

Abstract
Background
Acetylation of lysine residues is a reversible post-translational modification conserved from bacteria to humans. Several recent studies have revealed hundreds of lysine-acetylated proteins in various bacteria; however, the physiological role of these modifications remains largely unknown. Since lysine acetylation changes the size and charge of proteins and thereby may affect their conformation, we assumed that lysine acetylation can stimulate aggregation of proteins, especially for overproduced recombinant proteins that form inclusion bodies.
Results
To verify this assumption, we used Escherichia coli strains that overproduce aggregation-prone VP1GFP protein. We found that in ΔackA-pta cells, which display diminished protein acetylation, inclusion bodies were formed with a delay and processed faster than in the wild-type cells. Moreover, in ΔackA-pta cells, inclusion bodies exhibited significantly increased specific GFP fluorescence. In CobB deacetylase-deficient cells, in which protein acetylation was enhanced, the formation of inclusion bodies was increased and their processing was significantly inhibited. Similar results were obtained with regard to endogenous protein aggregates formed during the late stationary phase in ΔackA-pta and ΔcobB cells.
Conclusions
Our studies revealed that protein acetylation affected the aggregation of endogenous E. coli proteins and the yield, solubility, and biological activity of a model recombinant protein. In general, decreased lysine acetylation inhibited the formation of protein aggregates, whereas increased lysine acetylation stabilized protein aggregates. These findings should be considered during the designing of efficient strategies for the production of recombinant proteins in E. coli cells.
Electronic supplementary material
The online version of this article (doi:10.1186/s12934-016-0590-8) contains supplementary material, which is available to authorized users.
Keywords: Lysine acetylation, Recombinant proteins, Inclusion bodies, Protein aggregates
Background
Escherichia coli is by far the most widely used host for the production of recombinant proteins. There are many protocols, a great number of expression plasmids, and engineered strains that can be used to obtain high protein yields [1]. Despite numerous advantages, biotechnological potential of E. coli also has its limitations including the risk of protein aggregation and accumulation of acetate as by-product. Upon high-level production, recombinant proteins may aggregate and form inclusion bodies (IBs) in E. coli cells. Formation of IBs is a complex and dynamic process affected by various factors involving molecular chaperones that control protein folding. The vast increase in misfolded recombinant proteins exhausts the capacity of molecular chaperones leading to the formation of IBs [2, 3]. To recover biologically active proteins from IBs, additional steps in purification procedure such as solubilization of IBs and subsequent refolding of desired proteins are required. Coexpression of molecular chaperones or lower production rate can prevent aggregation and facilitate the proper folding of recombinant proteins [1–3]. A whole spectrum of polypeptides with different conformations, including partially or even fully native structures, can be found in IBs [4–6]. Therefore, in some cases, insoluble but active proteins sequestered in IBs can be the preferred form of the product [7].
Another obstacle in recombinant protein production in E. coli cells is accumulation of acetate during extensive aerobic fermentation in cultures supplemented with glucose. At higher glucose concentrations, the carbon flux into the cells exceeds tricarboxylic acid cycle capacity, and acetyl-CoA is converted into acetate, which is easily excreted from the cell. The excretion of acetate into the environment results also from the need to regenerate the NAD+ consumed by glycolysis and to recycle the CoA that is required to convert pyruvate to acetyl-CoA [8]. A high concentration of acetate, externally added or excreted from the cell, limits the growth of E. coli. Undissociated or acidic forms of acetate permeate the membranes and dissipate the transmembrane pH gradient. After acetate dissociation, the proton acidifies the cytoplasm, while the anion increases the internal osmotic pressure. Acetate toxicity results also from the depletion of the intracellular methionine pool, with the concomitant accumulation of the toxic intermediate homocysteine [9]. Various strategies have been developed to limit acetate accumulation or reduce its negative effects [10]. One of such strategies is reduction of acetate concentration and improvement of protein production achieved by deletion or downregulation of the ackA-pta pathway using antisense-RNA strategy [11, 12]. The ackA-pta operon encodes phosphate acetyltransferase (Pta), which converts acetyl-CoA and inorganic phosphate to acetyl-phosphate (AcP), and acetate phosphotransferase (AckA), which converts acetyl phosphate to acetate and ATP. Downregulation or deletion of the ackA-pta reduces both acetate level and also the concentration of AcP. Interestingly, it has been demonstrated that the ΔackA-pta strain was defective in the degradation of model unstable proteins and accumulated increased levels of protein aggregates formed upon heat stress [13]. Furthermore, it was found that the ΔackA-pta mutation resulted in reduced refolding and disaggregation of heat-denatured luciferase [14]. The authors proposed that the presence of AcP is required for efficient proteolysis and function of molecular chaperones in E. coli cells. The exact role of AcP in the protein quality-control system remains, however, unknown. Recent studies have demonstrated that AcP participates in non-enzymatic Nε-lysine acetylation in E. coli [15]. Acetylation of lysine residues is a conserved post-translational reversible modification that occurs in all three kingdoms. In eukaryotes protein lysine acetylation regulates diverse physiological processes such as cell cycle, cell morphology, protein synthesis, mRNA splicing, and central metabolism. Contrary to eukaryotes, the impact of protein acetylation on bacterial physiology is poorly understood; however, global proteomic studies revealed acetylation of thousands of lysine residues in hundreds of E. coli proteins involved in various cellular processes [16]. Apart from non-enzymatic acetylation by AcP, E. coli proteins can be modified by lysine acetyltransferase PatZ (the pka gene product), which transfers acetyl group from acetyl-CoA to Nε lysine residues. Nε-lysine acetylation can be reversed by the deacetylase CobB regardless of acetylation mechanism [17].
Nε-lysine protein acetylation is stimulated in media supplemented with glucose or acetate [16, 18]. We demonstrated previously that acetate or glucose enhanced protein aggregation in stationary E. coli cultures [19]. We assume that increased protein aggregation in cells exposed to a high concentration of glucose or acetate can result not only from the aforementioned toxic effects of acetate but also from N-lysine acetylation. Several lines of evidence exist to support this notion. Neutralization of the charge on lysine residues by acetylation has been shown to induce aggregation of modified lysozyme in vitro [20]. Whereas acetylation of the microtubule-associated protein tau inhibited tau function via impaired tau–microtubule interactions and promotes pathological tau aggregation in neurons [21].
Altogether, these data suggest that the formation of inclusion bodies and biological activities of proteins sequestered in the aggregates can be affected by acetylation. This is an important issue to address because recombinant proteins are often produced under acetylation- or deacetylation-favorable conditions (e.g., glucose-supplemented media or acetate-deficient engineered strains); however, the effect of acetylation on recombinant products has not been investigated yet. To verify our hypothesis, we analyzed the formation and processing of IBs and aggregates of endogenous proteins in two bacterial strains: ΔackA-pta cells, which are characterized by diminished protein acetylation, and in CobB-deficient cells (ΔcobB) with enhanced acetylation.
Results
Overproduction of VP1GFP and formation of IBs are enhanced in ΔcobB cells and inhibited in the ΔackA-pta strain
First, we analyzed the formation and processing of IBs containing the fusion VP1GFP protein, which consists of GFP and foot-and-mouth disease virus VP1 capsid protein, and retains its fluorescence in IBs [22]. We found that the overproduction of VP1GFP was enhanced in the ΔcobB strain and inhibited in ΔackA-pta cells compared to wild-type (wt) cells (Fig. 1a). VP1GFP synthesis inhibited the growth of wt and ΔcobB but not ΔackA-pta cells. Just before isopropyl β-d-1-thiogalactopyranoside (IPTG) induction, ΔackA-pta cells were found to grow slower than wt strain (Fig. 1b). After IPTG induction, the generation time increased from 33 to 44 min in the wt culture and, surprisingly, decreased from 53 to 44 min in the ΔackA-pta culture. Overproduction of VP1GFP affected the growth of ΔcobB cells—its generation time increased after IPTG induction from 33 to 54 min. It should be noted, however, that the ΔcobB culture reached the stationary phase later than wt cells (Fig. 1b). Interestingly, specific fluorescence of VP1GFP in the ΔackA-pta strain was threefold higher than in wt and ΔcobB cells (Fig. 1c). Presumably, slower VP1GFP synthesis in ΔackA-pta cells gives the recombinant protein time to fold properly and resulted in higher fluorescence. In accordance with these data, we found that in cells with impaired AckA-Pta pathway, the formation of IBs was postponed. On the other hand, faster accumulation of IBs was observed in ΔcobB cells (Fig. 1d).
Fig. 1.
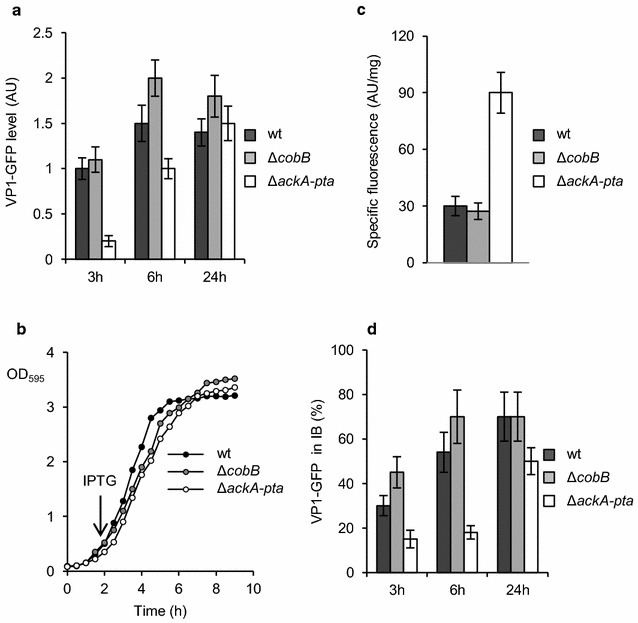
Overproduction of VP1GFP in mutant strains defective in deacetylation (ΔcobB) or acetylation (ΔackA-pta). Bacteria were grown at 37 °C in LB medium supplemented with 100 µg/ml ampicillin. 1 mM IPTG was added at an OD595 = 0.5 to induce the synthesis of VP1GFP. a After 3, 6, and 24 h, whole-cell extracts were resolved by SDS-PAGE, subjected to immunodetection using anti-GFP antibodies and analyzed by densitometry to estimate the relative level of VP1GFP. b Representative growth curves of wt, ΔcobB, and ΔackA-pta cells. c GFP fluorescence in whole-cell extracts was recorded after 3 h of induction using EnSpire plate reader (PerkinElmer). Specific fluorescence emission of VP1GFP was estimated as described in the “Methods” section. d The amounts of VP1GFP in IBs were calculated in relation to the total VP1GFP level in bacterial extracts (100%). IBs were isolated from E. coli cells after induction of VP1GFP synthesis by 1 mM IPTG at the times indicated in the figure. Error bars represent the standard deviation of three values. AU-arbitrary units
ΔcobB and ΔackA-pta mutations affect the size, fluorescence, and processing of IBs
To further characterize the IBs, we immunodetected acetylated lysines in whole cell extracts and in the aggregates (Fig. 2). To allow efficient IPTG induction, the bacteria were grown in Lysogeny broth (LB) without glucose; therefore, acetylated proteins were hardly detectable in whole cell extracts (Fig. 2). Nevertheless, we observed strong acetylation of aggregated VP1GFP in wt cells and significantly stronger acetylation of aggregated VP1GFP isolated from ΔcobB cells. According to our expectations, IBs in the ΔackA-pta strain were not acetylated. Since VP1GFP retains its fluorescence in IBs [22], it was possible to examine the size of aggregates by fluorescence microscopy (Fig. 3a). We found that the average diameters of IBs isolated from ΔcobB and ΔackA-pta cells were slightly higher and lower, respectively (Fig. 3b). The differences between strains were more noticeable when the distribution of IBs’ diameters was compared (Fig. 3c). The IBs showed wide ranges of diameters from 0.2 to 1.3 μm. The population of IBs from the CobB-deficient strain was enriched in larger particles (>0.8 μm), whereas smaller IBs (<0.4 μm) were found mainly in ΔackA-pta cells (Fig. 3c). In all tested strains, IBs isolated after 24 h were larger than the aggregates formed after 3 h of induction. Next, we found that the specific fluorescence of VP1GFP was significantly higher in IBs than in soluble fractions (Fig. 3d). This difference was particularly striking for the ΔackA-pta strain. After 3 h of IPTG induction, we detected ~320 fluorescence AU/mg protein in IBs versus ~20 fluorescence AU/mg protein in the soluble fraction. Specific fluorescence emission of IBs from ΔackA-pta cells was fourfold higher than that observed in wt IBs. This might be due to the fact that the IBs from ΔackA-pta cells were produced more slowly, were smaller in size, and probably contained more polypeptides with native structures than IBs from the other strains. In wt and ΔackA-pta cells, IBs appeared to become less fluorescent over time, whereas in the ΔcobB strain, the specific fluorescence remained relatively constant and at the end of the experiment was higher by 30% than in wt IBs. These results may suggest that acetylation could facilitate the retainment of VP1GFP fluorescence. Although specific fluorescence of IBs decreased in ΔackA-pta cells with the highest rate, at the end of the experiment, the specific fluorescence of insoluble VP1GFP in this strain was at least twofold higher than that in IBs from wt cells.
Fig. 2.
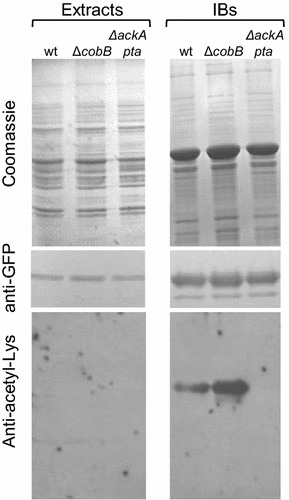
IBs from wt and ΔcobB cells contain acetylated VP1GFP. IBs were isolated after 24 h of IPTG induction from the bacteria cultivated, as described in the legend to Fig. 1. The whole-cell extracts and purified IBs were submitted to SDS-PAGE and immunodetection using anti-GFP (Sigma) and anti-acetyl lysine antibodies (Abcam). IBs isolated from equal amounts of cells were loaded onto the gel
Fig. 3.
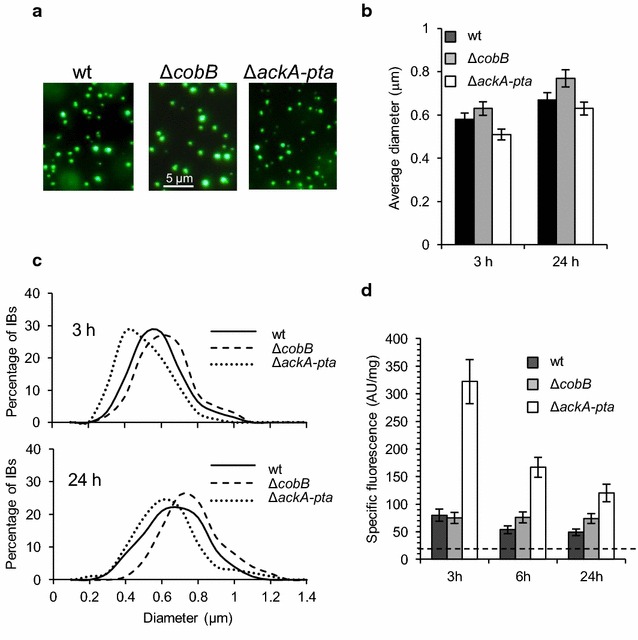
The size of IBs is affected by ΔcobB and ΔackA-pta mutations. a Fluorescence microscopy images of IBs purified from wt, ΔcobB, and ΔackA-pta cells after 24 h of IPTG-induction. b, c Average diameters of IBs isolated after 3 or 24 h of induction were estimated using Zen imaging software (Zeiss). d Specific fluorescence emission of VP1GFP in IBs. The dotted line indicates specific fluorescence of soluble VP1GFP. Error bars represent the standard deviation of three independent experiments
In further experiments, we analyzed the processing of IBs after the arrest of protein synthesis by chloramphenicol. We found that the removal of IBs was inhibited in ΔcobB cells, but occurred significantly faster in the ΔackA-pta strain when compared to wt cells (Fig. 4a). Three hours after the arrest of protein synthesis, 45, 72, and 20% of the initial amounts of IBs remained in wt, ΔcobB, and ΔackA-pta cells, respectively. In the beginning, an increase in soluble VP1GFP fraction was observed in wt and ΔcobB cells, but further incubation resulted in a decrease in the level of soluble VP1GFP due to the degradation of the protein (Fig. 4a). The degradation of soluble VP1GFP was significantly faster in ΔackA-pta cells. We observed that during the processing of IBs, the specific fluorescence of VP1GFP increased, particularly in soluble fractions (Fig. 4b). These data suggested that after the arrest of protein synthesis, unfolded but soluble VP1GFP accumulated in the cell was refolded. Again, this effect was drastically enhanced in the ΔackA-pta soluble fraction. In addition, a twofold increase in specific fluorescence was detected in IBs isolated from this strain.
Fig. 4.
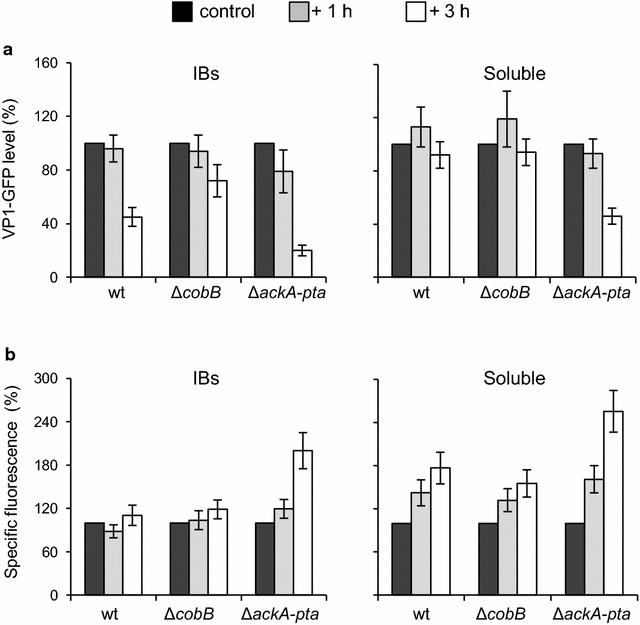
IBs processing is affected by the ΔcobB and ΔackA-pta mutations. The bacteria were grown and VP1GFP synthesis was induced as described in the legend to Fig. 1. At 3 h after IPTG induction (control), chloramphenicol (200 mg/L) was added to arrest protein synthesis and the cultures were incubated further at 28 °C. a After 1 and 3 h (+1 h, +3 h), cell samples were collected to isolate soluble fractions and IBs, which were subsequently analyzed by SDS-PAGE, immunodetection using anti-GFP antibodies, and densitometry. The level of VP1GFP after 3 h of induction (control) corresponds to 100%. b GFP fluorescence in soluble fractions and IBs was recorded in EnSpire plate reader (PerkinElmer). Specific fluorescence emission of VP1GFP was estimated as described in the “Methods” section. Error bars represent the standard deviation of three independent experiments
The key factors that determine the rate of protein aggregation are the net charge, hydrophobicity and propensity to form a secondary structure [23]. VP1GFP contains 28 lysine residues and at the intracellular pH (pH 7.6) [24] has a net charge of −4. Acetylation of all 28 lysine residues in VP1GFP increases the negative net charge of the protein to −32. This should generate electrostatic repulsions between VP1GFP molecules resulting in increased solubility [25, 26]. However, our results indicated that enhanced VP1GFP acetylation caused faster aggregation of the protein (Fig. 1d). This could be explained by the fact that acetylation increases the hydrophobicity of the lysine side chain which may in turn enhance protein aggregation. It seems that the electrostatic repulsions were not sufficient to completely prevent hydrophobic interactions between VP1GFP molecules. To support our assumption we analysed the tendency of acetylated VP1GFP to form aggregates by submitting modified VP1GFP sequence to Aggrescan software [27]. The algorithm identifies specific fragments in proteins that act as “hot spots” driving aggregation. Lysine residues in the VP1GFP sequence were changed for hydrophobic amino acids: phenylalanine, isoleucine or valine to mimic lysine acetylation. We found that in modified VP1GFP variants new hot-spot areas were created suggesting that acetylated VP1GFP exhibits enhanced tendency to form aggregates (Additional file 1: Figure S1).
ΔcobB and ΔackA-pta mutations affect the aggregation of endogenous proteins in stationary E. coli cells
Our studies revealed that acetylation or deacetylation can influence the formation and processing of IBs. However, it is not excluded that the observed effect can depend on an overproduced recombinant protein. To extend our studies, we investigated the effect of ΔcobB and ΔackA-pta mutations on the aggregation of endogenous E. coli proteins during the late stationary phase [28]. In this experiment, we used LB supplemented with 0.4% glucose to stimulate protein acetylation [16]. We found that in ΔcobB whole cell extract and aggregates, the overall levels of acetylated proteins were increased, whereas in ΔackA-pta extracts and aggregates, acetylated proteins were hardly detectable (Fig. 5a). The ΔcobB strain produced ~25% more aggregates than wt cells and the ΔackA-pta strain contained ~40% less aggregates than wt cells. Under deacetylation-favorable conditions, when the bacteria were transferred to fresh LB medium without glucose, the removal of aggregates was most effective in the ΔackA-pta strain (Fig. 5b). These results strongly resembled those obtained for IBs (Figs. 1, 2, 3, 4), suggesting that acetylation or deacetylation of proteins can generally affect the formation and processing of protein aggregates. Surprisingly, partial deacetylation of aggregated proteins was also observed in the ΔcobB strain (Fig. 5a). It should be noted that our results were not fully consistent with the data published by Mizrahi et al. which demonstrated that the aggregation of heat denatured proteins was enhanced in ΔackA-pta cells [13, 14] (see “Discussion” section).
Fig. 5.
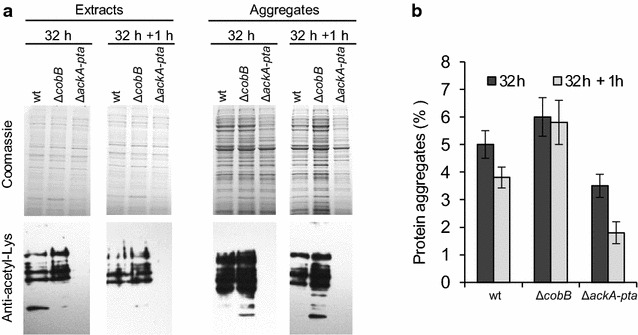
Aggregation of endogenous E. coli proteins in ΔcobB and ΔackA-pta strains. Bacteria were grown at 37 °C in LB medium supplemented with 0.4% glucose. After 32 h, the cells were collected, resuspended in fresh LB medium without glucose, and incubated for 1 h at 37 °C. a Protein aggregates were isolated as described in “Methods” section, resolved by SDS-PAGE, and visualized by Coomassie staining or immunodetected using anti-acetyl lysine antibodies (Abcam). Volumes corresponding to equal amounts of bacteria were loaded onto the gels. b The level of aggregated proteins in relation to total protein in cell extracts was calculated on the basis of densitometry. Error bars represent the standard deviation of three values
It was demonstrated that ΔackA-pta cells accumulate transcripts of heat shock protein (hsp) genes, including dnaK, groEL, groES, and clpB [8]. Therefore, we assumed that the faster removal of aggregated proteins in ΔackA-pta cells could at least partly result from chaperone activity of overexpressed heat shock proteins (Hsps). Indeed, we found that the ΔackA-pta cell extract and aggregates contained increased levels of IbpA/IbpB chaperones when compared to the wt and ΔcobB strains (Fig. 6a). Further experiments demonstrated, however, that IbpA/B were not required for the efficient removal of the aggregated proteins. In the stationary phase and under deacetylation conditions, the levels of protein aggregates were comparable in the ΔackA-pta-ibpAB and ΔackA-pta strains (Fig. 6b). We supposed that the lack of IbpA/B in ΔackA-pta-ibpAB cells could be compensated by the overproduction of DnaK or other molecular chaperones [29–31], but the levels of DnaK, DnaJ, ClpB, and GroEL were similar in all tested strains (Fig. 6a). These results suggest that the removal of aggregates in ΔackA-pta cells was enhanced due to the lack of protein acetylation rather than increased activities of chaperones.
Fig. 6.
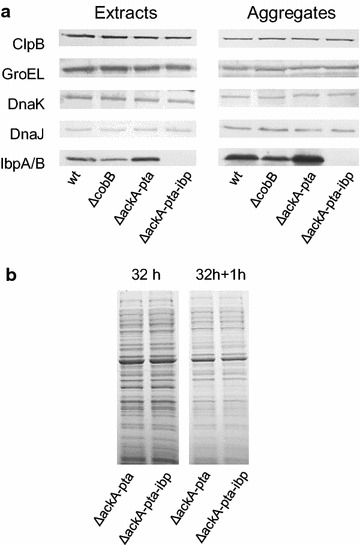
The IbpA/B molecular chaperones are overproduced in ΔackA-pta cells. Bacteria were grown as described in the legend to Fig. 5. a Whole-cell extracts and purified aggregates were resolved by SDS-PAGE and submitted to immunodetection using antibodies against ClpB, DnaK, GroEL, DnaJ, and IbpA/B molecular chaperones. Both IbpA and IbpB migrated in the polyacrylamide gel as one band. Volumes corresponding to equal amounts of bacteria were loaded onto the gels. b Purified aggregates were visualized by Coomassie staining
Discussion
In this study, we demonstrated that ΔackA-pta and ΔcobB mutations exerted different effects on the formation and processing of IBs. We found that IBs isolated from the ΔackA-pta strain showed fourfold higher specific VP1GFP fluorescence than their counterparts from wt cells. Previously, Garcia-Fruitos et al. [32] demonstrated that the specific GFP fluorescence can be increased up to threefold in the absence of selected molecular chaperones or proteases. The increase in fluorescence was often accompanied by enhanced aggregation of GFP. This was probably caused by the stabilization of the recombinant protein, due to impaired degradation, and the activity of the protein quality system which supported proper folding of insoluble GFP [32]. In our studies, it was observed that slower VP1GFP synthesis in ΔackA-pta cells probably gives the recombinant protein time to fold properly and resulted in postponed aggregation and higher fluorescence (Fig. 1a, c). The lack of toxic effects of acetate could also improve the solubility of VP1GFP and conformational quality of IBs in ΔackA-pta cells. This assumption can be supported by our previous studies, showing that externally added acetate strongly stimulated the aggregation of E. coli proteins [19]. Conformation of proteins can also be affected by acetylation, leading to the aggregation of modified polypeptides [20, 21]. Our results suggest that diminished acetylation (in ΔackA-pta) or enhanced acetylation of proteins (in ΔcobB cells) resulted in decreased or increased aggregation in E. coli, respectively. Although IBs from the ΔcobB strain were characterized by lower specific fluorescence than IBs from ΔackA-pta cells (Fig. 3d), they contained higher percentage of total VP1GFP (Fig. 1d), and they were more stable regarding protein level (Fig. 4a) and specific fluorescence (Fig. 3d). It is known that overproduction of recombinant proteins interferes with many physiological processes and causes induction of stress responses in E. coli cells [33]. It seems that ΔcobB cells coped with this stress better than wt cells and hence overproduced more VP1GFP. This observation is in agreement with the results by Ma and Wood [34], showing that the ΔcobB strain exhibits increased oxidative and heat stress resistance. It is believed that soluble misfolded proteins are more toxic for cells than final insoluble aggregates [35]. Therefore, we suppose that faster formation of IBs also enabled ΔcobB cells to withstand the stress induced by recombinant protein production. Our results indicated that in the ΔcobB strain potentially harmful soluble species of misfolded VP1GFP were sequestered in less toxic IBs faster than in wt cells (Fig. 1d).
The effects observed in the case of endogenous protein aggregates were similar to the results obtained for IBs. In ΔackA-pta cells, the formation of aggregates was diminished and the removal of aggregates was increased, whereas the opposite effect was found in ΔcobB cells. Surprisingly, under deacetylation-favorable conditions, the aggregates in ΔcobB cells were partly deacetylated (Fig. 5a). Recently, a new deacetylase named YcgG, which targets different substrates than CobB, has been identified [36]. Our results may suggest that the aggregates formed during the stationary phase contain CobB-specific as well as YcgG-specific substrates.
We cannot exclude that protein aggregation can be affected indirectly by other physiological changes caused by the loss of AckA-Pta pathway or CobB deacetylase. AcP has been proposed to act as a global transcription regulator, which phosphorylates a number of response regulators [37]. It has been also reported that CobB-controlled acetylation of isocitrate lyase affects the glyoxylate shunt, whereas acetylation of the transcription factor RcsB prevents DNA binding and activates flagella biosynthesis and motility [38].
In our experiments, IBs containing VP1GFP and endogenous protein aggregates formed in stationary E. coli cells were efficiently removed in the AcP-deficient strain (Figs. 4, 6). However, Mizrahi and co-workers demonstrated that AcP was required for the proteolysis of some proteins [13, 14]. This discrepancy can be explained by the fact that different proteins and growth conditions were used in our studies and in the experiments described previously [13, 14]. Mizrahi and co-workers found that AcP was required for the removal of truncated soluble forms of homoserine trans–succinylase and alkaline phosphatase, and puromycin-derived peptides [13] but not for the degradation of heat shock–denatured luciferase [14]. The Authors observed also enhanced aggregation of endogenous proteins in ΔackA-pta cells submitted to heat shock [13]. These studies were performed using exponentially growing cells, in contrast to our experiments in which stationary-phase cells were employed. The level of acetylated proteins in exponential-phase cells is very low but increases significantly upon entry into stationary phase [15]. Therefore, we suppose that in exponentially growing cells the effect of the ΔackA-pta mutation could result from the lack of AcP-dependent phosphorylation rather than impaired protein acetylation.
Conclusions
Recent studies have demonstrated that lysine acetylation is one of the prevalent post- translational modifications of bacterial proteins. Physiological functions of acetylation/deacetylation of proteins in bacteria are still being discovered. Our studies revealed that diminished and enhanced lysine acetylation can differentially affect the yield, solubility, and biological activity of recombinant proteins. The formation of endogenous protein aggregates during the late stationary phase in E. coli cells was also influenced by acetylation. In general, decreased acetylation postponed the formation of endogenous and recombinant protein aggregates, whereas decreased deacetylation stabilized the aggregates. We also found that non-acetylated IBs exhibited significantly higher biological activity than their acetylated counterparts. These findings can be useful for the optimization of the production of recombinant proteins in E. coli.
Methods
Strains and growth conditions
BW25113 [F−, Δ(araD-araB)567, lacZ4787(del)::rrnB-3, LAM−, rph-1, Δ(rhaD-rhaB)568, hsdR514], as a wild-type strain, and its derivatives BW25113 ΔcobB (Keio collection) and BW25113 ΔackA-pta were used in this study. The l Red system [39] was used for obtaining ΔackA-pta mutant strain. The presence of the deletion was confirmed by PCR. For the overproduction of VP1GFP, the strains were transformed with pTVP1GFP plasmid [22]. The strains were grown at 37 °C in LB medium with agitation (200 rpm.). The strains transformed with the pTVP1GFP plasmid were grown in LB medium supplemented with 100 mg/L ampicillin. To induce overproduction of VP1GFP, isopropyl β-d-thiogalactoside (IPTG) (1 mM) was added to the exponentially growing cultures. To arrest protein synthesis, cultures were treated with 200 mg/L chloramphenicol. Endogenous protein aggregates were isolated from bacterial cultures grown in LB supplemented with 0.4% glucose.
Isolation of IBs and endogenous protein aggregates
IBs were isolated with CelLytic B, a bacterial cell lysis extraction reagent (Sigma), according to the supplied protocol. For recording GFP fluorescence and fluorescence microscopy, IBs were resuspended in phosphate-buffered saline (PBS).
To isolate endogenous protein aggregates from stationary E. coli cultures, cells were pelleted, converted into spheroplasts, and lysed by sonication as described previously [19]. After removal of unbroken cells by centrifugation (15 min, 2000×g), the supernatant was incubated with 2% Triton X-100 for 15 min at room temperature. Subsequently, insoluble aggregates were pelleted after 30 min of centrifugation at 21,000×g and the remaining supernatant was collected as soluble fraction. Endogenous protein aggregates and whole cell extracts were resolved by SDS-PAGE and analyzed by densitometry to estimate the amounts of aggregated proteins in relation to the total protein content in whole cell extracts (set to 100%).
SDS-PAGE and western blotting
SDS-PAGE and Western blotting were performed according to Sambrook et al. [40]. Polyclonal rabbit antisera against ClpB, DnaK, DnaJ, GroEL, IbpA/B [29], GFP (Sigma), anti-rabbit peroxidase conjugate (Sigma), and substrates such as 4-chloro-1-naphtol and H2O2 (Sigma) were used for immunodetection. Acetylated proteins were detected using anti-acetyl-lysine antibodies (Abcam), anti-rabbit peroxidase conjugate (Sigma), and Clarity Western ECL Substrate (Bio-Rad).
Fluorescence determination
Fluorescence of VP1GFP was recorded in PerkinElmer EnSpire plate reader (excitation, 450 nm; emission, 510 nm). Appropriately diluted whole-cell extracts and soluble fractions and IBs resuspended in PBS were used for these measurements. To determine the specific fluorescence of VP1GFP, amount of VP1GFP protein in IBs, extracts and soluble fractions were estimated after SDS-PAGE and immunodetection.
Epifluorescence micrographs of IBs were taken by using a Zeiss Axio microscope at a spatial resolution of 0.2 µm (excitation wavelength at 450–490 nm and emission wavelength at 515 nm).
Authors’ contributions
DKW and EL designed the study. DKW, MMA and KSS performed most experiments and analyzed the results. EL analyzed the results and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Professor Antonio Villaverde for the generous gift of the plasmid pTVP1GFP and Professor Barbara Lipińska for helpful discussion.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated and analyzed during this study are included in this article and its Additional file.
Funding
This work was supported by the National Science Centre (Grant 2011/03/B/NZ1/01206).
Abbreviations
- AcP
acetyl phosphate
- AU
arbitrary units
- IBs
inclusion bodies
- Hsps
heat shock proteins
Additional file
Additional file 1: Figure S1. Hot-spot area plot predicted by Aggrescan software [27] for VP1GFP. New aggregation hot-spot areas were created in VP1GFP variants in which lysine residues were replaced by hydrophobic aminoacids (F, I or V) to mimic lysine acetylation.
Contributor Information
Dorota Kuczyńska-Wiśnik, Email: dorota.kuczynska@biol.ug.edu.pl.
María Moruno-Algara, Email: mma@biol.ug.edu.pl.
Karolina Stojowska-Swędrzyńska, Email: karolina.stojowska@biol.ug.edu.pl.
Ewa Laskowska, Email: ewa.laskowska@biol.ug.edu.pl.
References
- 1.Rosano GL, Ceccarelli EA. Recombinant protein expression in microbial systems. Front Microbiol. 2014;5:341. doi: 10.3389/fmicb.2014.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlieker C, Bukau B, Mogk A. Prevention and reversion of protein aggregation by molecular chaperones in the E. coli cytosol: implications for their applicability in biotechnology. J Biotechnol. 2002;96:13–21. doi: 10.1016/S0168-1656(02)00033-0. [DOI] [PubMed] [Google Scholar]
- 3.de Marco A, Deuerling E, Mogk A, Tomoyasu T, Bukau B. Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol. 2007;7:32. doi: 10.1186/1472-6750-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuczyńska-Wiśnik D, Żurawa-Janicka D, Narkiewicz J, Kwiatkowska J, Lipińska B, Laskowska E. Escherichia coli small heat shock proteins IbpA/B enhance activity of enzymes sequestered in inclusion bodies. Acta Biochim Polonica. 2004;51:925–931. [PubMed] [Google Scholar]
- 5.García-Fruitós E, Arís A, Villaverde A. Localization of functional polypeptides in bacterial inclusion bodies. Appl Environ Microbiol. 2007;73:289–294. doi: 10.1128/AEM.01952-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martínez-Alonso M, González-Montalbán N, García-Fruitós E, Villaverde A. Learning about protein solubility from bacterial inclusion bodies. Microb Cell Fact. 2009;8:4. doi: 10.1186/1475-2859-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hrabárová E, Achbergerová L, Nahálka J. Insoluble protein applications: the use of bacterial inclusion bodies as biocatalysts. Methods Mol Biol. 2015;1258:411–422. doi: 10.1007/978-1-4939-2205-5_24. [DOI] [PubMed] [Google Scholar]
- 8.Wolfe AJ. The acetate switch. Microbiol Mol Biol Rev. 2005;69:12–50. doi: 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roe AJ, O’Byrne C, McLaggan D, Booth IR. Inhibition of Escherichia coli growth by acetic acid: a problem with methionine biosynthesis and homocysteine toxicity. Microbiology. 2002;148:2215–2222. doi: 10.1099/00221287-148-7-2215. [DOI] [PubMed] [Google Scholar]
- 10.Eiteman MA, Altman E. Overcoming acetate in Escherichia coli recombinant protein fermentations. Trends Biotechnol. 2006;24:530–536. doi: 10.1016/j.tibtech.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Kim JY, Cha HJ. Down-regulation of acetate pathway through antisense strategy in Escherichia coli: improved foreign protein production. Biotechnol Bioeng. 2003;83:841–853. doi: 10.1002/bit.10735. [DOI] [PubMed] [Google Scholar]
- 12.Kim TS, Jung HM, Kim SY, Zhang L, Li J, Sigdel S, Park JH, Haw JR, Lee JK. Reduction of acetate and lactate contributed to enhancement of a recombinant protein production in E. coli BL21. J Microbiol Biotechnol. 2015;25:1093–1100. doi: 10.4014/jmb.1503.03023. [DOI] [PubMed] [Google Scholar]
- 13.Mizrahi I, Biran D, Ron EZ. Involvement of the Pta-AckA pathway in protein folding and aggregation. Res Microbiol. 2009;160:80–84. doi: 10.1016/j.resmic.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Mizrahi I, Biran D, Ron EZ. Requirement for the acetyl phosphate pathway in Escherichia coli ATP-dependent proteolysis. Mol Microbiol. 2006;62:201–211. doi: 10.1111/j.1365-2958.2006.05360.x. [DOI] [PubMed] [Google Scholar]
- 15.Weinert BT, Iesmantavicius V, Wagner SA, Schölz C, Gummesson B, Beli P, Nyström T, Choudhary C. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol Cell. 2013;51:265–272. doi: 10.1016/j.molcel.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, Minasov G, Lima BP, Scholle M, Mrksich M, Anderson WF, Gibson BW, Schilling B, Wolfe AJ. Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS ONE. 2014;9(4):e94816. doi: 10.1371/journal.pone.0094816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.AbouElfetouh A, Kuhn ML, Hu LI, Scholle MD, Sorensen DJ, Sahu AK, Becher D, Antelmann H, Mrksich M, Anderson WF, Gibson BW, Schilling B, Wolfe AJ. The E. coli sirtuin CobB shows no preference for enzymatic and nonenzymatic lysine acetylation substrate sites. Microbiology. 2015;4:66–83. doi: 10.1002/mbo3.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327(5968):1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leszczynska D, Matuszewska E, Kuczynska-Wisnik D, Furmanek-Blaszk B, Laskowska E. The formation of persister cells in stationary-phase cultures of Escherichia coli is associated with the aggregation of endogenous proteins. PLoS ONE. 2013;8(1):e54737. doi: 10.1371/journal.pone.0054737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morshedi D, Ebrahim-Habibi A, Moosavi-Movahedi AA, Nemat-Gorgani M. Chemical modification of lysine residues in lysozyme may dramatically influence its amyloid fibrillation. Biochim Biophys Acta. 2010;1804:714–722. doi: 10.1016/j.bbapap.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 21.Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.García-Fruitós E, González-Montalbán N, Morell M, Vera A, Ferraz RM, Arís A, Ventura S, Villaverde A. Aggregation as bacterial inclusion bodies does not imply inactivation of enzymes and fluorescent proteins. Microb Cell Fact. 2005;4:27. doi: 10.1186/1475-2859-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belli M, Ramazzotti M, Chiti F. Prediction of amyloid aggregation in vivo. EMBO Rep. 2011;12:657–663. doi: 10.1038/embor.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slonczewski JL, Fujisawa M, Dopson M, Krulwich TA. Cytoplasmic pH measurement and homeostasis in bacteria and archaea. Adv Microb Physiol. 2009;55:1–79. doi: 10.1016/S0065-2911(09)05501-5. [DOI] [PubMed] [Google Scholar]
- 25.Chiti F, Calamai M, Taddei N, Stefani M, Ramponi G, Dobson CM. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16419–16426. doi: 10.1073/pnas.212527999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawrence MS, Phillips KJ, Liu DR. Supercharging proteins can impart unusual resilience. J Am Chem Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conchillo-Solé O, de Groot NS, Avilés FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007;8:65. doi: 10.1186/1471-2105-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwiatkowska J, Matuszewska E, Kuczyńska-Wiśnik D, Laskowska E. Aggregation of Escherichia coli proteins during stationary phase depends on glucose and oxygen availability. Res Microbiol. 2008;159:651–657. doi: 10.1016/j.resmic.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 29.Kuczyńska-Wiśnik D, Kedzierska S, Matuszewska E, Lund P, Taylor A, Lipińska B, Laskowska E. The Escherichia coli small heat-shock proteins IbpA and IbpB prevent the aggregation of endogenous proteins denatured in vivo during extreme heat shock. Microbiology. 2002;148:1757–1765. doi: 10.1099/00221287-148-6-1757. [DOI] [PubMed] [Google Scholar]
- 30.Mogk A, Deuerling E, Vorderwülbecke S, Vierling E, Bukau B. Small heat shock proteins, ClpB and the DnaK system form a functional triade in reversing protein aggregation. Mol Microbiol. 2003;50:585–595. doi: 10.1046/j.1365-2958.2003.03710.x. [DOI] [PubMed] [Google Scholar]
- 31.Lethanh H, Neubauer P, Hoffmann F. The small heat-shock proteins IbpA and IbpB reduce the stress load of recombinant Escherichia coli and delay degradation of inclusion bodies. Microb Cell Fact. 2005;11(4):6. doi: 10.1186/1475-2859-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.García-Fruitós E, Martínez-Alonso M, Gonzàlez-Montalbán N, Valli M, Mattanovich D, Villaverde A. Divergent genetic control of protein solubility and conformational quality in Escherichia coli. J Mol Biol. 2007;374:195–205. doi: 10.1016/j.jmb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Hoffmann F, Rinas U. Stress induced by recombinant protein production in Escherichia coli. Adv Biochem Eng Biotechnol. 2004;89:73–92. doi: 10.1007/b93994. [DOI] [PubMed] [Google Scholar]
- 34.Ma Q, Wood TK. Protein acetylation in prokaryotes increases stress resistance. Biochem Biophys Res Commun. 2011;410:846–851. doi: 10.1016/j.bbrc.2011.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bednarska NG, Schymkowitz J, Rousseau F, Van Eldere J. Protein aggregation in bacteria: the thin boundary between functionality and toxicity. Microbiology. 2013;159:1795–1806. doi: 10.1099/mic.0.069575-0. [DOI] [PubMed] [Google Scholar]
- 36.Tu S, Guo SJ, Chen CS, Liu CX, Jiang HW, Ge F, Deng JY, Zhou YM, Czajkowsky DM, Li Y, Qi BR, Ahn YH, Cole PA, Zhu H, Tao SC. YcgC represents a new protein deacetylase family in prokaryotes. Elife. 2015;4:e05322. doi: 10.7554/eLife.05322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lima BP, Lennon CW, Ross W, Gourse RL, Wolfe AJ. In vitro evidence that RNA Polymerase acetylation and acetyl phosphate-dependent CpxR phosphorylation affect cpxP transcription regulation. FEMS Microbiol Lett. 2016;363(5):fnw011. doi: 10.1093/femsle/fnw011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castaño-Cerezo S, Bernal V, Post H, Fuhrer T, Cappadona S, Sánchez-Díaz NC, Sauer U, Heck AJ, Altelaar AF, Cánovas M. Protein acetylation affects acetate metabolism, motility and acid stress response in Escherichia coli. Mol Syst Biol. 2014;10:762. doi: 10.15252/msb.20145227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sambrook J, Fritsh EF, Maniatis F. Molecular cloning: a laboratory manual. 2. New York: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated and analyzed during this study are included in this article and its Additional file.