

Abstract
Small regulatory RNAs comprise critically important modulators of gene expression in bacteria, yet very little is known about their prevalence and functions in Rickettsia species. R. conorii, the causative agent of Mediterranean spotted fever, is a tick-borne pathogen that primarily infects microvascular endothelium in humans. We have determined the transcriptional landscape of R. conorii during infection of Human Microvascular Endothelial Cells (HMECs) by strand-specific RNA sequencing to identify 4 riboswitches, 13 trans-acting (intergenic), and 22 cis-acting (antisense) small RNAs (termed ‘Rc_sR’s). Independent expression of four novel trans-acting sRNAs (Rc_sR31, Rc_sR33, Rc_sR35, and Rc_sR42) and known bacterial sRNAs (6S, RNaseP_bact_a, ffs, and α-tmRNA) was next confirmed by Northern hybridization. Comparative analysis during infection of HMECs vis-à-vis tick AAE2 cells revealed significantly higher expression of Rc_sR35 and Rc_sR42 in HMECs, whereas Rc_sR31 and Rc_sR33 were expressed at similar levels in both cell types. We further predicted a total of 502 genes involved in all important biological processes as potential targets of Rc_sRs and validated the interaction of Rc_sR42 with cydA (cytochrome d ubiquinol oxidase subunit I). Our findings constitute the first evidence of the existence of post-transcriptional riboregulatory mechanisms in R. conorii and interactions between a novel Rc_sR and its target mRNA.
Over the past decade, bacterial post-transcriptional regulation is garnering significant interest due to discoveries related to the roles of small RNAs in modulating gene expression during growth and stress conditions in vitro and host interactions in vivo1,2. Bacterial small RNAs (sRNAs) are generally 50–500 bp long and fall into three categories, namely riboswitches which are located upstream of mRNAs, cis-acting sRNAs synthesized from the complementary strand of an open reading frame (ORF), and trans-acting sRNAs transcribed from the intergenic regions with only partial complementarity to their target genes. In contrast to eukaryotic microRNAs which only down-regulate their target mRNAs, bacterial sRNAs can both activate or inhibit translation by either stabilizing the mRNA and opening the ribosome binding site or by degrading the target mRNA, respectively3,4.
Mediterranean spotted fever (MSF) caused by Rickettsia conorii is an acute, febrile illness transmitted to humans through the bite of infected tick(s). R. conorii is a Gram-negative obligate intracellular bacterium exhibiting tropism for microvascular endothelium of the blood vessels in mammalian hosts5,6. Acquisition of a less virulent, dormant phenotype in infected ticks allowing for transovarial transmission to the progeny and transstadial transmission to the next stage during the ticks’ natural developmental lifecycle supports rickettsial persistence, survival, and maintenance in the arthropod vector. In contrast, infection of humans as the dead-end hosts is associated with significant morbidity/mortality attributed to a virulent phenotype7. Despite the disease prevalence and significant impact of MSF on public health, little is known about the mechanisms by which R. conorii adapts to different host environments and manifests serious disease sequelae such as ‘rickettsial vasculitis’ in the human host while persisting in its arthropod vector.
The seminal publication of the entire genome sequence for R. prowazekii, a typhus group Rickettsia species, revealed an AT-rich, highly reduced genome, presence of mobile elements and pseudogenes, low coding density compared to the genomes of other intracellular bacteria, and a close evolutionary relationship to eukaryotic mitochondria8. A number of other rickettsial genomes belonging to different species have since been sequenced. R. conorii genome harbors a single chromosome of 1268755 bp encoding for 1578 protein coding genes, 9 pseudogenes, 33 tRNAs, 2 rRNAs, and contains ~32% intergenic region9,10. This relatively high percentage of non-coding region in R. conorii and other rickettsial genomes has traditionally been considered to be the ‘junk DNA’ or defunct genes resulting from reductive evolution and pseudogenization7,11,12,13,14. However, recent advances in computational bioinformatics and bacterial molecular genetics have led to the appreciation that the intergenic regions, in addition to harboring transcription factor binding sites and mobile genetic elements, encode for small non-coding RNAs involved in the regulation of target genes. Indeed, the discovery of sRNAs has been a major cornerstone of investigations pertaining to their importance in almost every aspect of bacterial physiology, including pathogenesis, quorum sensing, developmental programming, and regulation of growth and replication. Accordingly, bacterial sRNAs are now well recognized as critically important post-transcriptional regulators in both free-living as well as pathogenic bacteria3.
In a recent study, we predicted the existence of ~1700 sRNAs in 13 different species of rickettsiae across all four groups, namely ancestral, spotted fever, transitional, and typhus, and confirmed the expression and biogenesis of six sRNAs in R. prowazekii15. The specific aims of this follow up study were to identify and catalogue R. conorii sRNAs expressed during host cell infection, to understand their conservation profile in different rickettsial species, to compare their expression during infection of human microvascular endothelium vis-à-vis tick vector cells as the host, and to demonstrate the potential riboregulatory roles of trans-acting sRNAs. Using an RNA-Seq based approach, we have identified 39 novel non-coding RNAs across the R. conorii chromosome in addition to four well-known bacterial sRNAs (ssrS, ssrA, RNaseP_bact_a, and ffs). Among these, two of the abundantly expressed candidate sRNAs, namely Rc_sR35 and Rc_sR42, display significant differences in their expression during human versus tick cell infection, whereas another two (Rc_sR31 and Rc_sR33) exhibit similar levels of expression. We have further predicted 502 target genes and 191 gene ontology (GO) functional categories that may be potentially regulated by newly identified trans-acting sRNAs and obtained evidence for Rc_sR42 interactions with cydA mRNA, implicating regulation of target mRNA transcripts by sRNAs in R. conorii. Finally, we have also identified four riboswitches upstream of hypothetical proteins with unknown functions and cis-acting sRNAs corresponding to important, functionally characterized rickettsial genes such as rickA and virB10, known to be involved in actin-based motility and type IV secretion system, respectively.
Results
Identification of novel riboswitches, cis- and trans-acting sRNAs
To explore the entire spectrum of R. conorii sRNAs expressed during the infection of human microvascular endothelial cells (HMECs), cDNA libraries from total cellular RNA subjected to enrichment for bacterial RNA were processed for Illumina sequencing resulting in an average of 23.76 and 22.20 million paired-end reads at 3 and 24 h post-infection, respectively. To avoid interference due to eukaryotic RNAs, all reads mapping to human genome version 38 were first eliminated and remaining unmapped reads were included in our analysis. To ensure quality control, only the reads with a Phred score of >15 were mapped to complete R. conorii genome (PATRIC Genome ID: 272944.4). Thus, approximately 7.26 and 14.29 million reads, accounting for 31% and 64% of the entire read sets at 3 and 24 h post-infection, respectively, mapped to the rickettsial genome (Supplementary Table S1). Among these, there was abundant expression of known bacterial sRNAs, namely, 6S, α-tmRNA, RNaseP_bact_a, and ffs (Supplementary Fig. S1). Importantly, we further identified 22 cis-acting sRNAs on the anti-sense strand of corresponding coding genes, 13 trans-acting sRNAs in the intergenic spacer regions, and four riboswitches within the 5′ leader regions of hypothetical proteins (Fig. 1 and Table 1). The novel cis- and trans-acting sRNAs as well as riboswitches thus identified are hereafter referred to as ‘Rickettsia conorii_small RNAs’ (Rc_sR).
Figure 1. Circular chromosome map of R. conorii genome showing the location of four well-known and all sRNAs expressed during the infection of HMECs in vitro.

The Rc_sRs are dispersed throughout the R. conorii genome. Complete R. conorii genome showing the location of all trans- and cis-acting Rc_sRs expressed during the infection of HMECs. The different circles with bars represent: (1) Dark green (outermost): R. conorii ORFs annotated on the sense strand, (2) Red: R. conorii ORFs annotated on the anti-sense strand, (3) Bright green: Genomic location of novel trans-acting sRNAs, (4) Blue: Genomic location of novel cis-acting sRNAs, (5): GC plot, (6) GC skew (innermost circle). The Black lines in the third circle (outside to inside) represent the 4 well known sRNAs namely, ssrS, ssrA, RNaseP_bact_a, and ffs. The red lines in the fourth circle (outside to inside) represent riboswitches namely, Rc_sR3, Rc_15, Rc_sR28 and Rc_sR30. The genome map was generated in dnaplotter using R. conorii annotated genome (PATRIC genome ID: 27944.4).
Table 1. List of R. conorii sRNAs expressed during the infection of human microvascular endothelial cells in vitro.
| sRNAa | Startb | Stopb | Size (bp) | Riboswitch/Cis/Transactingc | Upstream ORFd | Downstream ORFd | Cis-ORFd | Orientatione |
|---|---|---|---|---|---|---|---|---|
| Rc_sR1 | 466 | 838 | 373 | cis | — | — | peg.1 (RC0001) | </> |
| Rc_sR2 | 6026 | 6239 | 214 | trans | peg.5 (RC0005) | peg.6 (RC0007) | — | >/>/< |
| Rc_sR3 | 328935 | 329168 | 234 | riboswitch | — | peg.376 | — | >/> |
| Rc_sR4 | 351165 | 351368 | 204 | cis | — | — | peg.395 (RC0347) | </> |
| Rc_sR5 | 379393 | 379571 | 179 | trans | peg.432 (RC0377) | peg.433 (RC0378) | — | </>/< |
| Rc_sR6 | 384964 | 385196 | 233 | cis | — | — | peg.438 (RC0383) | >/< |
| Rc_sR7 | 389015 | 389183 | 169 | cis | — | — | peg.444 (RC0389) | </> |
| Rc_sR8 | 414438 | 414565 | 128 | trans | peg.467 (RC0412) | peg.468 (RC0413) | — | >/</> |
| Rc_sR9 | 420477 | 420741 | 265 | cis | — | — | peg.480 (TolA) | </> |
| Rc_sR10 | 435394 | 435697 | 304 | cis | — | — | peg.498 (RC0440) | </> |
| Rc_sR11 | 466022 | 466209 | 188 | cis | — | — | peg.537 (RC0469) | </> |
| Rc_sR12 | 531156 | 531375 | 220 | cis | — | — | peg.619 (RC0540) | >/< |
| Rc_sR13 | 541604 | 541785 | 182 | cis | — | — | peg.629 (RC0550) | >/< |
| Rc_sR14 | 545029 | 545234 | 206 | cis | — | — | peg.632 (RC0553) | >/< |
| Rc_sR15 | 555623 | 555757 | 135 | riboswitch | — | peg.643 (RC0563) | — | >/> |
| Rc_sR16 | 559197 | 559344 | 148 | trans | peg.648 (RC0567) | peg.649 (RC0568) | — | </>/< |
| Rc_sR17 | 591563 | 591758 | 196 | cis | — | — | peg.691 (RC0605) | >/< |
| Rc_sR18 | 672763 | 672934 | 172 | cis | — | — | peg.793 (RC0698) | </> |
| Rc_sR19 | 693856 | 694153 | 298 | cis | — | — | peg.823 (RC0723) | </> |
| Rc_sR20 | 732978 | 733122 | 145 | trans | peg.876 (RC0773) | peg.877 (RC0774) | — | </</< |
| Rc_sR21 (RNaseP_bact_a) | 739375 | 739855 | 481 | — | peg.887 (RC0782) | peg.888 (RC0783) | — | </</> |
| Rc_sR22 | 752032 | 752195 | 164 | cis | — | — | peg.907 (RC0798) | >/< |
| Rc_sR23 | 763895 | 764018 | 124 | cis | — | — | peg.918 (RC0808) | </> |
| Rc_sR24 | 770130 | 770278 | 149 | trans | peg.925 (RC0815) | peg.926 (RC0816) | — | </>/< |
| Rc_sR25 | 811219 | 811387 | 169 | trans | peg.978 (RC0860) | peg.979 (RC0862) | — | >/</> |
| Rc_sR26 | 853068 | 853320 | 253 | cis | — | — | peg.1033 (RC0909) | >/< |
| Rc_sR27 | 869414 | 869639 | 226 | cis | — | — | peg.1051 (RC0924) | >/< |
| Rc_sR28 | 880969 | 881142 | 174 | riboswitch | — | peg.1074 (RC0942) | — | </< |
| Rc_sR29 | 883911 | 884189 | 279 | trans | peg.1077 (RC0945) | RCRNA26 | — | </</< |
| Rc_sR30 | 969095 | 969353 | 259 | riboswitch | — | peg.1196 (RC1045) | — | </< |
| Rc_sR31 | 1020148 | 1020450 | 303 | trans | peg.1256 (RC1094) | peg.1257 (RC1095) | — | </</< |
| Rc_sR32 (α_tmRNA, ssrA) | 1030837 | 1031001 | 165 | — | peg.1278 (RC1109) | peg.1279 (RC1110) | — | </</> |
| Rc_sR33 | 1041477 | 1041770 | 294 | trans | peg.1290 (RC1120) | peg.1291 (RC1121) | — | >/</> |
| Rc_sR34f | 1042717 | 1042899 | 183 | cis | peg.1291 (RC1121) | peg.1292 (RC1122) | — | >/</> |
| Rc_sR35 | 1052516 | 1052743 | 228 | trans | peg.1310 (RC1138) | peg.1311 (RC1139) | — | >/</< |
| Rc_sR36 (6S, ssrS) | 1071875 | 1072033 | 159 | — | peg.1346 (RC1170) | peg.1347 (RC1171) | — | </</< |
| Rc_sR37 | 1100103 | 1100327 | 225 | cis | — | — | peg.1380 (RC1205) | </> |
| Rc_sR38 | 1121923 | 1122042 | 120 | cis | — | — | peg.1397 (RC1218) | </> |
| Rc_sR39 | 1132389 | 1132619 | 231 | trans | peg.1406 (RC1227) | peg.1407 (RC1228) | — | >/>/> |
| Rc_sR40 | 1146031 | 1146254 | 224 | cis | — | —— | peg.1419 (RC1238) | >/< |
| Rc_sR41 | 1153327 | 1153508 | 182 | cis | — | — | peg.1428 (RC1246) | >/< |
| Rc_sR42 | 1160321 | 1160565 | 245 | trans | peg.1439 (RC1255) | peg.1440 (RC1256) | — | </>/< |
| Rc_sR43 (ffs) | 1239529 | 1239651 | 123 | — | peg.1530 (RC1335) | peg.1531 (RC1336) | — | >/>/> |
aR. conorii sRNAs are numbered depending on their genomic location in the genome annotation available in PATRIC and starting from the 5′ of the genome. The names in parentheses are the other names reported in the databases for the respective sRNA.
bThe start and stop co-ordinates for the sRNAs corresponding the R. conorii genome annotation available in PATRIC.
cBased on the genomic location, the sRNAs are defined as riboswitches or as ‘cis-‘ (present on the anti-sense strand of an ORF) or ‘trans-acting’ (intergenic region).
dThe upstream, downstream and cis-ORFs are identified based on the R. conorii genome annotation available in PATRIC. The number in parentheses indicate known alternative name of the ORF.
eThe arrows ‘>’ and ‘<’ indicate ‘sense’ and ‘anti-sense’ orientation respectively. For all riboswitches, the orientation of the riboswitch and the downstream ORF, respectively, are shown. For trans-acting sRNAs, the orientation of upstream ORF, sRNA and downstream ORF are shown in order. The orientation of cis-acting sRNAs is shown as sRNA and cis-ORF, respectively.
fRc_sR35 is a cis-acting sRNA present on the positive strand and spanning across both up and downstream ORFs (RC1121 and RC1122) coding from the negative strand. So the orientation of upstream ORF, sRNA and downstream ORF are shown.
All novel sRNAs were found to be expressed as independent transcripts and exhibited an MEV of >2 when compared to 50 nucleotides up- and downstream of the sRNA transcript. As expected, their length ranged from 100 to 400 bp and the average length of cis- and trans-acting sRNAs was 213 bp and 209 bp, respectively (Table 1). No candidate rickettsial sRNA was longer than Rc_sR1 (373 bp) and Rc_sR38 (120 bp) was the smallest. Also, a majority of both cis-acting (20/22) and trans-acting (12/13) sRNAs in R. conorii ranged between 100–300 bp. No significant difference in strand bias was evident since 12 cis-acting and 7 trans-acting sRNAs were located on the anti-sense strand, while the remaining sRNAs were present on the positive strand (Table 1). The genomic context and location of all trans-acting sRNAs was next ascertained by comparing the orientation of up and downstream ORFs with respect to the transcribed sRNA. Seven (Rc_sR5, Rc_sR8, Rc_sR16, Rc_sR24, Rc_sR25, Rc_sR33, and Rc_sR42) out of 13 trans-acting sRNAs were transcribed in the opposite orientation with respect to both adjacent genes, and one each was processed in the same direction as their respective upstream (Rc_sR2) and downstream (Rc_sR35) gene (Table 1). The remaining 4 sRNAs (Rc_sR20, Rc_sR29, Rc_sR31, and Rc_sR39) and their respective adjacent up and downstream genes were located on the same strand and transcribed in the same orientation (Table 1). In addition, four riboswitches (Rc_sR3, Rc_sR15, Rc_sR28 and Rc_sR30) were positioned upstream of hypothetical proteins. As expected, most of the cis-acting sRNAs originated from the anti-sense strand of an ORF (Table 1), except for Rc_sR34 that was found to be overlapping two ORFs (RC1121 and RC1122) as well as the intergenic region between these ORFs (Table 1 and Supplementary Fig. S2). Representative read coverage plots of cis- acting Rc_sRs and riboswitches are presented in Fig. 2.
Figure 2. Representative graphs showing the read coverage of novel sRNAs in R. conorii during the infection of HMECs.

HMECs were infected with R. conorii (MOI = 20) and total RNA was harvested at 3 and 24 h post-infection. High throughput RNA sequencing was performed on total RNA enriched for bacterial transcripts (see materials and methods). The strand specific reads were mapped onto R. conorii genome (PATRIC genome ID: 27944.4). The read coverage plots of two cis-acting sRNAs (a) Rc_sR19 and (b) Rc_sR26, and two riboswitches (c) Rc_sR3 and (d) Rc_sR15 are presented. The reads above and below the x-axis represent the forward (green) and reverse strand (blue), respectively. The identified sRNAs are indicated by red arrow. The ORFs up- and downstream, and cis-acting ORF are shown by black arrow. The orientation and genomic location coordinates correspond to the R. conorii genome annotation in PATRIC.
BLAST analysis revealed the presence of sequences homologous (>90% query coverage and identity) to a majority of R. conorii trans-acting sRNAs in other spotted fever Rickettsia species. Of note, although Rc_sR35 was conserved among spotted fever rickettsiae in general, R. rickettsii displayed only partially conserved sequence with 47% query coverage. Also, the first 44 bases of Rc_sR31 exhibited low level similarity despite excellent overall homology in several SFG rickettsiae. None of the R. conorii sRNAs were found to have homologs in typhus group rickettsiae, but the homologs of Rc_sR15, Rc_sR16, Rc_sR29, and Rc_sR31 were present with >90% query coverage in at least one of the transitional group species, which include R. felis, R. akari, and R. australis. Intriguingly, we noticed significant sequence homology (90% identity) between Rc_sR3 (328935–329168), a potential riboswitch upstream of a hypothetical protein (peg.376), and Rc_sR33 (1041477–1041770), a trans-acting sRNA (Supplementary Fig. S3). In-depth analysis of the genomic context, orientation, and read depth revealed that Rc_sR3 was located in the 5′ leader region of peg.376 and transcribed from the forward strand, whereas Rc_sR33 was encoded on the reverse strand and located in the intergenic region between RC1120 and RC1121. The average number of Rc_sR33 reads were >3-fold higher than those for Rc_sR3.
In contrast to trans-acting sRNAs, ORFs coding for all cis-acting sRNAs in R. conorii were present in a majority of rickettsial species across different groups and most (17/22) of these ORFs had known function. Notably, genes involved in key functions such as actin based motility (rickA), type IV secretion system (virB10), bicyclomycin resistance (bcr1), cell division (ftsY), and LPS biosynthesis (IpxB), were identified as possible regulatory targets of cis-acting sRNAs. Only five cis-acting sRNAs were present on the anti-sense strand of hypothetical proteins, some of which may have been independently lost in a few rickettsial species/strains (Supplementary Fig. S4). Rc_sR34, spanning across ORFs RC1121 and RC1122, was absent in R. prowazekii, but homologous sequences mapped partially to dnaK ORF in other Rickettsia species, including R. typhi (Supplementary Fig. S5). RC1122, coding for a 60 amino acid long hypothetical protein in R. conorii, exhibited partial sequence homology to full length dnaK in other species. Based on genome organization in the RC1121-RC1122 locus, which is unique in R. conorii genome with only limited homology to dnaK in other rickettsiae, it appears that the genesis of two shorter ORFs (RC1121-RC1122) and Rc_sR34 may be a consequence of gene degradation in this region (Supplementary Figs 2 and 5).
The regions upstream of transcriptional start sites (TSSs) of all sRNAs were next analyzed to identify putative promoters and consensus RpoD (σ70) binding sites. All R. conorii sRNAs were found to carry a highly consensus −10 box with a typically conserved TATAAT motif that was often preceded by a ‘TT’ dinucleotide in at least 50% of the sRNAs (Fig. 3a). Similar to many other prokaryotes, the −35 motif of sRNAs was relatively less conserved and deviated from the typical E. coli housekeeping promoter motif (TTGACA) (Fig. 3b). Interestingly, our attempts to identify Rc_sRs with similarity to other known sRNAs in the Rfam non-coding RNA database did not yield any hits, precluding prediction of potential function(s) and/or classification and suggesting that Rc_sRs likely represent a unique group of transcriptional regulators in rickettsiae.
Figure 3. Web logos showing the conservation of −10 and −35 motifs upstream of all novel R. conorii sRNAs identified.

The 150 bp upstream genomic sequence of all novel Rc_sRs identified was subjected to promoter prediction using BPROM66. The −10 and −35 motifs were detected upstream of sRNAs and all motif sequences were used to generate consensus sequence based on nucleotide position. The consensus nucleotides for −10 motif (a) and −35 motif (b) are presented above. A relatively conserved −10 motif was seen upstream of all sRNAs while the −35 motif is less conserved.
Validation of sRNAs by Northern blot analysis
To confirm the expression of both novel and known bacterial sRNAs in R. conorii, we performed Northern blot analysis. Independent expression of Rc_sR31, Rc_sR33, Rc_sR35 and Rc_sR42 was clearly evident during R. conorii infection of HMECs (Fig. 4). Interestingly, two transcripts of ~300 and ~250 bp were detected for Rc_sR33, of which ~300bp was more prominent, suggesting that smaller transcript of ~250 bp likely represents the processed transcript. In agreement with our RNA-Seq data, abundant and independent expression of Rc_sR35 was also seen (Fig. 4). Based on its genomic location and read coverage in RNA-Seq, the transcript size for Rc_sR42 was estimated to be ~245 bp. Northern blot analysis demonstrated the presence of two bands of approximately 250 and 200 bp, suggesting that the smaller transcript may be the outcome of either processing or degradation of the primary transcript. For Rc_sR31, a single transcript of ~300 bp was also detected in R. conorii at 24 h post-infection.
Figure 4. Northern blots showing the expression of selected R. conorii novel trans-acting and known bacterial sRNAs during the infection of HMECs.

HMECs were infected with R. conorii (MOI = 20) and total RNA was extracted at 24 h post-infection. The DNase I treated total RNA was enriched for bacterial transcripts using MICROBEnrich and MICROBExpress kits (Ambion). Enriched RNA was size separated on 1.5% agarose-formaldehyde gel and transferred onto nylon membranes (BioRad). The membranes were probed with [α-32P] UTP-labeled strand-specific RNA probes generated by in vitro transcription. The membranes were washed following the NorthernMax kit manufacturer’s protocol (Ambion) and developed by autoradiography. All sRNAs were expressed as independent transcripts in R. conorii during the infection of HMECs. The scanned images for four novel trans-acting identified in this study (Rc_sR31, Rc_sR33, Rc_sR35 and Rc_sR42) and four well-known sRNAs (6S, a_tmRNA, RNaseP_bact_a and ffs) are shown. Two bands of varying transcript sizes were detected in Rc_sR33 and Rc_sR42 which may represent both primary and processed transcripts. Asterisk indicates a processed transcript of lower size and a non-specific band in Rc_sR33 is shown by arrow. The adjacent up and downstream genes of each sRNA are shown in green arrows. The sRNA is indicated by red arrow and the estimated size is shown above the arrow. The orientation of all sRNA and ORFs are based on R. conorii genome annotation available in PATRIC. Total RNA from uninfected HMECs was used as a control (C).
To ascertain the expression of known sRNAs evolutionarily conserved in bacteria, we also determined the expression of 6S, α-tmRNA, RNaseP_bact_a, and ffs in R. conorii during infection of HMECs. All sRNAs were expressed as independent transcripts as evidenced by the presence of a single band of the expected size (Fig. 4). Although expression of 6S and RNaseP_bact_a was clearly abundant as shown, relatively lower level of expression of α-tmRNA was observed, which is consistent with the read coverages in our RNA-Seq data (Fig. 4 and Supplementary Fig. S1).
Expression profile of R. conorii sRNAs in human and vector host cells
To conduct a comparative analysis of Rc_sRs in different host niches, we infected HMECs and tick AAE2 cells from A. americanum nymphs with R. conorii (MOI = 20). Although Rhipicephalus sanguineus is the natural vector of R. conorii, A. americanum ticks can also acquire and transmit R. conorii16. All four novel Rc_sRs, identified by RNA-Seq and confirmed by Northern blots above, were expressed during the infection of both HMECs and AAE2 cells, indicating their presence and biogenesis in R. conorii. In HMECs, the most notable change of ~4 fold up-regulation of Rc_sR35 was observed at 24 h post-infection. Expression levels of Rc_sR31 and Rc_sR33 were apparently higher at 24 h, albeit these differences in transcript levels were not significant (Fig. 5a). Interestingly, Rc_sR31 and Rc_sR33 showed similar levels of expression in both cell types (Fig. 5a), whereas Rc_sR35 and Rc_sR42 were expressed at significantly higher levels in HMECs as compared to tick cells (Fig. 5b).
Figure 5. Expression profile of R. conorii novel small RNAs, Rc_sR31, Rc_sR33, Rc_sR35 and Rc_sR42, during the infection of human endothelium and tick cells in vitro.
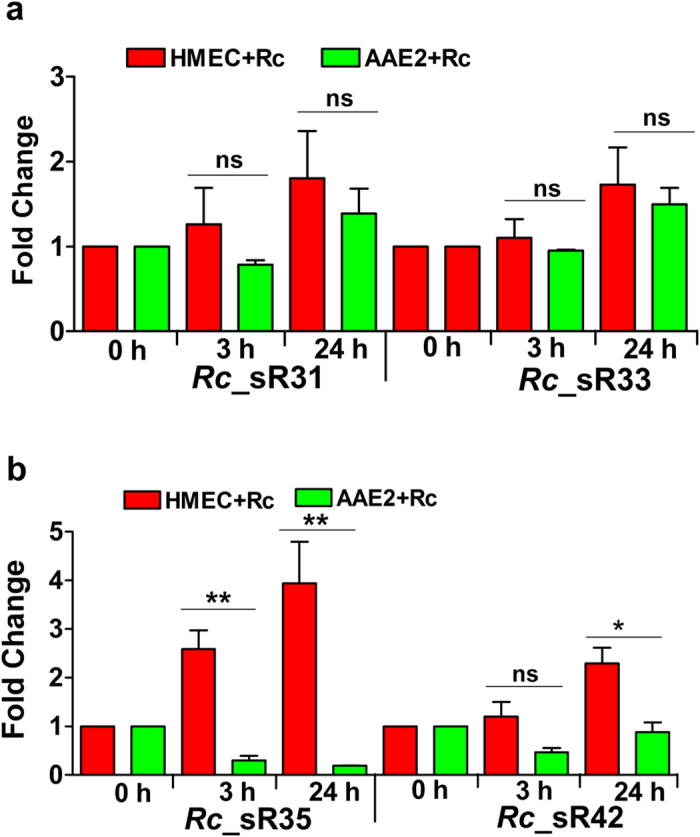
Confluent monolayer of HMECs and AAE2 cells were infected with R. conorii (MOI = 20) for 3 and 24 h. Total RNA was extracted by Tri-reagent® method and genomic DNA contamination was eliminated by DNase I treatment. Complementary DNA was generated and the sRNA transcript abundance was assessed by quantitative PCR using sRNA specific primers listed in Supplementary table S2. R. conorii 16S rRNA was used as housekeeping control and HMECs or AAE2 cells infected with R. conorii for 15 minutes were used as baseline. The data from a minimum of three independent experiments were analyzed by ∆∆CT method and presented as mean ± SEM. Significant differences in fold change of Rc_sR35 and Rc_sR42 were observed depending on the host (bottom panel), while the expression of Rc_sR31 and Rc_sR33 were similar (top panel) during human and tick cell infection in vitro. Legend: Red: Expression of R. conorii sRNAs in human cell line (HMECs); Green: Expression of R. conorii sRNAs in tick cell line (AAE2). * = p < 0.05, ** = p < 0.005.
To further ensure the expression of sRNAs as independent transcripts, we next performed qRT-PCR analysis of Rc_sR42 and its respective upstream (RC1255) and downstream (RC1256) ORFs at 3 and 24 h post-infection. Both the flanking genes were highly up-regulated in comparison to Rc_sR42 and significant differences in their expression profile were evident clearly indicating that these transcripts are transcribed independently of each other (Supplementary Fig. S6).
Prediction of Rc_sRs’ target genes in R. conorii
To determine the functional role(s) of rickettsial trans-acting sRNAs, we employed two independent algorithms, namely IntaRNA and CopraRNA, to identify their target genes17 and to further categorize these genes based on their involvement in biological processes such as pathogenesis and virulence using STRING 9.1. Both IntaRNA and CopraRNA, despite identifying a varying number of potential target genes based upon the probability of sRNA-mRNA seed interactions (p < 0.05), predicted several common target mRNAs for each sRNA. The lowest and highest number of targets were predicted to be regulated by Rc_sR16 and Rc_sR24 (20 vs. 53), respectively (Table 2). We further identified a plethora of important biological processes that could be regulated by Rc_sR24, including single-organism metabolic and cellular processes, tetrahydrofolate interconversion and metabolic process, response to stress, proteolysis, and other critical cellular and metabolic activities (Supplementary Table S3). In addition, Rc_sR31 is predictably involved in the regulation of type IV secretion and protein transport; Rc_sR33 is a potential regulator of genes involved in translation and type IV secretion system; the target genes for Rc_sR35 are required for porphyrin biosynthesis and heme metabolism; and Rc_sR42 may be uniquely involved in RNA and tRNA modification and processing (Supplementary Table S3). Overall, our data reveals that a total of 191 biological processes may be regulated by 13 R. conorii sRNAs and regulation of a majority (104) of these functions by only one sRNA. Protein secretion by the type IV secretion system (GO:0030255) is likely regulated by several sRNAs (Rc_sR8, Rc_sR20, Rc_sR31 and Rc_sR33). Notably, genes involved in nucleobase-containing compound metabolic process (GO:0006139), protein localization (GO:0008104), protein secretion (GO:0009306), establishment of protein localization (GO:0045184), transport (GO:0006810) and nitrogen compound metabolic process (GO:0006807) are candidates for regulation by 3 different sRNAs indicating their importance in disease pathogenesis and survival mechanisms utilized by Rickettsia inside the host cytosol (Supplementary Table S3). Additionally, the predicted secondary fold of 6S sRNA (Rc_sR36) exhibits structural similarity to that of R. prowazekii and E. coli18, and the secondary structures of Rc_sR31, 33, 35 and 42 display several stems and loops indicating that different regions of the sRNA may be involved in regulating different target genes (Supplementary Fig. S8).
Table 2. List of targets gene regulated by R. conorii trans-acting sRNAs.
| sRNA | Number of predicted target genesa |
Number of common target genes predicted by both programsb | |
|---|---|---|---|
| CopraRNA | IntaRNA | ||
| Rc_sR2 | 60 | 56 | 39 |
| Rc_sR5 | 36 | 33 | 21 |
| Rc_sR8 | 61 | 49 | 44 |
| Rc_sR16 | 46 | 30 | 20 |
| Rc_sR20 | 77 | 69 | 41 |
| Rc_sR24 | 71 | 57 | 53 |
| Rc_sR25 | 59 | 45 | 31 |
| Rc_sR29 | 62 | 55 | 32 |
| Rc_sR31 | 65 | 67 | 42 |
| Rc_sR33 | 60 | 66 | 49 |
| Rc_sR35 | 62 | 62 | 48 |
| Rc_sR39 | 74 | 73 | 49 |
| Rc_sR42 | 72 | 53 | 33 |
aOnly target genes having a significant p-value (p < 0.05) for the predicted seed region are included in the analysis.
bTargets predicted by both CopraRNA and IntraRNA and having significant p-value (p < 0.05) for the predicted seed region.
Validation of sRNA-target mRNA interactions
For experimental validation of target gene predictions, we investigated the interactions between Rc_sR42 and four putative target genes, namely, RC0288 (cydA), RC0822 (tlyA), RC0977 (grpE) and RC1333 (pntAB). Rc_sR42 sRNA was chosen in light of its differential modulation during infection of HMECs versus AAE2 host cells (Fig. 5b) and evidence for its expression as an independent transcript in R. conorii (Fig. 4 and Supplementary Fig. S6). Target genes exhibiting significant p value (p < 0.05) for the seed region and encoding conserved proteins such as hemolysin A (tlyA), heat shock protein (grpE), NAD(P) transhydrogenase α-subunit (pntAB), and cytochrome d ubiquinol oxidase subunit I (cydA) were chosen to determine riboregulation in R. conorii. Gel-shift mobility assays were conducted using in vitro generated, isotopically-labeled transcripts of sRNA and target mRNAs in an optimized binding reaction. Interestingly, Rc_sR42 formed stable RNA duplex with RC0288 (cydA) mRNA (Fig. 6a), which was effectively competed off in the presence of excess unlabeled sRNA, suggesting the specificity of Rc_sR42 and cydA mRNA interaction. No interactions were observed between Rc_sR42 and other chosen target mRNAs (Fig. 6a). To further validate Rc_sR42-cydA interaction, labeled sRNA was incubated with increasing concentrations of cydA mRNA (sRNA to mRNA ratio of 1:1 to 1:20). As shown in Fig. 6b, the intensity of gel shifted complex increased in direct correlation with mRNA concentrations, further ascertaining the specificity and efficacy of target mRNA binding to Rc_sR42. Using IntaRNA, we next predicted the seed region (293359–293402) for Rc_sR42 binding to be located at the 3′ end of cydA. To experimentally authenticate whether the predicted seed region indeed holds true, we systematically performed mobility shift assays using different cydA mRNA fragments (schematics presented in Supplementary Fig. S9). Interestingly, the seed region for Rc_sR42 interactions with cydA mRNA was located slightly upstream of the prediction and between bases 293225 and 293283 of R. conorii genome (PATRIC Genome ID: 272944.4) (Fig. 6c).
Figure 6. EMSAs showing the interaction of Rc_sR42 with cydA target mRNA.
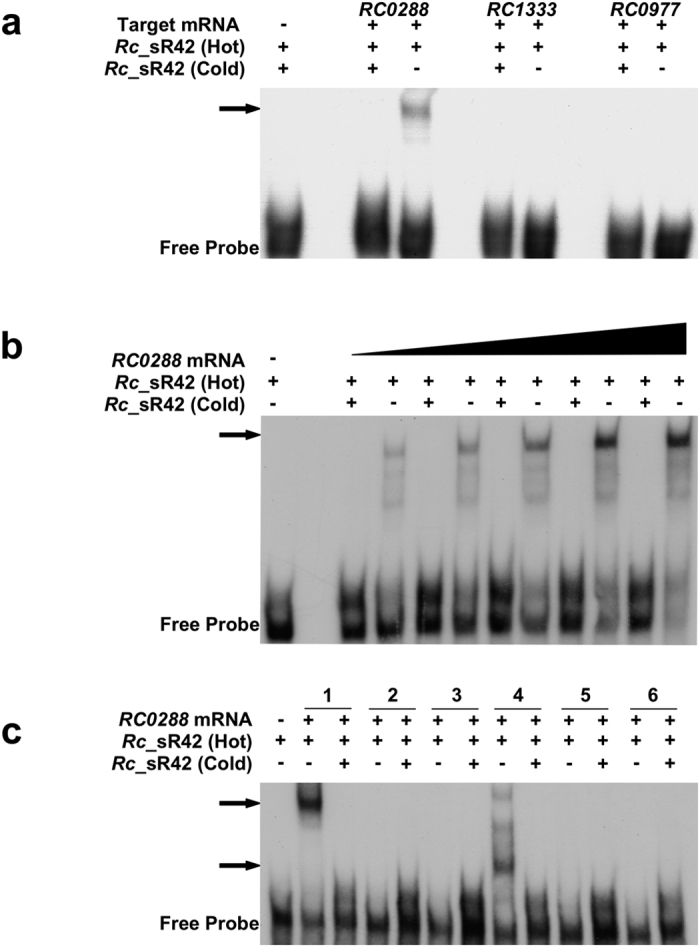
In vitro transcripts of Rc_sR42 and target mRNAs were generated from PCR templates amplified from R. conorii genomic DNA using primers containing T7 promoter (Supplementary table S2). Only the full length Rc_sR42 transcript was labeled with [α-32P] UTP. For cold competition, unlabeled full length Rc_sR42 was used. The samples were separated on native 4% polyacrylamide gels, vacuum dried, and developed by autoradiography. The photomicrographs shown are a representative gel from three independent experiments. (a) EMSA showing the interaction of R. conorii Rc_sR42 with RC0288 (cydA) target mRNA in vitro. The arrow indicates gel-shifted Rc_sR42-cydA mRNA complex. The other target mRNAs (RC0977 and RC1333) did not show any interaction. (b) EMSA showing the interaction of R. conorii Rc_sR42 with increasing concentrations of RC0288 (cydA) target mRNA in vitro. An increase in the complex intensity was observed with increasing concentrations of RC0288 mRNA. (c) EMSA showing the identification of seed region of Rc_sR42 and cydA interaction. Different regions of cydA mRNA fragment (see Supplementary Fig. S9) were used for incubation with full length Rc_sR42 to identify the seed region. The genomic locations for in vitro transcribed fragments of cydA are as follows: 1: 293225–293587 (363 bases); 2: 293284–293587 (304 bases); 3: 293311–293587 (277 bases); 4: 293225–293464 (240 bases); 5: 293284–293464 (181 bases); and 6: 293311–293430 (120 bases). A gel shifted complex (indicated by arrow) was observed only in 1 and 4 indicating that the seed region in cydA mRNA is located between bases 293225–293283 corresponding to R. conorii genome (PATRIC genome ID: 27944.4).
Discussion
Reductive evolution owing to progressive gene decay/loss is now widely accepted as a major driving force resulting in smaller genomes in Rickettsia species as well as other obligate intracellular bacteria12,19. Since intracellular pathogens tend to coevolve with their respective hosts, the transcriptional landscape of these organisms varies and several pathways are either independently lost or retained to meet their nutritional and survival requirements depending on the host19. For instance, Rickettsia lack several important enzymes required for the pathways of amino acid and sugar metabolism, nucleotide synthesis, and lipid biosynthesis, but encode for ATP/ADP translocases to fulfill their energy requirements via exchange of ADP for ATP from the host cytosol8,20. In contrast, riboflavin synthesis genes in Buchnera infer this endosymbiont with the ability to provide riboflavin to its host21.
As well-recognized and emerging mediators of gene regulation, bacterial sRNAs are now garnering considerable attention due primarily to their pivotal roles in the transcriptional control of a number of regulatory, enzymatic, and structural mechanisms1. Despite the prevalence of sRNAs in many bacteria, there exists only limited evolutionary conservation and both species- and strain-specific sRNA catalogues are now documented22. A search of 400 transcriptomic datasets belonging to 40 different strains of bacteria and archaea has revealed that the ‘Goldilocks Zone’ (where species are neither too close nor too distant phylogenetically) for non-coding RNAs is rather narrow, indicating independent evolution of lineage-specific post-transcriptional machinery23. As further confirmatory evidence supporting this notion, a search for orthologs of 2208 non-coding RNAs within 1156 bacterial genomes reported in Rfam (a collection of non-coding RNA families)24, including members of Rickettsiales, also reveals limited taxonomic distribution and suggests a low degree of evolutionary conservation in a majority of these ncRNA families25.
To define the non-coding transcriptional landscape of R. conorii, we first exploited a global high throughput sequencing approach to identify novel sRNAs expressed during the infection of human endothelial cells in vitro. Our underlying rationale here was that to be fully virulent during human infections, vector-borne pathogenic rickettsiae, including R. conorii, primarily target microvascular endothelium, rapidly escape from the phagosome, and subvert normal host cell functions to promote their replication and intracellular dissemination. Based on the location of the reads mapping to R. conorii genome, four highly conserved (ssrS, ssrA, ffs and RNaseP_bact_a) and 39 (4 riboswitches, 13 trans- and 22 cis-acting) novel sRNAs were identified to be expressed at 3 h (early stage of infection) and 24 h (established infection) post-infection. All sRNAs displayed an MEV of 2-fold or higher in relation to the respective 50 flanking nucleotides, indicating their independent expression, an observation further ascertained by Northern blot analysis and computational identification of σ70 promoters. We have further characterized the expression profile of four trans-acting sRNAs during host-pathogen and vector-pathogen interactions. Importantly, two sRNAs (Rc_sR35 and Rc_sR42) were differentially expressed in human endothelial cells when compared directly with tick vector cells, suggesting their regulation depending on the host niche. We recently predicted 126 candidate sRNAs to be encoded by R. conorii genome using SIPHT/sRNAPredict3, a web-based program based on promoter, transcriptional terminator, and RNA secondary structure prediction tools15, but only five of these predicted sRNAs (MEV >2) were identified in this study. It is likely that computational prediction approach reported the presence of potential transcripts based on the conservation of IGRs containing stable RNA secondary structures with defined promoter and terminator sequences, and that several of these predicted sRNAs are either not expressed during infection of host cells or may be expressed under different growth conditions. It is now well appreciated that several bacterial sRNAs are uniquely expressed under conditions of oxygen limitation, iron homeostasis, stress, quorum sensing, and virulence3,26. For example, sRNA RyhB is highly up-regulated during iron starvation leading to the down-regulation of its target genes sodB and sdhC/A in E. coli27. More recently, induction and upregulation of PinT, a PhoP activated Salmonella sRNA, has been shown to be necessary for transition from invasion to intracellular replication and survival during in vivo infection28. It is also possible that several sRNA candidates predicted by SIPHT and based on RefSeq annotation, represent unannotated ORFs resulting from differences in genome annotation. A comparison of RAST (Rapid Annotations using Subsystems Technology)29 and RefSeq based R. conorii genome annotation revealed the presence of 204 ORFs annotated only by RAST, and four SIPHT predicted sRNAs mapped to the genomic location of these uncharacterized ORFs. Thus, in light of prediction versus experimental determination, our results validate the use of ‘strand specific deep-sequencing of enriched bacterial transcripts’ approach for identification of novel sRNAs in R. conorii and demonstrate its applicability for the evaluation of novel transcripts in other obligate intracellular pathogens.
While some of the R. conorii IGRs encoding trans-acting sRNAs share limited homology in other species belonging to the SFG, the ORFs harboring cis-acting sRNAs are conserved in all rickettsial groups including typhus. An intriguing finding in this study is the presence of a riboswitch (Rc_sR3) and a novel trans-acting sRNA (Rc_sR33) sharing >90% sequence homology (Supplementary Fig. S3). Independent expression of Rc_sR33 was confirmed by Northern blot analysis and similar levels of induction were evident during the infection of human endothelial and tick cells in vitro (Figs 4 and 5a). Further comparative analysis of R. conorii and other rickettsial genomes revealed that only 6 genomes (R. conorii, R. parkeri, R. slovaca, R. africae, R. peacockii, and R. amblyommii) carry both of these homologous IGRs in their genomes. Interestingly, RC1120 as an ORF upstream of Rc_sR33 with partial overlap with the 3′ end of this sRNA was present only in the genomes harboring both the riboswitch and the trans-acting sRNA. Furthermore, the location of Rc_sR3 was upstream of an ORF (peg.376) annotated only by RAST (PATRIC), but not RefSeq (NCBI). The synteny of genomic region adjacent to Rc_sR3 was highly conserved, but greater divergence was observed in the genomic location harboring Rc_sR33, indicating that Rc_sR33 might have originated and coevolved with RC1120 ORF in few rickettsial genomes (Supplementary Fig. S7). It is rather intriguing that R. conorii expresses two unique sRNAs with significant homology, of which one likely functions as a riboswitch upstream of a short ORF (peg.376) and the other is confined to an intergenic region as a trans-acting sRNA. Pseudomonas aeruginosa encodes PrrF1 and PrrF2, homologs of a Fur-repressed sRNA RyhB with >95% homology. Both of these sRNAs are expressed during iron starvation and their suppression/deletion is required for the regulation of iron metabolism, indicating similarities in their function as well30. On a similar note, multiple copies of Pxr sRNA have been reported in Cystobacter and it is hypothesized that Pxr paralogs retain functional similarities resulting in the tight regulation of target genes31. In contrary, functional divergence in sRNA paralogs/multiple copies has also been reported. Legionella pneumophila and few other bacterial genomes have two copies of 6S RNA, a chelator of σ70 RNA polymerase. Of these, while one copy of 6S regulates the similar set of genes as reported in E. coli, another copy of 6S RNA has been shown to regulate distant and restricted set of genes32.
The inference of this study that sequences homologous to most R. conorii trans-acting sRNAs are confined to only a few other rickettsial genomes excluding those from the typhus group, is expected owing to the genetic diversity resulting from gene degradation, transposon mutagenesis, repetitive and insertion sequences, mobile genetic elements, and lateral gene transfer13,33. Computational comparison of rickettsial genomes reveals that nearly 50% of the genes encoded by each species/strain are unique to its genome with approximately 700 protein coding genes shared across all genomes11,12. Even greater genetic divergence is evident for R. felis, the etiological agent of a typhus-like flea borne rickettsiosis, whose genome is overrun by mobile genetic elements. Despite coding for ~1600–1800 ORFs, only 300 of these belong to the core set of genes shared by other rickettsial species14. It has also been suggested that typhus group genomes have a faster divergence rate in comparison to spotted fever group (SFG) species. Specifically, core proteins in typhus species have 2.43 times higher rate of substitution11, indicating that noticeable differences in the rate of evolution and extensive genetic diversity among different species is responsible for an altered and unique transcriptional landscape. Furthermore, single nucleotide polymorphisms (SNPs) in promoter regions can result in the loss of transcription as shown in E. coli and Campylobacter jejuni22. R. rickettsii strains Sheila Smith (virulent) and Iowa (avirulent), despite sharing 99% sequence identity, have 492 SNPs and 143 deletions between them34. A comparison of sRNAs in 27 E. coli and Shigella genomes reveals that despite sharing a core set of sRNAs, several sRNAs are highly variable, indicating that the secondary loss of sRNAs, but not horizontal gene transfer, may be the reason for variable distribution even among phylogenetically close organisms35. Accordingly, we anticipate that although a few IGRs are shared among the SFG species, some sRNAs may potentially be inactivated or lost leading to differences in sRNA repertoire, thus necessitating the need for identification and characterization of sRNAs in a species- and strain-specific manner to better understand their roles in post-transcriptional regulation.
In general, antisense transcription in bacteria varies between 3–50%36. For example, while E. coli encodes ~20% of antisense sRNAs, only ~1.5% Salmonella sRNAs are cis-acting and a much higher proportion of cis-regulatory elements (27%) are reported in the sRNA repertoire of Helicobacter37,38,39. In this study, we have identified 22 cis-acting sRNAs originating from the antisense strand of key regulatory genes such as rickA, virB10, and ftsY (Table 1, Fig. 2 and Supplementary Fig. S2). Rickettsia species belonging to SFG are known to hijack the host actin assembly for cell-to-cell spread. RickA is critical for the activation of host Arp2/3 complex and required for early stages of motility after invasion. The absence or deletion of rickA results in erratic movements or non-motile forms40. Bacteria encode for several systems to secrete genetic material, metabolites, and proteins into their extracellular milieu. Type IV secretion system, composed of several Vir proteins, is one of the most thoroughly characterized secretory systems and a majority of these proteins, except for VirB5, are present and conserved in all rickettsial genomes41,42. Sec-TolC is another secretory system that has been reported in rickettsiae and RARP1, an ankyrin repeat protein, is shown to be secreted in a TolC-dependent manner43. Interestingly, apart from a cis-acting sRNA as a potential regulator of virB10, we have also identified a sRNA antisense to ftsY, encoding a signal recognition particle-like protein. FtsY is an essential component of machinery required for the biogenesis and insertion of proteins into the membrane and a role for this protein has been implicated in Sec translocation system44. Additionally, based on the prediction of target genes regulated by trans-acting sRNAs, we project the possibility of regulation of virD4, virB3 and virB6 by four different trans-acting sRNAs (Rc_sR8, Rc_sR20, Rc_sR31, and Rc_sR33) (Supplementary Table S3). Our findings, thus, present a premise for tight regulation of bacterial motility and secretory systems in R. conorii. An important consideration in this context, however, is that approximately 44% of top predictions by CopraRNA turn out to be true and the success rates for IntaRNA and TargetRNA as target prediction tools are 28 and 11%, respectively45. Experimental verification and repudiation of R. conorii genes predicted as potential targets of Rc_sRs by such algorithms would, therefore, be necessary and is currently ongoing.
Several bacterial sRNAs are known to contain tandem repeats and other repeat regions that are critical for their regulation. One classical example is E. coli CsrB sRNA, present in several bacterial species. It is known to contain 7 repeats and 18 sites required for binding to CsrA mRNA regulating its translation and stability3. Repeat regions have been found in both intergenic spacers and ORFs of coding genes present in Rickettsia species13,46. We have identified two trans-acting sRNAs (Rc_sR5 and Rc_sR8) and one cis-acting sRNA (Rc_sR26, anti-sense to rickA) harboring repeat regions in their transcripts. Our strand-specific RNA-Seq showed that both Rc_sR26 sRNA and rickA (corresponding ORF) are abundantly expressed in R. conorii during the infection of HMECs (Fig. 2). It is likely that Rc_sR26 expression might be a requisite for stabilizing rickA mRNA resulting in its translation. In obligate intracellular bacteria, Wolbachia trans-acting sRNA WsnRNA-46 is shown to specifically interact with the palindromic sequences in the murD ORF resulting in its down regulation47. It is, therefore, possible that trans-acting sRNAs with repeat regions in their transcripts may interact with other complementary repeat regions in coding ORFs resulting in regulation of their expression by direct base pairing. Experimental validation of the interactions between R. conorii trans-acting sRNAs and target mRNAs is currently under progress and expected to further illuminate sRNA mediated riboregulatory mechanisms in pathogenic rickettsiae.
Bacterial riboswitches are defined as non-coding RNA elements located within the 5′ UTR of mRNA and exert their regulatory control on the downstream gene in a cis-fashion by directly binding to trans-acting ligand(s)48. Our results illustrate the presence of four riboswitches in 5′ non-coding regions upstream of hypothetical proteins with as yet uncharacterized functions. Further, secondary structure analysis of R. conorii riboswitches (Rc_sR3, sR28, and sR30) reveals terminator/anti-terminator hairpin like structures with a central bulge and Rc_sR15 exhibits a single hairpin structure closely resembling that of ThiC riboswitch in Sinorhizobium meliloti49 (Supplementary Fig. S10). Importantly, the 3′ end of Rc_sR3 riboswitch is positioned 233 bp upstream of the 5′end of the downstream coding gene (peg.376). Albeit not a common occurrence, the presence of riboswitches >200 nucleotides upstream of the 5′ region of the downstream gene has been documented for several bacterial species, for example three B12 riboswitches in Listeria monocytogenes. In addition, there is precedence that such riboswitches can regulate trans-acting sRNAs rather than the downstream ORF50,51.
Although several conserved bacterial riboswitches and their respective metabolites have been identified by sequence comparison, our attempts to identify potential ligands that may interact with R. conorii riboswitches revealed no hits, indicating that riboswitches identified in this study are likely unique and species-specific. Recently, application of ‘Term-seq’ has revealed the presence of 18 new riboswitch candidates as determinants of antibiotic resistance in Bacillus subtilis52.
Small RNA mediated post-transcriptional regulation can result from different modes of action. Among the well characterized sRNAs, ssrS (6S RNA), ubiquitously present in bacterial genomes, is shown to specifically bind to σ70 holozyme resulting in transcriptional regulation of genes containing σ70 promoters during the stationary phase of growth53. The ssrA (α-tmRNA) acts by releasing the stalled ribosomes during translation, while ffs (4.5S RNA) is involved in the targeting of proteins to the membranes immediately after translation54,55. Cis-acting sRNAs, originating from the anti-sense strand of a coding gene potentially bind to their counterparts by base pairing of complementary sequences resulting in transcriptional regulation of the ORF. Complexities of regulation by trans-acting sRNAs have also been reported. In several bacteria, the interactions of trans-acting sRNAs with their target mRNAs are facilitated by RNA chaperones and the location of seed region in the target mRNA determines the fate of the mRNA. In some instances, trans-acting sRNAs may stabilize the transcript by initiation of translation, while in other cases, the target mRNA may be degraded by ribonucleases. Using two independent algorithms, we predict the possibility of regulation of a total of 502 target genes by R. conorii trans-acting sRNAs. Key regulatory pathways involved in LPS biosynthesis, nucleotide metabolism, secretion, protein biosynthesis and metabolism, and carbohydrate biosynthesis are predicted to be regulated by these intergenic sRNAs (Supplementary Table S3). Furthermore, we have experimentally validated the interaction of Rc_sR42 with cydA mRNA (Fig. 6). In Rickettsia, cydA encodes for cytochrome d ubiquinol oxidase subunit I, a terminal oxidase required for aerobic respiration. The cytochrome bd oxidase encoded by cydAB operon in Coxiella is shown to exhibit high affinity to oxygen and is required for ATP synthesis during microaerophilic intracellular growth56. The cydA and cydB are synthesized as polycistronic mRNAs in R. conorii and the sRNA-mRNA seed region lies at the 3′ end of the cydA transcript at genomic position 293225–293283, slightly upstream of the location predicted by IntaRNA (293359–293402) (Fig. 6). We reason that this may be because predictive algorithms often ignore the sRNA secondary structure complexity, pseudoknots, and double-kissing hairpin complexes45. Since sRNA-mRNA interactions tend to occur over a short and imperfect complementarity, further investigations to decipher critical base(s) within the validated 60 bp seed region (293225–293283) are now in progress. The regulatory outcomes of such an interaction may likely be that Rc_sR42 is either involved in stabilizing the cydA transcript as a result of the cleavage of polycistronic cydAB transcript or in the degradation of cydAB transcript by forming a double stranded RNA. Rickettsial genomes encode ATP/ADP translocases and can also synthesize ATP during later stages of infection when ATP supply in the host cytosol is exhausted. Intriguingly, Rc_sR42 is highly expressed at 24 h post-infection (Fig. 5b), indicating that regulation of cydA mRNA at later stages of infection may facilitate R. conorii survival in the intracellular niche. Furthermore, cydA mutants in several bacteria are unable to survive indicating the key functional role of this protein in survival. Comprehensive molecular studies employing appropriate heterologous model systems and aimed at generating knock-out mutants should reveal the functional implications of such regulatory sRNAs.
Although a role for hfq, an RNA chaperone, in facilitating mRNA interactions with trans-acting sRNAs is well established, the homologs of hfq are absent in rickettsial genomes. As of now, it is not clear if trans-acting sRNAs identified in this study would function in a chaperone-dependent or –independent manner. Hypothetical proteins such as HP1334 in H. pylori and several other RNA binding proteins such as YbeY in Sinorhizobium meliloti have been implicated with chaperone activity57,58. More recently, a ProQ/FinO domain containing protein Lpp0148 has been reported to function as a RNA chaperone in Legionella pneumophila59. It is, therefore, possible that R. conorii proteins with unknown function may play a role in sRNA-mediated regulation. At this stage, our preliminary investigations of this aspect reveal that total protein extracts from R. conorii enhance Rc_sR42 interaction with cydA, indicating chaperone-like activity of as yet unidentified rickettsial protein(s) (Narra et al., unpublished data). Alternatively, the trans-acting sRNAs may bind to their target mRNA by direct base pairing at the seed regions in a chaperone-independent manner.
In conclusion, our high resolution transcriptomic profiling revealed the presence of novel non-coding RNAs in R. conorii during host-pathogen interactions. We have further shown that two trans-acting sRNAs (Rc_sR35 and Rc_sR42) are differentially expressed during host-pathogen and vector-pathogen interactions, indicating their role in survival and transovarial/transstadial transmission in arthropod vectors and regulation of virulence in the human host. Most notably, we also provide the first experimental evidence for riboregulation in rickettsiae. Investigations are underway to further validate the importance of post-transcriptional regulatory network in the mechanisms of rickettsial survival, adaptation, and pathogenesis.
Material And Methods
Preparation of R. conorii stocks
R. conorii (Malish 7 strain) was grown in cultured Vero cells, purified by differential centrifugation as described previously60, and stored frozen at −80 °C as aliquots to avoid repeated freeze-thaw cycles. The infectivity titers of purified stocks were estimated by citrate synthase (gltA)-based quantitative PCR following a published protocol61.
Cell culture and infection
Human microvascular endothelial cells (HMECs) were cultured at 37 °C in MCDB131 medium supplemented with 10% fetal bovine serum, 10 mM L-glutamine, 1 μg/ml hydrocortisone, and 10 ng/ml epidermal growth factor in an atmosphere of 95%O2:5%CO2. Cells were infected with R. conorii at an MOI of 20 following our established protocol62. Briefly, cell monolayers were incubated with R. conorii in a minimum volume of culture medium to ensure efficient adhesion and internalization. After 15 minutes, cells were placed in fresh medium and incubated at 37 °C for 3 and 24 h. For comparative analysis, the condition in which R. conorii was incubated with the host cells for 15 minutes only was designated as the ‘baseline’.
Amblyomma americanum tick cells (AAE2) were grown in L-15B complete medium (pH 7.5) at 34 °C as described63. The AAE2 cells were infected with R. conorii (MOI = 20) at 34 °C for 15 minutes. At this point, the medium was gently aspirated off and centrifuged to collect any viable semi-adherent cells in culture. The pellet was resuspended in fresh L-15B infection medium and added back to the culture flasks for further incubation for 3 and 24 h. Similar to the HMECs above, AAE2 cells infected with R. conorii for the first 15 minutes to simply allow sufficient time for adhesion and invasion were employed as the baseline control.
RNA extraction and library preparation
Total RNA from R. conorii-infected HMECs was extracted by Tri-reagent® method64. The RNA thus obtained was treated with DNase I (0.5 units/μg RNA) at 37 °C for 1 h to remove any genomic DNA. The RNA quality was then verified for its integrity on an Agilent 2100 BioAnalyzer (Agilent Technologies) and samples with a RIN score of ≥9.0 were used in our experiments. Enrichment for bacterial transcripts was next performed using Dynabeads® Oligo (dT)25 (ThermoFisher Scientific) to capture eukaryotic polyadenylated mRNAs and a Ribo-Zero™ kit (Illumina) to remove eukaryotic (18S/28S) rRNAs and bacterial (16S/23S) rRNAs. The enriched RNA was reverse transcribed and subjected to the preparation of strand-specific cDNA libraries using TruSeq RNA Sample Prep Kit (Illumina). A minimum of two independent samples meeting our quality-control criteria were processed for the preparation of cDNA libraries belonging to complementary strands and deep sequencing as outlined below.
RNA Sequencing and identification of small RNAs
Strand-specific cDNA libraries were sequenced as 50 base long, paired-end reads on Illumina Hi-Seq 1000 at the Next Generation Sequencing core facility, UTMB. The sequencing read statistics for each library are presented in Supplementary Table S1. The reads from each library were analyzed for their base quality and any base with a PHRED score of ≤15 was excluded from the analysis. The reads were then mapped to the complete, annotated genome of R. conorii (Malish 7) available in Pathosystems Resource Intergration Center (PATRIC Genome ID: 272944.4) allowing up to two mismatches per read using Bowtie 265. The coverage for each nucleotide was visualized in Integrated Genome Viewer (IGV: Broad Institute) and bacterial sRNAs were identified depending on the origin of reads, i.e. the reads mapping either to the intergenic region (trans-acting) or to the complementary strand of a coding open reading frame (cis-acting), or to the 5′ regions upstream of mRNA (riboswitches). Expression levels were determined by normalizing the number of reads mapping to the genomic region corresponding to the sRNA against total number of reads mapping to R. conorii genome (excluding those mapping to rRNAs and known tRNAs). The average expression values from independent libraries were then calculated and designated as the Mean Expression Value (MEV). A small RNA was considered to be bona fide, if its MEV was ≥1.5 fold in direct comparison to the same for 50 flanking nucleotides and it did not correspond to or contain an ORF within the sequence based on R. conorii genome in PATRIC. Coverage plots for select sRNAs and their respective up and downstream genes were generated using GraphPad Prism. The presence of consensus sigma-70 (σ70) promoters upstream of all small RNAs was identified using BPROM66. The minimum free energy based secondary structures of Rc_sR31, 33, 35, 36 (6S), and 42 were predicted by RNAfold webserver (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) using default parameter settings.
Quantitative real-time PCR
The primers for real-time PCR (Supplementary Table S2) were designed using Primer Express 3.0.1 (Applied Biosystems). To determine the expression of R. conorii sRNAs and their adjacent upstream and downstream genes, total RNA from HMECs and AAE2 cells infected with R. conorii was reverse transcribed (1 μg RNA as the input) using high capacity cDNA reverse transcription kit (Life Technologies). Quantitative PCR (qRT-PCR) was performed using SYBR Green-based assay with rickettsial 16S rRNA as the housekeeping control. The Ct values for 3 and 24 h post-infection were normalized to the baseline control, which was assigned a value of 1, and analyzed by ∆∆Ct method64. The data sets were calculated as the mean ± SEM from a minimum of three independent experiments and statistical analysis was performed using GraphPad Prism with statistical significance set to a threshold P-value of ≤ 0.05.
Northern blot analysis
To confirm the expression of R. conorii sRNAs identified in the RNA-Seq analysis, Northern blot analysis was performed using enriched RNA preparations and NorthernMax® kit reagents (Ambion). Briefly, total RNA was enriched for bacterial transcripts using MICROBEnrich and MICROBExpress kits (Ambion) and resultant enriched RNA was size separated on 1.5% formaldehyde-agarose gels and transferred onto positively-charged nylon membranes (Bio-Rad). For hybridization, [α-32P] UTP-labeled strand-specific RNA probes were generated by in vitro transcription using sRNA-specific primers (Supplementary Table S2) and MAXIscript® kit (Ambion). The radioactively labeled probes were purified using Sephadex microspin columns (GE Healthcare) and used for overnight hybridization according to the NorthernMax® Kit (Ambion). Finally, the blots were exposed to autoradiography films (BioExpress) and images were scanned and saved as TIFF files.
Prediction and functional enrichment of target genes regulated by trans-acting sRNAs
Two independent programs, IntaRNA and CopraRNA17, were used to identify target genes for R. conorii sRNAs. The predictions by IntaRNA are based on minimum hybridization energy between two RNA molecules taking the accessibility and length of seed region into consideration, while CopraRNA integrates phylogenetic information to predict sRNA targets at the genomic scale and reconstructs regulatory networks employing functional enrichment and network analysis, allowing for high confidence target prediction and efficient classification of sRNAs. For both programs, default settings were employed with the only exception that the region under interrogation was adjusted to include −150 to +100 base region with respect to the transcription start site of the target gene. All target genes exhibiting significant (p < 0.05) interaction in the sRNA-mRNA seed regions were considered for further analysis. To gain insight into their functional roles, target genes common to both programs and displaying a seed region p-value of <0.05 were used for functional categorization using STRING 9.167 and only the gene ontology (GO) functional categories showing a significant p-value of <0.05 were included in the analysis.
Electrophoretic mobility shift assay (EMSA)
To validate the binding interactions between sRNA and mRNA, EMSA were performed following a standard protocol68. All primers are listed in Supplementary Table S2. Specifically, [α-32P]UTP-labeled full length Rc_sR42 (1160321-1160565: 245 bp) and target mRNA transcripts were generated by in vitro transcription using T7 polymerase and MAXIscript® kit following the manufacturer’s instructions (Ambion). The mRNA fragments used in the study, based on the PATRIC annotation of R. conorii genome, were as follows: RC0288 (cydA): 293225–293587 (363 bp), RC0822 (tlyA): 777540–777819 (280 bp), RC0977 (grpE): 915855–916054 (200 bp), and RC1333 (pntAB): 1237558–1237877 (320 bp). To identify seed region in the cydA fragment used above, fragments of varying legths were generated by in vitro transcription. The genomic locations for in vitro transcribed fragments of cydA were: 293225–293587 (363 bases); 293284–293587 (304 bases); 293311–293587 (277 bases); 293225–293464 (240 bases); 293284–293464 (181 bases); and 293311–293430 (120 bases). The schematic of different cydA mRNA fragments used in this study is presented in the Supplementary Fig. S9. The in vitro transcribed sRNA and mRNA were mixed in a binding buffer (Promega) and incubated at 70 °C for 5 minutes followed by 30 °C for 15 minutes. Since RNAs are known to exhibit complex structures, incubation at 70–90 °C for 1–5 minutes was found to be optimal to achieve RNA population with homogeneous fold and has been extensively followed while performing EMSA studies to identify the sRNA-mRNA interactions69,70,71. Further incubation at 30 °C for 15 minutes was chosen for this study as Rickettsia species while present in tick vectors are exposed to temperatures ranging from 28–30 °C. A five-fold excess of unlabeled sRNA was used to ensure specificity of interactions. The samples were separated by electrophoresis on a native 4% polyacrylamide gel, which was vacuum-dried on a Whatman filter and subjected to autoradiographic exposures. The images were scanned and saved in TIFF format.
Additional Information
How to cite this article: Narra, H. P. et al. Small Regulatory RNAs of Rickettsia conorii. Sci. Rep. 6, 36728; doi: 10.1038/srep36728 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We thank Drs. Thomas Wood and Steve Widen for their help with library preparation and RNA sequencing. We also thank Victor Luciano for his kind assistance with the scanning of the autoradiographs and preparation of the figures. This work was supported by National Institute of Allergy and Infectious Diseases at the National Institutes of Health through exploratory research grant R21 AI115231, and in part by a pilot project and a mini-center grant from the Institute for Human Infections and Immunity, University of Texas Medical Branch, Galveston, USA.
Footnotes
Author Contributions H.P.N. performed all bioinformatics analyses, in vitro experiments and data analysis, and prepared the draft of the manuscript; C.L.C.S. assisted with the performance and analysis of all aspects; M.R., K.K. and Y.F. provided assistance with the details of bioinformatics and calculation of MEVs; A.S. assisted with the preparation of rickettsial stocks and cultures of endothelial cells; and S.K.S. conceived of the study and participated in its design and coordination and helped to finalize the manuscript. All authors read and approved the final manuscript.
References
- Caldelari I., Chao Y., Romby P. & Vogel J. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb. Perspect. Med. 3, a010298, doi: 10.1101/cshperspect.a010298 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pabic H., Germain-Amiot N., Bordeau V. & Felden B. A bacterial regulatory RNA attenuates virulence, spread and human host cell phagocytosis. Nucleic Acids Res. 43, 9232–9248, doi: 10.1093/nar/gkv783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G., Vogel J. & Wassarman K. M. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 43, 880–891, doi: 10.1016/j.molcel.2011.08.022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updegrove T. B., Shabalina S. A. & Storz G. How do base-pairing small RNAs evolve? FEMS Microbiol. Rev. 39, 379–391, doi: 10.1093/femsre/fuv014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni S. K. & Rydkina E. Host-cell interactions with pathogenic Rickettsia species. Future Microbiol. 4, 323–339, doi: 10.2217/fmb.09.6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydkina E., Sahni A., Baggs R. B., Silverman D. J. & Sahni S. K. Infection of human endothelial cells with spotted fever group rickettsiae stimulates cyclooxygenase 2 expression and release of vasoactive prostaglandins. Infect. Immun. 74, 5067–5074, doi: 10.1128/IAI.00182-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni S. K., Narra H. P., Sahni A. & Walker D. H. Recent molecular insights into rickettsial pathogenesis and immunity. Future Microbiol. 8, 1265–1288, doi: 10.2217/fmb.13.102 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson S. G. et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396, 133–140, doi: 10.1038/24094 (1998). [DOI] [PubMed] [Google Scholar]
- Ogata H. et al. Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 293, 2093–2098, doi: 10.1126/science.1061471 (2001). [DOI] [PubMed] [Google Scholar]
- Wattam A. R. et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591, doi: 10.1093/nar/gkt1099 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G. et al. Reductive genome evolution from the mother of Rickettsia. Plos Genet. 3, e14, doi: 10.1371/journal.pgen.0030014 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie J. J. et al. Rickettsia phylogenomics: unwinding the intricacies of obligate intracellular life. Plos One 3, e2018, doi: 10.1371/journal.pone.0002018 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie J. J. et al. A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle. J. Bacteriol. 194, 376–394, doi: 10.1128/JB.06244-11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie J. J. et al. Genomic diversification in strains of Rickettsia felis Isolated from different arthropods. Genome Biol. Evol. 7, 35–56, doi: 10.1093/gbe/evu262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder C. L. et al. Bacterial small RNAs in the Genus Rickettsia. BMC Genomics 16, 1075, doi: 10.1186/s12864-015-2293-7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakumanu M. L. et al. Development and validation of an improved PCR method using the 23S-5S intergenic spacer for detection of rickettsiae in Dermacentor variabilis ticks and tissue samples from humans and laboratory animals. J. Clin. Microbiol. 54, 972–979, doi: 10.1128/JCM.02605-15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P. R. et al. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 42, W119–W123, doi: 10.1093/nar/gku359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder C. L. et al. Identification and characterization of novel small RNAs in Rickettsia prowazekii. Front. Microbiol. 7, 859, doi: 10.3389/fmicb.2016.00859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran N. A. & Bennett G. M. The tiniest tiny genomes. Annu. Rev. Microbiol. 68, 195–215, doi: 10.1146/annurev-micro-091213-112901 (2014). [DOI] [PubMed] [Google Scholar]
- Walker D. H. & Yu X. J. Progress in rickettsial genome analysis from pioneering of Rickettsia prowazekii to the recent Rickettsia typhi. Ann. N Y Acad. Sci. 1063, 13–25, doi: 10.1196/annals.1355.003 (2005). [DOI] [PubMed] [Google Scholar]
- Moran N. A., McCutcheon J. P. & Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42, 165–190, doi: 10.1146/annurev.genet.41.110306.130119 (2008). [DOI] [PubMed] [Google Scholar]
- Dugar G. et al. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. Plos Genet. 9, e1003495, doi: 10.1371/journal.pgen.1003495 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgreen S. et al. Robust identification of noncoding RNA from transcriptomes requires phylogenetically-informed sampling. Plos Comput. Biol. 10, e1003907, doi: 10.1371/journal.pcbi.1003907 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Bateman A., Marshall M., Khanna A. & Eddy S. R. Rfam: an RNA family database. Nucleic Acids Res. 31, 439–441 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matelska D., Kurkowska M., Purta E., Bujnicki J. M. & Dunin-Horkawicz S. Loss of conserved noncoding RNAs in genomes of bacterial endosymbionts. Genome Biol. Evol. 8, 426–438, doi: 10.1093/gbe/evw007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Puyvelde S., Vanderleyden J. & De Keersmaecker S. C. Experimental approaches to identify small RNAs and their diverse roles in bacteria–what we have learnt in one decade of MicA research. Microbiology Open 4, 699–711, doi: 10.1002/mbo3.263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masse E. & Gottesman S. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl. Acad. Sci. USA 99, 4620–4625, doi: 10.1073/pnas.032066599 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann A. J. et al. Dual RNA-seq unveils noncoding RNA functions in host-pathogen interactions. Nature 529, 496–501, doi: 10.1038/nature16547 (2016). [DOI] [PubMed] [Google Scholar]
- Aziz R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75, doi: 10.1186/1471-2164-9-75 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilderman P. J. et al. Identification of tandem duplicate regulatory small RNAs in Pseudomonas aeruginosa involved in iron homeostasis. Proc. Natl. Acad. Sci. USA 101, 9792–9797, doi: 10.1073/pnas.0403423101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I. C., Griesenauer B., Yu Y. T. & Velicer G. J. A recent evolutionary origin of a bacterial small RNA that controls multicellular fruiting body development. Mol. Phylogenet. Evol. 73, 1–9, doi: 10.1016/j.ympev.2014.01.001 (2014). [DOI] [PubMed] [Google Scholar]
- Faucher S. P. & Shuman H. A. Small Regulatory RNA and Legionella pneumophila. Front. Microbiol. 2, 98, doi: 10.3389/fmicb.2011.00098 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades K., Merhej V., El Karkouri K., Raoult D. & Pontarotti P. Gene gain and loss events in Rickettsia and Orientia species. Biol. Direct. 6, 6, doi: 10.1186/1745-6150-6-6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T. R. et al. Comparative genome sequencing of Rickettsia rickettsii strains that differ in virulence. Infect. Immun. 83, 1568–1576, doi: 10.1128/IAI.03140-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skippington E. & Ragan M. A. Evolutionary dynamics of small RNAs in 27 Escherichia coli and Shigella genomes. Genome Biol. Evol. 4, 330–345, doi: 10.1093/gbe/evs001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasa I., Toledo-Arana A. & Gingeras T. R. An effort to make sense of antisense transcription in bacteria. RNA Biol. 9, 1039–1044, doi: 10.4161/rna.21167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroger C. et al. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. USA 109, E1277–E1286, doi: 10.1073/pnas.1201061109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma C. M. et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255, doi: 10.1038/nature08756 (2010). [DOI] [PubMed] [Google Scholar]
- Georg J. & Hess W. R. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 75, 286–300, doi: 10.1128/MMBR.00032-10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed S. C., Lamason R. L., Risca V. I., Abernathy E. & Welch M. D. Rickettsia actin-based motility occurs in distinct phases mediated by different actin nucleators. Curr. Biol. 24, 98–103, doi: 10.1016/j.cub.2013.11.025 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie J. J. et al. An anomalous type IV secretion system in Rickettsia is evolutionarily conserved. Plos One 4, e4833, doi: 10.1371/journal.pone.0004833 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie J. J. et al. Structural insight into how bacteria prevent interference between multiple divergent type IV secretion systems. mBio 6, e01867–01815, doi: 10.1128/mBio.01867-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S. J. et al. TolC-dependent secretion of an ankyrin repeat-containing protein of Rickettsia typhi. J. Bacteriol. 194, 4920–4932, doi: 10.1128/JB.00793-12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovits A. A. et al. Evidence for coupling of membrane targeting and function of the signal recognition particle (SRP) receptor FtsY. EMBO Rep. 2, 1040–1046, doi: 10.1093/embo-reports/kve226 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P. R. et al. Comparative genomics boosts target prediction for bacterial small RNAs. Proc. Natl. Acad. Sci. USA. 110, E3487–E3496, doi: 10.1073/pnas.1303248110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson J. O. & Andersson S. G. Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol. Biol. Evol. 18, 829–839 (2001). [DOI] [PubMed] [Google Scholar]
- Mayoral J. G. et al. Wolbachia small noncoding RNAs and their role in cross-kingdom communications. Proc. Natl. Acad. Sci. USA 111, 18721–18726, doi: 10.1073/pnas.1420131112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahvi A. et al. Genetic control by a metabolite binding mRNA. Chemistry & Biology 9, 1043 (2002). [DOI] [PubMed] [Google Scholar]
- Sauerwine B. & Widom M. Folding kinetics of riboswitch transcriptional terminators and sequesterers. Entropy-Switz 15, 3088–3099, doi: 10.3390/e15083088 (2013). [DOI] [Google Scholar]
- Mellin J. R. & Cossart P. Unexpected versatility in bacterial riboswitches. Trends in Genetics 31, 150–156, doi: 10.1016/j.tig.2015.01.005 (2015). [DOI] [PubMed] [Google Scholar]
- Mellin J. R. et al. Sequestration of a two-component response regulator by a riboswitch-regulated noncoding RNA. Science 345, 940–943, doi: 10.1126/science.1255083 (2014). [DOI] [PubMed] [Google Scholar]
- Dar D. et al. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science 352, aad9822, doi: 10.1126/science.aad9822 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh A. T. & Wassarman K. M. 6S RNA, a global regulator of transcription in Escherichia coli, Bacillus subtilis, and beyond. Annu. Rev. Microbiol. 68, 45–60, doi: 10.1146/annurev-micro-092611-150135 (2014). [DOI] [PubMed] [Google Scholar]
- Janssen B. D. & Hayes C. S. The tmRNA ribosome-rescue system. Adv. Protein Chem. Struct. Biol. 86, 151–191, doi: 10.1016/B978-0-12-386497-0.00005-0 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekkes P. & Driessen A. J. Protein targeting to the bacterial cytoplasmic membrane. Microbiol. Mol. Biol. Rev. 63, 161–173 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omsland A. et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc. Natl. Acad. Sci. USA 106, 4430–4434, doi: 10.1073/pnas.0812074106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S. P., Minesinger B. K., Kumar J. & Walker G. C. A highly conserved protein of unknown function in Sinorhizobium meliloti affects sRNA regulation similar to Hfq. Nucleic Acids Res. 39, 4691–4708, doi: 10.1093/nar/gkr060 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder R., Reinhardt R., Sharma C. & Vogel J. Experimental tools to identify RNA-protein interactions in Helicobacter pylori. RNA Biol. 9, 520–531, doi: 10.4161/rna.20331 (2012). [DOI] [PubMed] [Google Scholar]
- Attaiech L. et al. Silencing of natural transformation by an RNA chaperone and a multitarget small RNA. Proc. Natl. Acad. Sci. USA 113, 8813–8818, doi: 10.1073/pnas.1601626113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydkina E., Silverman D. J. & Sahni S. K. Activation of p38 stress-activated protein kinase during Rickettsia rickettsii infection of human endothelial cells: role in the induction of chemokine response. Cell Microbiol. 7, 1519–1530, doi: 10.1111/j.1462-5822.2005.00574.x (2005). [DOI] [PubMed] [Google Scholar]
- Labruna M. B. et al. Rickettsia species infecting Amblyomma cooperi ticks from an area in the state of Sao Paulo, Brazil, where Brazilian spotted fever is endemic. J. Clin. Microbiol. 42, 90–98 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydkina E., Turpin L. C. & Sahni S. K. Rickettsia rickettsii infection of human macrovascular and microvascular endothelial cells reveals activation of both common and cell type-specific host response mechanisms. Infect. Immun. 78, 2599–2606, doi: 10.1128/IAI.01335-09 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munderloh U. G. & Kurtti T. J. Formulation of medium for tick cell culture. Exp. Appl. Acarol. 7, 219–229 (1989). [DOI] [PubMed] [Google Scholar]
- Schmittgen T. D. & Livak K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008). [DOI] [PubMed] [Google Scholar]
- Langmead B. & Salzberg S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359, doi: 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev V. & Salamov A. In Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies Vol. Nova Science Publishers 61–78 (2011). [Google Scholar]
- Franceschini A. et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815, doi: 10.1093/nar/gks1094 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T., Maki K. & Aiba H. Detection of sRNA-mRNA interactions by electrophoretic mobility shift assay. Methods Mol. Biol. 905, 235–244, doi: 10.1007/978-1-61779-949-5_15 (2012). [DOI] [PubMed] [Google Scholar]
- Maki K., Uno K., Morita T. & Aiba H. RNA, but not protein partners, is directly responsible for translational silencing by a bacterial Hfq-binding small RNA. Proc. Natl. Acad. Sci. USA 105, 10332–10337, doi: 10.1073/pnas.0803106105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faner M. A. & Feig A. L. Identifying and characterizing Hfq-RNA interactions. Methods 63, 144–159, doi: 10.1016/j.ymeth.2013.04.023 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pabic H., Germain-Amiot N., Bordeau V. & Felden B. A bacterial regulatory RNA attenuates virulence, spread and human host cell phagocytosis. Nucleic Acids Res. 43, 9232–9248, doi: 10.1093/nar/gkv783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.