

ABSTRACT
Herpes simplex virus 1 (HSV-1) envelope glycoprotein D (gD) plays an essential role in viral entry. The functional regions of gD responsible for viral entry have been mapped to its extracellular domain, whereas the gD cytoplasmic domain plays no obvious role in viral entry. Thus far, the role(s) of the gD cytoplasmic domain in HSV-1 replication has remained to be elucidated. In this study, we show that ectopic expression of gD induces microvillus-like tubular structures at the plasma membrane which resemble the reported projection structures of the plasma membrane induced in HSV-1-infected cells. Mutations in the arginine cluster (residues 365 to 367) in the gD cytoplasmic domain greatly reduced gD-induced plasma membrane remodeling. In agreement with this, the mutations in the arginine cluster in the gD cytoplasmic domain reduced the number of microvillus-like tubular structures at the plasma membrane in HSV-1-infected cells. In addition, the mutations produced an accumulation of unenveloped nucleocapsids in the cytoplasm and reduced viral replication and cell-cell spread. These results suggest that the arginine cluster in the gD cytoplasmic domain is required for the efficient induction of plasma membrane projections and viral final envelopment, and these functions of the gD domain may lead to efficient viral replication and cell-cell spread.
IMPORTANCE The cytoplasmic domain of HSV-1 gD, an envelope glycoprotein essential for viral entry, was reported to promote viral replication and cell-cell spread, but the role(s) of the domain during HSV-1 infection has remained unknown. In this study, we clarify two functions of the arginine cluster in the HSV-1 gD cytoplasmic domain, both of which require host cell membrane remodeling, i.e., the formation of microvillus-like projections at the plasma membrane and viral final envelopment in HSV-1-infected cells. We also show that the gD arginine cluster is required for efficient HSV-1 replication and cell-cell spread. This is the first report clarifying not only the functions of the gD cytoplasmic domain but also identifying the gD arginine cluster to be the HSV-1 factor responsible for the induction of plasma membrane projections in HSV-1-infected cells. Our results elucidate some of the functions of this multifunctional envelope glycoprotein during HSV-1 infection.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is classified in the Alphaherpesvirinae subfamily of the Herpesviridae family and is one of the best-studied members in the family (1). It is well-known that HSV-1 infection induces the deformation of various host cell membranes (2). In general, membrane deformation is necessary for the initiation of enveloped virus budding, in which infected cell membranes are deformed to wrap around nascent nucleocapsids. Herpesviruses acquire envelopes twice in their life cycle. Nascent progeny nucleocapsids become enveloped by budding through the inner nuclear membrane (INM) into the perinuclear space between the INM and the outer nuclear membrane (ONM), a process designated primary envelopment, and the enveloped nucleocapsids subsequently fuse with the ONM to release deenveloped nucleocapsids into the cytoplasm (2). In the cytoplasm, the nucleocapsids acquire final envelopes by budding into cytoplasmic membranes, likely those derived from the trans-Golgi network and/or endosomes, a process designated secondary envelopment (2). Therefore, herpesviruses induce the deformation of INMs and cytoplasmic membranes during primary and secondary envelopment, respectively. In addition, it has long been recognized that infection of cells with HSV-1, as well as other members of the alpha-, beta-, and gammaherpesviruses, generally induces projections of the plasma membrane (3–12). A heterodimer complex of the HSV-1 UL31 and UL34 proteins has been reported to be involved in the regulation of INM deformation during viral primary envelopment (13, 14). However, the factors that deform cytoplasmic membranes for secondary envelopment and induce plasma membrane projections in infected cells have not yet been identified.
HSV-1 envelope glycoprotein D (gD), the subject of this study, is essential for the virus-cell membrane fusion step in viral entry. Viral entry depends on gD binding to one of the HSV-1 host cell surface receptors, including (i) herpesvirus entry mediator (HVEM), a cell adhesion molecule in the immunoglobulin (Ig) superfamily; (ii) nectin-1 and nectin-2; and (iii) heparin sulfate (3-O-S HS) with specific modifications catalyzed by an isoform of 3-O-sulfotransferase (15). Binding of gD to its receptor activates HSV-1 gH/gL glycoproteins to bind to and activate the fusogenic activity of HSV-1 gB (16–18). However, it has also been reported that deletion of gD and either gE or gB produces a significant reduction in HSV-1 secondary envelopment (19, 20). The regions of gD required for viral entry have been mapped to its extracellular domain (15), and the transmembrane (TM) and cytoplasmic domains of gD have been reported to play no obvious roles in viral entry (21, 22). Recombinant viruses lacking the gD cytoplasmic domain have reduced progeny virus titers and plaque sizes in cell cultures, indicating that the domain is required for efficient HSV-1 replication and cell-cell spread (21–23). However, the mechanism(s) by which the gD cytoplasmic domain acts in HSV-1 replication and cell-cell spread has remained to be elucidated.
In this study, we attempted to clarify the function(s) of the gD cytoplasmic domain during HSV-1 replication. In particular, we focused on the arginine cluster in the gD cytoplasmic domain, since positively charged amino acid clusters in proteins have been reported to be involved in various protein functions, including binding to other proteins, membrane lipids, RNA, and DNA and regulation of subcellular localization (24–27). We report here that the arginine cluster in the gD cytoplasmic domain is required for the efficient formation of microvillus-like projections at the plasma membrane of HSV-1-infected cells and for viral secondary envelopment in HSV-1-infected cells.
MATERIALS AND METHODS
Cells and viruses.
Vero, HEK293T, and VD60 cells were described previously (28–30). Wild-type HSV-1(F) was described previously (31). The gD-deficient virus F-BAC gD− (Fig. 1) was kindly provided by D. Johnson and was described previously (20). In experiments with F-BAC gD−, VD60 cells were used to propagate and assay wild-type HSV-1(F), F-BAC gD−, and F-BAC gD−-repair.
FIG 1.
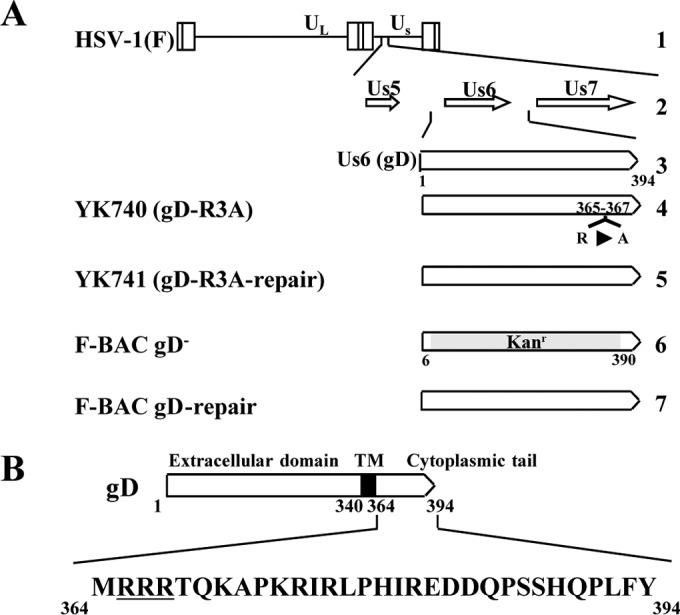
Schematic diagrams of the genome structure of wild-type HSV-1(F) and the relevant domains of the recombinant viruses used in this study. (A) Line 1, wild-type HSV-1(F) genome (UL and Us, unique long and unique short domains, respectively); line 2, domains of the Us5 to Us7 genes; line 3, domain of the Us6 (gD) gene; lines 4 to 7, recombinant viruses with mutations in the gD gene. (B) Amino acid sequence of the cytoplasmic domain of gD. The arginine cluster discussed in this paper is underlined.
Plasmids.
Plasmid pPEP99-gD was used for expression of HSV-1 envelope glycoprotein gD, as described previously (32). Plasmids pEGFP-gD and pEGFP-gG, carrying, respectively, a fusion protein of gD or gG and the enhanced green fluorescent protein (EGFP) (designated gD-EGFP and gG-EGFP, respectively), were constructed by amplifying and cloning the HSV-1 Us6 gene encoding gD and the HSV-1 Us4 gene encoding gG from pYEbac102 (33), containing a full-length infectious HSV-1(F) clone, into pEGFP-N1 (Clontech) in frame with EGFP. Plasmid pEGFP-gD-CTD, carrying a fusion protein of EGFP and the TM and cytoplasmic domains of gD (codons 340 to 394), was constructed by amplifying and cloning gD codons 340 to 394 into pEGFP-N1 in frame with EGFP. Plasmid pEGFP-gD-R3A was constructed by replacing gD codons R365, R366, and R367 in pEGFP-gD with alanine codons as described previously (34). Plasmid pEGFP-gD1–339-gB770–864 was constructed by replacing the TM and cytoplasmic domains of gD (codons 340 to 394) in pEGFP-gD with gB codons 770 to 864. Transfer plasmid pBS-gD-rep, which was used for generating recombinant F-BAC gD−-repair, in which the gD deletion in F-BAC gD− was repaired (Fig. 1), was constructed by amplifying and cloning the domain containing the gD open reading frame (ORF; the 385 bp upstream sequence flanking the gD start codon, the gD ORF, and the 429 bp downstream sequence flanking the gD stop codon) from pYEbac102 into pBluescript II KS(+) (Stratagene).
Mutagenesis of viral genomes and generation of recombinant HSV-1.
Recombinant virus YK740 (gD-R3A) containing gD in which the arginines at residues 365, 366, and 367 (R365, R366 and R367, respectively) were replaced by alanines (Fig. 1) was generated by the two-step bacteriophage lambda Red-mediated mutagenesis procedure (35, 36) using Escherichia coli GS1783 containing pYEbac102 and primers 5′-CTGGCAGCCCTGGTCATTTGCGGAATTGTGTACTGGATGGCCGCCGCCACTCAAAAAGCCAGGATGACGACGATAAGTAGGG-3′ and 5′-GGGAGGCGTATGCGCTTTGGGGCTTTTTGAGTGGCGGCGGCCATCCAGTACACAATTCCGCAACCAATTAACCAATTCTGATTAG-3′. Recombinant virus YK741, in which the gD-R3A mutation in YK740 was repaired (Fig. 1), was generated as described above, except that primers 5′-CTGGCAGCCCTGGTCATTTGCGGAATTGTGTACTGGATGGCCGCCGCCACTCAAAAAGCCAGGATGACGACGATAAGTAGGG-3′ and 5′-GGGAGGCGTATGCGCTTTGGGGCTTTTTGAGTGGCGGCGGCCATCCAGTACACAATTCCGCAACCAATTAACCAATTCTGATTAG-3′ were used. F-BAC gD−-repair (Fig. 1) was generated by transfection of HEK293T cells with pBS-gD-rep using polyethylenimine (37) and subsequent superinfection of the transfected cells with F-BAC gD− as described previously (34).
Antibodies and immunoblotting.
Mouse monoclonal antibodies to α-tubulin (antibody DM1A; Sigma), gD (antibody DL6; Santa Cruz Biotechnology), gB (antibody 1105, Goodwin Institute), and VP5 (antibody 3B6; Virusys) were used in this study. Rabbit polyclonal antibody to green fluorescent protein (GFP) was purchased from MBL. Rabbit polyclonal antibody to VP22 was described previously (30). Immunoblotting was performed as described previously (38).
Determination of plaque size.
Vero cells were infected with 100 PFU of each of the recombinant viruses. After adsorption for 1 h, the inoculum was removed and the cell monolayers were overlaid with medium 199 containing 1% fetal calf serum and 160 μg pooled human immunoglobulin (Sigma)/ml. At 2 days postinfection, 20 plaques produced by each of the recombinant viruses were analyzed using a microscope equipped with a digital DP80 camera (Olympus) and CellSens software (Olympus).
Virion purification.
Vero cells were infected with the viruses indicated below at a multiplicity of infection (MOI) of 0.1 for 48 h. Cell culture supernatants were harvested and clarified by low-speed centrifugation. The virus-containing supernatants were centrifuged at 22,000 rpm for 1 h in a P28S rotor (Hitachi). The pellets were resuspended and laid onto a discontinuous sucrose gradient (30, 40, and 50% [wt/vol] sucrose) at 22,000 rpm for 2 h in a P40ST rotor. The virions accumulating at the boundary between the 40 and 50% sucrose layers were collected, pelleted by centrifugation at 22,000 rpm for 1 h in a 40ST rotor, and subjected to immunoblotting.
Live cell imaging.
HEK293T and Vero cells cultured on 35-mm-diameter glass-bottom dishes (MatTek or Matsunami) were transfected with the plasmid expressing gD-EGFP or gG-EGFP using polyethylenimine, as described previously (37). The transfected cells were examined with an LSM5 or LSM800 inverted confocal microscope (Carl Zeiss) at 24 h posttransfection.
Immunofluorescence.
Vero cells were mock infected or infected with the viruses indicated below at an MOI of 5 and for the infection time noted below for each experiment, fixed with 4% formaldehyde in phosphate-buffered saline (PBS), permeabilized with 0.1% Triton X-100 in PBS, blocked with 10% human serum in PBS, reacted with anti-gD antibody, reacted with anti-mouse IgG-Alexa Flour 488 (Invitrogen), and examined with an LSM5 or LSM800 inverted confocal microscope (Carl Zeiss). For cell surface staining, infected cells were reacted with wheat germ agglutinin (WGA) conjugated with Alexa Fluor 555 (Thermo Fisher scientific), fixed with 4% formaldehyde in PBS, blocked as described above, reacted with anti-gD antibody, reacted with anti-mouse IgG-Alexa Flour 488, and examined with an LSM5 or LSM800 inverted confocal microscope (Carl Zeiss).
Electron microscopy.
HEK293T cells were transfected with a plasmid expressing gD or a control plasmid using the Lipofectamine 2000 reagent (Invitrogen) and grown for 48 h. Vero cells were infected with wild-type HSV-1(F), YK740 (gD-R3A), YK741 (gD-R3A-repair), F-BAC gD−, or F-BAC gD−-repair at an MOI of 5 and grown for 18 h. At the times indicated below for each experiment, cells were fixed, embedded, sectioned, strained with uranyl acetate and lead citrate, and examined by transmission electron microscopy as described previously (39).
RESULTS
Effect of gD on plasma membrane deformation.
To clarify the function(s) of HSV-1 gD, we investigated the effect(s) of ectopic expression of gD in transfected cells. For this study, HEK293T cells were transfected with the expression plasmid for EGFP or gD (Fig. 2A and B) and for gD-EGFP or gG-EFGP (Fig. 2C and D) and examined by electron microscopy. As shown in Fig. 2A and C, cells transfected with the gD or gD-EGFP expression plasmid showed the formation of microvillus-like tubular structures at the plasma membrane. In contrast, these tubular structures were rarely observed in cells transfected with the expression plasmid for EGFP or gG-EGFP (Fig. 2A and C). These plasma membrane projections were approximately 100 nm in diameter and 0.5 to 2 μm in length. Expression of gD, EGFP, gD-EGFP, and gG-EGFP in the transfected HEK293T cells was confirmed by immunoblotting (Fig. 2B and D).
FIG 2.
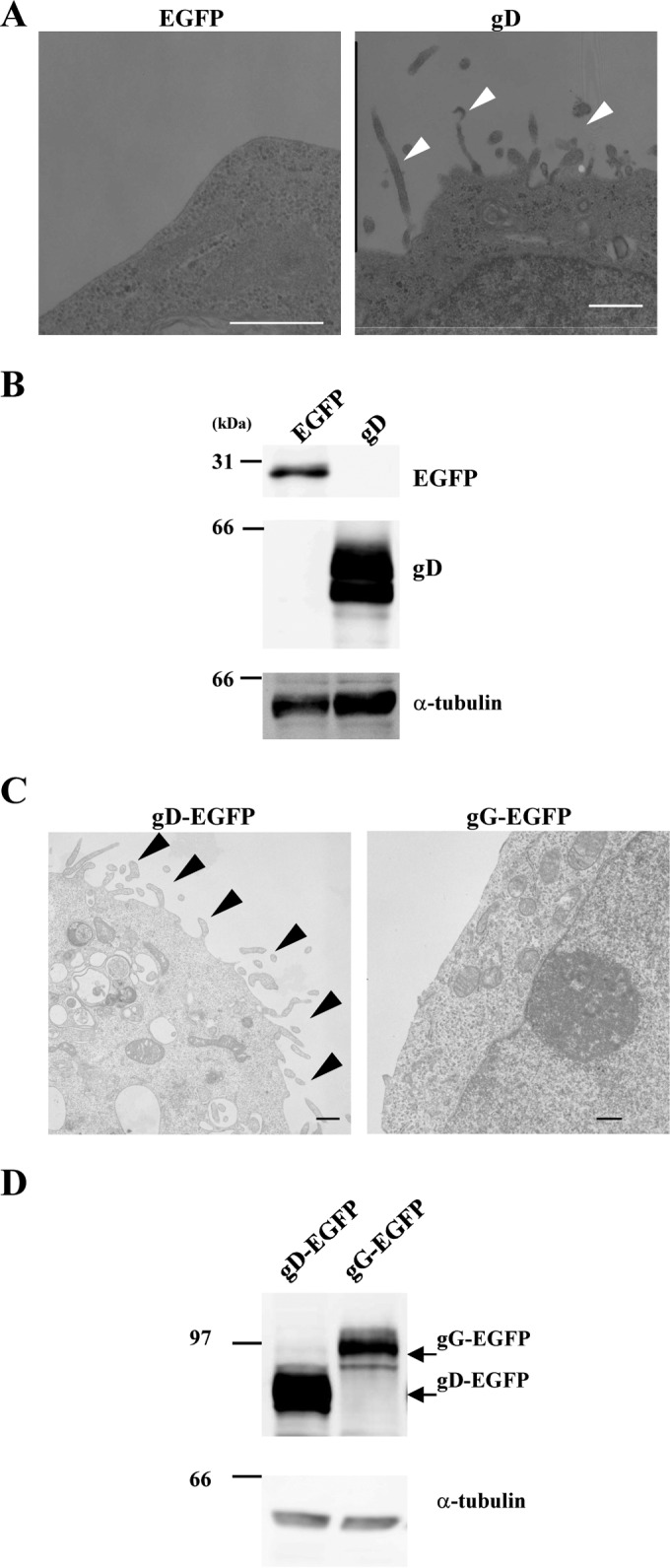
Ultrastructure of HEK293T cells transfected with the gD expression plasmids. HEK293T cells were transfected with the EGFP or gD expression plasmid (A and B) or the gD-EGFP or gG-EGFP expression plasmid (C and D). At 48 h posttransfection, the cells were analyzed by transmission electron microscopy (A and C) and immunoblotting with anti-gD, anti-GFP, and anti-α-tubulin antibodies (B and D). Arrowheads, microvillus-like projections at the plasma membrane. Bars, 500 nm.
In addition, HEK293T and Vero cells were transfected with the gD-EGFP or gG-EGFP expression plasmid and examined by fluorescence microscopy. As shown in Fig. 3A, gD-positive projection structures were induced at the plasma membrane in HEK293T cells transfected with the gD-EGFP expression plasmid, but gG-positive projection structures were barely detectable in cells transfected with the gG-EGFP expression plasmid (Fig. 3A and C). Similar results were obtained with Vero cells transfected with the gD-EGFP or gG-EGFP expression plasmid (Fig. 3B to D). These results are in agreement with the electron microscopy results, described above, indicating that projection structures were observed at the plasma membrane in cells transfected with the gD-EGFP expression plasmid but not in cells transfected with the gG-EGFP expression plasmid (Fig. 2).
FIG 3.
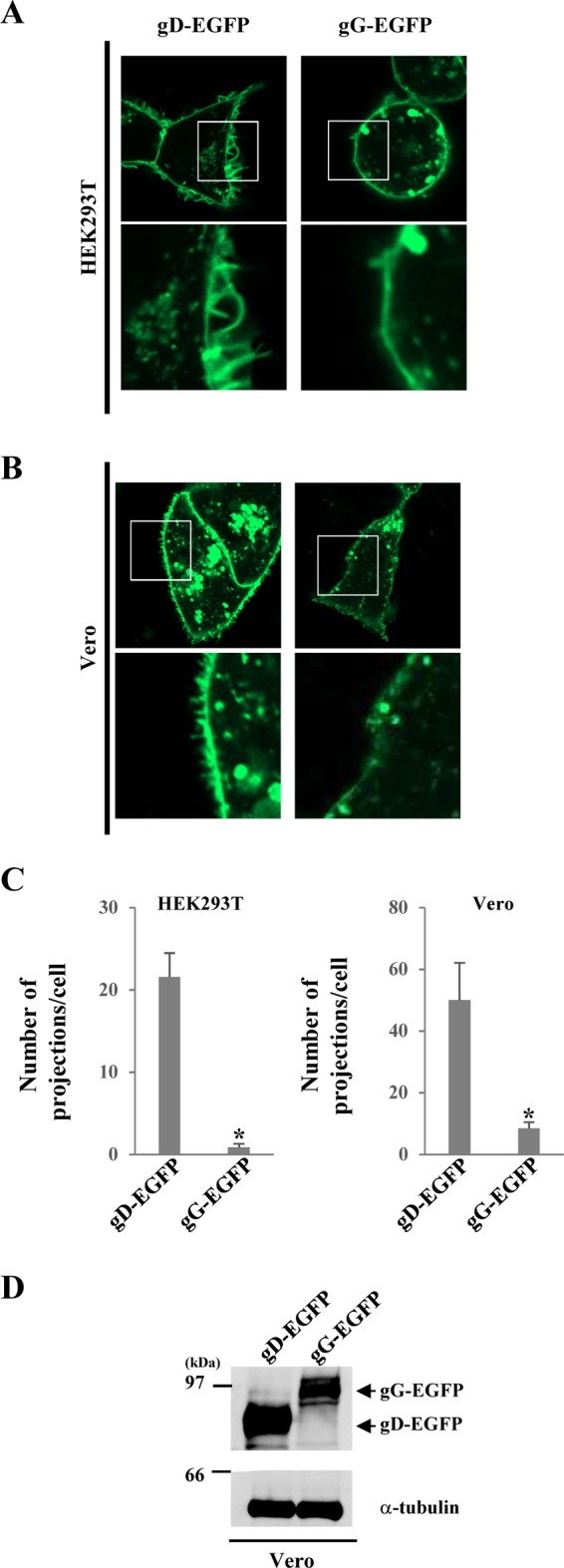
Fluorescence microscopy of HEK293T and Vero cells transfected with the gD-EGFP expression plasmid. (A and B) HEK293T (A) and Vero (B) cells were transfected with the gD-EGFP or gG-EGFP expression plasmid. At 24 h posttransfection, the cells were analyzed by confocal microscopy. The boxed area in each upper panel is shown in the panel below it. (C) Transfected cells were examined by confocal microscopy, as described in the legends to panels A and B, and the number of EGFP-positive projections at the plasma membranes of 7 cells was quantitated. Statistical analysis was performed by one-way analysis of variance with the Tukey test. The asterisks indicate statistically significant differences, as follows: *, P < 0.01. (D) Vero cells were transfected with the indicated expression plasmid. At 24 h posttransfection, cell lysates were analyzed by immunoblotting with anti-EGFP and anti-α-tubulin antibodies.
These results suggest that gD is able to induce microvillus-like projections at the plasma membrane in the absence of any other viral protein and that gD localizes at the projections induced by ectopic gD expression.
Effect of the arginine cluster in the gD cytoplasmic domain on gD-induced microvillus-like projections.
To determine the gD domain required for projection formation, we examined the effects of ectopic expression of gD-EGFP mutants (Fig. 4A) on the plasma membrane projections in HEK293T cells by fluorescence microscopy. As shown in Fig. 4B, expression of gD-CTD-EGFP, which contained only the gD TM and cytoplasmic domains, induced gD-positive projection structures at the plasma membrane at a level slightly lower than the level at which they were induced by expression of gD-EGFP. We next tested a gD-EGFP mutant in which the TM and cytoplasmic domains of gD were replaced by those of gB (gD1–339-gB770–864-EGFP). As shown in Fig. 4B and C, in HEK293T cells transfected with the gD1–339-gB770–864-EGFP expression plasmid, gD-positive projection structures were induced at the plasma membrane, but the number of structures was much less than that in cells transfected with the gD-EGFP expression plasmid. Expression of gD-EGFP, gD-CTD-EGFP, and gD1–339-gB770–864-EGFP in the transfected cells was confirmed by immunoblotting (Fig. 4D).
FIG 4.
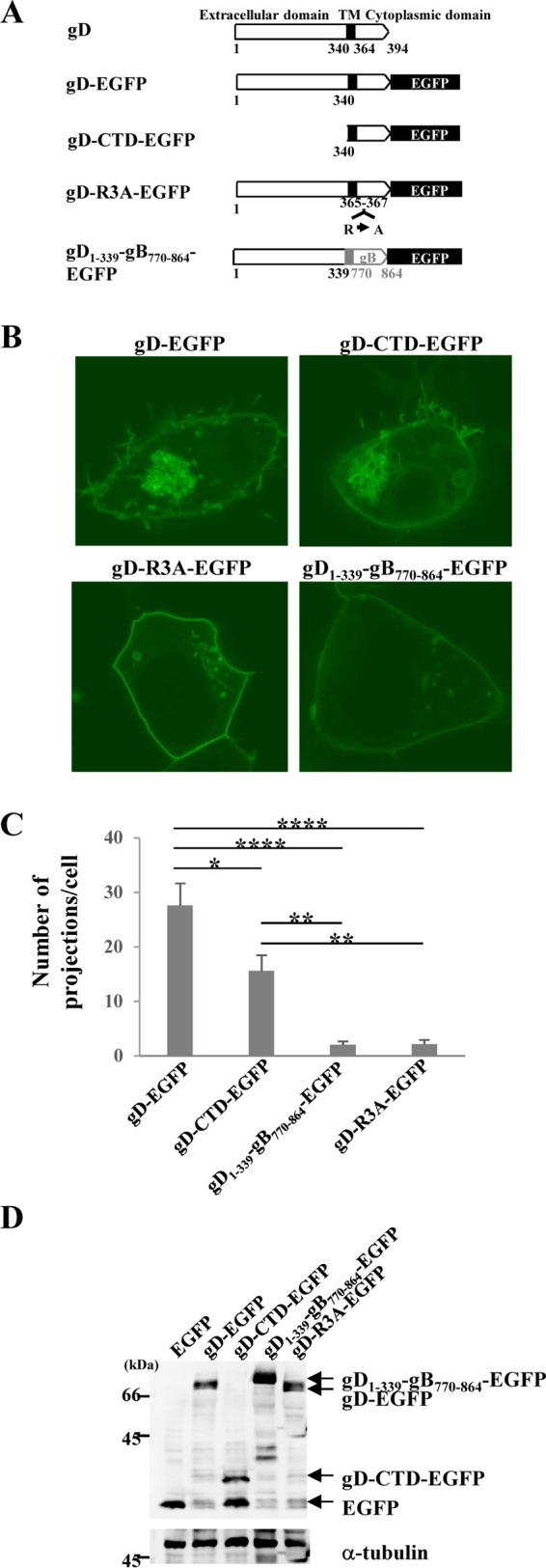
Mapping the gD domain(s) responsible for gD-induced projections at the plasma membrane in HEK293T cells. (A) Schematic diagrams of wild-type and mutant gD expression plasmids. (B) HEK293T cells were transfected with the indicated gD expression plasmid. At 24 h posttransfection, the cells were analyzed by confocal microscopy. (C) Transfected cells were examined by confocal microscopy as described in the legend to panel B, and the number of EGFP-positive projections at the plasma membranes of 7 cells was quantitated. Statistical analysis was performed by one-way analysis of variance with the Tukey test. The asterisks indicate statistically significant differences, as follows: *, P < 0.05; **, P < 0.001; ****, P < 0.0001. (D) HEK293T cells were transfected with the indicated expression plasmid. At 24 h posttransfection, cell lysates were analyzed by immunoblotting with anti-GFP and anti-α-tubulin antibodies.
The gD cytoplasmic domain contains an arginine cluster (Fig. 4A). Since arginine clusters have been reported to be involved in some protein-lipid interactions (40, 41), we tested a gD-EGFP mutant in which the arginines at gD residues 365, 366, and 367 were replaced by alanines (gD-R3A-EGFP). As observed with the expression of gD1–339-gB770–864-EGFP, the expression of gD-R3A-EGFP induced gD-positive projection structures at the plasma membrane, but at significantly levels lower than the levels at which they were induced by expression of wild-type gD-EGFP (Fig. 4B and C). In agreement with the findings of the fluorescence microscopy analyses of HEK293T cells transfected with the gD-EGFP or gD-R3A-EGFP expression plasmid, the ultrastructure of HEK293T cells transfected with the gD-R3A-EGFP expression plasmid showed fewer projection structures at the plasma membrane than the number of projection structures found in cells transfected with the wild-type gD-EGFP expression plasmid (Fig. 5). Expression of the gD-R3A-EGFP mutant in HEK293T cells transfected with the gD-R3A-EGFP expression plasmid was confirmed by immunoblotting (Fig. 4D).
FIG 5.
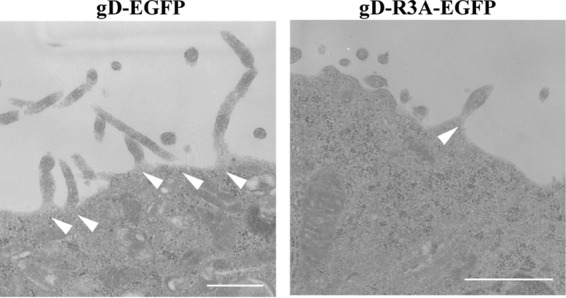
Effect of mutations in the gD arginine cluster on gD-induced projections at the plasma membrane in HEK293T cells. HEK293T cells were transfected with the gD-EGFP or gD-R3A-EGFP expression plasmid. At 48 h posttransfection, the cells were examined by transmission electron microscopy. Arrowheads, microvillus-like projections. Bars, 500 nm.
These results suggest that the gD TM and cytoplasmic domains are necessary and sufficient for gD-induced microvillus-like projection formation at the plasma membrane and that the arginine cluster in the gD cytoplasmic domain is required for efficient gD-induced projection formation.
Effect of the arginine cluster in the gD cytoplasmic domain on viral growth and cell-cell spread.
To clarify the role(s) of the arginine cluster in the gD cytoplasmic domain during HSV-1 replication, we generated recombinant viruses YK740 (gD-R3A), encoding a mutant gD in which the arginines in gD residues 365, 366, and 367 were replaced by alanines, and YK741 (gD-R3A-repair), the repaired YK740 virus in which the alanines in gD residues 365, 366, and 367 in YK740 (gD-R3A) were replaced by arginines (Fig. 1A). These recombinant viruses were characterized by the following results: (i) Vero cells infected with YK740 (gD-R3A) produced gD at levels similar to those for Vero cells infected with wild-type HSV-1(F) or YK741 (gD-R3A-repair) (Fig. 6A), (ii) the amount of gD-R3A incorporated into YK740 (gD-R3A) virions was similar to the amount of gD incorporated into wild-type HSV-1(F) and YK741 (gD-R3A-repair) virions (Fig. 6B), and (iii) the pattern of gD-R3A localization in Vero cells infected with YK740 (gD-R3A) was similar to that of gD localization in Vero cells infected with wild-type HSV1(F) or YK741 (gD-R3A-repair) (Fig. 6C). These results indicate that the R3A mutation in gD has no effect on (i) the accumulation of gD in Vero cells, (ii) the incorporation of gD into virions, or (iii) the subcellular localization of gD in infected Vero cells.
FIG 6.
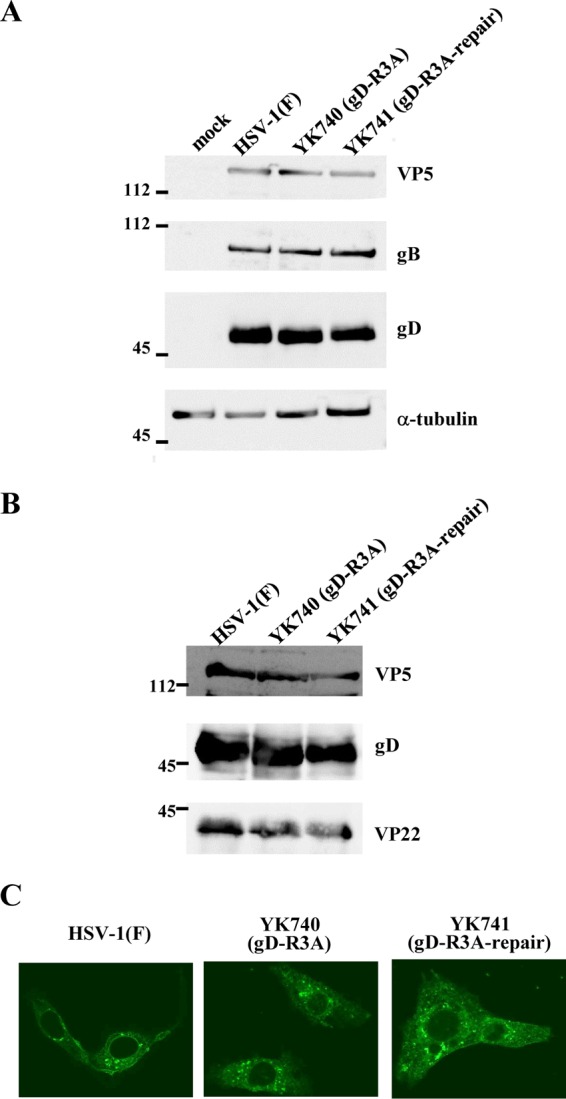
Effect of mutations in the gD arginine cluster on gD expression and subcellular localization in infected cells and on virion incorporation of gD. (A) Vero cells were mock infected or infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 5, harvested at 18 h postinfection, lysed, and analyzed by immunoblotting with antibodies against the indicated proteins. (B) Vero cells were infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 0.1. At 48 h postinfection, extracellular virions were purified and analyzed by immunoblotting with antibodies against the indicated proteins. Numbers to the left of the gels in panels A and B are molecular masses (in kilodaltons). (C) Vero cells were infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 3 and at 18 h postinfection were fixed, permeabilized, stained with anti-gD antibody, and examined by confocal microscopy.
To examine the effect of the arginine cluster in the gD cytoplasmic domain on viral replication, Vero cells were infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at a multiplicity of infection (MOI) of 0.005 or 5, and viral titers were assayed at various times postinfection. As shown in Fig. 7A, in cells infected at an MOI of 0.005, YK740 (gD-R3A) growth was less than that of wild-type HSV-1(F) and YK741 (gD-R3A-repair). The progeny virus titer of YK740 (gD-R3A) at 48 h postinfection was significantly less than that of wild-type HSV-1(F) and YK741 (gD-R3A-repair) (20- and 15-fold, respectively). In cells infected at an MOI of 5, the progeny virus titer of YK740 (gD-R3A) at 12 and 18 h postinfection was significantly less than that of wild-type HSV-1(F) and YK741 (gD-R3A-repair): 9.7- and 18-fold, respectively, at 12 h postinfection, and 2.5- and 2.8-fold, respectively, at 18 h postinfection (Fig. 7B). In contrast, the YK740 (gD-R3A) progeny virus titer was similar to that of wild-type HSV-1(F) and YK741 (gD-R3A-repair) at 24 h postinfection (Fig. 7B).
FIG 7.
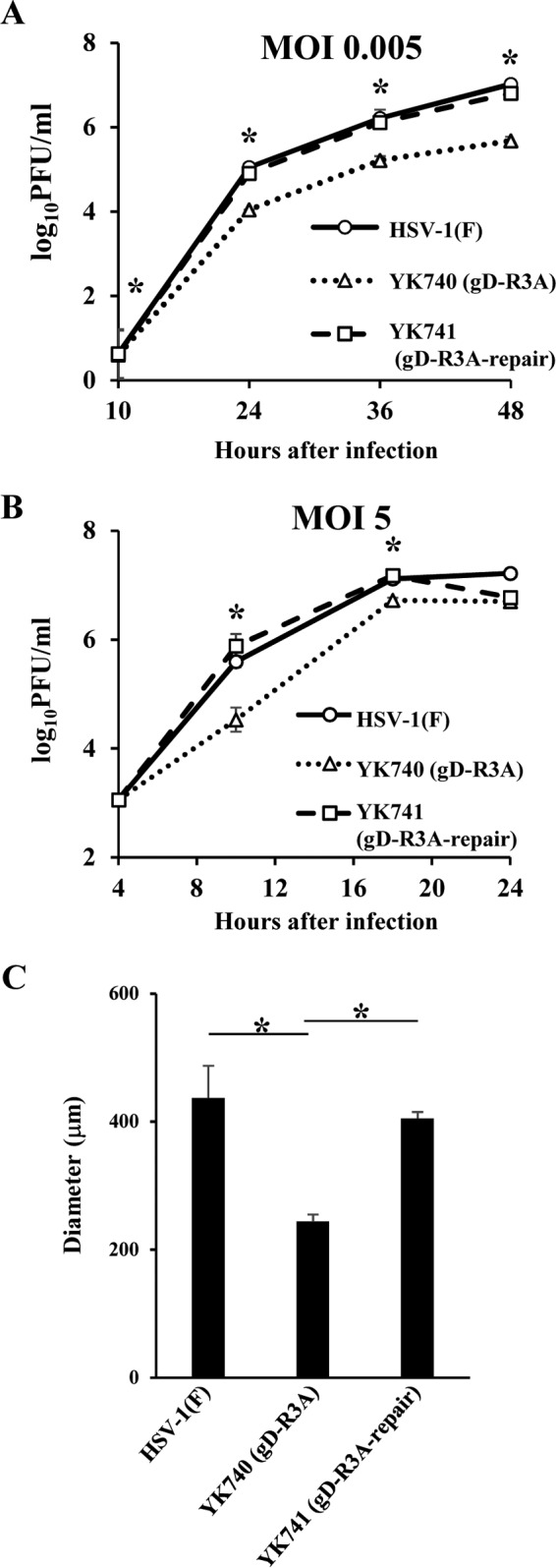
Effect of mutations in the gD arginine cluster on viral growth and cell-cell spread. (A and B) Vero cells were infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 0.005 (A) or 5 (B), harvested at the indicated times postinfection, and assayed for determination of the number of PFU on Vero cells. Each data point is the mean ± standard error from 3 independent experiments. (C) Vero cells in 35-mm-diameter dishes were infected with 100 PFU wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) under plaque assay conditions. For each of the indicated viruses, the diameters of 20 single plaques were measured at 48 h postinfection. Each data point is the mean measured plaque size ± standard error. Statistical analysis was performed by one-way analysis of variance with the Tukey test. The asterisks indicate statistically significant differences, as follows: *, P < 0.01 for YK740 (gD-R3A) versus HSV-1(F) and for YK740 (gD-R3A) versus YK741 (gD-R3A-repair).
To examine the significance of the gD arginine cluster on viral cell-cell spread, we analyzed the plaque sizes on Vero cell cultures infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair). YK740 (gD-R3A) produced significantly smaller plaques than wild-type HSV-1(F) and YK741 (gD-R3A-repair) (Fig. 7C). The effects of these mutations in the gD arginine cluster on viral plaque size were in agreement with those of the truncated gD cytoplasmic domain reported previously (21, 22, 42).
Collectively, these results suggest that the arginine cluster in the gD cytoplasmic domain is required for efficient viral growth and cell-cell spread.
Effect of the arginine cluster in the gD cytoplasmic domain on the plasma membrane projections and virion morphogenesis in HSV-1-infected cells.
To determine whether the arginine cluster in the gD cytoplasmic domain mediated microvillus-like projection formation at the plasma membrane in HSV-1-infected cells, Vero cells were mock infected or infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 5 for 18 h and then examined by electron microscopy. As reported previously (5–7), microvillus-like structures were observed at the plasma membrane in cells infected with wild-type HSV-1(F) or YK741 (gD-R3A-repair) but were rarely observed in mock-infected cells (Fig. 8A). The microvillus-like projections induced by HSV-1 infection appeared to be similar in size to those induced by ectopic expression of gD (Fig. 2A and C and 5). In cells infected with YK740 (gD-R3A), the number of projection structures induced at the plasma membrane was significantly less than the number induced in cells infected with wild-type HSV-1(F) or YK741 (gD-R3A-repair) (Fig. 8A and B).
FIG 8.
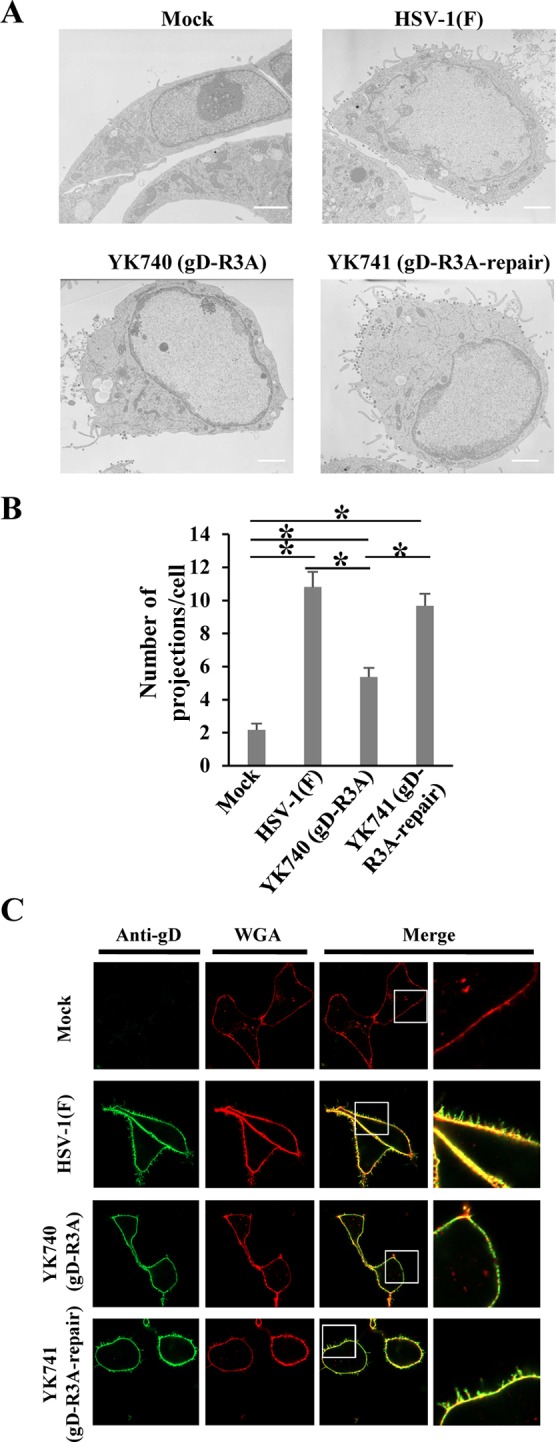
Effect of mutations in the gD arginine cluster on projection formation at the plasma membrane in HSV-1-infected cells. (A) Vero cells were mock infected or infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 5, fixed at 18 h postinfection, and examined by transmission electron microscopy. Bars, 2 μm. (B) Infected cells were examined by transmission electron microscopy as described in the legend to panel A, and the number of projections at the plasma membranes of 30 cells were quantitated. Statistical analysis was performed by one-way analysis of variance with the Tukey test. The asterisks indicate statistically significant differences, as follows: *, P < 0.05. (C) Vero cells were mock infected or infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 3. At 18 h postinfection, infected cells were stained with WGA and anti-gD antibody without permeabilization and examined by confocal microscopy. Each image in the far right column is the magnified image of the boxed area in the image to its left.
In addition, Vero cells were mock infected or infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 5 for 18 h, and their cell surfaces were then stained with anti-gD antibody and WGA and examined by fluorescence microscopy. WGA is a lectin probe that can be used for selective plasma membrane labeling. In agreement with the findings of the electron microscopy analyses of infected Vero cells described above, (i) in cells infected with wild-type HSV-1(F) or YK741 (gD-R3A-repair), gD- and WGA-positive projection structures were induced at the plasma membrane, (ii) these projection structures were rarely observed at the plasma membrane in mock-infected cells, and (iii) in cells infected with YK740 (gD-R3A), gD- and WGA-positive projection structures were induced at the plasma membrane but were induced at significantly lower levels than they were in cells infected with wild-type HSV-1(F) or YK741 (gD-R3A-repair) (Fig. 8C). These results indicate that the arginine cluster in the gD cytoplasmic domain is required for efficient microvillus-like projection formation at the plasma membrane in HSV-1-infected cells.
During the course of the electron microscopic analyses described above, we noted that nucleocapsids and partially enveloped virions appeared to be observed more frequently in cells infected with YK740 (gD-R3A) than in cells infected with wild-type HSV-1(F) or YK741 (gD-R3A-repair) (Fig. 9). Therefore, we quantitated the number of virus particles at different morphogenic stages in Vero cells infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 5 at 18 h postinfection by electron microscopy. As shown in Table 1, in cells infected with wild-type HSV-1(F) or YK741 (gD-R3A-repair), 3.6 and 4.2% of virus particles, respectively, were nucleocapsids in the cytoplasm, but in cells infected with YK740 (gD-R3A), the percentage of virus particles that were nucleocapsids in the cytoplasm increased to 24.4%, which was 6.8- and 5.8-fold more than that in cells infected with wild-type HSV-1(F) or YK741 (gD-R3A-repair), respectively. Also, in cells infected with wild-type HSV-1(F) or YK741 (gD-R3A-repair), 36.5 and 37.1% of virus particles, respectively, were enveloped virions in the cytoplasm and extracellular space, but in cells infected with YK740 (gD-R3A), only 17.7% of virus particles were enveloped virions in the cytoplasm and extracellular space. In contrast, the fraction of total virus particles in the nucleus and perinuclear space in cells infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) was similar (59.9, 57.9, and 58.6%, respectively). These results indicate that the arginine clusters in the gD cytoplasmic domain are required for proper virion morphogenesis in the cytoplasm. In particular, virion morphogenesis in YK740 (gD-R3A)-infected cells, including the aberrant accumulation of unenveloped nucleocapsids in the cytoplasm and the decrease in the amount of enveloped virions in the cytoplasm and extracellular space, may reflect a block in a process(es) for viral secondary envelopment (19, 20, 43). Therefore, our results indicate that the arginine clusters in the gD cytoplasmic domain are required for efficient HSV-1 secondary envelopment.
FIG 9.
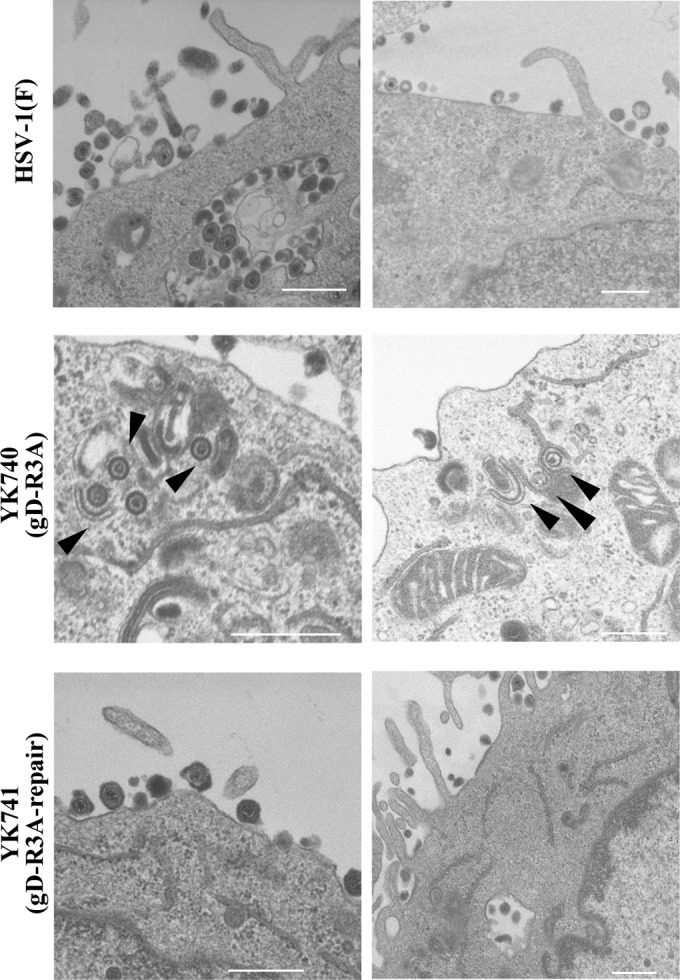
Effect of mutations in the gD arginine cluster on HSV-1 secondary envelopment. Vero cells were infected with wild-type HSV-1(F), YK740 (gD-R3A), or YK741 (gD-R3A-repair) at an MOI of 5, fixed at 18 h postinfection, and examined by transmission electron microscopy. Arrowheads, partially enveloped nucleocapsids. Bars, 500 nm.
TABLE 1.
Effect of gD R3A mutation on distribution of virus particles in infected Vero cells
| Virus | Mean ± SE % virus particles in each morphogenetic stagea |
Total no. of particles counted/total no. of cells | ||||
|---|---|---|---|---|---|---|
| Nucleocapsids in nucleus | Enveloped virions in perinuclear space | Nucleocapsids in cytoplasm | Enveloped virions in cytoplasm | Extracellular enveloped virions | ||
| HSV-1(F) | 55.6 ± 9.3 (827) | 4.3 ± 0.9 (64) | 3.6 ± 1.3b (54) | 3.7 ± 0.8 (55) | 32.8 ± 5.3b (487) | 1,487/9 |
| YK740 (gD-R3A) | 52.3 ± 2.1 (774) | 5.6 ± 1.6 (83) | 24.4 ± 4.9 (362) | 10.3 ± 3.1 (153) | 7.4 ± 1.3 (109) | 1,481/9 |
| YK741 (gD-R3A-repair) | 54.5 ± 6.4 (822) | 4.1 ± 0.9 (62) | 4.2 ± 0.8b (64) | 4.8 ± 0.8 (73) | 32.3 ± 7.4b (487) | 1,508/9 |
Numbers in parentheses are the numbers of virus particles.
The difference between this value and that for the YK740 (gD-R3A) mutant was statistically significant according to one-way analysis of variance with the Tukey test (P < 0.05).
We also examined the effect of the gD-null mutation on microvillus-like projection formation at the plasma membrane and on virion morphogenesis in HSV-1-infected cells. For this study, Vero cells were mock infected or infected with wild-type HSV-1(F), HSV-1 with the gD-null mutation (F-BAC gD−), or the repaired F-BAC gD− virus (F-BAC gD−-repair) at an MOI of 5 for 18 h and examined by electron microscopy. These results confirm that cells infected with wild-type HSV-1(F) or F-BAC gD−-repair produce gD but that cells infected with F-BAC gD− do not (Fig. 10A). In addition, cells infected with HSV-1 with the gD-null mutation induced fewer microvillus-like projection structures at the plasma membrane (Fig. 10B and C), accumulated cytoplasmic nucleocapsids, and produced fewer extracellular enveloped virions (Fig. 11 and Table 2), as was observed in cells infected with HSV-1 with the gD R3A mutation. These results confirm that gD with its arginine cluster is required for efficient microvillus-like projection formation at the plasma membrane and for viral secondary envelopment in HSV-1-infected cells.
FIG 10.
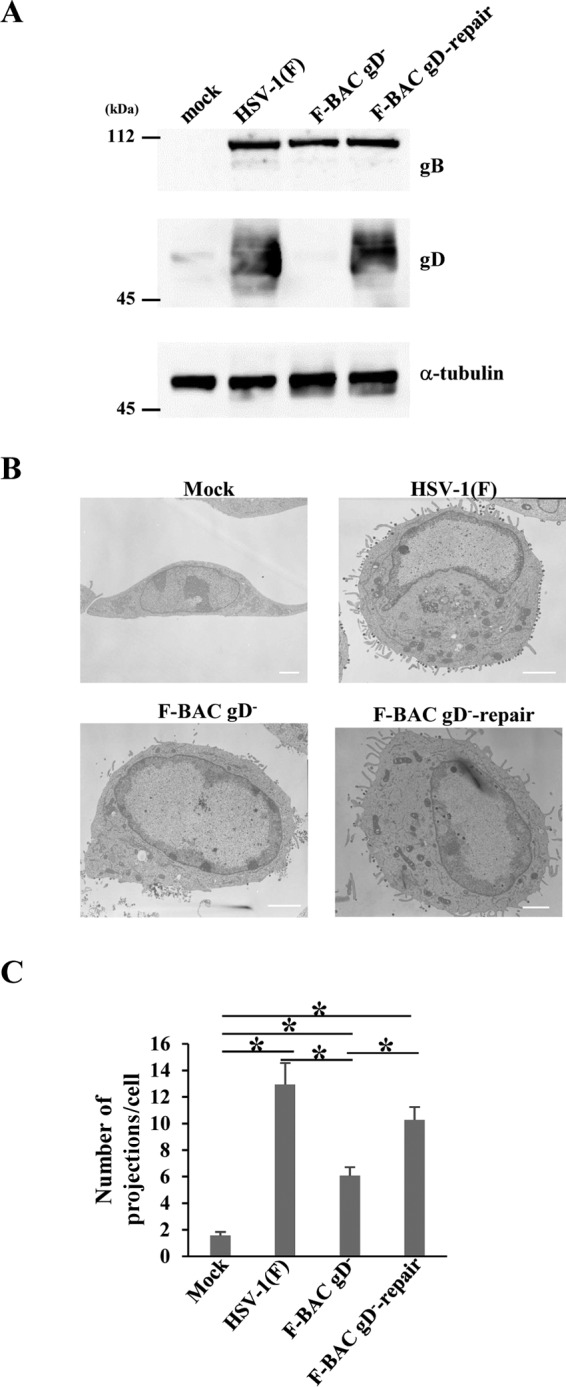
Effect of the gD-null mutation on projection formation at the plasma membrane in HSV-1-infected cells. (A) Vero cells were mock infected or infected with wild-type HSV-1(F), F-BAC gD−, or F-BAC gD−-repair at an MOI of 5, harvested at 18 h postinfection, lysed, and analyzed by immunoblotting with antibodies against the indicated proteins. (B) Vero cells were mock infected or infected with wild-type HSV-1(F), F-BAC gD−, or F-BAC gD−-repair at an MOI of 5, fixed at 18 h postinfection, and examined by transmission electron microscopy. Bars, 2 μm. (C) Infected cells were examined by transmission electron microscopy as described in the legend to panel B, and the number of projections at the plasma membranes of 30 cells was quantitated. Statistical analysis was performed by one-way analysis of variance with the Tukey test. The asterisks indicate statistically significant differences, as follows: *, P < 0.05.
FIG 11.
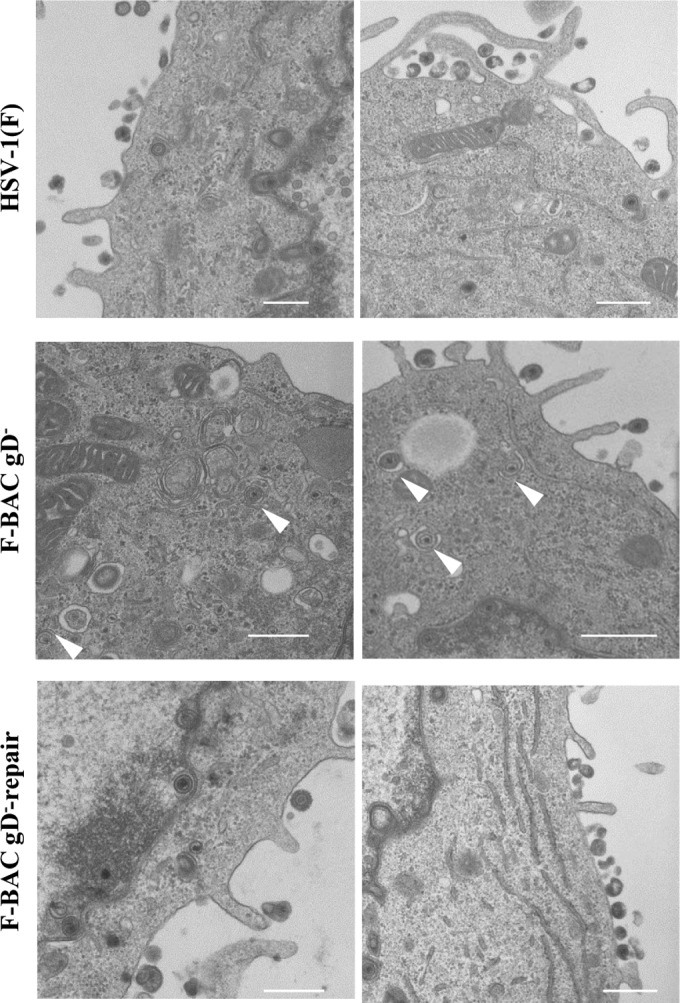
Effect of the gD-null mutation on HSV-1 secondary envelopment. Vero cells infected with wild-type HSV-1(F), F-BAC gD−, or F-BAC gD−-repair at an MOI of 5 were fixed at 18 h postinfection and examined by transmission electron microscopy. Arrowheads, partially enveloped nucleocapsids. Bars, 500 nm.
TABLE 2.
Effect of gD-null mutation on distribution of virus particles in infected Vero cells
| Virus | Mean ± SE % virus particles in each morphogenetic stagea |
Total no. of particles counted/total no. of cells | ||||
|---|---|---|---|---|---|---|
| Nucleocapsids in nucleus | Enveloped virions in perinuclear space | Nucleocapsids in cytoplasm | Enveloped virions in cytoplasm | Extracellular enveloped virions | ||
| HSV-1(F) | 51.7 ± 11.8 (800) | 8.0 ± 3.6 (124) | 7.8 ± 1.5b (121) | 3.8 ± 0.5 (59) | 30.3 ± 3.2b (876) | 1,546/12 |
| F-BAC gD− | 58.5 ± 10.2 (683) | 6.4 ± 1.3 (75) | 19.2 ± 4.1 (224) | 4.6 ± 0.9 (52) | 18.9 ± 3.0 (508) | 1,167/12 |
| F-BAC gD−-repair | 51.7 ± 9.7 (652) | 7.0 ± 2.9 (88) | 6.8 ± 2.1b (86) | 5.0 ± 1.9 (63) | 25.6 ± 3.4b (783) | 1,261/12 |
Numbers in parentheses are the numbers of virus particles.
The difference between this value and that for the F-BAC gD− mutant was statistically significant according to one-way analysis of variance with the Tukey test (P < 0.05).
DISCUSSION
As described above, the role(s) of the cytoplasmic domain of gD during HSV-1 replication and cell-cell spread has remained to be elucidated, although the gD extracellular domain has been reported to be necessary for recognition of gD receptors, virus-cell membrane fusion, and HSV-1 entry (15, 22, 28). In this study, analyses of mutations in the HSV-1 gD cytoplasmic domain identified two functions of the arginine cluster in the gD cytoplasmic domain, i.e., the formation of microvillus-like projections at the plasma membrane and the promotion of viral secondary envelopment, both of which require host cell membrane deformation (44, 45). These results suggest that the gD functions in projection formation at the plasma membrane and in viral secondary envelopment are distinct from the reported gD function in viral entry. Phosphatidyl lipids, which are membrane components, have been reported to be involved in membrane deformation (46) and to have a high affinity for positively charged peptides, including the arginine cluster (40, 41). Although at present there are no data indicating that gD directly deforms membranes during plasma membrane remodeling and viral secondary envelopment, the interaction between the gD arginine cluster and phosphatidyl lipid may be involved in gD-mediated membrane deformation.
Herpesviruses appear to have evolved mechanisms to remodel the host cell plasma membranes of infected cells. For example, protein kinase Us3 encoded by pseudorabiesvirus (PRV), an alphaherpesvirus, induces long thick projections on the surfaces of infected cells that can contact adjacent cells: PRV capsids move inside the projections and are transferred to adjacent cells (3). Similar cellular projections were also reported in cells infected with human cytomegalovirus (HCMV), a human betaherpesvirus (12). In addition, membrane glycoprotein gp48 encoded by murine gammaherpesvirus 68 (MHV-68) was reported to localize on the cell surface in infected cells and to promote viral cell-cell spread (9, 47). Also, microvillus-like projections at the plasma membrane in MHV-68-infected cells were induced by the cytoplasmic domain of gp48, and MHV-68 virions move in these projections (9). Similar microvillus-like projections at the plasma membrane were reported for cells infected with the alphaherpesvirus varicella-zoster virus (VZV), HCMV (4, 8), and the gammaherpesvirus Kaposi's sarcoma-associated herpesvirus (KSHV) (11). The HSV-1-induced microvillus-like projections at the plasma membrane shown in this study and previous studies (5–7) seem to be similar in appearance to those observed with the MHV-68-infected cells described above. In agreement with these reports, we show here that HSV-1 virions are observed on the projection structures but not inside the structures and that the cytoplasmic domain of gD, which was reported to localize at the cell surface in infected cells and promote viral cell-cell spread, like MHV-68 gp48 (9, 47), mediates the induction of microvillus-like projections at the plasma membrane in HSV-1-infected cells. In addition, a previous report demonstrated that HSV-1 virions can move on HSV-1-induced microvillus-like projections at the plasma membrane (10). Thus far, although there have been no direct data, the accumulating results described above suggest that the plasma membrane remodeling induced by various herpesviruses may be involved in efficient viral spread. The induction of microvillus-like projections at the plasma membrane by the HSV-1 gD cytoplasmic domain in HSV-1-infected cells, as shown in this study, may be one of the membrane-remodeling mechanisms. In support of this hypothesis, it has been reported that a retrovirus infection establishes filopodial bridges between an infected cell and adjacent cells and that retroviral virions travel along the outer surface of the filopodial bridges to adjacent cells to promote viral spread (48, 49).
We have also presented here data indicating that the arginine cluster in the gD cytoplasmic domain is required for efficient viral secondary envelopment. This result is in agreement with a previous report (20) showing that the gD-null mutation caused a reduction in viral secondary envelopment. However, at present, the mechanism by which the arginine cluster acts in viral secondary envelopment is not known. It has been thought that the protein-protein interaction network between tegument proteins, capsid proteins, and envelope proteins facilitates herpesviral secondary envelopment by bridging the nucleocapsids and the cytoplasmic membrane (2, 50). The gD arginine cluster may interact with tegument, capsid, and/or other envelope proteins to regulate viral secondary envelopment. Alternatively, the gD arginine cluster may interact with a viral structural protein(s) that affects gD transport. It has been reported that HSV-1 tegument proteins VP22 and UL11 and envelope glycoproteins gB and gH/gL complex, which were reported to be involved in viral secondary envelopment, interact with gD and that gD transport is regulated by the UL20/gK envelope protein complex or by gM, which may interact with gD like gB and gH do (16, 43, 51–56). Moreover, the gD arginine cluster may directly regulate the deformation of cytoplasmic membranes, probably by interacting with the cytoplasmic membranes through phosphatidyl lipids, as described above. The ability of gD to induce remodeling of cellular membranes, as observed in this study when gD was ectopically expressed, may be related to a gD role in viral secondary envelopment in infected cells. Further studies will be needed to clarify the role of the arginine cluster in the gD cytoplasmic domain in viral secondary envelopment.
gD is conserved in most viruses in the Alphaherpesvirinae subfamily. However, the gD cytoplasmic domain varies among members of the Alphaherpesvirinae, and some members, like varicella-zoster virus, do not have gD (57). These facts indicate that the contribution of gD to plasma membrane remodeling and viral secondary envelopment may vary in alphaherpesviruses. In support of this hypothesis, the PRV gD had no effect on viral secondary envelopment, unlike the HSV-1 gD (58, 59). However, it has been reported that PRV gE and gM are both needed for PRV secondary envelopment, although deletion of both of these glycoprotein homologs in HSV-1 was reported to have no effect on secondary envelopment (60, 61). The genomes of herpesviruses encode more than 10 envelope glycoproteins (1), and, similar to gD, many of these glycoproteins have arginine-rich regions in their cytoplasmic domains. Therefore, multiple herpesvirus envelope glycoproteins may be involved redundantly in the regulation of plasma membrane remodeling and/or viral secondary envelopment.
ACKNOWLEDGMENTS
We thank David C. Johnson for providing the gD-deficient virus F-BAC gD−. We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers and Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS); Grants-in-Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (16H06433, 16H06429, and 16K21723); a contract research fund for the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from MEXT and the Japan Agency for Medical Research and Development (AMED); and grants from the Takeda Science Foundation, the Mitsubishi Foundation, the Ichiro Kanehara Foundation, and the Mochida Memorial Foundation.
Funding Statement
This work, including the efforts of Yasushi Kawaguchi, was funded by Ministry of Education, Culture, Sports, Science, and Technology (MEXT) Grants-in-Aid for Scientific Research on Innovative Areas (16H06433, 16H06429, and 16K21723). This work, including the efforts of Yasushi Kawaguchi, was funded by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the Japan Agency for Medical Research and Development (AMED), a contract research fund for the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID). This work, including the efforts of Yasushi Kawaguchi, was funded by the Japan Society for the Promotion of Science (JSPS) (Funding Program for Next Generation World-Leading Researchers). This work, including the efforts of Jun Arii, Akihisa Kato, and Yasushi Kawaguchi, was funded by the Japan Society for the Promotion of Science (JSPS) (Grants-in-Aid for Scientific Research). This work, including the efforts of Jun Arii, Akihisa Kato, and Yasushi Kawaguchi, was funded by the Takeda Science Foundation. This work, including the efforts of Jun Arii and Akihisa Kato, was funded by the Mochida Memorial Foundation for Medical and Pharmaceutical Research. This work, including the efforts of Yasushi Kawaguchi, was funded by the Mitsubishi Foundation. This work, including the efforts of Jun Arii and Akihisa Kato, was funded by the Ichiro Kanehara Foundation for the Promotion of Medical Sciences and Medical Care (Ichiro Kanehara Foundation).
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 3.Favoreel HW, Van Minnebruggen G, Adriaensen D, Nauwynck HJ. 2005. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc Natl Acad Sci U S A 102:8990–8995. doi: 10.1073/pnas.0409099102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter JE, Hutchinson JA, Jackson W, Grose C. 2008. Egress of light particles among filopodia on the surface of varicella-zoster virus-infected cells. J Virol 82:2821–2835. doi: 10.1128/JVI.01821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Archer S, Morgan H, Rixon FJ. 1999. Electrorotation studies of baby hamster kidney fibroblasts infected with herpes simplex virus type 1. Biophys J 76:2833–2842. doi: 10.1016/S0006-3495(99)77437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katsumoto T, Hirano A, Kurimura T, Takagi A. 1981. In situ electron microscopical observation of cells infected with herpes simplex virus. J Gen Virol 52:267–278. doi: 10.1099/0022-1317-52-2-267. [DOI] [PubMed] [Google Scholar]
- 7.Krempien U, Jockusch BM, Jungwirth C. 1984. Herpes simplex virus-induced cell surface protrusions. Intervirology 22:156–163. doi: 10.1159/000149545. [DOI] [PubMed] [Google Scholar]
- 8.Garnett HM. 1979. Early alterations in the morphology of cytomegalovirus-infected human fibroblasts. Intervirology 11:359–362. doi: 10.1159/000149058. [DOI] [PubMed] [Google Scholar]
- 9.Gill MB, Edgar R, May JS, Stevenson PG. 2008. A gamma-herpesvirus glycoprotein complex manipulates actin to promote viral spread. PLoS One 3:e1808. doi: 10.1371/journal.pone.0001808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oh MJ, Akhtar J, Desai P, Shukla D. 2010. A role for heparan sulfate in viral surfing. Biochem Biophys Res Commun 391:176–181. doi: 10.1016/j.bbrc.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raghu H, Sharma-Walia N, Veettil MV, Sadagopan S, Chandran B. 2009. Kaposi's sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J Virol 83:4895–4911. doi: 10.1128/JVI.02498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goulidaki N, Alarifi S, Alkahtani SH, Al-Qahtani A, Spandidos DA, Stournaras C, Sourvinos G. 2015. RhoB is a component of the human cytomegalovirus assembly complex and is required for efficient viral production. Cell Cycle 14:2748–2763. doi: 10.1080/15384101.2015.1066535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci U S A 104:7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spear PG, Manoj S, Yoon M, Jogger CR, Zago A, Myscofski D. 2006. Different receptors binding to distinct interfaces on herpes simplex virus gD can trigger events leading to cell fusion and viral entry. Virology 344:17–24. doi: 10.1016/j.virol.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 16.Avitabile E, Forghieri C, Campadelli-Fiume G. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J Virol 81:11532–11537. doi: 10.1128/JVI.01343-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84:3825–3834. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farnsworth A, Goldsmith K, Johnson DC. 2003. Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J Virol 77:8481–8494. doi: 10.1128/JVI.77.15.8481-8494.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson DC, Wisner TW, Wright CC. 2011. Herpes simplex virus glycoproteins gB and gD function in a redundant fashion to promote secondary envelopment. J Virol 85:4910–4926. doi: 10.1128/JVI.00011-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chouljenko DV, Kim IJ, Chouljenko VN, Subramanian R, Walker JD, Kousoulas KG. 2012. Functional hierarchy of herpes simplex virus 1 viral glycoproteins in cytoplasmic virion envelopment and egress. J Virol 86:4262–4270. doi: 10.1128/JVI.06766-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feenstra V, Hodaie M, Johnson DC. 1990. Deletions in herpes simplex virus glycoprotein D define nonessential and essential domains. J Virol 64:2096–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee HC, Chouljenko VN, Chouljenko DV, Boudreaux MJ, Kousoulas KG. 2009. The herpes simplex virus type 1 glycoprotein D (gD) cytoplasmic terminus and full-length gE are not essential and do not function in a redundant manner for cytoplasmic virion envelopment and egress. J Virol 83:6115–6124. doi: 10.1128/JVI.00128-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Long JC, Caceres JF. 2009. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 25.Bjerke SL, Cowan JM, Kerr JK, Reynolds AE, Baines JD, Roller RJ. 2003. Effects of charged cluster mutations on the function of herpes simplex virus type 1 UL34 protein. J Virol 77:7601–7610. doi: 10.1128/JVI.77.13.7601-7610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paßvogel L, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC. 2015. Functional characterization of nuclear trafficking signals in pseudorabies virus pUL31. J Virol 89:2002–2012. doi: 10.1128/JVI.03143-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia-Saez AJ, Mettenleiter TC, Antonin W. 2015. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem 290:6962–6974. doi: 10.1074/jbc.M114.627521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ligas MW, Johnson DC. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J Virol 62:1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor alpha. J Virol 83:4520–4527. doi: 10.1128/JVI.02601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 32.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka M, Kodaira H, Nishiyama Y, Sata T, Kawaguchi Y. 2004. Construction of recombinant herpes simplex virus type I expressing green fluorescent protein without loss of any viral genes. Microbes Infect 6:485–493. doi: 10.1016/j.micinf.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 34.Arii J, Wang J, Morimoto T, Suenaga T, Akashi H, Arase H, Kawaguchi Y. 2010. A single-amino-acid substitution in herpes simplex virus 1 envelope glycoprotein B at a site required for binding to the paired immunoglobulin-like type 2 receptor alpha (PILRalpha) abrogates PILRalpha-dependent viral entry and reduces pathogenesis. J Virol 84:10773–10783. doi: 10.1128/JVI.01166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jarosinski KW, Margulis NG, Kamil JP, Spatz SJ, Nair VK, Osterrieder N. 2007. Horizontal transmission of Marek's disease virus requires US2, the UL13 protein kinase, and gC. J Virol 81:10575–10587. doi: 10.1128/JVI.01065-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. 2013. Roles of p53 in herpes simplex virus 1 replication. J Virol 87:9323–9332. doi: 10.1128/JVI.01581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawaguchi Y, Nakajima K, Igarashi M, Morita T, Tanaka M, Suzuki M, Yokoyama A, Matsuda G, Kato K, Kanamori M, Hirai K. 2000. Interaction of Epstein-Barr virus nuclear antigen leader protein (EBNA-LP) with HS1-associated protein X-1: implication of cytoplasmic function of EBNA-LP. J Virol 74:10104–10111. doi: 10.1128/JVI.74.21.10104-10111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83:11624–11634. doi: 10.1128/JVI.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mazerik JN, Tyska MJ. 2012. Myosin-1A targets to microvilli using multiple membrane binding motifs in the tail homology 1 (TH1) domain. J Biol Chem 287:13104–13115. doi: 10.1074/jbc.M111.336313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takahashi S, Pryciak PM. 2007. Identification of novel membrane-binding domains in multiple yeast Cdc42 effectors. Mol Biol Cell 18:4945–4956. doi: 10.1091/mbc.E07-07-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones NA, Geraghty RJ. 2004. Fusion activity of lipid-anchored envelope glycoproteins of herpes simplex virus type 1. Virology 324:213–228. doi: 10.1016/j.virol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 43.Farnsworth A, Wisner TW, Johnson DC. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J Virol 81:319–331. doi: 10.1128/JVI.01842-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Votteler J, Sundquist WI. 2013. Virus budding and the ESCRT pathway. Cell Host Microbe 14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mattila PK, Lappalainen P. 2008. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol 9:446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- 46.Bohdanowicz M, Grinstein S. 2013. Role of phospholipids in endocytosis, phagocytosis, and macropinocytosis. Physiol Rev 93:69–106. doi: 10.1152/physrev.00002.2012. [DOI] [PubMed] [Google Scholar]
- 47.May JS, Walker J, Colaco S, Stevenson PG. 2005. The murine gammaherpesvirus 68 ORF27 gene product contributes to intercellular viral spread. J Virol 79:5059–5068. doi: 10.1128/JVI.79.8.5059-5068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. 2005. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol 170:317–325. doi: 10.1083/jcb.200503059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. 2007. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat Cell Biol 9:310–315. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res 143:222–234. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 51.Ren Y, Bell S, Zenner HL, Lau SY, Crump CM. 2012. Glycoprotein M is important for the efficient incorporation of glycoprotein H-L into herpes simplex virus type 1 particles. J Gen Virol 93:319–329. doi: 10.1099/vir.0.035444-0. [DOI] [PubMed] [Google Scholar]
- 52.Chi JH, Harley CA, Mukhopadhyay A, Wilson DW. 2005. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J Gen Virol 86:253–261. doi: 10.1099/vir.0.80444-0. [DOI] [PubMed] [Google Scholar]
- 53.Lau SY, Crump CM. 2015. HSV-1 gM and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Viruses 7:915–938. doi: 10.3390/v7030915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chouljenko VN, Iyer AV, Chowdhury S, Kim J, Kousoulas KG. 2010. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J Virol 84:8596–8606. doi: 10.1128/JVI.00298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rogalin HB, Heldwein EE. 2015. Interplay between the herpes simplex virus 1 gB cytodomain and the gH cytotail during cell-cell fusion. J Virol 89:12262–12272. doi: 10.1128/JVI.02391-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A 104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klupp B, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J Virol 82:6299–6309. doi: 10.1128/JVI.00386-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mettenleiter TC, Klupp BG, Weiland F, Visser N. 1994. Characterization of a quadruple glycoprotein-deleted pseudorabies virus mutant for use as a biologically safe live virus vaccine. J Gen Virol 75(Pt 7):1723–1733. [DOI] [PubMed] [Google Scholar]
- 60.Browne H, Bell S, Minson T. 2004. Analysis of the requirement for glycoprotein M in herpes simplex virus type 1 morphogenesis. J Virol 78:1039–1041. doi: 10.1128/JVI.78.2.1039-1041.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brack AR, Dijkstra JM, Granzow H, Klupp BG, Mettenleiter TC. 1999. Inhibition of virion maturation by simultaneous deletion of glycoproteins E, I, and M of pseudorabies virus. J Virol 73:5364–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]