or the dimer model alone (χ > 5.4). Better fits were obtained from monomer-dimer ensembles (χ = 0.86–0.99) (Fig. 5A). The optimal monomer:dimer ratios for the three higher concentration samples were estimated to be 75:25, 69:31, and 54:46 (Fig. 5A). This analysis is consistent with SmPutA exhibiting a monomer-dimer equilibrium. Furthermore, the calculations suggest that the two-body assembly in the crystal is the dimer formed in solution.
Novel ALDH Superfamily Dimer
The SmPutA dimer is unique among ALDHs that have been structurally characterized, and fittingly, the CTD features prominently in the dimer interface. The β-flap of the CTD does not participate in dimerization. The 2-fold axis of the dimer passes next to Leu1147 of the CTD (Fig. 6A). A large section of the interface is formed by the extended polypeptide chain at the N terminus of the CTD and the first two helices of the CTD (Fig. 6B). These elements interact with helices of the catalytic modules. Each chain of SmPutA contributes 1800 Å2 of surface area to the interface.
FIGURE 6.
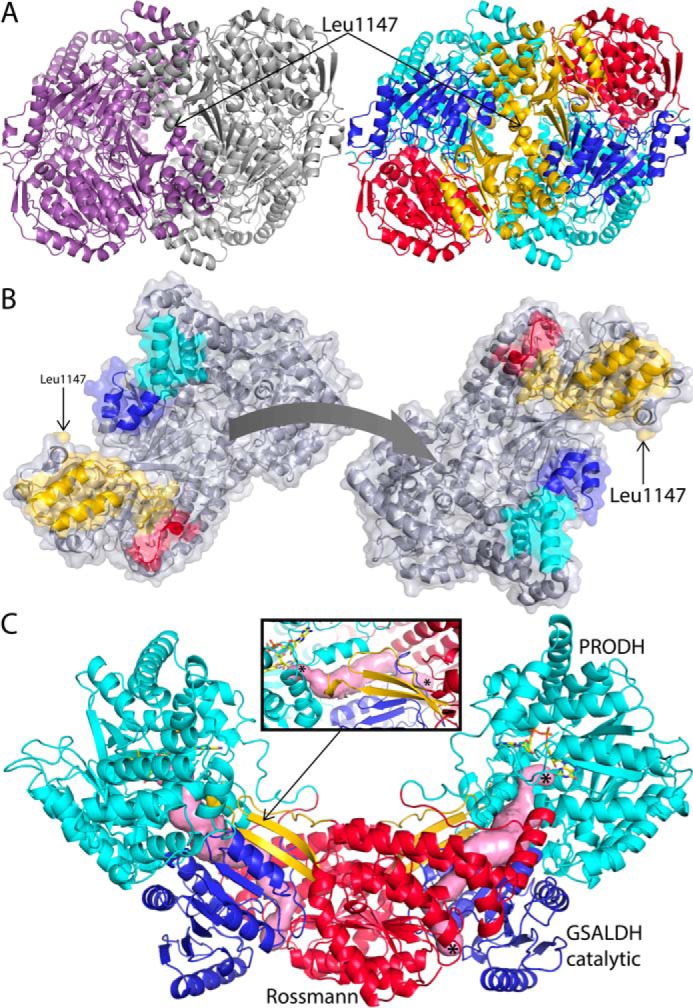
A novel ALDH dimer. A, cartoon representation of the SmPutA dimer viewed down the 2-fold axis. On the left, the two protomers have different colors. On the right, the protein is color-coded by modules/domains as in Fig. 2A: PRODH, cyan; Rossmann 1, red; GSALDH catalytic, blue; and CTD, gold. B, the separated protomers of the SmPutA dimer. The interaction surfaces are color-coded according to modules/domains: PRODH, cyan; Rossmann 1, red; GSALDH catalytic, blue; and CTD, gold. C, the traditional ALDH mode of dimerization as seen in BjPutA (a type A PutA, PDB code 3HAZ). The domains are colored as follows: PRODH, cyan; Rossmann NAD+-binding domain, red; GSALDH catalytic, blue; and oligomerization flap, gold. The pink surface represents the substrate-channeling tunnels. The inset shows a close-up view of the oligomerization flap of one protomer covering the substrate-channeling tunnel of the opposite protomer. The asterisks indicate the locations of the two active sites. Note that the quaternary structural interactions in BjPutA resemble the tertiary structural interactions of the β-flap in SmPutA (Fig. 2A).
The SmPutA dimer is unlike any other ALDH dimer that has been structurally characterized. Typically, ALDHs form domain swapped dimers in which the oligomerization flap of one protomer contacts the catalytic domain of the opposing protomer. For example, BjPutA exhibits the traditional mode (Fig. 6C). Note that the Rossmann dinucleotide-binding domains are close together in the traditional dimer (red domains in Fig. 6C). In contrast, the dinucleotide-binding domains are far apart in the SmPutA dimer (Fig. 6A, right panel).
Kinetic Evidence for Substrate Channeling
Further insight into the function of the CTD was gained by inspecting its tertiary structural interactions with other domains within the protomer. Substrate channeling is one possible role. The β-flap of the CTD extends toward the PRODH site and covers a large section of the tunnel that connects the two active sites (Fig. 2A). If SmPutA has a substrate-channeling mechanism, a likely role for the CTD is to help sequester the intermediate in the tunnel.
We investigated substrate channeling by measuring the kinetics of the PRODH-GSALDH coupled reaction of SmPutA. The time dependence of NADH production was monitored in an assay containing proline, an electron acceptor for the flavin (ubiquinone-1, CoQ1), and NAD+. The progress curve for NADH is approximately linear with negligible lag (Fig. 7A). In contrast, a free diffusion kinetic model of SmPutA predicts a time lag of 6 min under the conditions of the assay (Fig. 7A). The substantially shorter time lag observed for SmPutA compared with the one predicted for the uncoupled enzyme model is a good indicator of substrate channeling (21, 22).
FIGURE 7.

Kinetics of substrate channeling. A, transient time analysis of SmPutA. The circles show NADH production from SmPutA (0.25 μm) with 40 mm proline, 200 μm CoQ1, and 200 μm NAD+, pH 7.5. The solid curve shows the predicted NADH formation using a two-enzyme nonchanneling model of the SmPutA PRODH-GSALDH coupled reaction (Equation 1). Linear extrapolation of the nonchanneling model as shown by the dashed line yields a transient time of 6 min. B, initial velocity of the coupled PRODH-GSALDH reaction with varied proline concentration and fixed CoQ1 (300 μm) and NAD+ (200 μm). Non-linear least squares fit of the data to a substrate inhibition model as shown gave best fit parameters of kcat = 1.6 ± 0.1 s−1, Km = 7 ± 1 mm, and Ki = 263 ± 36 mm proline.
The PRODH-GSADH coupled reaction was also characterized by measuring the rate of NADH production while varying the proline concentration at fixed CoQ1 concentration (Fig. 7B). The data were fit to a model that includes the inhibition of the GSADH site by proline. The kinetic parameters of the coupled reaction are kcat of 1.6 s−1, Km for proline of 7 mm, and Ki of 263 mm proline. The kcat/Km for the overall reaction is 229 m−1 s−1, which is nearly 7-fold higher than for PutA from Escherichia coli (23).
Discussion
We have shown that the ALDH superfamily CTD consists of two subdomains: the five-stranded ALDH superfamily variation of the Rossmann dinucleotide-binding domain and a β-flap that resembles the ALDH oligomerization domain. Because the CTD neither binds NAD+ nor mediates traditional ALDH dimerization, this structural annotation belies function. However, the tertiary and quaternary structural interactions formed by the CTD imply hypotheses for the functions of the CTD.
Facilitating substrate channeling is one likely function of the CTD. The steady-state kinetic data for SmPutA are consistent with efficient substrate channeling (Fig. 7). Presumably, the tunnel connecting the two active sites is the channeling pathway. We note that the analogous pathway in BjPutA, a type A PutA, has been validated by tunnel-blocking mutagenesis (24). Type A PutAs lack the CTD (Fig. 1B) and form traditional ALDH domain-swapped dimers in which the oligomerization flap of one protomer seals the substrate-channeling tunnel of the opposite protomer (Fig. 6C). Interestingly, the quaternary structural interactions of the oligomerization flap in the type A PutA dimer resemble the tertiary structural interactions of the CTD in the SmPutA monomer (compare the inset in Fig. 6C with Fig. 2A). Because the CTD seals the tunnel from the bulk medium in SmPutA, it is reasonable to conclude that the CTD facilitates channeling by helping to sequester the intermediate in the tunnel.
The biological rationale for substrate channeling in proline catabolism is based in part on the chemical properties of the intermediates P5C and GSAL. P5C/GSAL is a labile intermediate (21) that has damaging reactions with other metabolites, such as deactivation of pyridoxal phosphate in type II hyperprolinemia patients who have a deficiency in GSALDH (25). P5C/GSA also inhibits enzymes such as glucosamine-6-phosphate synthase (26). Thus, a channeling mechanism helps protect against unwanted and potentially damaging effects of P5C/GSA. Furthermore, P5C/GSAL is also the intermediate of proline biosynthesis, so channeling provides a mechanism for segregating catabolism and biosynthesis (21).
The SmPutA structure also implies an indirect role for the CTD in GSALDH activity. The flap of the CTD interacts with the β-sheet of the GSALDH catalytic domain, effectively extending the sheet from seven strands to ten (Fig. 8). An analogous interaction is formed in conventional ALDHs, except that extension of the sheet is achieved through quaternary structural interactions of the oligomerization flap rather than tertiary interactions within a monomer. It is unlikely that the GSALDH active site can adopt the correct conformation without the β-sheet interactions provided by the CTD. The observed proximity of the CTD flap to the GSALDH domain in SmPutA explains why deletion of the CTD in a type C PutA eliminated GSALDH activity, which was a perplexing observation at the time (27).
FIGURE 8.
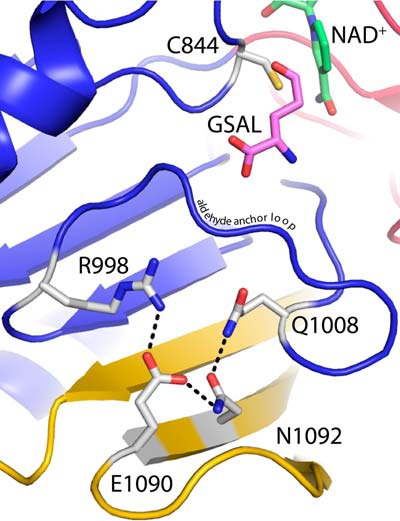
Proximity of the β-flap of the CTD to the GSALDH active site. The cartoon is color-coded according to domains: Rossmann 1, red; GSALDH catalytic, blue; and CTD, gold. A model of GSAL has been docked into SmPutA based on a structure of GSALDH complexed with glutamate (PDB code 3V9K).
Specific side chains of the CTD are also predicted to facilitate GSALDH activity. Glu1090 and Asn1092 of the β-flap interact with Arg998 and Gln1008 of the aldehyde anchor loop (Fig. 8). The anchor loop is a conserved element of GSALDH active sites that binds the α-carboxylate of GSAL (28, 29). The interactions provided by the β-flap presumably help stabilize the active conformation of the anchor loop. We note that the mutation of residues analogous to Glu1090 and Asn1092 in another type B PutA eliminated GSALDH activity (30).
It is interesting that the CTD does not bind NAD+ despite adopting the Rossmann dinucleotide-binding fold. Inspection of the structure reveals features that are inconsistent with NAD+ binding. Docking of NAD+ into the expected binding site predicts severe clashes with the protein. In particular, the nicotinamide riboside clashes with the β4-αd-β5 motif of the Rossmann fold (Fig. 9, A and B). Also, side chains invade the space that would be occupied by the adenine ribose (Fig. 9A). Thus, the surface of the CTD where NAD+ would be expected to bind is unlike that of a bona fide Rossmann NAD+-binding domain. For example, the true NAD+-binding site in Rossmann 1 has a pocket that is complementary in shape to the active conformation of NAD+ (Fig. 9C). In particular, a groove bordered by αC and αD is the binding site for the adenine, whereas a deeper hole provides the site for the nicotinamide (Fig. 9C). These features are conspicuously absent in the CTD Rossmann domain (Fig. 9B), where the adenine groove is replaced by a flat surface and the aforementioned steric clash residues eliminate the nicotinamide hole.
FIGURE 9.
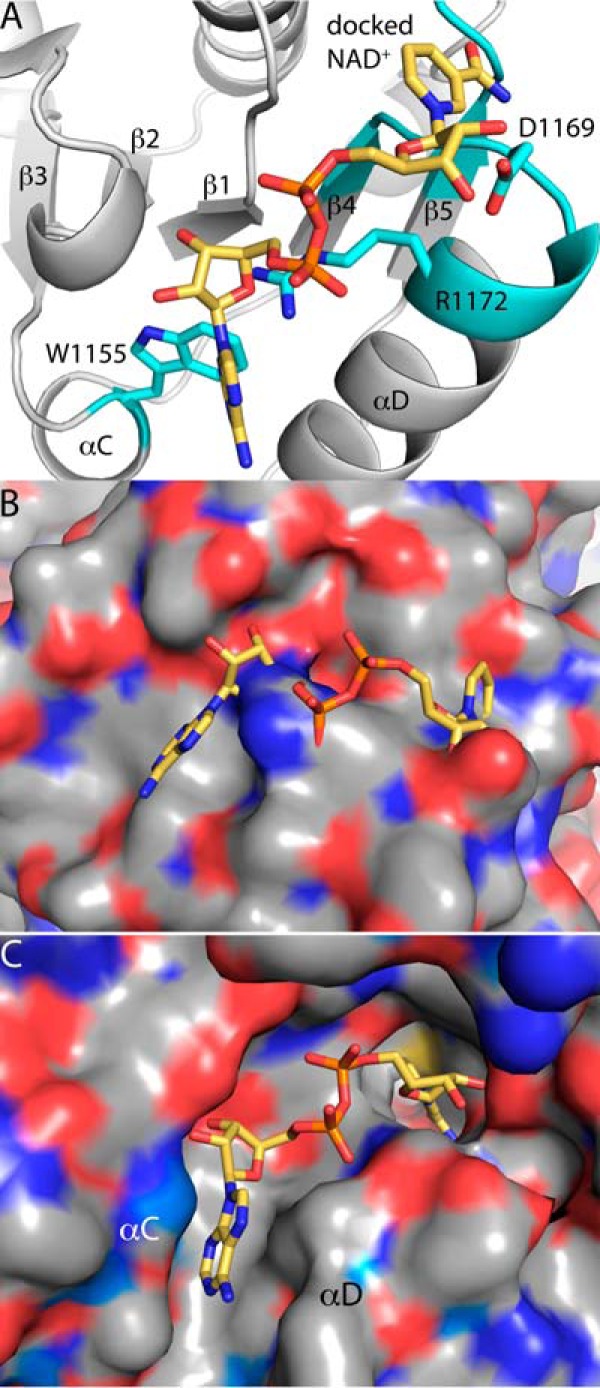
Structural explanation for why the CTD does not bind NAD+. A, a model of NAD+ from the GSALDH domain has been docked to the CTD in the canonical position for Rossmann dinucleotide-binding domains. Residues predicted to clash NAD+ are in cyan. B, the surface of the CTD where NAD+ would be expected to bind. NAD+ is docked into the canonical position for Rossmann dinucleotide-binding domains. Note the absence of a well defined pocket for NAD+. C, the surface of the bona fide NAD+-binding site of the Rossmann 1 domain of SmPutA. Note the presence of a pocket that is complementary in shape to the active conformation of NAD+.
Although unusual, Rossmann domains that do not bind dinucleotides have been observed in other enzymes. Haloalcohol dehalogenase (also called halohydrin dehalogenases) is one prominent example. These bacterial enzymes catalyze the co-factor-independent dehalogenation of haloalcohols and show low yet significant sequence similarity to short chain dehydrogenase/reductase NAD(P)(H)-dependent oxidoreductases (31, 32). Although this relationship may seem paradoxical, the two enzyme families are connected mechanistically by a conserved Ser-Tyr-Lys/Arg catalytic triad. Indeed, the first structure of a haloalcohol dehalogenase revealed the characteristic α/β short-chain dehydrogenase/reductase fold, which features a Rossmann dinucleotide-binding domain (33). The Rossmann fold in this case functions as a structural scaffold for the catalytic Ser and other motifs needed for catalysis.
The SmPutA structure also provides an interesting example of structural homology whereby the tertiary structure of one protein mimics the quaternary structure of another protein. The tertiary structural interactions formed by the CTD resemble quaternary structural interactions found in traditional ALDH dimers. For example, Rossmann 2 packs against Rossmann 1 in the SmPutA monomer (Fig. 3C). This relationship mimics the intermolecular contacts between the Rossmann domains from opposing protomers in the traditional ALDH dimer (Fig. 6C). Also, the packing of the β-flap against the GSALDH catalytic domain in SmPutA (Fig. 2A) is strikingly similar to the domain swapping of the oligomerization flap in the classic ALDH dimer (Fig. 6C). In PutA, the structural mimicry leads to an interesting functional similarity. The β-flap of the CTD forms an intramolecular lid over the substrate-channeling tunnel in SmPutA that is analogous to the intermolecular lid formed by the dimerization flap in type A PutAs.
The SmPutA structure provides a template for modeling ALDH16A1, a poorly characterized member of the ALDH superfamily whose biological function is unknown. Humans have two isoforms of ALDH16A1 that differ in polypeptide chain length. Isoform 1 (NCBI RefSeq NP_699160.2) has 802 residues, whereas isoform 2 (NP_001138868.1) lacks an exon encoding 50 residues in the predicted catalytic domain. The common residues of the two isoforms are 100% identical. ALDH16A1 genes have also been identified in fish, amphibians, protists, and bacteria but not in archaea, fungi, and plants (8).
ALDH16A1 is predicted to have ALDH superfamily Rossmann dinucleotide-binding and catalytic domains in residues ∼1–500, followed by the CTD (Fig. 1B). An atypical feature of ALDH16A1 in humans, mouse, and zebrafish is that the catalytic domain apparently lacks the essential Cys residue found in all other ALDHs (8). This observation motivated the idea of human ALDH16A1 being a pseudo-enzyme that functions through protein-protein interactions. For example, it has been proposed that ALDH16A1 associates with the uric acid metabolic enzyme HPRT1 to regulate serum uric acid levels to prevent gout (8, 34). Also the interaction of ALDH16A1 with maspardin is proposed to play a role in the pathogenesis of mast syndrome (SPG21), an autosomal-recessive form of hereditary spastic paraplegia characterized by dementia and other brain abnormalities (7). The substrates for ALDH16A1s having the catalytic Cys are unknown.
Reliable homology models would aid the study of ALDH16A1 function. However, modeling the structure has been challenging because the PDB lacks a structural template that describes the tertiary structural interactions of the CTD with the other domains of the protein. The SmPutA structure provides this missing information. We predict that ALDH16A1 resembles the GSALDH-CTD part of SmPutA, in which the two Rossmann domains interact across a pseudo 2-fold axis, whereas the β-flap contacts the catalytic domain (Fig. 3C). We note that the models that have been proposed for ALDH16A1 show a substantially different tertiary structure (8). The new structural information reported here should aid the modeling of an enigmatic member of the ALDH superfamily.
Finally, the ALDH superfamily provides an example of protein structure evolution through domain repeats. Domain repeats are common in multidomain eukaryotic proteins; familiar examples include zinc fingers, immunoglobulin domains, and EF hands (35). In type B/C PutAs and ALDH16A1, the Rossmann dinucleotide-binding and associated oligomerization domains have been repeated. Unlike typical repeats, the duplicated domains have different functions than the original ones. The repetition of domain structure without conservation of function is an intriguing aspect of multidomain protein architecture.
Experimental Procedures
Expression and Purification of SmPutA
The gene encoding full-length SmPutA (1233 residues; National Center for Biotechnology Information RefSeq WP_010968598.1) in plasmid pNIC28-Bsa4 was obtained from the New York Structural Genomics Research Consortium. The expressed protein has an N-terminal His6 tag that is cleavable with tobacco etch virus protease.
SmPutA was expressed in E. coli BL21 (DE3) pLysS. Starter cultures of 10 ml were grown in LB medium containing 50 μg/ml kanamycin overnight and used to inoculate 2 liters of LB broth containing 35 μg/ml kanamycin. After the culture reached A600 of 0.8, 0.3 mm isopropyl β-d-1-thiogalactopyranoside was added to induce protein expression for 12 h at 18 °C. The cells were collected by centrifugation; resuspended in 50 mm Tris, 300 mm NaCl, 10 mm imidazole, and 5% glycerol at pH 7.5; and stored at −80 °C.
SmPutA was purified as follows. The frozen cells were thawed at 4 °C in the presence of protease inhibitors (0.5 mm phenylmethylsulfonyl fluoride, 0.1 μm pepstatin, and 0.01 mm leupeptin) and broken using sonication with 0.2% Triton X-100 included. The mixture was centrifuged at 16,500 rpm in an SS34 rotor for 1 h at 4 °C, filtered through a 0.45-μm filter, and applied to a 5-ml HisTrap HP column that had been charged with NiCl2 and equilibrated in 50 mm Tris, 300 mm NaCl, 10 mm imidazole, and 5% glycerol, pH 7.8. Washing steps were performed with 20 mm imidazole followed by 40 mm imidazole. The target protein was eluted with 250 mm imidazole. The His tag was removed by incubating SmPutA with tobacco etch virus protease at a ratio of 1 mg of protease/10 mg of SmPutA for 1 h at 28 °C, followed by dialysis overnight in the dark at 4 °C into 50 mm Tris, 0.5 mm EDTA, 0.5 mm Tris(hydroxypropyl)phosphine (THP), and 5% glycerol at pH 7.8. The dialyzed sample was injected onto the HisTrap HP column, and the flow-through was collected. The protein was then purified using anion exchange chromatography (HiTrap Q). The column was equilibrated with a buffer similar to the dialysis buffer and eluted with a linear 0–1 m NaCl gradient. The protein was then supplemented with 50 μm mm FAD and 50 μm NAD+. Finally, size exclusion chromatography (SEC; Superdex 200) or dialysis was used to exchange the protein into the precrystallization buffer of 50 mm NaCl, 50 mm Tris, 0.5 mm THP, 5% glycerol, and 0.5 mm EDTA, pH 7.8. The protein concentration was estimated using the bicinchoninic acid method (Pierce kit) with bovine serum albumin as the standard.
SAXS
SAXS experiments were performed at Beamline 12.3.1 of the Advanced Light Source via the mail-in program (17, 36, 37). Prior to data collection, all protein samples were subjected to SEC (Superdex 200). The column buffer was 50 mm Tris, 1% (v/v) glycerol, 0.5 mm THP, and 50 mm NaCl, pH 7.8. For some samples, the protein was then dialyzed into the SEC buffer. Scattering intensities were measured at four nominal protein concentrations in the range of 1–4 mg/ml using exposure times of 0.5, 1.0, 3.0, and 6.0 s. Scattering curves collected from the protein samples were corrected for background scattering using intensity data collected from the SEC effluent or dialysis buffer.
Crystallization of SmPutA
Crystals were grown at 295 K using sitting drop vapor diffusion in CrystalEX 96 plates (Hampton Research) with drops formed by mixing equal volumes (2.5–3.0 μl) of protein and reservoir solution. The protein stock solution contained 5 mg/ml SmPutA in a buffer containing 50 mm Tris, pH 7.8, 0.5 mm THP, 0.5 mm EDTA, 50 mm NaCl, and 5% glycerol. Initial conditions were identified using commercially available crystal screening kits (Hampton Research). Optimization of these hits resulted in monoclinic and trigonal crystal forms.
Monoclinic crystals were grown using a reservoir of 0.1 m Tris, pH 8.0, 0.1 m ammonium sulfate, 0.1 m lithium sulfate monohydrate, and 22% (w/v) PEG 3350. These crystals were used as microseeds to co-crystallize SmPutA complexed with the proline analog THFA and NAD+. The pH of a stock solution of THFA was adjusted to 7.2 and then added to SmPutA (5 mg/ml) to a final concentration of 50 mm THFA. NAD+ was also included in the enzyme solution (1 mm). Crystals suitable for high resolution diffraction data collection were grown using a reservoir of 0.1 m Hepes, pH 7.5, 0.1 m ammonium sulfate, 0.1 m lithium sulfate monohydrate, 50 mm MgCl2, and 25% (w/v) PEG 3350. The crystals were cryoprotected using the reservoir supplemented with 20 mm THFA, 0.05 mm NAD+, and 16% (v/v) PEG 200, picked up with Hampton nylon loops, and flash-cooled in liquid nitrogen. The space group is P21 with unit cell dimensions a = 101 Å, b = 102 Å, c = 126 Å, and β = 107°. The asymmetric unit contains a protein dimer, which implies 48% solvent and VM of 2.36 Å3/Da (38).
Trigonal crystals were grown using a reservoir solution of 0.1 m Hepes, pH 7.8, 0.2 m ammonium sulfate, 0.1 m sodium formate, 0.1 m MgCl2, and 22% (w/v) PEG 3350. The crystals were soaked for a few minutes in a cryobuffer containing 50 mm THFA, 0.5 mm NAD+, 0.1 mm Hepes, pH 7.8, 0.2 m ammonium sulfate, 0.1 m sodium formate, 0.1 m MgCl2, 22% (w/v) PEG 3350, and 18% (v/v) PEG 200. The space group is P3121 with unit cell parameters of a = 129 Å and c = 150 Å. The asymmetric unit includes 1 protein molecule, which implies 55% solvent and VM of 2.74 Å3/Da.
X-ray Diffraction Data Collection, Phasing, and Refinement
X-ray diffraction data were collected at Beamline 4.2.2 of Advanced Light Source using a Taurus-1 CMOS detector in shutterless mode. The 1.70 Å resolution P21 data set consisted of 180° of data recorded in 900 frames during a total exposure time of 450 s with the detector at 220 mm. The 1.90 Å resolution P3121 data set consisted of 140° of data recorded in 700 frames during a total exposure time of 350 s with the detector at 240 mm. The data were processed with XDS (39) and AIMLESS (40, 41). Data collection statistics are listed in Table 1.
The phase problem was solved using molecular replacement as implemented in PHASER (42). Two search models were used to phase the P21 data. One model represented the PRODH barrel and consisted of a polyalanine representation of residues 264–543 from the structure of an E. coli PutA PRODH domain construct (PDB entry 1TJ1 (9)). The second model represented the GSALDH module and consisted of a polyalanine representation of residues 547–975 of BjPutA (PDB code 3HAZ (4)). A protomer resulting from molecular replacement was used as the search model in a second round of molecular replacement.
The second generation molecular replacement solution was refined with PHENIX (43) and adjusted manually with COOT (44). The resulting model included ∼56% of the expected residues. This model, which lacked the CTD, was input to PHENIX AutoBuild for automated model building. This calculation increased the model completeness to ∼80%. The model was completed with iterative cycles of model building in COOT and refinement in PHENIX. The B-factor model in PHENIX consisted of 1 TLS group per protein chain and isotropic atomic B-factors. The initial phases for the trigonal structure were estimated with molecular replacement using a protomer from the P21 structure. Table 1 lists refinement statistics.
Kinetic Measurements
Steady-state kinetic assays of the coupled PRODH-GSALDH reaction were performed in 50 mm potassium phosphate, pH 7.5, and 25 mm NaCl at 20 °C with 0.25 μm SmPutA enzyme, 200 μm NAD+, 300 μm CoQ1, and varying concentrations of proline (0.1–450 mm). Progress of the reaction was monitored at 340 nm by following the production of NADH (ϵ340 = 6,220 m−1 cm−1). The reaction velocity was observed to decrease at high proline concentration; thus the parameters Km and kcat were determined by fitting the data to a substrate inhibition model of the Michaelis-Menten equation as previously described (24) in which a KI for proline was derived.
Transient time analysis (4, 5, 22) of the coupled PRODH-GSALDH reaction was performed in 50 mm potassium phosphate (pH 7.5, 425 mm NaCl) at 20 °C with 0.25 μm SmPutA enzyme, 200 μm NAD+, 200 μm CoQ1, and 40 mm proline. The nonchanneling reaction was simulated using a free diffusion two-enzyme model as described previously for PutAs (4, 5). To simulate the nonchanneling reaction, the parameters Km and kcat for GSALDH activity and the rate of PRODH activity under the same experimental conditions are required. GSALDH activity assays were performed in 50 mm potassium phosphate, pH 7.5, and 425 mm NaCl at 20 °C with 200 μm NAD+ and varying dl-P5C (0.05–10 mm). Km and kcat for GSALDH activity were estimated from non-linear regression of the initial reaction velocity versus l-P5C concentration using the Michaelis-Menten equation. SmPutA PRODH activity was determined under the same assay conditions except NAD+ was excluded and 1 mm o-aminobenzaldehdye, which reacts with P5C to produce a dihydroquinazolinium compound (ϵ443 nm = 2900 m−1 cm−1), was added to monitor P5C production (45). All kinetic data were collected using a 96-well plate reader (Powerwave XS 96-well plate reader, Biotek). The concentration of SmPutA was determined spectrophotometrically using a molar extinction coefficient of 12,700 m−1 cm−1 at 451 nm (46). The progress curve for a two enzyme nonchanneling reaction of PRODH and GSALDH was then simulated using the free diffusion model described by Equation 1 (47).
| (Eq. 1) |
In Equation 1, v1 is the rate of PRODH activity under the reaction conditions (2.8 μm min−1). The parameters v2 (30.8 μm min−1) and Km (320 μm) are the steady-state kinetic parameters Vmax and the Michaelis constant, respectively, for GSALDH activity of SmPutA.
Author Contributions
M. L. and T. T. G. generated protein and performed X-ray crystallography experiments and structure refinements. B. W. A. and K. N. S. performed kinetics experiments. D. F. B. and J. J. T. designed experiments and interpreted data. J. J. T. and D. F. B. wrote the paper.
Acknowledgments
We thank Prof. Steve Almo and the NYSGRC for the SmPutA clone. The NYSGRC is supported by NIGMS Grant U54GM094662. We thank Dr. Jay Nix for help with X-ray diffraction data collection and Kevin Dyer for collecting the SAXS data through the SIBYLS mail-in program at the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231. Additional support for the SYBILS beamline comes from the National Institute of Health project MINOS (R01GM105404).
This work was supported by NIGMS, National Institutes of Health Grants R01GM065546, R01GM061068, and P30GM103335 and National Science Foundation Grant DBI-1156692. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5KF6 and 5KF7) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ALDH
- aldehyde dehydrogenase
- DUF
- domain of unknown function
- CTD
- C-terminal domain
- PRODH
- proline dehydrogenase
- P5C
- Δ1-pyrroline-5-carboxylate
- GSAL
- l-glutamate-γ-semialdehyde
- GSALDH
- l-glutamate-γ-semialdehyde dehydrogenase
- PutA
- proline utilization A
- SmPutA
- S. meliloti proline utilization A
- PDB
- Protein Data Bank
- BjPutA
- B. japonicum proline utilization A
- SAXS
- small angle X-ray scattering
- THFA
- l-tetrahydrofuroic acid
- CoQ1
- ubiquinone-1
- THP
- Tris(hydroxypropyl)phosphine
- rmsd
- root mean square deviation
- SEC
- size exclusion chromatography.
References
- 1. Liu Z. J., Sun Y. J., Rose J., Chung Y. J., Hsiao C. D., Chang W. R., Kuo I., Perozich J., Lindahl R., Hempel J., and Wang B. C. (1997) The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat. Struct. Biol. 4, 317–326 [DOI] [PubMed] [Google Scholar]
- 2. Tanner J. J. (2008) Structural biology of proline catabolism. Amino Acids 35, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanner J. J., and Becker D. F. (2013) PutA and proline metabolism. In Handbook of Flavoproteins: Volume 1. Oxidases, Dehydrogenases and Related Systems (Hille R., Miller S. M., and Palfey B. A., eds) pp. 31–56, De Gruyter, Berlin [Google Scholar]
- 4. Srivastava D., Schuermann J. P., White T. A., Krishnan N., Sanyal N., Hura G. L., Tan A., Henzl M. T., Becker D. F., and Tanner J. J. (2010) Crystal structure of the bifunctional proline utilization A flavoenzyme from Bradyrhizobium japonicum. Proc. Natl. Acad. Sci. U.S.A. 107, 2878–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh H., Arentson B. W., Becker D. F., and Tanner J. J. (2014) Structures of the PutA peripheral membrane flavoenzyme reveal a dynamic substrate-channeling tunnel and the quinone-binding site. Proc. Natl. Acad. Sci. U.S.A. 111, 3389–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou Y., Larson J. D., Bottoms C. A., Arturo E. C., Henzl M. T., Jenkins J. L., Nix J. C., Becker D. F., and Tanner J. J. (2008) Structural basis of the transcriptional regulation of the proline utilization regulon by multifunctional PutA. J. Mol. Biol. 381, 174–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanna M. C., and Blackstone C. (2009) Interaction of the SPG21 protein ACP33/maspardin with the aldehyde dehydrogenase ALDH16A1. Neurogenetics 10, 217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vasiliou V., Sandoval M., Backos D. S., Jackson B. C., Chen Y., Reigan P., Lanaspa M. A., Johnson R. J., Koppaka V., and Thompson D. C. (2013) ALDH16A1 is a novel non-catalytic enzyme that may be involved in the etiology of gout via protein-protein interactions with HPRT1. Chem. Biol. Interact 202, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang M., White T. A., Schuermann J. P., Baban B. A., Becker D. F., and Tanner J. J. (2004) Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors. Biochemistry 43, 12539–12548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luo M., Arentson B. W., Srivastava D., Becker D. F., and Tanner J. J. (2012) Crystal structures and kinetics of monofunctional proline dehydrogenase provide insight into substrate recognition and conformational changes associated with flavin reduction and product release. Biochemistry 51, 10099–10108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adams M. J., Ford G. C., Koekoek R., Lentz P. J., McPherson A. Jr, Rossmann M. G., Smiley I. E., Schevitz R. W., and Wonacott A. J. (1970) Structure of lactate dehydrogenase at 2–8 A resolution. Nature 227, 1098–1103 [DOI] [PubMed] [Google Scholar]
- 12. Rossmann M. G., Moras D., and Olsen K. W. (1974) Chemical and biological evolution of nucleotide-binding protein. Nature 250, 194–199 [DOI] [PubMed] [Google Scholar]
- 13. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 14. Koppaka V., Thompson D. C., Chen Y., Ellermann M., Nicolaou K. C., Juvonen R. O., Petersen D., Deitrich R. A., Hurley T. D., and Vasiliou V. (2012) Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 64, 520–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lagautriere T., Bashiri G., Paterson N. G., Berney M., Cook G. M., and Baker E. N. (2014) Characterization of the proline-utilization pathway in Mycobacterium tuberculosis through structural and functional studies. Acta Crystallogr. D Biol. Crystallogr. 70, 968–980 [DOI] [PubMed] [Google Scholar]
- 16. Srivastava D., Singh R. K., Moxley M. A., Henzl M. T., Becker D. F., and Tanner J. J. (2012) The three-dimensional structural basis of type II hyperprolinemia. J. Mol. Biol. 420, 176–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dyer K. N., Hammel M., Rambo R. P., Tsutakawa S. E., Rodic I., Classen S., Tainer J. A., and Hura G. L. (2014) High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol. Biol. 1091, 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schneidman-Duhovny D., Hammel M., and Sali A. (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schneidman-Duhovny D., Hammel M., Tainer J. A., and Sali A. (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 105, 962–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 21. Arentson B. W., Sanyal N., and Becker D. F. (2012) Substrate channeling in proline metabolism. Front. Biosci. (Landmark Ed.) 17, 375–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anderson K. S. (1999) Fundamental mechanisms of substrate channeling. Methods Enzymol. 308, 111–145 [DOI] [PubMed] [Google Scholar]
- 23. Moxley M. A., Sanyal N., Krishnan N., Tanner J. J., and Becker D. F. (2014) Evidence for hysteretic substrate channeling in the proline dehydrogenase and Δ1-pyrroline-5-carboxylate dehydrogenase coupled reaction of proline utilization A (PutA). J. Biol. Chem. 289, 3639–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arentson B. W., Luo M., Pemberton T. A., Tanner J. J., and Becker D. F. (2014) Kinetic and structural characterization of tunnel-perturbing mutants in Bradyrhizobium japonicum proline utilization A. Biochemistry 53, 5150–5161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Farrant R. D., Walker V., Mills G. A., Mellor J. M., and Langley G. J. (2001) Pyridoxal phosphate de-activation by pyrroline-5-carboxylic acid. Increased risk of vitamin B6 deficiency and seizures in hyperprolinemia type II. J. Biol. Chem. 276, 15107–15116 [DOI] [PubMed] [Google Scholar]
- 26. Bearne S. L., and Wolfenden R. (1995) Glutamate γ-semialdehyde as a natural transition state analogue inhibitor of Escherichia coli glucosamine-6-phosphate synthase. Biochemistry 34, 11515–11520 [DOI] [PubMed] [Google Scholar]
- 27. Singh R. K., Larson J. D., Zhu W., Rambo R. P., Hura G. L., Becker D. F., and Tanner J. J. (2011) Small-angle X-ray scattering studies of the oligomeric state and quaternary structure of the trifunctional proline utilization A (PutA) flavoprotein from Escherichia coli. J. Biol. Chem. 286, 43144–43153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pemberton T. A., and Tanner J. J. (2013) Structural basis of substrate selectivity of Δ1-pyrroline-5-carboxylate dehydrogenase (ALDH4A1): semialdehyde chain length. Arch Biochem. Biophys. 538, 34–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo M., and Tanner J. J. (2015) Structural basis of substrate recognition by aldehyde dehydrogenase 7A1. Biochemistry 54, 5513–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luo M., Christgen S., Sanyal N., Arentson B. W., Becker D. F., and Tanner J. J. (2014) Evidence that the C-terminal domain of a type B PutA protein contributes to aldehyde dehydrogenase activity and substrate channeling. Biochemistry 53, 5661–5673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kavanagh K. L., Jörnvall H., Persson B., and Oppermann U. (2008) Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol. Life Sci. 65, 3895–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Hylckama Vlieg J. E., Tang L., Lutje Spelberg J. H., Smilda T., Poelarends G. J., Bosma T., van Merode A. E., Fraaije M. W., and Janssen D. B. (2001) Halohydrin dehalogenases are structurally and mechanistically related to short-chain dehydrogenases/reductases. J. Bacteriol. 183, 5058–5066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Jong R. M., Tiesinga J. J., Rozeboom H. J., Kalk K. H., Tang L., Janssen D. B., and Dijkstra B. W. (2003) Structure and mechanism of a bacterial haloalcohol dehalogenase: a new variation of the short-chain dehydrogenase/reductase fold without an NAD(P)H binding site. EMBO J. 22, 4933–4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sulem P., Gudbjartsson D. F., Walters G. B., Helgadottir H. T., Helgason A., Gudjonsson S. A., Zanon C., Besenbacher S., Bjornsdottir G., Magnusson O. T., Magnusson G., Hjartarson E., Saemundsdottir J., Gylfason A., Jonasdottir A., et al. (2011) Identification of low-frequency variants associated with gout and serum uric acid levels. Nat. Genet. 43, 1127–1130 [DOI] [PubMed] [Google Scholar]
- 35. Björklund A. K., Ekman D., and Elofsson A. (2006) Expansion of protein domain repeats. PLoS Comput. Biol. 2, e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Classen S., Hura G. L., Holton J. M., Rambo R. P., Rodic I., McGuire P. J., Dyer K., Hammel M., Meigs G., Frankel K. A., and Tainer J. A. (2013) Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. J. Appl. Crystallogr. 46, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hura G. L., Menon A. L., Hammel M., Rambo R. P., Poole F. L. 2nd, Tsutakawa S. E., Jenney F. E. Jr, Classen S., Frankel K. A., Hopkins R. C., Yang S. J., Scott J. W., Dillard B. D., Adams M. W., and Tainer J. A. (2009) Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 6, 606–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 39. Kabsch W. (2010) XDS. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Potterton E., Briggs P., Turkenburg M., and Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 42. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mezl V. A., and Knox W. E. (1976) Properties and analysis of a stable derivative of pyrroline-5-carboxylic acid for use in metabolic studies. Anal. Biochem. 74, 430–440 [DOI] [PubMed] [Google Scholar]
- 46. Becker D. F., and Thomas E. A. (2001) Redox properties of the PutA protein from Escherichia coli and the influence of the flavin redox state on PutA-DNA interactions. Biochemistry 40, 4714–4721 [DOI] [PubMed] [Google Scholar]
- 47. Meek T. D., Garvey E. P., and Santi D. V. (1985) Purification and characterization of the bifunctional thymidylate synthetase-dihydrofolate reductase from methotrexate-resistant Leishmania tropica. Biochemistry 24, 678–686 [DOI] [PubMed] [Google Scholar]