

Abstract
The Wnt/β-catenin signaling pathway is instrumental in successful differentiation and proliferation of mammalian cells. It is therefore not surprising that the herpesvirus family has developed mechanisms to interact with and manipulate this pathway. Successful coexistence with the host requires that herpesviruses establish a lifelong infection that includes periods of latency and reactivation or persistence. Many herpesviruses establish latency in progenitor cells and viral reactivation is linked to host-cell proliferation and differentiation status. Importantly, Wnt/β-catenin is tightly connected to stem/progenitor cell maintenance and differentiation. Numerous studies have linked Wnt/β-catenin signaling to a variety of cancers, emphasizing the importance of Wnt/β-catenin pathways in development, tissue homeostasis and disease. This review details how the alpha-, beta-, and gammaherpesviruses interact and manipulate the Wnt/β-catenin pathway to promote a virus-centric agenda.
Keywords: Herpesvirus, Herpes simplex virus-1, Varicella zoster virus, Cytomegalovirus, Epstein-Barr virus, Kaposi’s sarcoma-associated herpesvirus, Wnt/β-catenin, Glycogen synthase kinase-3, Axin
Core tip: The Wnt/β-catenin signaling pathway is essential for many host cell functions. Herpesviruses have evolved to manipulate and control this vital pathway to promote viral propagation, evade host immune recognition and maintain latency.
INTRODUCTION
Herpesviruses have been coevolving with vertebrates for millions of years and have developed multiple mechanisms to avoid immune recognition and manipulate host signaling pathways to promote efficient viral replication. This is evident in the ability of herpesviruses to persist for the lifetime of the host while causing limited adverse effects[1]. In general, severe symptoms are only seen in those individuals who are immunocompromised[2]. Accumulating evidence suggests that herpesviruses interact with the Wnt/β-catenin pathway to regulate viral gene expression and alter host cell gene expression by manipulating downstream signaling components during both active infection and latency.
The Wnt/β-catenin pathway is responsible for a signaling cascade that is required during embryonic development and continues throughout the life of an organism. Nearly every tissue and organ depends on this signaling cascade for normal function. Correct Wnt/β-catenin signaling is crucial in the development of many organs including the brain, heart, lung, bone, liver, kidney and gut among others[3,4]. Many of these essential roles continue in adulthood in relation to tissue homeostasis, regeneration, maintenance and repair functions. Additionally, Wnt/β-catenin has been shown to be important in cell migration, genetic stability and apoptosis[5-8]. With such widespread influence on many diverse signaling cascades, dysfunctional Wnt/β-catenin signaling can have deleterious effects. Unregulated Wnt/β-catenin signaling was first linked to human disease in the 1990s when adenomatous polyposis coli (APC) protein was found to interact with β-catenin[9,10]. Since then, Wnt/β-catenin signaling has been implicated in many cancers[11-15], fibrosis[16,17], and metabolic disease[18].
Although conclusive data on the importance of Wnt/β-catenin signaling during the complete replication cycle of all herpesvirus members are lacking, accumulating data are beginning to reveal the importance of this pathway to viral replication, latency and pathogenesis. The potential to target the Wnt/β-catenin pathway for therapeutic intervention is enormous but is compounded by the complexity of the signaling cascade, the number of potential players involved during signaling activation and its importance to cellular homeostasis. Understanding how herpesviruses manipulate this pathway has increased our knowledge of this important pathway and may ultimately lead to novel antiviral therapies.
THE WNT/β-CATENIN SIGNALING CASCADE
Wnts are lipid-modified glycoprotein ligands that act in an autocrine or paracrine manner. Wnt signaling can be divided into three main signaling cascades: Canonical Wnt and two β-catenin-independent pathways, the non-canonical planar cell pathway[19] and the non-canonical Wnt/calcium pathway[20,21]. This review will focus on the canonical Wnt pathway but crosstalk of the three signaling cascades has been reported and is therefore unavoidable. Briefly, in the absence of Wnt stimulation, cytoplasmic β-catenin is phosphorylated and degraded by the ubiquitin-proteasome system (Figure 1). Upon binding of Wnt, phosphorylation of β-catenin is blocked allowing it to translocate to the nucleus where it complexes with transcription factors to upregulate Wnt target gene transcription (Figure 1). Canonical Wnt signaling is initiated when Wnts bind to a heterodimeric transmembrane receptor complex consisting of Frizzled (Fz) receptor and the co-receptors low-density lipoprotein receptor-related protein 5 (LRP5) and LRP6. The ligand interaction induces conformational changes and subsequent phosphorylation of target proteins. This results in recruitment and signaling through the scaffold protein Dishevelled promoting the inhibition of the destruction complex, which contains Axin, APC, β-catenin, casein kinase Iα/β (CKI Iα/β), and glycogen synthase kinase-3α/β (GSK-3α/β). APC directly interacts with β-catenin and Axin. Axin binds to the cytoplasmic tail of LRP6 and this complex is regulated through phosphorylation by GSK-3 and CK1. When the destruction complex is intact, Axin associated β-catenin is phosphorylated by CKI and GSK-3β at N-terminal Ser/Thr residues. Phosphorylated β-catenin is then recognized by the E3 ubiquitin ligase complex β-TrCP (Beta-Transducin Repeat Containing E3 Ubiquitin Protein Ligase) and targeted for degradation by the proteasome. In the presence of Wnt ligand, signaling results in the dissociation of the destruction complex and loss of GSK-3 mediated phosphorylation of β-catenin. Axin is recruited to the phosphorylated tail of LRP preventing β-catenin phosphorylation and ubiquitination. As a result, β-catenin is free to accumulate and translocate to the nucleus where it interacts with members of the T cell factor/lymphoid enhancer-binding factor (TCF/LEF) family of transcription factors and transcriptional coactivators such as CREB-binding protein (CBP), E1A-associated protein p300, and Pygopus to initiate Wnt target gene expression[22]. β-catenin can also interact with many other transcription factors not linked to the TCF/LEF family but that do play important roles in cell maintenance and differentiation[23-25]. For more in depth reviews on Wnt/β-catenin signaling, the reader is referred to many of the excellent reviews available[23,26-29].
Figure 1.
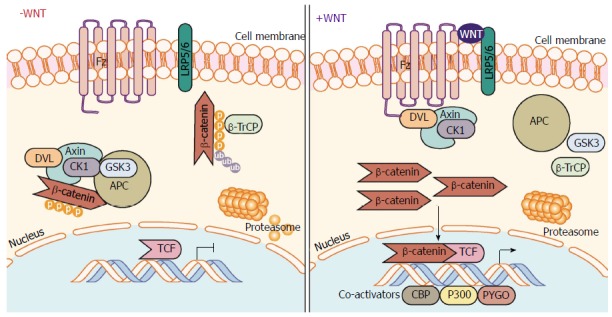
Canonical Wnt/β-catenin signaling pathway. In the absence of Wnt ligand stimulation, the β-catenin destruction complex - consisting of the proteins Axin, CK1, GSK-3α/β, APC, and DVL - phosphorylate β-catenin allowing β-TrCP to ubiquitinate β-catenin marking it for proteasomal degradation. When stimulated by Wnt ligands, engagement of the Fz receptor and co-receptors LRP5/6, induces signaling through DVL inhibiting the action of the destruction complex. This frees β-catenin from degradation pathways allowing β-catenin to translocate and accumulate in the nucleus. β-catenin mediated interaction with TCF family transcription factors and co-activators (CBP, etc.) and initiates transcription of target genes. APC: Adenomatous polyposis coli; β-TrCP: Beta-transducin repeat containing E3 ubiquitin protein ligase; CBP: CREB-binding protein; CK1: Casein kinase 1; DVL: Dishevelled; Fz: Frizzled receptor; GSK-3: Glycogen synthase kinase 3; LRP: Low-density lipoprotein receptor-related protein; TCF/LEF-1: T-cell factor/lymphoid enhancer-binding factor 1.
HERPESVIRUSES
The taxonomic order Herpesvirales includes over 130 herpesviruses divided into three virus families: Herpesviridae that can infect mammals, birds and reptiles; Alloherpesviridae that infect amphibians and bony fish; and Malacoherpesviridae that infects some invertebrates, including molluscs[30-32]. These classifications are based on genome size/structure and biological function. Herpesviridae is a family of enveloped, DNA viruses that is further divided into 3 subfamilies (Alphaherpesvirinae, Betaherpesvirinae and Gammaherpesvirinae). A criterion for inclusion in the Herpesviridae family morphologically is centered on the virion structure[33]. The virion is spherical in shape and includes a core, capsid, tegument and envelope. The core contains the viral genome, which is a linear, double-stranded DNA molecule. The core is surrounded by an icosahedral capsid that is enclosed within a proteinaceous layer called the tegument. Finally, a lipid bilayer envelope surrounds the exterior of the tegument and completes the structure of the virion.
Humans can be infected by eight different herpesviruses. Herpesvirus infections are typically systemic, although some may be localized. Gene expression is tightly regulated and orchestrated in a temporal manner. Simplistically, immediate-early genes encoding regulatory proteins are expressed soon after infection, followed by expression of early genes that are important for replication of viral DNA. Finally, late genes encoding structural proteins are expressed. Due to various host immune evasion strategies, herpesviruses establish life-long latent infections in infected individuals. In an oversimplified model in regards to human infection, Alphaherpesvirinae establish latency in neurons, Betaherpesvirinae in monocytes and Gammaherpesvirinae in lymphocytes, monocytes, and macrophages[1,32,34].
HUMAN ALPHAHERPESVIRUSES
The subfamily Alphaherpesvirinae includes three members. The human herpesviruses 1 and 2 (HHV-1/2) also known as herpes simplex virus (HSV) (type 1/2) belong in the genus Simplexvirus while HHV-3 or Varicella-zoster virus (VZV) is classified in the genus Varicellovirus[32,33]. Infection can result in skin vesicles or mucosal ulcers and on rare occasions meningitis and encephalitis[2].
HHV-1 (HSV-1)
To date there have been no focused, thorough investigations of the role of Wnt/β-catenin on HSV-1/2. The studies that have been completed implicate individual members of the Wnt/β-catenin signaling cascade in viral pathogenesis. An example of this is the upregulation of the antiviral cytokine interferon-β (IFN-β) during HSV-1 infection. In adult immunocompetent mice, macrophages are essential for clearing HSV-1 from the blood; however, it was observed that macrophages from Akt-/- mice display poor clearance of HSV-1. The Akt1 family of serine/threonine kinases was shown to phosphorylate β-catenin at serine 552 allowing accumulation and β-catenin mediated induction of IFN-β[35]. Akt1 classically has been described as a β-catenin transcriptional promoter, exerting its effects by repressing GSK-3 mediated β-catenin proteasomal degradation[36]. Interestingly, the serine 552-phosphorylation site is distinct from the site typically targeted by GSK-3. The authors conclude that Akt1 is responsible for inhibiting GSK-3 phosphorylation of β-catenin on Ser9 and also for direct phosphorylation of β-catenin at serine 552 allowing for stabilization, enhanced nuclear translocation and transcriptional activity of β-catenin (Figure 2).
Figure 2.
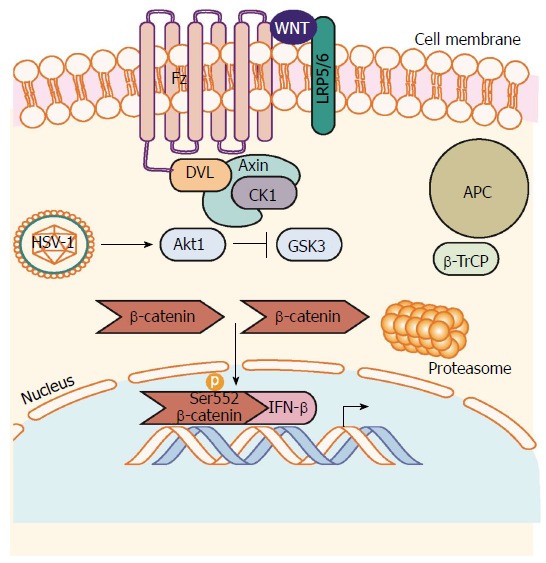
β-catenin mediated antiviral interferon response during herpes simplex virus 1 infection. HSV-1 infection induces activation of Akt1 activity. Akt1 phosphorylates β-catenin on Serine 552 inhibiting degradation signaling through GSK-3-mediated phosphorylation of β-catenin on Serine 9. β-catenin can then accumulate in the nucleus to induce transcription of β-catenin target genes such as the antiviral cytokine IFN-β. Akt1: Protein kinase B; HSV-1: Herpes simplex virus 1.
In a second study, Choi et al[37] observed that HSV-1 infection and replication was more efficient in a fibroblast-like murine cell line, L929. Knocking down Axin or treatment with Wnt3a conditioned media reduces HSV-1 replication in L929 cells. They further showed that Axin expression minimalizes HSV-1 induced cell death, which in turn promotes increased HSV-1 replication. In a follow up study, this group observed that HSV-1 infection also induced autophagy but this is delayed in L929 cells ectopically expressing L-Axin[38]. The authors concluded that delay in induction of autophagy favors HSV-1 viral replication likely by suppression of HSV-1 mediated cell death. The implication is that HSV-1 replication is inversely related to Wnt signaling.
Lastly, Piacentini et al[39] demonstrated that HSV-1 infection disrupts synaptic function in cultured murine cortical neurons through GSK-3 activation and intracellular accumulation of amyloid-β protein. In a previous study this group showed that HSV-1 mediated increases in intracellular Ca2+ is the main mechanism for activation of GSK-3 in this model[39]. These studies suggest a possible link between HSV-1 pathogenesis and Alzheimer’s disease.
To date, the involvement of Wnt/β-catenin signaling during VZV infection has been underinvestigated. Markus et al[40] observed an increase in canonical Wnt pathway transcription in infection of neurons derived from human embryonic stem cells. The Wnt pathway was unaffected during late VZV infection of fibroblasts. Intriguingly, like HSV-1 and -2, VZV will enter latency in neurons but will lytically replicate in fibroblasts suggesting a differing need for Wnt pathway modification by the virus in different stages of the viral life cycle[40]. Given the limited studies on Wnt/β-catenin signaling during alphaherpesvirus infections, how vital Wnt/β-catenin signaling is to viral replication and pathogenesis remains unknown. The studies mentioned above seem to portray a conflicting role of β-catenin in viral replication. More thorough studies using defined cell types and carefully delineated “branches” of the Wnt pathway will provide a clearer understanding.
HUMAN BETAHERPESVIRUS
The human Betaherpesvirinae subfamily consists of the three viruses: HHV-5 known as human cytomegalovirus (HCMV), HHV-6A/B, and HHV-7 (the latter two are commonly referred to as Roseolovirus)[32,33]. Infection is usually asymptomatic but infectious mononucleosis like symptoms are seen in HCMV infections and the development of a rash is associated with Roseolovirus. In immunocompromised individuals (organ transplant patients, HIV positive individuals, etc.) or during pregnancy, infection and/or reactivation of β-herpesvirus can have life-threatening consequences. Of these three viruses, HCMV is the most studied and is considered the prototypical betaherpesvirus. As little is known about Wnt/β-catenin regulation during infection by the polyphyletic Roseolovirus group, this portion of the review will focus exclusively on HCMV.
HHV-5 (HCMV)
The Wnt/β-catenin pathway is one of the many cellular pathways manipulated by HCMV to likely facilitate lytic viral replication. By dysregulating the physiological condition of the Wnt/β-catenin pathway, HCMV inhibits or severely hampers the processes of cellular replication, movement/migration, and differentiation among others[41,42].
HCMV infection of the placenta may cause impaired invasion of placental-derived cells toward maternal spiral arteries leading to shallow placentation and a deficit in oxygen/nutrient flow to the developing fetus[43]. The Wnt/β-catenin pathway is important in the differentiation of placental cytotrophoblasts into extravillous trophoblasts, the invasive lineage of cells that remodel maternal spiral arteries to establish blood flow to the placenta[44-46]. Using an in vitro model of first trimester cytotrophoblasts (SGHPL-4) infected with HCMV, Angelova et al[41] demonstrated that β-catenin protein levels decrease significantly during the late stages of infection roughly corresponding to expression of late proteins and packaging of nucleocapsids into an envelope to produce mature virions. This decrease in β-catenin protein is dependent on proteasomal degradation and occurs in all cellular pools including membrane, cytoplasm and nucleus. Remaining β-catenin, aggregates near the viral assembly compartment, a juxtanuclear region present during infection involved in virion assembly and egress; however, the reasons for this are currently unclear. Transcriptional targets of β-catenin, such as Dickkopf-related protein 1 (Dkk1) and Cyclin D, also exhibit transcriptional repression as a result. However, β-catenin mRNA levels actually increase in the same timeframe[41]. Consistent with these results, Ueland et al[47] showed that plasma levels of DKK-1 were significantly lower in solid organ transplant patients with HCMV DNAemia. In contrast, Langemeijer et al[48] reported that HCMV infection increases transcriptional activation of β-catenin in a glioblastoma cell line that is dependent on expression of the virally encoded G-protein coupled receptor, US28. These different results may be explained by the use of different cell types and methods to detect β-catenin activity.
The mechanism by which HCMV depletes membrane stores of β-catenin is currently unknown although infection extensively remodels cellular membranes[49]. As for cytoplasmic and nuclear stores of β-catenin, HCMV exerts control at the level of the β-catenin destruction complex as disruption of this complex with lithium chloride (LiCl), a GSK-3β inhibitor can inhibit the degradation and depletion of β-catenin during infection. It should be noted that inhibition of β-catenin degradation does not rescue transcriptional function of β-catenin[41]. This may be due to further regulation of transcriptional activity of β-catenin, for example through regulation of β-catenin coactivators like TCF/LEF-1, by the virus or due to undetected post-translational modification of β-catenin. Viral regulation of the destruction complex appears to be mostly mediated at Axin1, the rate-limiting protein in the β-catenin destruction complex in the cytoplasm. PolyADP Ribose Polymerase 5a and 5b (PARP5a/b), also called Tankyrase (TNKS as a combination of isoforms 1 and 2), PARsylates Axin1 leading to degradation through the ubiquitin proteasome pathway. During HCMV infection, TNKS PARsylation activity is inhibited allowing for stabilization of Axin1 and stabilization of the β-catenin destruction complex leading to the degradation of β-catenin seen during infection (Figure 3)[50]. This suggests that HCMV requires a complete and competent β-catenin destruction complex for degradation of β-catenin.
Figure 3.
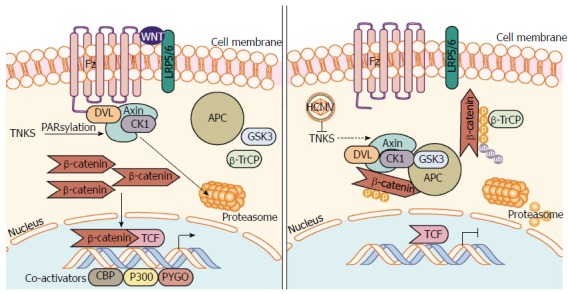
Human cytomegalovirus inhibits PARsylation activity of tankyrase 1 and 2 (PARP5a/b) to enhance infection. Regulation of Axin is the rate-limiting step in the assembly and function of the β-catenin destruction complex. PARsylation, a process driven by NAD+, marks Axin for proteasomal degradation, inhibiting β-catenin destruction complex formation resulting in β-catenin accumulation and transcription in the nucleus. HCMV infection causes inhibition of TNKS (PARP5a/b) PARsylation activity, which inhibits PARsylation of Axin and increases its stability. Further inhibition of TNKS PARsylation activity or knockdown of TNKS significantly aids in HCMV replication. An increase in stable Axin permits β-catenin destruction complex formation, increased β-catenin degradation, and subsequent inhibition of β-catenin-mediated transcription. HCMV: Human cytomegalovirus; NAD+: Nicotinamide adenine dinucleotide; PARsylation: Poly-ADP ribose modification; PARP5a/b: Poly-ADP ribose polymerase 5a/b; TNKS: Tankyrase 1 and 2 (PARP5a/b).
The non-canonical pathways of Wnt signaling, although lacking direct β-catenin regulation, seem to play a role in regulation of the canonical Wnt/β-catenin pathway during HCMV infection. Wnt5a interacts with the tyrosine-like orphan kinase 2 ROR2 and physiologically activates the Wnt/Planar Cell Polarity pathway and Wnt/Ca2+ pathway[42]. During HCMV infection, infected cells become insensitive to normal Wnt5a ligand signaling but ROR2 expression is significantly increased. Uninfected trophoblasts invade toward a Wnt5a gradient in vitro but are incapable of doing so when infected despite the increased presence of ROR2. The increase in ROR2 expression inhibits canonical signaling by repressing β-catenin TCF/LEF-1 transcriptional activity. Knockdown of non-canonical ROR2 that is overexpressed during infection can rescue some function of the canonical Wnt/β-catenin pathway in trophoblasts suggesting that the canonical and non-canonical Wnt pathways are deeply intertwined, especially during HCMV infection[42].
Targeting of Wnt/β-catenin signaling with select pharmacological inhibitors can inhibit viral replication suggesting that some level of β-catenin or a member of the canonical Wnt pathway may be necessary for viral replication[51]. Why HCMV infection overrides normal Wnt/β-catenin signaling is unknown, but some research indicates involvement of repurposing the molecular members of the pathway to further HCMV replication. Activity of GSK-3 has been implicated in assembly of the viral nucleocapsid in simian CMV (infecting Chimpanzees and Orangutans). Phosphorylation of the viral assembly protein precursor (pAP) by GSK-3 may induce conformational changes in the protein and stabilize pAP interaction with the major capsid protein during capsid assembly[52]. Additionally GSK-3 (along with other members of the β-catenin destruction complex) has been identified as a target for phosphorylation by the viral kinase UL97[53]. However, inhibition of UL97 activity during infection does not seem to rescue β-catenin degradation suggesting that UL97 phosphorylation of GSK-3 is not the primary mechanism by which HCMV depletes β-catenin stores (our unpublished data). Further research must be conducted to determine the importance of molecular mechanisms of Wnt/β-catenin on viral replication itself.
HCMV infection has recently been associated with a diverse array of diseases and disorders such as diabetes[54], atherosclerosis[55], and some cancers (reviewed in[56,57]), along with the abovementioned issues with infection during pregnancy on the placenta and developing fetus. As data show that HCMV infection undermines normal functioning of canonical Wnt/β-catenin and non-canonical Wnt signaling in diverse ways, differing perhaps by infection of a multitude of diverse cell types, it becomes key to better characterize this viral regulation.
HUMAN GAMMAHERPESVIRUSES
The human Gammaherpesvirinae family includes two members: Human herpesvirus 4 (HHV-4) commonly known as Epstein-Barr virus (EBV) and HHV-8 or Kaposi’s sarcoma-associated herpesvirus (KSHV)[32,33]. They are further classified under the genera Lymphocryptovirus and Rhadinovirus, respectively. EBV was one of the first viruses to be associated with human cancer when it was originally identified in Burkitt’s lymphoma. Since then, it has become associated with B cell malignancies and epithelial cell associated cancers. KSHV was discovered in 1994 when samples from AIDS-associated Kaposi’s sarcoma came back positive for viral DNA sequences[58]. Diseases associated with KSHV include B cell malignancy primary effusion lymphoma (PEL), Castleman’s disease and the endothelial lesion, Kaposi’s sarcoma.
HHV-4 (EBV)
The accumulation of β-catenin is seen in EBV-infected epithelial and B cells. In the earliest report, Shackelford et al[59] reported that β-catenin was not degraded in lymphoid cells during an EBV type III latent infection. The authors postulate that these observations may be due to ubiquitinating enzymes or the dysregulation of other oncogenes. Interestingly, this effect was not observed during EBV type I latency infection[59]. Shortly after, a second group showed that telomerase-immortalized human foreskin keratinocytes have increased β-catenin accumulation after infection with EBV[60]. The mechanism was shown to be dependent on latent membrane protein 2A (LMP2A) activation of Akt and Akt-mediated inactivation of GSK-3, independent of phosphorylation at Ser9. Treatment with LiCl led to β-catenin accumulation in the cytoplasm, translocation into the nucleus and activation of a TCF-responsive reporter. In a follow-up study, the immunoreceptor tyrosine-based activation and PY motifs of LMP2A were found to mediate the accumulation and nuclear translocation of β-catenin[61]. Using LMP2A ΔPY mutants, they showed that β-catenin levels and translocation to the nucleus decreased along with epithelial cell differentiation. The authors concluded that LMP2A mediated epithelial cell differentiation appears to be inversely correlated with β-catenin activation in this model.
EBV latent membrane protein 1 (LMP1) has also been associated with an increase in β-catenin levels in EBV-infected BL cells[62]. Jang et al[63] reported that an E3 ubiquitin ligase, a human homolog of Drosophilia seven in absentia (Siah-1), is repressed by LMP1. Siah-1 binds APC and in a GSK-3 independent manner, degrades β-catenin. However, another study using transient and stable expression of LMP1 sequences failed to find evidence that LMP1 induces Wnt/β-catenin signaling or promotes the accumulation of β-catenin[64]. To further verify their observations, they proceeded to show that there was little evidence for interactions between LMP1 and β-catenin. The authors proposed that differences in cell lines and LMP1 sequences used may account for the conflicting results in these two studies.
Lastly, EBV-mediated dysregulation of Wnt/β-catenin was associated with idiopathic pulmonary fibrosis (IPF)[65]. EBV detection in alveolar epithelial cells has been associated with poor prognosis. Pathogenesis is believed to occur in IPF due to repetitive epithelial cell injury that may be mediated by EBV. Using transcriptomic data, the authors identified altered Wnt/β-catenin pathway transcripts. Specifically, Wnt5b expression was altered. The authors conclude that EBV may be using a non-canonical Wnt/β-catenin pathway that includes CUX1 and the EBV early gene Rta.
HHV-8 (KSHV)
Fujimuro et al[66] first observed the association between Wnt/β-catenin and KSHV in 2003. They made the observation that in latently KSHV-infected B cell lines derived from PEL, β-catenin accumulated at high levels in the cytoplasm. KSHV infection of PEL cells results in a high KSHV latency rate suggesting that the increased levels of β-catenin may be linked to expression of KSHV latency associated proteins. The latency-associated nuclear antigen (LANA) protein proved to be the protein responsible, as siRNA transient knockdown specific to LANA, decreased levels of LANA and β-catenin[67]. LANA was originally shown to be involved in the tethering of KSHV episomal genomes to host chromosomes to aid in viral DNA replication[68,69]. Using a yeast-two hybrid system, paired with coimmunoprecipitation assays, LANA was also found to possess the ability to bind to GSK-3α and GSK-3β[67].
In addition to mediating the phosphorylation of β-catenin as part of the destruction complex in the cytoplasm, GSK-3 also translocates to the nucleus during apoptotic stimuli in a cell cycle-dependent manner. Nuclear levels of GSK-3 protein increase in the nucleus of PEL cells specifically during the S phase of the cell cycle[66]. This results in lower GSK-3 within the destruction complex and more unphosphoryated β-catenin that translocates to the nucleus and activates target gene expression (Figure 4). The authors proposed that LANA promotes the accumulation of GSK-3 in the nucleus, reducing the total amount of GSK-3 in the cytoplasm.
Figure 4.
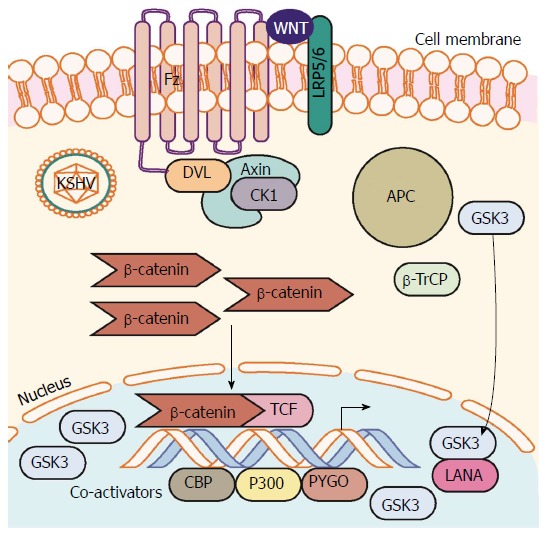
Kaposi’s sarcoma-associated herpesvirus latency upregulates β-catenin. Establishment of KSHV latency involves expression of the latency associated protein LANA. LANA binds with and translocates GSK-3 into the nucleus after phosphorylation by GSK-3. This translocation of cytoplasmic pools of GSK-3 prevents β-catenin destruction complex formation and stability of cytoplasmic β-catenin. β-catenin translocation to the nucleus occurs resulting in increased β-catenin-mediated transcription. LANA: Latency-associated nuclear antigen; KSHV: Kaposi’s sarcoma-associated herpesvirus.
Further studies revealed that the C-terminal region of LANA displayed limited homology to the domain of Axin that binds GSK-3β and is functionally similar to Axin[67]. LANA protein mutants were used to study the binding potential between LANA and GSK-3β[70]. These studies showed that changing Phe291 in the coding sequence of LANA to Leu (F291L mutant), leads to a reduction in binding to GSK-3 by 90%. The interaction of the various components of the destruction complex is mediated by phosphorylation, which also mediates the interaction of LANA and GSK-3. GSK-3 and LANA interactions require the LANA C-terminal GSK-3 interacting domain and GSK-3 phosphorylation of the LANA N-terminus. Within this region are four consensus GSK-3 phosphorylation sites [(Ser/Thr)xxx(Ser/Thr)p]. Mutation of the four consensus sites prevented GSK-3 binding to LANA, suggesting that this is a phosphorylation mediated event[70]. Additionally, as GSK-3 substrates typically must be primed prior to phosphorylation by GSK-3, a mutant (R96A) was used to determine if GSK-3 phosphorylation of LANA could proceed without priming. Results showed that, under in vitro conditions, GSK-3 phosphorylation of LANA requires priming kinases. The reader is referred to two comprehensive reviews detailing the manipulation of GSK-3 by KSHV[71,72].
Additional studies revealed that CKI and mitogen-activated protein kinase could each function as priming kinases for GSK-3 phosphorylation of LANA[73]. To summarize, KSHV latency protein LANA, promotes nuclear accumulation of GSK-3 to promote dysregulation of β-catenin. Functionally, there is increased expression of cyclin D1, and when β-catenin reporters have been tested, there is increased activity[74]. Surprisingly, it was also determined that most of the GSK-3 in the nucleus of LANA-expressing cells is in an inactive phosphorylated form suggesting that despite increased GSK-3 present in the nucleus, there is a decrease in nuclear GSK-3 activity. Inhibitor of the MyoD family a (I-mfa) and the human I-mfa domain-containing protein (HIC) has been shown to be negative inhibitors of the Wnt pathway. Kusano et al[75] showed that LANA interacts with HIC and I-mfa in the 995-1102 amino acid region of LANA. This site is located near the GSK-3 binding site and inhibits the LANA mediated transactivation of a β-catenin construct. Furthermore, this interaction decreases LANA-GSK-3 complex formation resulting in a decrease in Wnt/β-catenin signaling associated transcription. Thus manipulation of the Wnt/β-catenin pathway may play a key role in LANA-mediated oncogenesis in KSHV-infected cells.
Lastly, a recent paper reports that KSHV viral IFN regulatory factor 4 (vIRF4) targets the β-catenin/TCF transcription complex[76]. Using a TOPFlash system, the data suggests that LANA and vIRF4 are negative regulators of each other. Expression of LANA alone resulted in increased β-catenin protein and transcriptional levels, but introducing vIRF4 reduced the levels of LANA-mediated β-catenin/TCF activation. The authors also observed that that this effect was not dependent on β-catenin protein stability. In conclusion, the study suggests that KSHV employs vIRF4 to block the progression of the cell cycle at the G1-S phase to aid in viral replication.
It has been proposed that dysregulation of the viral gene program leads to nonlytic expression[4]. Angelova et al[77] show a novel pathway that KSHV uses to upregulate the Wnt/β-catenin pathway. The KSHV virally-encoded G-protein coupled receptor (vGPCR) inserted into a retroviral vector was transduced into endothelial cells. The authors observed increased cyclin D1, Wnt7A and pygopus 1 (Pygo) expression in vGPCR expressing cells as compared to non-expressing control cells. Additionally, β-catenin was found to accumulate in the nucleus of vGPCR expressing cells and β-catenin/LEF1-dependent TOPFlash reporter constructs displayed increased activity. Initial data suggests that vGPCR-induced activation of the Wnt/β-catenin is through the PI3K/Akt pathway, similar to what is seen in HSV and EBV. This conclusion was contrary to prior work suggesting that this effect may be mediated through COX2 activity; it was found that PI3K/Akt inhibition potently inhibited Wnt/β-catenin activity in endothelial cells and prevented formation of capillary endothelial tubes in vitro[77].
SPECULATION AND QUESTIONS
Despite numerous studies addressing the role of Wnt/β-catenin signaling in herpesviruses, there are still many questions to address. It seems at odds that the gammaherpesviruses would institute a program promoting the accumulation of β-catenin whilst the other family members inhibit the accumulation of β-catenin. The range and complexity of the Wnt/β-catenin pathway makes a simple answer unlikely; but factors such as stage of viral infection and cell type are obvious candidates. As we understand more about herpesviruses, it is conceivable that the herpesvirus family can change the regulation and function of such an important pathway at different times during the viral life cycle. Control over apoptosis, cytoskeletal rearrangement, migration and differentiation are all vital components of viral control over the host cell that would be required at different times post infection.
As mentioned previously, dysregulation of the Wnt/β-catenin pathway is tightly associated with numerous human cancers. In fact, most of the human herpesviruses can be thought of as oncomodulators, whether in a direct manner such as in the expression of oncogenic viral proteins in gammaherpesvirinae infection or through indirect generation of oncostimulatory microenvironments by virally induced inflammation or cellular metabolic shifts caused by alpha- and betaherpesvirinae infection. Why would an evolutionarily successful viral family induce cancer in its host? Ultimately, herpesviruses are successful because they coexist with their host. The development of cancer due to the persistence of a herpesvirus is likely an infrequent event that is complicated by others factors such as altered host cell metabolism and possibly the presence of other pathogens. For example, HCMV is now known to alter host cell metabolism during infection[78-81]. The changes are very similar to the Warburg-effect first identified in cancer cells.
Interestingly, a recent publication may bridge the different actions of viruses on the Wnt/β-catenin pathway during different stages of infection. Data from Marcato et al[82] suggests that the TCF/β-catenin complex is instrumental in mounting an effective antiviral response. They linked two observations, namely, that IFN-β is needed during the innate antiviral response and that murine models lacking IFN-β are susceptible to viral infections. In this paper, the authors show that inhibiting GSK-3 using LiCl increases IFN-β expression if β-catenin interacts with the IFN-β promoter by recruitment of TCF/β-catenin complexes to the promoter region. Using Rift Valley fever virus, a RNA virus belonging to the Bunyaviridae family, they showed pathogenicity is correlated to viral targeting of the β-catenin pathway.
Viral manipulation of Wnt/β-catenin signaling may be impeded using small molecules inhibitors that target the Wnt/β-catenin pathway. In fact, Chan et al[83] have shown results displaying the potential of this treatment. The authors used ICG-001, a small molecular Wnt modulator (CBP/β-catenin antagonist) to inhibit the growth of tumor spheres in a model of nasopharyngeal carcinoma. This epithelial malignancy is associated with EBV latent infection. It is hypothesized that ICG-001 targets the cancer stem cells within the tumor reducing growth due to alterations in signaling cascades. To date, no studies have looked at the direct effects of small molecule inhibitors as antivirals, but targeting the Wnt pathway is being explored in many other diseases and should be examined in the context of herpesvirus infection (reviewed in[28]).
CONCLUSION
Human herpesviruses exploit the Wnt/β-catenin signaling pathway to ensure successful replication and survival in host cells (Table 1). The manipulation of such an important signaling cascade by herpesviruses should not be surprising as this pathway dictates the expression of many essential transcriptional pathways. The current literature provides an incomplete picture of why herpesviruses alter the Wnt/β-catenin pathway when they do. A deeper understanding of why herpesviruses induce changes in the Wnt/β-catenin pathway when they do, would provide vital information about the viral purpose of manipulating this pathway and how to interfere with this host manipulation controlled by the virus. As we understand more about virally induced aberrant Wnt/β-catenin we can develop better antivirals and possibly apply this knowledge to other human diseases associated with the Wnt/β-catenin pathway.
Table 1.
Wnt/β-catenin molecular manipulations by human Herpesviridae
| Virus | Pathway component | Stabilization, activation or inhibition of pathway component | Outcome | Ref. |
| Alphaherpesvirinae | ||||
| HSV-1 | β-catenin | Stabilized | β-catenin stabilized, increased transcriptional activity of β-catenin | [35] |
| Axin | Stabilized | Reduced host cell apoptosis | [37,38] | |
| GSK-3 | Stabilized | Phosphorylation of APP | [39] | |
| Betaherpesvirinae | ||||
| HCMV | β-catenin | Inhibited | β-catenin degradation, decrease in β-catenin transcriptional targets | [41] |
| Axin | Stabilized | TNKS PARsylation activity inhibited resulting in β-catenin degradation | [50] | |
| ROR2 | Activated | Repression of β-catenin TCF/LEF-1 transcriptional activity | [42] | |
| GSK-3 | Stabilized | Stabilization of pAP and promotion of HCMV replication | [52,53] | |
| Gammaherpesvirinae | ||||
| EBV | β-catenin | Stabilized | Accumulation of β-catenin in type III latency | [59] |
| GSK-3 | Inhibited | LMP2A activation of Akt inactivates GSK-3 resulting in β-catenin accumulation | [60,61] | |
| APC | Activated/inhibited (conflicting results) | LMP1 represses Siah-1 promoting β-catenin accumulation. LMP1 does not promote β-catenin stabilization | [63,64] | |
| KSHV | β-catenin | Stabilized/inhibited (dependent on viral stage?) | Increased transcriptional activity, induction of viral latency/inhibition of LANA mediated transactivation of β-catenin | [66,76,77] |
| GSK-3 | Inhibited | LANA promotes nuclear accumulation of GSK-3 | [67,70] | |
GSK-3: Glycogen synthase kinase 3; HCMV: Human cytomegalovirus; TNKS: Tankyrase 1 and 2 (PARP5a/b); TCF/LEF-1: T-cell factor/lymphoid enhancer-binding factor 1; pAP: Protein precursor; LMP2A: Latent membrane protein 2A; APC: Adenomatous polyposis coli; LMP1: Latent membrane protein 1; LANA: Latency-associated nuclear antigen; KSHV: Kaposi’s sarcoma-associated herpesvirus.
ACKNOWLEDGMENTS
We would like to thank Robin Baudier for assistance with the production of the figures.
Footnotes
Conflict-of-interest statement: The authors have no conflicts to disclose.
Manuscript source: Invited manuscript
Specialty type: Virology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: May 30, 2016
First decision: July 6, 2016
Article in press: August 8, 2016
P- Reviewer: Davis DA, Diefenbach R, Qin ZQ, Rajcani J S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Grinde B. Herpesviruses: latency and reactivation - viral strategies and host response. J Oral Microbiol. 2013:5. doi: 10.3402/jom.v5i0.22766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans CM, Kudesia G, McKendrick M. Management of herpesvirus infections. Int J Antimicrob Agents. 2013;42:119–128. doi: 10.1016/j.ijantimicag.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 3.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 4.van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 5.Webster MR, Weeraratna AT. A Wnt-er migration: the confusing role of β-catenin in melanoma metastasis. Sci Signal. 2013;6:pe11. doi: 10.1126/scisignal.2004114. [DOI] [PubMed] [Google Scholar]
- 6.Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336:1549–1554. doi: 10.1126/science.1218370. [DOI] [PubMed] [Google Scholar]
- 7.Alberici P, Fodde R. The role of the APC tumor suppressor in chromosomal instability. Genome Dyn. 2006;1:149–170. doi: 10.1159/000092506. [DOI] [PubMed] [Google Scholar]
- 8.Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci. 2013;126:21–29. doi: 10.1242/jcs.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubinfeld B, Souza B, Albert I, Müller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with beta-catenin. Science. 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- 10.Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science. 1993;262:1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- 11.Porfiri E, Rubinfeld B, Albert I, Hovanes K, Waterman M, Polakis P. Induction of a beta-catenin-LEF-1 complex by wnt-1 and transforming mutants of beta-catenin. Oncogene. 1997;15:2833–2839. doi: 10.1038/sj.onc.1201462. [DOI] [PubMed] [Google Scholar]
- 12.Webster MR, Kugel CH, Weeraratna AT. The Wnts of change: How Wnts regulate phenotype switching in melanoma. Biochim Biophys Acta. 2015;1856:244–251. doi: 10.1016/j.bbcan.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 14.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 15.Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 16.Chilosi M, Poletti V, Zamò A, Lestani M, Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol. 2003;162:1495–1502. doi: 10.1016/s0002-9440(10)64282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dees C, Distler JH. Canonical Wnt signalling as a key regulator of fibrogenesis - implications for targeted therapies? Exp Dermatol. 2013;22:710–713. doi: 10.1111/exd.12255. [DOI] [PubMed] [Google Scholar]
- 18.Schinner S. Wnt-signalling and the metabolic syndrome. Horm Metab Res. 2009;41:159–163. [Google Scholar]
- 19.Wang Y, Nathans J. Tissue/planar cell polarity in vertebrates: new insights and new questions. Development. 2007;134:647–658. doi: 10.1242/dev.02772. [DOI] [PubMed] [Google Scholar]
- 20.Veeman MT, Axelrod JD, Moon RT. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev Cell. 2003;5:367–377. doi: 10.1016/s1534-5807(03)00266-1. [DOI] [PubMed] [Google Scholar]
- 21.Kohn AD, Moon RT. Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium. 2005;38:439–446. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 22.Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T, et al. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245–1256. doi: 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. EMBO J. 2012;31:2714–2736. doi: 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang Y, Liu Z, Zhao L, Clemens TL, Cao X. Smad7 stabilizes beta-catenin binding to E-cadherin complex and promotes cell-cell adhesion. J Biol Chem. 2008;283:23956–23963. doi: 10.1074/jbc.M800351200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholtysek C, Katzenbeisser J, Fu H, Uderhardt S, Ipseiz N, Stoll C, Zaiss MM, Stock M, Donhauser L, Böhm C, et al. PPARβ/δ governs Wnt signaling and bone turnover. Nat Med. 2013;19:608–613. doi: 10.1038/nm.3146. [DOI] [PubMed] [Google Scholar]
- 26.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 27.Cruciat CM. Casein kinase 1 and Wnt/β-catenin signaling. Curr Opin Cell Biol. 2014;31:46–55. doi: 10.1016/j.ceb.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 28.Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513–532. doi: 10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGeoch DJ, Rixon FJ, Davison AJ. Topics in herpesvirus genomics and evolution. Virus Res. 2006;117:90–104. doi: 10.1016/j.virusres.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Brown JC, Newcomb WW. Herpesvirus capsid assembly: insights from structural analysis. Curr Opin Virol. 2011;1:142–149. doi: 10.1016/j.coviro.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knipe DM, Howley PM. Fields virology. 6th ed. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins Health; 2013. p. 1. [Google Scholar]
- 33.Human herpesviruses: Biology, therapy, and immunoprophylaxis. In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Cambridge: Cambridge University Press; 2007. [PubMed] [Google Scholar]
- 34.Monini P, Colombini S, Stürzl M, Goletti D, Cafaro A, Sgadari C, Buttò S, Franco M, Leone P, Fais S, et al. Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi’s sarcoma. Blood. 1999;93:4044–4058. [PubMed] [Google Scholar]
- 35.Gantner BN, Jin H, Qian F, Hay N, He B, Ye RD. The Akt1 isoform is required for optimal IFN-β transcription through direct phosphorylation of β-catenin. J Immunol. 2012;189:3104–3111. doi: 10.4049/jimmunol.1201669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hay N. Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta. 2011;1813:1965–1970. doi: 10.1016/j.bbamcr.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi EJ, Kim S, Jho EH, Song KJ, Kee SH. Axin expression enhances herpes simplex virus type 1 replication by inhibiting virus-mediated cell death in L929 cells. J Gen Virol. 2013;94:1636–1646. doi: 10.1099/vir.0.051540-0. [DOI] [PubMed] [Google Scholar]
- 38.Choi EJ, Kee SH. Axin expression delays herpes simplex virus-induced autophagy and enhances viral replication in L929 cells. Microbiol Immunol. 2014;58:103–111. doi: 10.1111/1348-0421.12123. [DOI] [PubMed] [Google Scholar]
- 39.Piacentini R, Li Puma DD, Ripoli C, Marcocci ME, De Chiara G, Garaci E, Palamara AT, Grassi C. Herpes Simplex Virus type-1 infection induces synaptic dysfunction in cultured cortical neurons via GSK-3 activation and intraneuronal amyloid-β protein accumulation. Sci Rep. 2015;5:15444. doi: 10.1038/srep15444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markus A, Waldman Ben-Asher H, Kinchington PR, Goldstein RS. Cellular transcriptome analysis reveals differential expression of pro- and antiapoptosis genes by varicella-zoster virus-infected neurons and fibroblasts. J Virol. 2014;88:7674–7677. doi: 10.1128/JVI.00500-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angelova M, Zwezdaryk K, Ferris M, Shan B, Morris CA, Sullivan DE. Human cytomegalovirus infection dysregulates the canonical Wnt/β-catenin signaling pathway. PLoS Pathog. 2012;8:e1002959. doi: 10.1371/journal.ppat.1002959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Zuylen WJ, Ford CE, Wong DD, Rawlinson WD. Human Cytomegalovirus Modulates Expression of Noncanonical Wnt Receptor ROR2 To Alter Trophoblast Migration. J Virol. 2015;90:1108–1115. doi: 10.1128/JVI.02588-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tabata T, Petitt M, Zydek M, Fang-Hoover J, Larocque N, Tsuge M, Gormley M, Kauvar LM, Pereira L. Human cytomegalovirus infection interferes with the maintenance and differentiation of trophoblast progenitor cells of the human placenta. J Virol. 2015;89:5134–5147. doi: 10.1128/JVI.03674-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pollheimer J, Loregger T, Sonderegger S, Saleh L, Bauer S, Bilban M, Czerwenka K, Husslein P, Knöfler M. Activation of the canonical wingless/T-cell factor signaling pathway promotes invasive differentiation of human trophoblast. Am J Pathol. 2006;168:1134–1147. doi: 10.2353/ajpath.2006.050686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sonderegger S, Haslinger P, Sabri A, Leisser C, Otten JV, Fiala C, Knöfler M. Wingless (Wnt)-3A induces trophoblast migration and matrix metalloproteinase-2 secretion through canonical Wnt signaling and protein kinase B/AKT activation. Endocrinology. 2010;151:211–220. doi: 10.1210/en.2009-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knöfler M, Pollheimer J. Human placental trophoblast invasion and differentiation: a particular focus on Wnt signaling. Front Genet. 2013;4:190. doi: 10.3389/fgene.2013.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ueland T, Rollag H, Hartmann A, Jardine AG, Humar A, Michelsen AE, Bignamini AA, Åsberg A, Aukrust P. Secreted Wnt antagonists during eradication of cytomegalovirus infection in solid organ transplant recipients. Am J Transplant. 2014;14:210–215. doi: 10.1111/ajt.12506. [DOI] [PubMed] [Google Scholar]
- 48.Langemeijer EV, Slinger E, de Munnik S, Schreiber A, Maussang D, Vischer H, Verkaar F, Leurs R, Siderius M, Smit MJ. Constitutive β-catenin signaling by the viral chemokine receptor US28. PLoS One. 2012;7:e48935. doi: 10.1371/journal.pone.0048935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tabata T, McDonagh S, Kawakatsu H, Pereira L. Cytotrophoblasts infected with a pathogenic human cytomegalovirus strain dysregulate cell-matrix and cell-cell adhesion molecules: a quantitative analysis. Placenta. 2007;28:527–537. doi: 10.1016/j.placenta.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 50.Roy S, Liu F, Arav-Boger R. Human Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase--A Potential Strategy for Suppression of the Wnt Pathway. Viruses. 2015;8:pii: E8. doi: 10.3390/v8010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kapoor A, He R, Venkatadri R, Forman M, Arav-Boger R. Wnt modulating agents inhibit human cytomegalovirus replication. Antimicrob Agents Chemother. 2013;57:2761–2767. doi: 10.1128/AAC.00029-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Casaday RJ, Bailey JR, Kalb SR, Brignole EJ, Loveland AN, Cotter RJ, Gibson W. Assembly protein precursor (pUL80.5 homolog) of simian cytomegalovirus is phosphorylated at a glycogen synthase kinase 3 site and its downstream “priming” site: phosphorylation affects interactions of protein with itself and with major capsid protein. J Virol. 2004;78:13501–13511. doi: 10.1128/JVI.78.24.13501-13511.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oberstein A, Perlman DH, Shenk T, Terry LJ. Human cytomegalovirus pUL97 kinase induces global changes in the infected cell phosphoproteome. Proteomics. 2015;15:2006–2022. doi: 10.1002/pmic.201400607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mohammad AA, Rahbar A, Lui WO, Davoudi B, Catrina A, Stragliotto G, Mellbin L, Hamsten A, Rydén L, Yaiw KC, et al. Detection of circulating hcmv-miR-UL112-3p in patients with glioblastoma, rheumatoid arthritis, diabetes mellitus and healthy controls. PLoS One. 2014;9:e113740. doi: 10.1371/journal.pone.0113740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simanek AM, Dowd JB, Pawelec G, Melzer D, Dutta A, Aiello AE. Seropositivity to cytomegalovirus, inflammation, all-cause and cardiovascular disease-related mortality in the United States. PLoS One. 2011;6:e16103. doi: 10.1371/journal.pone.0016103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herbein G, Kumar A. The oncogenic potential of human cytomegalovirus and breast cancer. Front Oncol. 2014;4:230. doi: 10.3389/fonc.2014.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen HP, Chan YJ. The oncomodulatory role of human cytomegalovirus in colorectal cancer: implications for clinical trials. Front Oncol. 2014;4:314. doi: 10.3389/fonc.2014.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 59.Shackelford J, Maier C, Pagano JS. Epstein-Barr virus activates beta-catenin in type III latently infected B lymphocyte lines: association with deubiquitinating enzymes. Proc Natl Acad Sci USA. 2003;100:15572–15576. doi: 10.1073/pnas.2636947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morrison JA, Klingelhutz AJ, Raab-Traub N. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J Virol. 2003;77:12276–12284. doi: 10.1128/JVI.77.22.12276-12284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morrison JA, Raab-Traub N. Roles of the ITAM and PY motifs of Epstein-Barr virus latent membrane protein 2A in the inhibition of epithelial cell differentiation and activation of {beta}-catenin signaling. J Virol. 2005;79:2375–2382. doi: 10.1128/JVI.79.4.2375-2382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Everly DN, Kusano S, Raab-Traub N. Accumulation of cytoplasmic beta-catenin and nuclear glycogen synthase kinase 3beta in Epstein-Barr virus-infected cells. J Virol. 2004;78:11648–11655. doi: 10.1128/JVI.78.21.11648-11655.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jang KL, Shackelford J, Seo SY, Pagano JS. Up-regulation of beta-catenin by a viral oncogene correlates with inhibition of the seven in absentia homolog 1 in B lymphoma cells. Proc Natl Acad Sci USA. 2005;102:18431–18436. doi: 10.1073/pnas.0504054102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Webb N, Connolly G, Tellam J, Yap AS, Khanna R. Epstein-Barr virus associated modulation of Wnt pathway is not dependent on latent membrane protein-1. PLoS One. 2008;3:e3254. doi: 10.1371/journal.pone.0003254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Malizia AP, Lacey N, Walls D, Egan JJ, Doran PP. CUX1/Wnt signaling regulates epithelial mesenchymal transition in EBV infected epithelial cells. Exp Cell Res. 2009;315:1819–1831. doi: 10.1016/j.yexcr.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 66.Fujimuro M, Wu FY, ApRhys C, Kajumbula H, Young DB, Hayward GS, Hayward SD. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat Med. 2003;9:300–306. doi: 10.1038/nm829. [DOI] [PubMed] [Google Scholar]
- 67.Fujimuro M, Hayward SD. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J Virol. 2003;77:8019–8030. doi: 10.1128/JVI.77.14.8019-8030.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ballestas ME, Chatis PA, Kaye KM. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science. 1999;284:641–644. doi: 10.1126/science.284.5414.641. [DOI] [PubMed] [Google Scholar]
- 69.Cotter MA, Robertson ES. The latency-associated nuclear antigen tethers the Kaposi’s sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology. 1999;264:254–264. doi: 10.1006/viro.1999.9999. [DOI] [PubMed] [Google Scholar]
- 70.Fujimuro M, Liu J, Zhu J, Yokosawa H, Hayward SD. Regulation of the interaction between glycogen synthase kinase 3 and the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen. J Virol. 2005;79:10429–10441. doi: 10.1128/JVI.79.16.10429-10441.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujimuro M, Hayward SD. Manipulation of glycogen-synthase kinase-3 activity in KSHV-associated cancers. J Mol Med (Berl) 2004;82:223–231. doi: 10.1007/s00109-003-0519-7. [DOI] [PubMed] [Google Scholar]
- 72.Hayward SD, Liu J, Fujimuro M. Notch and Wnt signaling: mimicry and manipulation by gamma herpesviruses. Sci STKE. 2006;2006:re4. doi: 10.1126/stke.3352006re4. [DOI] [PubMed] [Google Scholar]
- 73.Price MA. CKI, there’s more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 2006;20:399–410. doi: 10.1101/gad.1394306. [DOI] [PubMed] [Google Scholar]
- 74.An FQ, Compitello N, Horwitz E, Sramkoski M, Knudsen ES, Renne R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J Biol Chem. 2005;280:3862–3874. doi: 10.1074/jbc.M407435200. [DOI] [PubMed] [Google Scholar]
- 75.Kusano S, Eizuru Y. Human I-mfa domain proteins specifically interact with KSHV LANA and affect its regulation of Wnt signaling-dependent transcription. Biochem Biophys Res Commun. 2010;396:608–613. doi: 10.1016/j.bbrc.2010.04.111. [DOI] [PubMed] [Google Scholar]
- 76.Lee HR, Mitra J, Lee S, Gao SJ, Oh TK, Kim MH, Ha T, Jung JU. Kaposi’s Sarcoma-Associated Herpesvirus Viral Interferon Regulatory Factor 4 (vIRF4) Perturbs the G1-S Cell Cycle Progression via Deregulation of the cyclin D1 Gene. J Virol. 2015;90:1139–1143. doi: 10.1128/JVI.01897-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Angelova M, Ferris M, Swan KF, McFerrin HE, Pridjian G, Morris CA, Sullivan DE. Kaposi’s sarcoma-associated herpesvirus G-protein coupled receptor activates the canonical Wnt/β-catenin signaling pathway. Virol J. 2014;11:218. doi: 10.1186/s12985-014-0218-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006;2:e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol. 2008;26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chambers JW, Maguire TG, Alwine JC. Glutamine metabolism is essential for human cytomegalovirus infection. J Virol. 2010;84:1867–1873. doi: 10.1128/JVI.02123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu Y, Maguire TG, Alwine JC. ChREBP, a glucose-responsive transcriptional factor, enhances glucose metabolism to support biosynthesis in human cytomegalovirus-infected cells. Proc Natl Acad Sci USA. 2014;111:1951–1956. doi: 10.1073/pnas.1310779111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marcato V, Luron L, Laqueuvre LM, Simon D, Mansuroglu Z, Flamand M, Panthier JJ, Souès S, Massaad C, Bonnefoy E. β-Catenin Upregulates the Constitutive and Virus-Induced Transcriptional Capacity of the Interferon Beta Promoter through T-Cell Factor Binding Sites. Mol Cell Biol. 2016;36:13–29. doi: 10.1128/MCB.00641-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chan KC, Chan LS, Ip JC, Lo C, Yip TT, Ngan RK, Wong RN, Lo KW, Ng WT, Lee AW, et al. Therapeutic targeting of CBP/β-catenin signaling reduces cancer stem-like population and synergistically suppresses growth of EBV-positive nasopharyngeal carcinoma cells with cisplatin. Sci Rep. 2015;5:9979. doi: 10.1038/srep09979. [DOI] [PMC free article] [PubMed] [Google Scholar]