

Abstract
Transgenic mice expressing a tamoxifen-inducible Cre recombinase specifically in cardiomyocytes were generated in 2001 and are in widespread use, having been employed in >150 published studies. However, several groups recently have reported that tamoxifen administration to these mice can have off-target effects that include cardiac dysfunction, fibrosis, and death. For this reason, among others, we considered it important to better characterize the transgene (termed herein, CM-MCM) and its chromosomal location(s). Cytogenetic analysis positioned the CM-MCM transgene within the C band of chromosome 19, and more precise mapping, using genome walking and DNA sequencing, showed that transgene insertion is in the C1 region. Using the genome walking data, we have developed PCR assays that not only identify mice that carry the transgene, but also distinguish homozygous animals (CM-MCMTg/Tg) from hemizygous (CM-MCMTg/0), permitting the rapid assessment of transgene zygosity and, thereby, helping to minimize off-target tamoxifen-induced effects. Substantial rearrangement / duplication of transgene elements is present, and transgene integration was accompanied by the deletion of a 19,500 bp fragment of genomic DNA that contains the promoter, exon 1 and part of intron 1 of the APOBEC1 complementation factor (A1cf) gene, as well as several elements that are predicted to regulate chromosomal architecture. A1cf protein expression is ablated by the deletion and, therefore, homozygous mice are functionally A1cf knockout. The implications of this unexpected finding are discussed.
Keywords: cardiomyocytes, heart, tamoxifen, MerCreMer, A1CF
INTRODUCTION
In 2001, Sohal et al. described mice bearing a transgene that encoded a modified Cre recombinase (MerCreMer; MCM) in which Cre was flanked by copies of a modified estrogen receptor (Mer) that is insensitive to its normal ligand, but sensitive to tamoxifen; drug injection leads to nuclear translocation of the MCM protein, permitting the enzymatic activity of Cre to act on loxP sequences in nuclear DNA. Transgene transcription was driven by the promoter sequences of the alpha myosin heavy chain (Myh6) locus; MCM expression was, therefore, strictly limited to cardiomyocytes (Sohal et al., 2001). These two attributes – cardiomyocyte-specific expression and inducible enzymatic activity – together conferred the ability to delete a “floxed” sequence only in cardiomyocytes, and at a time selected by the investigator. In the ensuing years, these mice have been used by many groups – including our own (Althof et al., 2014) – to disable a broad variety of genes in cardiomyocytes, thereby determining their importance to cellular and organismal function. The original paper has been cited more than 300 times, and >150 published articles have employed these mice (Mouse genome informatics, 2015). However, to date, the transgene insertion has not been well-characterized.
For several reasons, we considered it worthwhile to identify the location of the transgene, and to better characterize it. First, several reports have shown that, in this mouse line, excessive Cre activity can have negative effects that include transient myocardial dysfunction, myocardial fibrosis, and even death (Bersell et al., 2013; Hall et al., 2011; Hougen et al., 2010; Koitabashi et al., 2009; Lexow et al., 2013; Molkentin and Robbins, 2009). The general approach to minimizing these apparent off-target effects has been to use a single injection of tamoxifen, at as low a dose as possible while still achieving widespread knockout of the target gene in cardiomyocytes. However, the copy number of the inserted transgene is likely to affect Cre activity and, therefore, the extent to which these undesirable effects occur, so it is useful to be able to rapidly distinguish mice carrying two copies (homozygous, CM-MCMTg/Tg) from those carrying a single copy (hemizygous, CM-MCMTg/0). Second, one would anticipate that MCM gene dosage also would impact the efficiency of tamoxifen-induced deletion of the floxed target DNA. Thus, to ensure uniformity within “MCM+” experimental groups, animals should be characterized not merely by screening for MCM, but also by screening for zygosity. Third, the location at which the MCM transgene is inserted may have either physically interrupted a gene, or disrupted regulation of a nearby gene, possibly contributing to the tamoxifen-sensitivity that has been observed in this mouse line. In vitro studies have shown that some of the aspects of tamoxifen-related cardiomyocyte toxicity are observed following transduction of primary cardiomyocytes with Cre-expressing plasmids (Bersell et al., 2013), indicating that some of the deleterious effects do not absolutely require insertion of a transgene into the genome. However, if transgene insertion in this mouse line has disrupted local or global gene expression, this might influence cardiomyocyte function and thereby exacerbate the in vivo impact of tamoxifen. Fourth, when breeding mouse lines, the possibility of operator error cannot be discounted. For example, if homozygous mice were bred against hemizygous, instead of against wt, this error would not be revealed by the standard PCR recommended by JAX, which would merely identify all offspring as MCM+; consequently, the investigator would consider all the F1 mice to be hemizygous, and the fact that ~50% of the offspring were homozygous would go unnoticed.
Several techniques can be employed to assess the genotype of transgenic mice. Progeny analysis is the classical approach to determine parental zygosity and, using those data, relatively reliable breeding strategies can be developed. For lines in which transgene homozygosity is deleterious, breeders must be hemizygous, in which case ~75% of offspring will be transgene-positive. However, standard PCR analysis (i.e., testing only for presence of absence of the transgene) cannot distinguish homozygous mice (~⅓rd of the transgene-positive F1s) from hemizygous (~⅔rds). These two transgene-positive groups could be distinguished using in situ hybridization (e.g., with fluorescent probes – FISH) or, potentially, by Southern blot analyses. However, both of these approaches are too laborious for routine progeny screening, and are better applied (in combination with other approaches) early in transgenesis, to characterize founder animals. qPCR has been used to measure transgene copy number, but interpretation becomes more challenging as the differences between samples diminishes, and the identification of small differences – such as the 2-fold difference required here – is problematic (Aldhous et al., 2010; Armour et al., 2007); it has been shown that a change in amplification efficiency as small as 4% can result in a 400% error in ΔCt calculation (Guescini et al., 2008). More recently, high-throughput sequencing has been applied to transgene mapping, but this approach often has a poor signal:noise ratio (Srivastava et al., 2014).
Perhaps the simplest way to determine transgene zygosity is by using a PCR assay that can determine if the chromosomal insertion site is intact (wt chromosome) or disrupted (transgenic chromosome). This requires the precise mapping of the transgene insertion site and, herein, we have taken two approaches to localize and characterize the CM-MCM transgene. First, FISH (in combination with spectral karyotyping, SKY), which provides an overview of copy number and approximate chromosomal location(s). Second, genome walking, to identify the precise insertion site. This allowed us to develop straightforward PCR assays that assess the status of the chromosomal site where the transgene is inserted; these assays clearly and reliably distinguish between the three possible genotypes (CM-MCMTg/Tg, CM-MCMTg/0, and wt) of this widely-used mouse line. Our studies identified rearrangements of the transgenic DNA and, unexpectedly, a deletion of a 19,500bp fragment of genomic DNA that results in the loss of expression of at least one host gene.
MATERIALS & METHODS
CM-MCM mice were obtained from JAX laboratories [JAX line 005657; B6.FVB(129)-Tg(Myh6-cre/Esr1*)1Jmk/J], and were maintained by interbreeding. C57BL/6J mice (originating from JAX line 000664) were obtained from the mouse breeding facility at The Scripps Research Institute. In all experiments involving mice, all applicable international, national, and institutional guidelines for the care and use of animals were followed. The majority of the analyses reported herein were carried out using DNA from a single male Cre+ CM-MCM mouse that was known to be hemizygous, because, when crossed with a wt C57BL/6J female, ~50% of the F1 progeny were Cre+; this frequency was true for F1s of both sexes, indicating that the transgene resided on an autosome. FISH / SKY was carried out by J. Koeman CG(ASCP)CM at the Van Andel Institute (Grand Rapids, MI), on a fee-for-service basis. Analyses were carried out as described (VanBrocklin et al., 2009) on twenty metaphase cells isolated from a spleen from a hemizygous male CM-MCM mouse. The FISH probe was prepared by nick-translation (with green dUTP; Abbott Molecular Inc., Des Plaines IL) of a plasmid carrying a 2936bp PCR fragment spanning the αMHC promoter (~879 bp), Mer (~989 bp), and Cre (~1068 bp). Template for genome walking was prepared from tail DNA of this mouse. Genome walking was carried out using the Universal GenomeWalker 2.0 kit from Clontech (Mountain View, CA), following the manufacturer’s instructions. In brief: Genomic DNA was prepared from a male CM-MCM transgenic mouse, and from a male C57BL/6J mouse (control). For both DNAs, five 2.5 μg aliquots were prepared, and each was subjected to complete digestion with one of the following five restriction enzymes: DraI, EcoRV, PvuII, StuI or XmnI. The resulting blunt-ended DNA fragments were then ligated with GenomeWalker adaptors. Primary PCR was performed using an adaptor primer (AP1) and a primer specific for a sequence within the transgene (gene-specific primer, GSP), using the conditions recommended by the manufacturer: (i) 94°C for 25 seconds then 72°C for 3 mins (7 cycles); (ii) 94°C for 25 seconds then 67°C for 3 mins (32 cycles); then (iii) 67°C for 7 minutes. The reaction products were diluted 1:50 and 1μl of this material was subjected to a second, nested, PCR with a different adaptor primer (AP2) in conjunction with a second GSP [94°C for 25 seconds then 72°C for 3 mins (5 cycles); 94°C for 25 seconds then 67°C for 3 mins (20 cycles); then 67°C for 7 minutes]. The products were separated by gel electrophoresis, and bands unique to the CM-MCM libraries were isolated, then sequenced using the relevant primers. In this way, nucleotide sequences were identified that extended ~800–1400bp from the GSP primers, representing a single step along the genome. This knowledge was exploited to design new gene-specific primers that bind close to the end of the newly-identified sequence, and these primers were used (together with the AP1 / AP2 primers) to continue the genome walk. This process was repeated until mouse chromosomal DNA was encountered. Western blot analyses: Protein extracts were prepared from the liver and heart, and were subjected to polyacrylamide gel electrophoresis. Following transfer to PVDF membrane, A1cf was detected using a polyclonal goat primary antibody followed by a donkey anti-goat secondary antibody (both from Santa Cruz Biotechnology, Dallas TX). A mouse primary antibody (EMD Millipore, San Diego, CA) and a donkey anti-mouse secondary antibody (GE Amersham, Piscataway, NJ) were used to identify GAPDH. The secondary antibodies carried horseradish peroxidase, and bands were visualized using the enhanced chemiluminescence kit (GE Amersham).
RESULTS AND DISCUSSION
Mice have 40 chromosomes (19 autosomal pairs, plus 2 sex chromosomes) and all but the Y chromosome are telocentric. SKY and FISH analysis of 20 metaphase spleen cells from a heterozygous CM-MCM mouse were carried out, and representative images are shown in Figure 1. All cells had normal karyotype, and FISH identified a single insertion site for the transgene, just beyond the center point of chromosome 19, in the 19C region; no signal was observed in cells from control (C57BL/6J) mice. Mouse chromosome 19 contains ~61,431,566 base pairs (NCBI accession: NC_000085 GPC_000000792), placing the probable insertion site at ~31 megabases from the centromere. To determine the precise insertion site, we undertook a genome walk.
Figure 1. FISH/SKY shows single transgene insertion site, on chromosome 19.
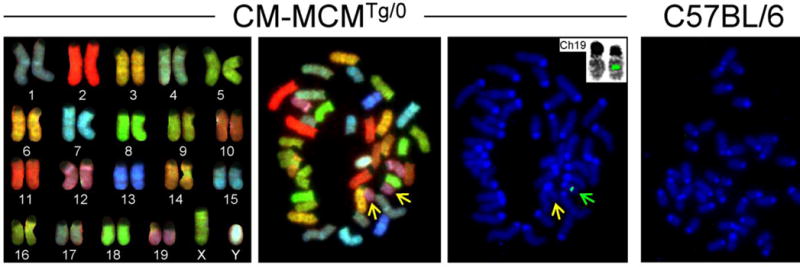
Representative metaphase images are shown, from spleen cells of male mice. For the CM-MCMTg/0 mouse, the left panel shows the spectral karyotypes of all chromosomes. The central panel shows that the cell has a normal karyotype. Both copies of chromosome 19 are indicated by yellow arrows. The right hand panel shows DAPI staining of DNA (blue) together with a FISH signal (green) that shows that the MCM transgene is located on chromosome 19 (green arrow). No FISH signal is present on the non-transgenic copy of this chromosome (yellow arrow), attesting to the specificity of the probe. The top-right inset (FISH + reverse DAPI) shows the two copies of chromosome 19 side by side: the FISH signal is just beyond the center point of the transgenic chromosome. The rightmost panel shows the result of FISH using a cell from a C57BL/6J mouse, and confirms the specificity of the FISH probe.
The sequences of the eight genome walking primers are tabulated in Figure 2. We chose to first walk in the “downstream” direction, to identify the junction between the 3′ end of the transgene and chromosomal DNA. The first GSP (GSP1) was designed to bind within the Cre sequence, close to the junction with the downstream Mer. We chose not to use a GSP within Mer, because that sequence is duplicated and could, potentially, have complicated data interpretation. Primary PCR was carried out using the GSP1 and AP1 primers, and the products of this reaction were used as templates for a second PCR using a nested adaptor primer (AP2) and a nested Cre-specific primer (GSP2). PCR bands were absent from the C57BL/6J library, as expected, but the DraI library of the CM-MCM DNA yielded a ~1580bp PCR product which was sequenced, revealing ~1040bp fragment comprising the C-terminus of Cre and the second copy of Mer. This was followed by a 60bp multiple cloning site, then ~480 bp sequence that was identified, by blast search, as a portion of the human growth hormone gene; this motif, which was not described by Sohal et al., is commonly used as a transcriptional terminator (HGHt). Thus, the CM-MCM transgene has three components: αMHC promoter, MCM coding region, and HGHt terminator. Mouse chromosomal sequences had not been identified during this first genome walking step, so two new GSPs were designed that were located in the HGHt sequence (GSP1A and GSP2A). Using the EcoRV libraries, a second genome walking step was taken using AP1 + GSP1A, followed by AP2 + GSP2A. PCR bands were again absent from the C57BL/6J library. In contrast, using the GSP2A / AP2 primers, the CM-MCM EcoRV library yielded two products, of ~1240bp and ~2000bp. From this, we deduced: (i) that the CM-MCM genome must contain at least two copies of the GSP2A sequence, which suggested that the HGHt region might be duplicated and (ii) EcoRV sites must exist both between the duplicate copies, and beyond the more distal copy. Sequencing of the two fragments using the GSP2A or AP2 primers revealed a number of transgene rearrangements and duplications. Following the first HGHt sequence there is a complex set of short duplicated regions from the αMHC promoter sequence (RptsProm in Figure 2). Five rearranged promoter tracts were identified, varying in length from 70–142 bp; one was in direct orientation, and the remainder were inverted. These promoter duplications / inversions were immediately followed by another duplication, encompassing the latter 321bp of Mer (RptMer), a multiple cloning sequence containing the predicted EcoRV site, and the predicted duplicated copy of HGHt, (RptHGHt). This was followed by another, different, multiple cloning site, then a short (60 bp) repeat from Mer that constitutes the 3′ terminus of the inserted transgene. To ensure that the observed duplications / rearrangements were not PCR artifacts generated during the second step of the genome walk, a separate nested PCR was carried out on the DNA libraries using AP1/AP2 primers along with new primers (MCMRPTS1 and MCMRPTS2) from the region that contained the promoter duplications. Bands of the predicted sizes were generated, and sequencing confirmed the existence of the various rearrangements and duplications that lie downstream of the CM-MCM-HGHt. Beyond the short (60bp) Mer repeat lay sequences that were shown, by blast search, to reside in mouse chromosome 19; the 3′ end of the CM-MCM transgene abuts chromosome 19 at bp 31,872,392. Next, using our existing genomic libraries, we attempted to locate the junction between chromosome 19 and the 5′ end of the transgene (i.e., the αMHC promoter). In so doing we identified, but did not fully characterize, a number of upstream transgene rearrangements and duplications; this less well-defined region is illustrated by a break symbol in Figure 2. Importantly, these upstream rearrangements do not appear to have diminished the cell specificity of transgene expression: Using these mice, we recently demonstrated (Althof et al., 2014) that a Cre reporter protein was expressed only in the heart and, moreover, only in cardiomyocytes (adjacent vascular cells were devoid of reporter protein). The high numbers of EcoRV and DraI sites in the transgenic DNA complicated our identification of the 5′ junction, leading us to prepare, from both C57BL/6J and CM-MCM DNA, new genomic libraries using XmnI. These libraries were subjected to nested PCR using AP1/AP2 primers with “antisense” GSPs close to the end of the αMHC promoter (PSP1 and P1P, Figure 2). A band of ~1150 bp, unique to the MCM DNA, was isolated and sequenced; this fragment contained the junctional sequence between chromosome 19 and the expected 5′ end of the αMHC promoter. Surprisingly, the final nucleotide of the adjacent chromosomal DNA lay at bp 31,852,891, some 19,500 bp upstream of the 3′ chromosomal insertion site, suggesting that transgene insertion had been accompanied by a large chromosomal deletion. Taken together, the observations of (i) significant rearrangement / duplication of transgenic DNA and (ii) concomitant deletion of a large genome segment suggest that transgene insertion may not have occurred by the relatively ordered method of homologous recombination, but rather by chromothripsis. This process, which involves local DNA “shattering” and reorganization, was first identified (and named) in studies of cancer cells (Stephens et al., 2011), but also has been invoked to explain some instances of transgene insertion in animals (Chiang et al., 2012).
Figure 2. Precise localization of the CM-MCM transgene on chromosome 19.
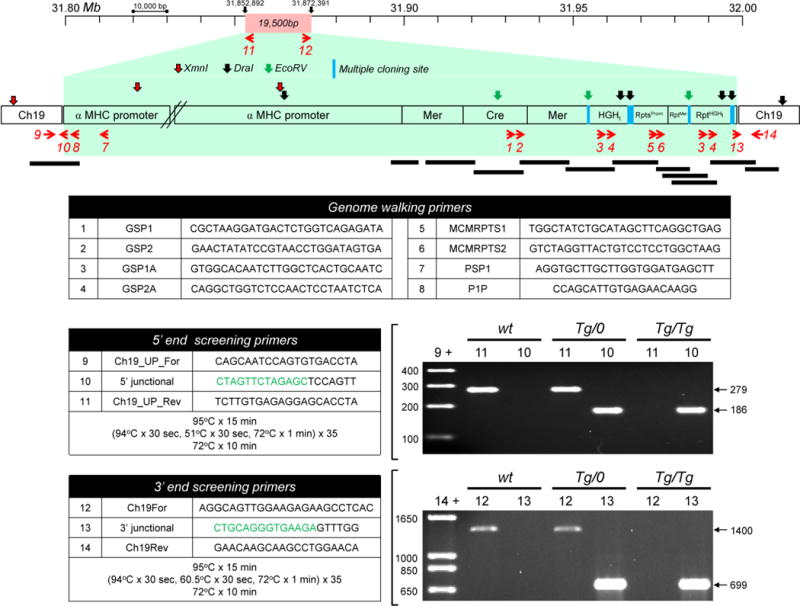
A 200,000bp fragment of wt mouse chromosome 19 is shown. The red rectangle at the top of the figure illustrates the 19,500 bp DNA fragment that is absent from a transgene-carrying copy of the chromosome. The general structure of the MCM transgene is enclosed in a green box, and the locations of XmnI, DraI and EcoRv sites are indicated by colored arrows. Some of the repeated motifs described in the text are shown (RptsProm, RptMer, RptHGHt), and regions of DNA that have been sequenced during these studies are illustrated by black bars. The upstream (promoter) region of the transgenic DNA has not been extensively sequenced, but it appears to contain additional duplications and rearrangements that are illustrated by the break symbol in this diagram. As stated in the text, this does not detrimentally affect the cardiomyocyte-specificity of MCM expression. Red arrows indicate the PCR primers that were used in genome walking, and in screening the DNA at the 5′ and 3′ transgene insertion sites. Sequences of all primers are tabulated, written 5′ to 3′. The “junctional” primers (#10 & #13) span either the 5′ or the 3′ intersection between chromosome 19 and the transgene. For each of the junctional primers, the green nucleotides are complementary to the end of the transgene insert, and the black bases are complementary to the first few bases of the adjacent chromosomal DNA. Two PCR panels are shown, with their reaction conditions outlined in the adjacent table. The data demonstrate that both the 5′ and 3′ junctional screening strategies can clearly distinguish among the three possible genotypes [wt, hemizygous (CM-MCMTg/0) and homozygous (CM-MCMTg/Tg)].
Identification of the 5′ and 3′ junctions allowed us to design two PCR strategies, one to probe the 5′ junctional DNA, and the other to evaluate the region around the 3′ junction. Each of the assays can detect both the wt and Tg-bearing versions of chromosome 19, thereby differentiating among the three possible genotypes (CM-MCMTg/Tg, CM-MCMTg/0, and wt) of this widely-used mouse line. For both assays, three primers were designed: one in the chromosomal DNA close to the transgene insertion site, one that spans the chromosome/transgene junction, and one that lies within the genomic DNA fragment that is deleted by transgene insertion. As shown in Figure 2, both assays provide clear readouts of mouse genotype. The 5′ assay (primers 9+11 or 9+10) generates bands of 279 bp (wt chromosome 19) or 186 bp (transgenic chromosome 19), while the 3′ assay (primers 14+12 or 14+13) yields bands of 1400bp (wt chromosome 19) or 699 bp (transgenic chromosome 19). The results of both of these PCR reactions are shown in Figure 2, for C57BL/6J mice, and for two MCM mice from our colony: one CM-MCMTg/0, and one CM-MCMTg/Tg. Note that, when hemizygous parents are crossed, the F1 progeny that score negative for the chromosome 19 transgene insertion using our assays are invariably also PCR-negative when screened using standard Cre primers, confirming that, as indicated by FISH / SKY, additional MCM insertions are not present at other chromosomal locations.
The study was undertaken primarily to identify the genomic location of the transgene, allowing us to develop the above PCR assays to determine zygosity. However, having unexpectedly identified a large genomic deletion, we have carried out a limited analysis thereof. In wt chromosome 19, this DNA fragment contains the promoter, the first exon, and part of the first intron of the gene encoding APOBEC1 complementation factor (A1cf; Ensembl genome browser ENSMUSG00000052595). In addition, several other computer-predicted regulatory sites are present in the 19,500bp fragment. These key sequences are listed in Table 1, and include an enhancer region of unknown function, as well as four predicted binding sites for CCCTC-binding factor (CTCF, see Lobanenkov et al., 1990), a protein that regulates three-dimensional genome architecture and plays a part in controlling transcriptional and post-transcriptional events (Ong and Corces, 2014). A fifth predicted CTCF binding site lies 9 bp downstream of the 3′ end of the transgene (Ensembl genome regulatory feature ENSMUSR00000586260, Ch19: 31,872,401–31,872,800). The 5′ and 3′ junctional analyses (Figure 2) show clearly that the termini of the 19,500 bp fragment are absent from the genome of Tg/Tg mice, so we next used PCR to probe for sequences that are contained within the body of the 19,500 bp fragment. We chose to focus on three of the sequences that are listed in Table 1, which (i) are of predicted biological relevance and (ii) are distributed approximately equidistantly within the deleted DNA, thereby spanning the 19,500 bp fragment. Primers were designed to probe for CTCF binding sites #1 and #4; and for the A1cf promoter / exon 1. As shown in Figure 3, all three sites are present in the genomes of wt mice and of Tg/0 mice; and all three are absent from genomic DNA extracted from Tg/Tg animals. Because our CM-MCM mouse colony has been on site for several years, we considered it important to confirm that the genotype reported above also applied to mice currently available from JAX laboratories. To that end, a homozygous MCM mouse was purchased, and analyzed by PCR. The results (not shown) were identical to those obtained for our mouse colony, supporting the observation (Chiang et al., 2012) that transgenic DNA that has been rearranged by chromothripsis is thereafter passed stably on to subsequent generations.
Table 1.
Sequences in the deleted 19,500 bp DNA fragment whose absence may alter local and/or global gene expression in Tg/Tg mice
| Ensemble reference # | Proposed function | bp position in C57BL/6J ch19 |
|---|---|---|
| ENSMUSR00000586254 | CTCF binding site #1 | 31,855,601–31,856,000 |
| ENSMUSR00000461760 | CTCF binding site #2 | 31,860,201–31,860,800 |
| ENSMUSR00000586255 | CTCF binding site #3 | 31,861,801–31,862,000 |
| ENSMUSR00000586256 | ENHANCER | 31,862,001–31,862,200 |
| ENSMUSR00000586258 | CTCF binding site #4 | 31,862,801–31,863,200 |
| ENSMUSR00000586259 | A1cf promoter | 31,868,601–31,869,001 |
| ENSMUSE00000620078 | A1cf exon 1 | 31,868,761–31,868,859 |
Figure 3. The 19,500bp deletion includes sequences that regulate A1cf gene expression.
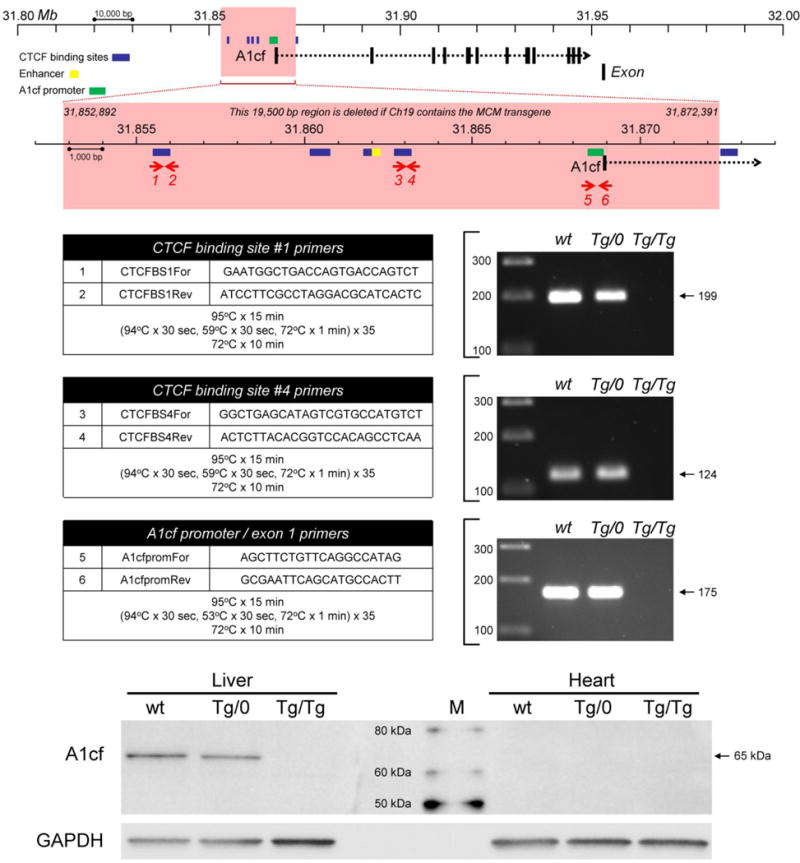
A 200,000bp fragment of chromosome 19 is shown. The 19,500bp fragment that is deleted in transgene-containing chromosome 19 is enclosed in a red box, together with its location with respect to the entire A1cf coding region. Computer-predicted key regulatory sequences that lie within the fragment are illustrated (four CTCF binding sites, dark blue boxes; an enhancer, yellow box), as well as sequences whose functions have been verified experimentally (the A1cf promoter, green box; exon 1 and part of intron 1). Three PCR reactions were carried out for three of these key sequences, and confirmed that all of them are absent from the genome of Tg/Tg animals. The primers and reaction conditions are tabulated for each PCR. A western blot (lower panels) of liver and heart extracts from mice of the indicated genotypes shows that expression of the 65 kDa A1cf protein is ablated by the genomic deletion reported herein. M = protein markers; GAPDH = loading control.
A1cf is a required cofactor for APOBEC1, an enzyme that edits the mRNA encoding apolipoprotein B. As such, A1cf is expressed in the same tissues as APOBEC1, but A1cf also is expressed in a number of tissues in which APOBEC1 appears to be absent, suggesting that A1cf may subserve additional, as yet undefined, roles (Mehta et al., 2000). Functional elements within the murine A1cf promoter have been mapped, and small deletions within the sequence appear to inactivate promoter activity, at least in tissue culture (Dur et al., 2004); thus, the complete ablation of the promoter sequence (in CM-MCMTg/Tg mice) would be expected to significantly disrupt gene expression, altering the abundance of the A1cf mRNA. Moreover, even if A1cf transcription were to be driven by an unidentified promoter, the primary RNA product would lack exon 1, and part of intron 1/2. Exon 1 is non-coding, i.e. it forms part of the 5′ untranslated region (5΄UTR) of the native A1cf mRNA, so its deletion should not directly affect the open reading frame of the mRNA. However, the observed deletion would prevent normal splicing, and would preclude the proper removal of intron 1/2 (which is 24,195 bases in length), resulting in an mRNA in which the wild type ATG codon (in the native exon 2) would be preceded by an abnormally-long 5′UTR, reducing the likelihood that the scanning ribosome would initiate translation at the native start codon. Therefore, the deletion reported herein could alter both the quantity and the quality of A1cf mRNA, thereby preventing, or substantially diminishing, expression of the A1cf protein. Others have reported that A1cf mRNA is highly-expressed in the liver, kidney and pancreas, but at much lower levels in several other tissues, including the heart (Mehta et al., 2000). To directly determine the impact of the observed deletion on A1cf protein levels, protein extracts were prepared from the liver and heart of three mice (C57BL/6J mice, CM-MCMTg/0 and CM-MCMTg/Tg), and were evaluated by western blot (Figure 3). When probed using an antibody specific for A1cf, a band of the expected size (65 kDa) was observed in the livers of wt mice and hemizygous mice, and their relative intensities are consistent with the predicted gene dosage (wt = 2 functional copies; hemizygous = single functional copy). In contrast, A1cf protein was undetectable in the liver of the CM-MCMTg/Tg mouse. Thus, as predicted, the observed deletion eradicates A1cf protein expression. The viability of CM-MCMTg/Tg mice indicates that A1cf protein expression is dispensable, a conclusion that contradicts the finding (Blanc et al., 2005) that targeted deletion of A1cf results in embryonic lethality. We have no explanation for this discrepancy, but our observations are supported by the fact that the International Mouse Phenotyping Consortium has recently reported that homozygous A1cf knockout mice are viable (http://www.mousephenotype.org/data/genes/MGI:1917115). In addition to having a local effect on gene expression (i.e., ablation of A1cf), we speculate that the deletion may have a more global impact on gene regulation throughout the 19C region, because chromosomal architecture and function may be altered in the absence of the predicted CTCF binding sites.
These findings raise an obvious question: might the functional deletion of A1cf modify the biological effect of Cre-induced gene deletion in cardiomyocytes? A1cf protein expression was difficult to detect in the hearts of any of the three mice analyzed (Figure 3), suggesting that this protein may not play a major role in cardiac function; this, in turn, implies that Cre-induced defects in the cardiomyocytes are, most probably, the direct consequence of deletion of the floxed target sequence. However, we cannot exclude the possibility that the absence of A1cf has some impact on studies in these mice. As noted above, A1cf is highly-expressed in the normal mouse liver, the organ in which many drugs – including tamoxifen (Rochat, 2005) – are metabolized by the cytochrome P450 enzyme family. Thus, it is possible that metabolism of the prodrug or its metabolites is aberrant in homozygous CM-MCM mice, potentially contributing to the vulnerability that these animals display to tamoxifen. Thus, while we believe that these mice remain a useful tool for assessing gene function in cardiomyocytes in vivo, our findings counsel caution when interpreting data obtained using this mouse line.
Acknowledgments
This work was supported by NIH grant R01 AI110621. This is manuscript number 29197 from the Scripps Research Institute.
References
- Aldhous MC, Abu BS, Prescott NJ, Palla R, Soo K, Mansfield JC, Mathew CG, Satsangi J, Armour JA. Measurement methods and accuracy in copy number variation: failure to replicate associations of beta-defensin copy number with Crohn’s disease. Hum Mol Genet. 2010;19:4930–4938. doi: 10.1093/hmg/ddq411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althof N, Harkins S, Kemball CC, Flynn CT, Alirezaei M, Whitton JL. In vivo ablation of type I interferon receptor from cardiomyocytes delays coxsackieviral clearance and accelerates myocardial disease. J Virol. 2014;88:5087–5099. doi: 10.1128/JVI.00184-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armour JA, Palla R, Zeeuwen PL, den HM, Schalkwijk J, Hollox EJ. Accurate, high-throughput typing of copy number variation using paralogue ratios from dispersed repeats. Nucleic Acids Res. 2007;35:e19. doi: 10.1093/nar/gkl1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersell K, Choudhury S, Mollova M, Polizzotti BD, Ganapathy B, Walsh S, Wadugu B, Arab S, Kuhn B. Moderate and high amounts of tamoxifen in αMHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis Model Mech. 2013;6:1459–1469. doi: 10.1242/dmm.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc V, Henderson JO, Newberry EP, Kennedy S, Luo J, Davidson NO. Targeted deletion of the murine apobec-1 complementation factor (acf) gene results in embryonic lethality. Mol Cell Biol. 2005;25:7260–7269. doi: 10.1128/MCB.25.16.7260-7269.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR, McLaughlan CJ, Bawden CS, Reid SJ, Faull RL, Snell RG, Hall IM, Shen Y, Ohsumi TK, Borowsky ML, Daly MJ, Lee C, Morton CC, MacDonald ME, Gusella JF, Talkowski ME. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet. 2012;44:390–7. doi: 10.1038/ng.2202. S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dur S, Krause K, Pluntke N, Greeve J. Gene structure and expression of the mouse APOBEC-1 complementation factor: multiple transcriptional initiation sites and a spliced variant with a premature stop translation codon. Biochim Biophys Acta. 2004;1680:11–23. doi: 10.1016/j.bbaexp.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Guescini M, Sisti D, Rocchi MB, Stocchi L, Stocchi V. A new real-time PCR method to overcome significant quantitative inaccuracy due to slight amplification inhibition. BMC Bioinformatics. 2008;9:326. doi: 10.1186/1471-2105-9-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ME, Smith G, Hall JE, Stec DE. Systolic dysfunction in cardiac-specific ligand-inducible MerCreMer transgenic mice. Am J Physiol Heart Circ Physiol. 2011;301:H253–H260. doi: 10.1152/ajpheart.00786.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougen K, Aronsen JM, Stokke MK, Enger U, Nygard S, Andersson KB, Christensen G, Sejersted OM, Sjaastad I. Cre-loxP DNA recombination is possible with only minimal unspecific transcriptional changes and without cardiomyopathy in Tg(αMHC-MerCreMer) mice. Am J Physiol Heart Circ Physiol. 2010;299:H1671–H1678. doi: 10.1152/ajpheart.01155.2009. [DOI] [PubMed] [Google Scholar]
- Koitabashi N, Bedja D, Zaiman AL, Pinto YM, Zhang M, Gabrielson KL, Takimoto E, Kass DA. Avoidance of Transient Cardiomyopathy in Cardiomyocyte-Targeted Tamoxifen-Induced MerCreMer Gene Deletion Models. Circ Res. 2009;105:12–15. doi: 10.1161/CIRCRESAHA.109.198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexow J, Poggioli T, Sarathchandra P, Santini MP, Rosenthal N. Cardiac fibrosis in mice expressing an inducible myocardial-specific Cre driver. Dis Model Mech. 2013;6:1470–1476. doi: 10.1242/dmm.010470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanenkov VV, Nicolas RH, Adler VV, Paterson H, Klenova EM, Polotskaja AV, Goodwin GH. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene. 1990;5:1743–1753. [PubMed] [Google Scholar]
- Mehta A, Kinter MT, Sherman NE, Driscoll DM. Molecular cloning of apobec-1 complementation factor, a novel RNA-binding protein involved in the editing of apolipoprotein B mRNA. Mol Cell Biol. 2000;20:1846–1854. doi: 10.1128/mcb.20.5.1846-1854.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Robbins J. With great power comes great responsibility: using mouse genetics to study cardiac hypertrophy and failure. J Mol Cell Cardiol. 2009;46:130–136. doi: 10.1016/j.yjmcc.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse genome informatics. References associated with the Tg(Myh6-cre/Esr1*)1Jmk allele. 2015 http://www.informatics.jax.org/reference/allele/MGI:3050453.
- Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochat B. Role of cytochrome P450 activity in the fate of anticancer agents and in drug resistance: focus on tamoxifen, paclitaxel and imatinib metabolism. Clin Pharmacokinet. 2005;44:349–366. doi: 10.2165/00003088-200544040-00002. [DOI] [PubMed] [Google Scholar]
- Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Philip VM, Greenstein I, Rowe LB, Barter M, Lutz C, Reinholdt LG. Discovery of transgene insertion sites by high throughput sequencing of mate pair libraries. BMC Genomics. 2014;15:367. doi: 10.1186/1471-2164-15-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, Varela I, Nik-Zainal S, Leroy C, Jia M, Menzies A, Butler AP, Teague JW, Quail MA, Burton J, Swerdlow H, Carter NP, Morsberger LA, Iacobuzio-Donahue C, Follows GA, Green AR, Flanagan AM, Stratton MR, Futreal PA, Campbell PJ. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBrocklin MW, Robinson JP, Whitwam T, Guilbeault AR, Koeman J, Swiatek PJ, Vande Woude GF, Khoury JD, Holmen SL. Met amplification and tumor progression in Cdkn2a-deficient melanocytes. Pigment Cell Melanoma Res. 2009;22:454–460. doi: 10.1111/j.1755-148X.2009.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]