

Abstract
Hyperemesis gravidarum-induced Wernicke's encephalopathy (WE) is an underestimated condition. The purpose of this study is to improve its awareness and early diagnosis. We report five cases of WE secondary to hyperemesis gravidarum. Classic triad of encephalopathy, ataxia, and ocular signs was seen in four out of five patients. Two unusual features noted in this series were papilledema in one patient and severe sensory-motor peripheral neuropathy in one patient. Magnetic resonance imaging (MRI) was abnormal in all the five patients, and high signal in medial thalamus and surrounding the aqueduct was the most common abnormality (5/5). Involvement of caudate nucleus was seen in two patients with severe psychosis, and two patients had bilateral cerebellar peduncle involvement. Median time delay between onset of neurological symptoms and diagnosis was 7 days. All patients improved with thiamine, but minor sequelae were seen in four patients at 12 months follow-up. One patient had a fetal demise. Hyperemesis gravidarum-induced WE is a common cause of maternal morbidity. Typical MRI findings of symmetric medial thalamic and periaqueductal signal changes may permit a specific diagnosis. A delay in diagnosis, therefore treatment, leads to worse prognosis.
KEY WORDS: Hyperemesis gravidarum, magnetic resonance imaging, Wernicke's encephalopathy
Introduction
Wernicke's encephalopathy (WE) is an acute neuropsychiatric syndrome resulting from thiamine deficiency, which is associated with significant morbidity and mortality. It is commonly encountered in alcoholics with malnourishment.[1] The association of WE with hyperemesis gravidarum was described way back in 1939 by Sheehan, but it remains an underrecognized condition. In this article, we report five women with WE due to hyperemesis of pregnancy and discuss the clinical presentation, utility of magnetic resonance imaging (MRI), and outcome.
Case Series
We enrolled five patients with WE in a setting of hyperemesis gravidarum during a 3-year period (2012–2015). Mean age was 28 years (range, 23–34 years). All patients were multigravida, of which four were second gravida and one patient was third gravida. All patients had onset of symptoms in the second trimester, following prolonged severe vomiting.
Neurological symptoms appeared generally within 14–40 days of vomiting. Median time delay between onset of neurological symptoms and diagnosis was 7 days. Initial central nervous system symptoms included disturbance in memory (three patients), psychotic symptoms (three patients), unsteadiness with walking (two patients), and diplopia (one patient). When acute WE was suspected, four patients had higher mental function disturbance including psychosis (3) and amnesia (3). Clinical features are summarized in Table 1. Nystagmus was the most common ocular sign. One patient had severe peripheral neuropathy-sensory-motor axonal type and another had papilledema.
Table 1.
Clinical characteristics, magnetic resonance imaging features, and outcome
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Age (years) | 28 | 23 | 26 | 29 | 34 |
| Psychosis | + | - | + | + | |
| Amnesia | + | + | + | + | |
| Ophthalmoplegia | - | + | + | + | - |
| Nystagmus | + | + | + | + | + |
| Ataxia | + | + | + | + | - |
| MRI findings | |||||
| Medial thalamus | + | + | + | + | + |
| Periaqueductal | - | + | + | + | + |
| Caudate nucleus | - | - | - | + | + |
| Cerebellar peduncles | - | - | - | + | + |
| Follow-up | |||||
| At 1 year | No deficits | Amnesia+ | Ataxia+ | Ataxia+ | Amnesia, weakness+ |
| Ataxia+ | Nystagmus+ | Nystagmus+ | |||
| Pregnancy outcome | Delivered by LSCS | Fetal demise | Normal delivery | Normal delivery | Normal delivery |
+: Means present, -: Means absent, MRI: Magnetic resonance imaging, LSCS: Lower segment cesarean section
MRI was done in all the five patients. All patients were examined with T1-weighted imaging, T2-weighted (T2W) imaging, T2 fluid-attenuated inversion recovery (FLAIR), and diffusion-weighted imaging (DWI). All patients had typical MRI of acute WE that showed areas of increased T2W and FLAIR signals, symmetrically involving medial thalamus and surrounding the aqueduct [Figures 1, 3–5]. DWI showed slightly increased signal in the above areas in four patients. Two patients with severe psychosis had caudate nucleus involvement, and two patients had cerebellar peduncle involvement [Figure 2]. None of the patients had abnormality in the cerebral cortex. No atrophy of cerebellar vermis and mammillary bodies was observed in any of the patients. One patient (patient no. 5) had an increased signal in mid pons with restricted diffusion, probably suggestive of central pontine myelinolysis.
Figure 1.
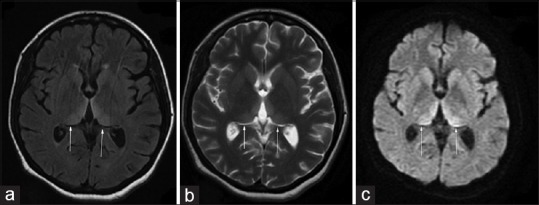
(a) Magnetic resonance imaging of the brain T2 fluid-attenuated inversion recovery sequence showing bilateral medial thalamic hyperintensities (b) T2 sequence showing same findings (c) diffusion sequence showing high signal in bilateral medial thalamus
Figure 3.
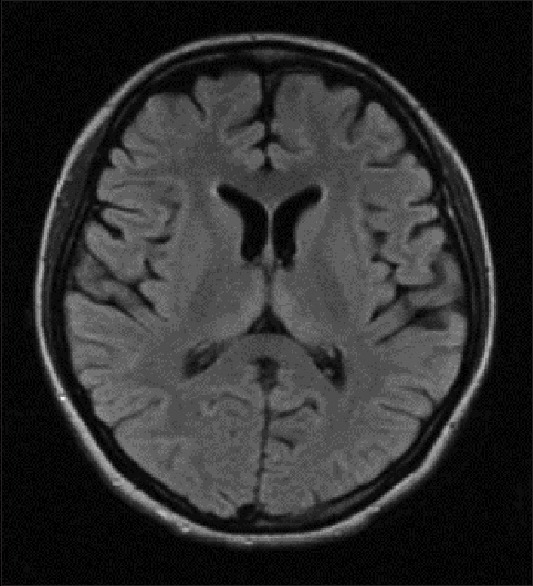
Magnetic resonance imaging of the brain T2 FLAIR sequence showing bilateral symmetrical medial thalamic hyperintensities (Case 1)
Figure 5.
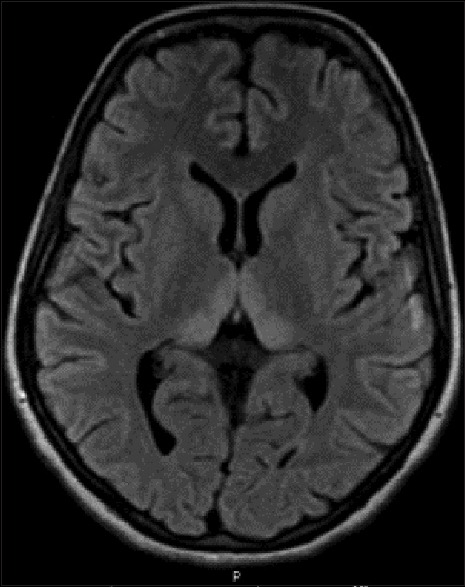
Magnetic resonance imaging of the brain T2 FLAIR sequence showing bilateral symmetrical medial thalamic hyperintensities (Case 3)
Figure 2.
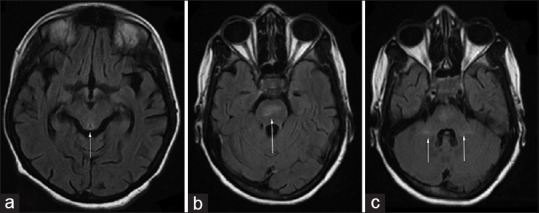
(a) Magnetic resonance imaging of the brain T2 fluid-attenuated inversion recovery sequence showing hyperintense signal in periaqueductal grey matter (b) magnetic resonance imaging of the brain T2 fluid-attenuated inversion recovery sequence showing hyperintense signal in pons (c) magnetic resonance imaging of the brain T2 fluid-attenuated inversion recovery sequence showing hyperintensities in both cerebellar peduncles
Figure 4.
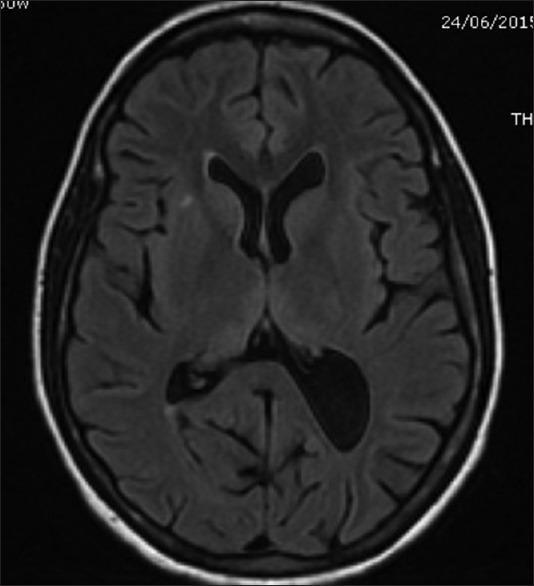
Magnetic resonance imaging of the brain T2 FLAIR sequence showing bilateral symmetrical hyperintensities in medial thalamus (Case 2)
All patients were treated with intravenous thiamine 500 mg every 8 h daily for initial 2 days, followed by 100 mg daily. Oral thiamine was continued up to 3 months. Follow-up at 3 months showed persisting signs with less severity in four out of five patients. At 1 year, overall four patients had persisting signs, two patients had mild amnesia and ocular signs, and three patients had ataxia. Four patients had a successful pregnancy and delivered at term, and one patient had a fetal demise.
Discussion
WE is a well-known neurological complication of thiamine deficiency that is most often seen in alcoholic individuals. Most of the early studies on WE have been described in alcoholics.[2] More than forty cases of WE associated with hyperemesis gravidarum have been described. Recent case studies reveal vulnerability in patients with thiamine deficiency secondary to anorexia nervosa or other psychiatric conditions, prolonged intravenous feeding without proper supplementation, gastrointestinal surgery (especially bariatric surgery), and systemic malignancy.[3,4] Thiamine is a cofactor for several key enzymes important in energy metabolism, including transketolase, alpha-ketoglutarate dehydrogenase, and pyruvate dehydrogenase. Thiamine requirements depend on metabolic rate, with the greatest need during periods of high metabolic demand and high glucose intake (including glucose-containing intravenous fluids). In pregnancy, thiamine stores are depleted due to excessive vomiting, poor intake, and increased metabolic demand.[5,6] Hypermetabolisms due to physiological pregnancy-related hyperthyroidism are also another risk factor. Chiossi et al. showed that on an average, WE occurred after 7 weeks of vomiting and around 14th week of the pregnancy.[7]
The classic triad of WE includes encephalopathy, oculomotor dysfunction, and gait ataxia. Detailed clinical and neuropathological data from 82 autopsies, and 245 patients examined over 10 years revealed that all features of the triad were recognized in only one-third of patients; in most, only one or two elements of the clinical triad were apparent.[1] Confusion was the most common presenting symptom, followed by staggering gait and ocular problems. The encephalopathy is characterized by profound disorientation, indifference, and inattentiveness. If these are less severe and permit higher cognitive testing, impaired memory and learning are also evident. Nystagmus is the most common ocular finding and is typically evoked by horizontal gaze to both sides. Ataxia primarily involves stance and gait and is likely due to a combination of polyneuropathy, cerebellar involvement, and vestibular dysfunction.[1] Although laboratory studies and neuroimaging studies can be helpful, WE is primarily a clinical diagnosis. All our patients presented in the second trimester of pregnancy with prolonged hyperemesis. In this series, four out of five patients had classical triad. Symptoms common to all the cases were behavioral disturbance. Two out of the four cases had ocular muscle involvement. In all cases, nystagmus and gait ataxia were common findings whereas dysarthria was found in two out of four cases. Papilledema was found in one of our patients. Papilledema is recorded very infrequently in literature. One of the explanations for optic nerve fiber swelling would be WE-associated optic neuritis or neuropathy, analogous to the nutritional retrobulbar neuropathies such as reported in pellagra.[8] One patient had severe sensory-motor peripheral neuropathy. Reversible axonal neuropathy with WE although rare has been reported. Physiological conduction failure with minimal conduction delay is postulated as an important mechanism.[9]
There are no laboratory studies that are diagnostic of WE. Thiamine deficiency can be most reliably detected by measurement of erythrocyte thiamine transketolase before and after the addition of thiamine pyrophosphate. This test is often not readily available, especially in the emergency setting. Hence, MRI plays an important role in diagnosis of WE. Reversible cytotoxic edema seems to be the most distinctive lesion in WE, and the most useful sequences to detect it are T2, FLAIR, and DWI. Periventricular regions of diencephalon, mesencephalon, brainstem, and superior vermis of the cerebellum are sensitive to thiamine deficiency due to cellular dependence on oxidative metabolism that causes T2W hyperintensities in the region.[10] Typical findings include areas of increased T2 and FLAIR signals, decreased T1 signal, and diffusion abnormality surrounding the aqueduct and third ventricle and within the medial thalamus, dorsal medulla, tectal plate, and mamillary bodies.[11,12] Atypical lesions can be observed in the cerebellum, red, dentate, caudate nuclei, and cerebral cortex. The atypical lesions have been described mostly in nonalcoholic patients while gadolinium uptake in thalamus and mammillary bodies is more common in alcoholics.[12] Two of our patients had atypical lesions at caudate and cerebellar peduncle. Pooled data from several studies show that conventional MRI was able to detect changes in two-thirds of alcoholic subjects with WE. T2 FLAIR and DWI images did not provide further information. In contrast, in nonalcoholic patients, MRI detected lesions in up to 97% in DWI, 99% in conventional MRI, and 100% in FLAIR sequences.[13] MRI was diagnostic in all the five cases with characteristic increased signal intensity in T2 and FLAIR images around the periventricular area.
The cornerstone of treatment remains timely administration of thiamine. However, how much and how long is an unanswered question. The traditional recommendation is a parenteral dose of 100 mg of thiamine per day. However, several British authors recommend a dosage of 500 mg although little evidence is available to support or refute this regimen. They recommend oral thiamine 200 mg/day after the parenteral dose for 3 months.[14]
Hyperemesis gravidarum is associated with considerable morbidity and mortality. WE can cause fetal loss, and Chiossi et al. in a review of 49 patients reported about 50% of fetal deaths through direct or indirect mechanisms.[7] In this series, only one case was associated with adverse fetal outcome which was IUD at 15 weeks of gestation. All the patients improved markedly with thiamine administration. Most common residual neurological deficit encountered was nystagmus which lasted for a maximum of 2 years in a subject, whereas in the other two patients, it lasted for a year. Gait disturbances were also found in two of the four patients as a residual neurological deficit. Behavioral disturbance was the symptom that responded well to therapy. Three out of five patients with behavioral disturbance were asymptomatic after a week of thiamine administration.
Conclusion
WE is an uncommon neurological disease complicating pregnancy in the setting of hyperemesis. MRI is the most useful investigation in diagnosis. Serious management of vomiting women during pregnancy and thiamine supplementation in patients who have acute neurological symptoms can preclude both maternal and fetal morbidity.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Victor M, Adams RA, Collins GH. The Wernicke. Korsakoff Syndrome: And Related Disorders Due to Alcoholism and Malnutrition. Philadelphia: FA Davis; 1989. [Google Scholar]
- 2.Harper C. The incidence of Wernicke's encephalopathy in Australia – A neuropathological study of 131 cases. J Neurol Neurosurg Psychiatry. 1983;46:593–8. doi: 10.1136/jnnp.46.7.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Netravathi M, Sinha S, Taly AB, Bindu PS, Bharath RD. Hyperemesis-gravidarum-induced Wernicke's encephalopathy: Serial clinical, electrophysiological and MR imaging observations. J Neurol Sci. 2009;284:214–6. doi: 10.1016/j.jns.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Merkin-Zaborsky H, Ifergane G, Frisher S, Valdman S, Herishanu Y, Wirguin I. Thiamine-responsive acute neurological disorders in nonalcoholic patients. Eur Neurol. 2001;45:34–7. doi: 10.1159/000052086. [DOI] [PubMed] [Google Scholar]
- 5.Gárdián G, Vörös E, Járdánházy T, Ungureán A, Vécsei L. Wernicke's encephalopathy induced by hyperemesis gravidarum. Acta Neurol Scand. 1999;99:196–8. doi: 10.1111/j.1600-0404.1999.tb07344.x. [DOI] [PubMed] [Google Scholar]
- 6.Sechi G, Serra A. Wernicke's encephalopathy: New clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6:442–55. doi: 10.1016/S1474-4422(07)70104-7. [DOI] [PubMed] [Google Scholar]
- 7.Chiossi G, Neri I, Cavazzuti M, Basso G, Facchinetti F. Hyperemesis gravidarum complicated by Wernicke encephalopathy: Background, case report, and review of the literature. Obstet Gynecol Surv. 2006;61:255–68. doi: 10.1097/01.ogx.0000206336.08794.65. [DOI] [PubMed] [Google Scholar]
- 8.Mumford CJ. Papilloedema delaying diagnosis of Wernicke's encephalopathy in a comatose patient. Postgrad Med J. 1989;65:371–3. doi: 10.1136/pgmj.65.764.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishibashi S, Yokota T, Shiojiri T, Matunaga T, Tanaka H, Nishina K, et al. Reversible acute axonal polyneuropathy associated with Wernicke-Korsakoff syndrome: Impaired physiological nerve conduction due to thiamine deficiency? J Neurol Neurosurg Psychiatry. 2003;74:674–6. doi: 10.1136/jnnp.74.5.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki S, Ichijo M, Fujii H, Matsuoka Y, Ogawa Y. Acute Wernicke's encephalopathy: Comparison of magnetic resonance images and autopsy findings. Intern Med. 1996;35:831–4. doi: 10.2169/internalmedicine.35.831. [DOI] [PubMed] [Google Scholar]
- 11.Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke's encephalopathy: Review of the literature. AJR Am J Roentgenol. 2009;192:501–8. doi: 10.2214/AJR.07.3959. [DOI] [PubMed] [Google Scholar]
- 12.Doherty MJ, Watson NF, Uchino K, Hallam DK, Cramer SC. Diffusion abnormalities in patients with Wernicke encephalopathy. Neurology. 2002;58:655–7. doi: 10.1212/wnl.58.4.655. [DOI] [PubMed] [Google Scholar]
- 13.Galvin R, Bråthen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA; EFNS. EFNS guidelines for diagnosis, therapy and prevention of Wernicke encephalopathy. Eur J Neurol. 2010;17:1408–18. doi: 10.1111/j.1468-1331.2010.03153.x. [DOI] [PubMed] [Google Scholar]
- 14.Chataway J, Hardman E. Thiamine in Wernicke's syndrome – How much and how long? Postgrad Med J. 1995;71:249. doi: 10.1136/pgmj.71.834.249. [DOI] [PMC free article] [PubMed] [Google Scholar]