

ABSTRACT
Lentiviruses have evolved the Vif protein to counteract APOBEC3 (A3) restriction factors by targeting them for proteasomal degradation. Previous studies have identified important residues in the interface of human immunodeficiency virus type 1 (HIV-1) Vif and human APOBEC3C (hA3C) or human APOBEC3F (hA3F). However, the interaction between primate A3C proteins and HIV-1 Vif or natural HIV-1 Vif variants is still poorly understood. Here, we report that HIV-1 Vif is inactive against A3Cs of rhesus macaques (rhA3C), sooty mangabey monkeys (smmA3C), and African green monkeys (agmA3C), while HIV-2, African green monkey simian immunodeficiency virus (SIVagm), and SIVmac Vif proteins efficiently mediate the depletion of all tested A3Cs. We identified that residues N/H130 and Q133 in rhA3C and smmA3C are determinants for this HIV-1 Vif-triggered counteraction. We also found that the HIV-1 Vif interaction sites in helix 4 of hA3C and hA3F differ. Vif alleles from diverse HIV-1 subtypes were tested for degradation activities related to hA3C. The subtype F-1 Vif was identified to be inactive for degradation of hA3C and hA3F. The residues that determined F-1 Vif inactivity in the degradation of A3C/A3F were located in the C-terminal region (K167 and D182). Structural analysis of F-1 Vif revealed that impairing the internal salt bridge of E171-K167 restored reduction capacities to A3C/A3F. Furthermore, we found that D101 could also form an internal interaction with K167. Replacing D101 with glycine and R167 with lysine in NL4-3 Vif impaired its counteractivity to A3F and A3C. This finding indicates that internal interactions outside the A3 binding region in HIV-1 Vif influence the capacity to induce degradation of A3C/A3F.
IMPORTANCE The APOBEC3 restriction factors can serve as potential barriers to lentiviral cross-species transmissions. Vif proteins from lentiviruses counteract APOBEC3 by proteasomal degradation. In this study, we found that monkey-derived A3C, rhA3C and smmA3C, were resistant to HIV-1 Vif. This was determined by A3C residues N/H130 and Q133. However, HIV-2, SIVagm, and SIVmac Vif proteins were found to be able to mediate the depletion of all tested primate A3C proteins. In addition, we identified a natural HIV-1 Vif (F-1 Vif) that was inactive in the degradation of hA3C/hA3F. Here, we provide for the first time a model that explains how an internal salt bridge of E171-K167-D101 influences Vif-mediated degradation of hA3C/hA3F. This finding provides a novel way to develop HIV-1 inhibitors by targeting the internal interactions of the Vif protein.
INTRODUCTION
Simian immunodeficiency virus (SIV) naturally infects many Old World primate species in Africa. The pandemic of human immunodeficiency virus (HIV) originated from cross-species transmission events of SIVs to humans. HIV-1 was introduced into the human population by multiple transmissions of a chimpanzee (cpz) virus, which is known as SIVcpz. The less virulent human lentivirus, HIV-2, was derived from SIVsmm, which was obtained from sooty mangabey monkeys (smm) (1).
The cellular restriction factors of the APOBEC3 (A3) family of DNA cytidine deaminases are an important arm of the innate immune defense system which can potentially serve as a barrier to lentiviral cross-species transmissions (recently reviewed in references 2 and 3). Human A3s include seven genes that contain either one (A3A, A3C, and A3H) or two (A3B, A3D, A3F, and A3G) zinc (Z)-binding domains with the conserved motifs of HXE(X)23–28CXXC (X can be any residue) (4, 5). Among these seven genes, A3D, A3F, A3G, and A3H inhibit HIV-1ΔVif replication by deamination of cytidines in the viral single-strand DNA that is formed during reverse transcription, thereby introducing G-to-A hypermutations in the coding strand (6–12). Additionally, some A3s inhibit virus replication by deaminase-independent mechanisms affecting reverse transcription and integration steps (13–18). Human A3A and A3C are not antiviral against HIV-1, but human A3C could effectively restrict SIVmacΔVif and SIVagmΔVif (11, 19–23), and both A3A and A3C could decrease human papillomavirus infectivity (24, 25). However, some studies found that A3C inhibited HIV-1ΔVif by around 50% (26–28). Human A3B is a potent inhibitor against HIV-1, SIV, and human T cell leukemia virus (HTLV) (19, 29–32). In addition, human A3B was reported to be upregulated in several cancer cells and found to be degraded by virion infectivity factor (Vif) from several SIV lineages (33–39).
To counteract the antiviral functions of A3, all lentiviruses except the equine infectious anemia virus encode the Vif that interacts with A3 proteins and then recruit them to an E3 ubiquitin ligase complex containing Cullin5 (CUL5), Elongin B/C (ELOB/C), RING-box protein RBX2, and CBFβ to induce degradation of the bound A3s by the proteasome (40–42). The Bet of foamy viruses, the nucleocapsid of HTLV-1, and the glycosylated Gag (glyco-Gag) of murine leukemia virus (MLV) are also shown to have the ability to counteract A3s (21, 43–47). In many cases, this counteraction is species specific and depends on several specific A3/Vif interfaces. For example, HIV-1 Vif efficiently neutralizes human A3G, but it does not inactivate African green monkey A3G (agmA3G) and rhesus macaque A3G (rhA3G) despite a sequence identity of almost 75% (10, 48–50). The amino acid 128 of A3G determines this species-specific counteraction: human A3G with D128 is sensitive to HIV-1 Vif, while A3G.K128 is susceptible to SIVagm Vif (48–50). However, residue 129 in human A3G, but not adjacent position 128, determines the sensitivity to degradation by SIVsmm and HIV-2 Vif proteins (51). Several other cross-species counteractions were also observed: SIVmac Vif mediates the degradation not only of human A3s and rhesus macaque A3s but also of cat A3Z2Z3 (52–57); maedi-visna virus (MVV) Vif can induce the degradation of both sheep and human A3Z3s (58).
Although hA3G, hA3H hapII, and hA3F share a conserved zinc coordination motif, HIV-1 Vif targets different sites in these A3 proteins for degradation. For example, the 128DPDY131 motif in hA3G is involved in direct interaction with the 14YRHHY17 domain of HIV-1 Vif (59, 60). The E121 residue in hA3H hapII determines its sensitivity to HIV-1 Vif derived from the NL4-3 strain (61, 62). hA3C and the C-terminal domain (CTD) of hA3F are conserved homologous Z2-typed A3s (4, 5), and 10 equivalent residues in these Z2-typed A3s are identified as being involved in HIV-1 Vif interaction (63). Additionally, A3F.E289 and HIV-1 Vif.R15 show a strong interaction by applying molecular docking (64). The equivalent residue E106 in A3C also determines A3-Vif binding (65). In contrast to this conserved A3-Vif interaction, it was also demonstrated that E324 in A3F is essential for HIV-1 Vif interaction but the equivalent residue E141 in A3C is not, which suggests that the Vif interaction interface differs between A3C and A3F (63, 66). In addition, previous studies have proved that these two glutamic acids vary in primate A3Fs and therefore determined the distinct sensitivities of primate A3F to HIV-1 Vif (67–69). However, the interaction between primate A3Cs and lentiviral Vifs is still less clear.
The N-terminal part of HIV-1 Vif is mainly involved in interaction with human A3s. For example, the 40YRHHY44 box is reported to be essential for A3G degradation, while the 14DRMR17 motif determines A3F degradation (70). Vif derived from HIV-1 clone LAI, but not that of NL4-3, could induce the degradation of hA3H hapII, which is determined by residues F39 and H48 (71). The C terminus of HIV-1 Vif consists of one zinc coordination motif that interacts with CUL5, one SLQ BC box that binds to ELOB/C, and one Vif dimerization domain (reviewed in reference 72). Previously, it was also reported that the 171EDRW175 motif in the C terminus of Vif determines the degradation of A3F (66, 73). However, A3 interaction sites in SIV Vif have not yet been identified. Recently, it was reported that the 16PXXME…PHXXV47 domain and G48 of HIV-2 Vif and SIVsmm Vif are involved in the interaction with A3F and A3G, respectively (74).
In this study, we tested the sensitivities of primate A3Cs to several primate lentiviral Vif proteins. HIV-1 Vif had a distinct and restricted degradation profile for primate A3C proteins, while HIV-2, SIVagm, and SIVmac Vif degraded all tested A3C proteins. Additionally, three residues (106, 130, and 133) were identified in rhA3C and smmA3C that determined their resistance to HIV-1 Vif. We demonstrated that the equivalent residues in A3F (313 and 316) were unimportant for HIV-1 Vif sensitivity. Furthermore, two additional residues in the C terminus of the HIV-1 F-1 subtype Vif were identified as being involved in the interaction between HIV-1 Vif and hA3C/F. These observations suggest that Vif proteins from diverse HIV/SIV lineages have distinct interaction interfaces with A3C which mediate their degradation.
MATERIALS AND METHODS
Plasmids.
HIV-1, HIV-2, SIVagm, and SIVmac Vif genes were inserted into pcWPRE containing a C-terminal V5 tag (75). SIVpts1 (Tan1; SIVCPZTAN1.910) and SIVpts2 (Tan2; SIVCPZTAN2.69) Vifs were amplified from a full-length molecular clone of SIVcpz (76). Amplicons were digested by EcoRI and NotI and inserted into pcWPRE containing a C-terminal V5 tag. The 21 different HIV-1 strain Vif expression plasmids were kindly provided by Viviana Simon (71). Recently described A3 expression plasmids for hA3C, hA3G, and hA3F (77) as well as rhA3C agmA3C (21) were used. According to the cpzA3C sequence in NCBI (NM_001251910.1), K85, D99, E103, and N115 in hA3C were replaced by N85, E99, K103, and K155 using overlapping PCR, which produced the cpzA3C expression plasmid. All A3s were inserted into pcDNA3.1(+) (Life Technologies, Darmstadt, Germany) expressing a C-terminal hemagglutinin (HA) tag. All A3 chimeras and mutants and Vif mutants were generated by overlapping PCR. PCR primers are shown in Table 1. A3 chimeras were inserted into pcDNA3.1(+), and Vif mutants were inserted into pCRV1 as described previously (78). The expression plasmid of smmA3C was generated by exon assembly from white-crowned mangabey (Cercocebus torquatus lunulatus) genomic DNA. Three fragments were amplified separately using the primer pairs SmA3C-1.fw (5′-TAAGCGGAATTCGAACATGAATCCACAGATCAGAAACCCG-3′) and SmA3C-2.rv (5′-GATCGACCTGGTTTCGGAAG-3′), SmA3C-3.fw (5′-CTTCCGAAACCAGGTCGATC-3′) and SmA3C-4.rv (5′-CAATATTTAAAATCTTCGTAGCCC-3′), and SmA3C-5.fw (5′-GGGCTACGAAGATTTTAAATATTG-3′) and SmA3C-6HA.rv (5′-AGGATATCTCAAGCGTAATCTGGAACATCGTATGGATACTCGAGAATCTCCTG-3′). The fragments were fused through overlapping extension PCR, and the final fragment was amplified by using the forward SmA3C-1.fw and reverse SmA3C-6HA.rv primers. The amplicon was cloned into restriction sites EcoRI and EcoRV of the pcDNA3.1(+) plasmid and the sequence was verified. SIVmac-Luc (R-E-) and SIVmac-Luc (R-E-)ΔVif were provided by N. R. Landau (10). The HIV-1-Luc reporter system was described previously (79).
TABLE 1.
Primers used in this study
| Primer name | Product | Sequence (5′–3′) |
|---|---|---|
| PrmrRhAPO3C | TATAAGCTTTGAAGAGGAATGAATCCACAGATCAGAAACC | |
| PrmrRhAPOend | AGCTCGAGTCAAGCGTAATCTGGAACATCGTATGGATACTGAAGAATCTCCCGTAGGCG | |
| CM28 | AGCTCGAGTCAAGCGTAATCTGGAACATCGTATGGATACTGGAGACTCTCCCGTAGCCTT | |
| PrmrRhehu 9.1 | rh/hA3C9 | AAGGGCTCCAAGATGTGTAC |
| PrmrRhehu 9.2 | GTACACATCTTGGAGCCCTT | |
| PrmrRhehu 11.1 | rh/hA3C11 | CCACCTCCCCTGCACA |
| PrmrRhehu 11.2 | TGTGCAGGGGAGGTGG | |
| PrmrRhehu 1.1 | rh/hA3C13 | TCCACGGTGAAGCACAGC |
| PrmrRhehu 1.2 | GCTGTGCTTCACCGTGGA | |
| PrmrRhehu 3.1 | GTGAGATTCACGTTGCTGTGCCTGGCCAGGAACTTGG | |
| PrmrRhehu 3.2 | CCAAGTTCCTGGCCAGGCACAGCAACGTGAATCTCAC | |
| PrmrRhehu 1.1 | rh/hA3C15 | TCCACGGTGAAGCACAGC |
| PrmrRhehu 1.2 | GCTGTGCTTCACCGTGGA | |
| PrmrRhehu 11.1 | CCACCTCCCCTGCACA | |
| PrmrRhehu 11.2 | TGTGCAGGGGAGGTGG | |
| PrmrRhehu 1.1 | rh/hA3C17 | TCCACGGTGAAGCACAGC |
| PrmrRhehu 1.2 | GCTGTGCTTCACCGTGGA | |
| PrmrRhehu 9.1 | AAGGGCTCCAAGATGTGTAC | |
| PrmrRhehu 9.2 | GTACACATCTTGGAGCCCTT | |
| PrmrRhehu 7.1 | rh/hA3C31 | AGGAAGCACCTTTCTGCATG |
| PrmrRhehu 7.2 | CATGCAGAAAGGTGCTTCCT | |
| PrmrRhehu 3.1 | GTGAGATTCACGTTGCTGTGCCTGGCCAGGAACTTGG | |
| PrmrRhehu 3.2 | CCAAGTTCCTGGCCAGGCACAGCAACGTGAATCTCAC | |
| HuA3C-EcoRI-F | ATGAATTCGCCACCATGAATCCACAGATCAGAAAC | |
| HA-NotI-R | ATGCGGCCGCTCAAGCGTAATCTGGAACATC | |
| HuSmmC1 + 2-F | smm/hA3C1 + 2 | TCGGAACGAAACTTGGCTGTGCTTC |
| HuSmmC1 + 2-R | GAAGCACAGCCAAGTTTCGTTCCGA | |
| HuSmmC3 + 4-F | smm/hA3C3 + 4 | CCATTGTCATGCAGAAAGGTGCTTCCTCTC |
| HuSmmC3 + 4-R | GAGAGGAAGCACCTTTCTGCATGACAATGG | |
| HuSmmC5 + 6-F | smm/hA3C5 + 6 | TGCAGGGGAGGTGGCCGAGTTCCTGGCCAGG |
| HuSmmC5 + 6-R | CCTGGCCAGGAACTCGGCCACCTCCCCTGCA | |
| HuSmmC7 + 8-F | smm/hA3C-7 + 8 | AGATTTTAAATATTGTTGGGAAAACTTTGTG |
| HuSmmC7 + 8-R | CACAAAGTTTTCCCAACAATATTTAAAATCT | |
| HuSmmC9 + 10-F | smm/hA3C-9 + 10 | ATGAGCCATTCAAGCCTTGGAAGGGA |
| HuSmmC9 + 10-R | TCCCTTCCAAGGCTTGAATGGCTCAT | |
| RhA3CCE+QE-F | rhA3C-CE+QE | CAGTATCCGTGTTACCAGGAGGGGCTCCGCAGCCTGAGTCAGGAAGGAGTC |
| RhA3CCE+QE-R | GACTCCTTCCTGACTCAGGCTGCGGAGCCCCTCCTGGTAACACGGATACTG | |
| SmmA3CCE+QE-F | smmA3C-CE+QE | GGGATACATGTTACCAGGAGGGGCTCCGCAGCCTGAGTCAGGAAGGG |
| SmmA3CCE+QE-R | CCCTTCCTGACTCAGGCTGCGGAGCCCCTCCTGGTAACATGTATCCC | |
| HuA3CE106K-F | hA3C E106K | GGAGGTGGCCAAGTTCCTGGC |
| HuA3CE106K-R | GCCAGGAACTTGGCCACCTCC | |
| HuA3CQE-EK-F | hA3C QE-EK | AGCCTGAGTGAGAAAGGGGTCGCTG |
| HuA3CQE-EK-R | CAGCGACCCCTTTCTCACTCAGGCT | |
| HuA3CNQ+EK-F | hA3C NQ+EK | CAGTATCCAAATTACCAGCAGGGGCTCCGCAGCCTGAGTGAGAAAGGG |
| HuA3CNQ+EK-R | CCCTTTCTCACTCAGGCTGCGGAGCCCCTGCTGGTAATTTGGATACTG | |
| HuA3FDE-HQ-F | hA3F DE-HQ | CTGGGATACACATTACCAGCAGGGGCTC |
| HuA3FDE-HQ-R | GAGCCCCTGCTGGTAATGTGTATCCCAG | |
| HuA3FQE-EK-F | hA3F QE-EK | GCAGCCTGAGTGAGAAAGGGGCCTCCG |
| HuA3FQE-EK-R | CGGAGGCCCCTTTCTCACTCAGGCTGC | |
| HIV1Vif-EcorI-F | ATGAATTCGCCACCATGGAAAACAGATGGCAGG | |
| HIV1Vif-NotI-R | ATGCGGCCGCCTAGTGTCCATTCATTGTATGG | |
| F1D-G-NotI-R | F-1 D-G | ATGCGGCCGCCTAGTGTCCATTCATTGTATGGCTCCCTCTGTGGCCCTTGGTC |
| F1KD-RG-F | F-1K D-RG | AAGCCACCTTTGCCCAGTGTTA |
| F1KD-RG-R | TAACACTGGGCAAAGGTGGCTT | |
| F1K-R-F | F-1 K-R | GCCCAGTGTTAGGAAACTGACAGAG |
| F1K-R-R | CTCTGTCAGTTTCCTAACACTGGGC | |
| HIV1VIFRR-AA-F | B-NL4-3 RR-AA | GCAAGTAGACGCGATGGCGATTAACACATGG |
| HIV1VIFRR-AA-R | CCATGTGTTAATCGCCATCGCGTCTACTTGC | |
| HIV1VIFED-AA-F | B-NL4-3 ED-AA | GAAACTGACAGCGGCCAGATGGAAC |
| HIV1VIFED-AA-R | GTTCCATCTGGCCGCTGTCAGTTTC | |
| HIV1VIFD101G-F | B-NL4-3 D-G | CACAAGTAGACCCTGGCCTAGCAGACC |
| HIV1VIFD101G-R | GGTCTGCTAGGCCAGGGTCTACTTGTG |
Cells, transfections, and infections.
HEK293T (293T; ATCC CRL-3216) cells were maintained in Dulbecco's high-glucose modified Eagle's medium (DMEM; Biochrom, Berlin, Germany) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). A3 degradation experiments were performed in 24-well plates, and 1 × 105 293T cells were transfected with 150 ng hA3G, 150 ng hA3F, or 50 ng hA3C expression plasmids together with 350 ng HIV-1, SIVagm, or SIVmac Vif; pcDNA3.1(+) was used to increase the total transfected plasmid DNA to 500 ng. To produce SIVmac-luciferase viruses, 293T cells were cotransfected with 650 ng SIVmac-Luc (R-E-) or SIVmac-Luc (R-E-)ΔVif, 150 ng A3C expression plasmid, 25 ng vesicular stomatitis virus glycoprotein (VSV-G) plasmid (pMD.G), or 200 ng HIV-1 Vif expression plasmid; in some experiments pcDNA3.1(+) was used instead of Vif or A3 expression plasmids. For HIV-1-luciferase viruses, transfection experiments in 6-well plates consisted of 300 ng of the HIV-1 packaging construct pMDLg/RRE, 130 ng of HIV-1 Rev expression plasmid pRSV-Rev, 300 ng of the HIV-1 reporter vector pSIN.PPT.CMV.Luc.IRES.GFP, 100 ng of the VSV-G expression plasmid pMD.G, and 800 ng HIV-1 Vif together with 800 ng hA3F expression plasmids. pcDNA3.1(+) was used instead of HIV-1 Vif or hA3F. At 48 h posttransfection, cells and supernatants were collected. The reverse transcriptase (RT) activity of SIVmac and HIV-1 was quantified by using the Cavidi HS lenti RT kit (Cavidi Tech, Uppsala, Sweden). For reporter virus infection, 293T cells were seeded in 96-well plates 1 day before transduction. After normalizing for RT activity, the same amounts of viruses were used for infection. Two days postransduction, firefly luciferase activity was measured with the Steadylite HTS reporter gene assay system (Perkin-Elmer, Cologne, Germany) according to the manufacturer's instructions on a MicroLumat Plus luminometer (Berthold Detection Systems, Pforzheim, Germany). Each sample was analyzed in triplicate; the error bars for each triplicate are shown.
Immunoblot analysis.
Transfected 293T cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-HCl [pH 8.0], 137 mM NaCl, 1% NP-40, 1% glycerol, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 2 mM EDTA, and protease inhibitor cocktail set III [Calbiochem, Darmstadt, Germany]). To pellet virions, culture supernatants were centrifuged at 12,000 rpm for 10 min, subjected to centrifugation through a 20% sucrose cushion at 35,000 rpm in an SW40 Ti rotor for 1.5 h, and resuspended in RIPA buffer. The solution was boiled at 95°C for 5 min with Roti load-reducing loading buffer (Carl Roth, Karlsruhe, Germany) and resolved on an SDS-PAGE gel. The expression of A3s and lentivirus Vif was detected by mouse anti-HA antibody (1:7,500 dilution; MMS-101P; Covance, Münster, Germany) and mouse anti-V5 antibody (1:4,500 dilution; MCA1360; ABD Serotec, Düsseldorf, Germany) separately. HIV-1 Vifs from different subtype strains were detected by rabbit anti-Vif polyclonal antibody (1:1,000 dilution; catalog no. 2221; NIH AIDS Research and Reference Reagent Program) (80). Tubulin, SIVmac, and HIV-1 capsid proteins were detected using mouse anti-α-tubulin antibody (1:4,000 dilution; clone B5-1-2; Sigma-Aldrich, Taufkirchen, Germany) and mouse anti-capsid p24/p27 monoclonal antibody AG3.0 (1:50 dilution) separately (81), followed by horseradish peroxidase-conjugated rabbit anti-mouse or donkey anti-rabbit antibody (α-mouse or rabbit-IgG-horseradish peroxidase; GE Healthcare, Munich, Germany), and developed with ECL chemiluminescence reagents (GE Healthcare).
IP.
To determine Vif and A3 binding, 293T cells were cotransfected with 1 μg HIV-1 Vif.SLQ-AAA and 1 μg wild-type or mutant A3 or with pcDNA3.1(+). Forty-eight hours later, the cells were lysed in immunoprecipitation (IP) lysis buffer (50 mM Tris-HCl pH 8, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10% glycerol, 0.8% NP-40, 150 mM NaCl, and complete protease inhibitor; Roche, Penzberg, Germany). The lysates were cleared by centrifugation. The supernatant were incubated with 20 μl α-HA affinity matrix beads (Roche) at 4°C for 2 h. The samples were washed 5 times with lysis buffer. Bound proteins were eluted by boiling the beads for 5 min at 95°C in SDS loading buffer. Immunoblot analysis and detection were done as described above.
Model structure.
To analyze the interaction surface between HIV-1 Vif and hA3C/A3F, the hA3C (PDB entry 3VOW), hA3F C-terminal (PDB entry 4J4J), and HIV-1 Vif (PDB entry 4N9F) structures were used. The structural models of F-1 Vif and mutants were built by the SWISS-MODEL online server (http://www.swissmodel.expasy.org/) (82) using B-NL4-3 Vif (PDB entry 4N9F) as a template. The graphical visualizations shown in Fig. 8 and 9 were constructed using PyMOL (PyMOL Molecular Graphics System, version 1.5.0.4; Schrödinger, Portland, OR).
FIG 8.

Identification of important residues in subtype F-1 Vif that determine its counteraction of hA3C/F. (A) Schematic structure of F-1 Vif mutants. The N-terminal hA3H hapII box and C-terminal hA3C/F box are shown. (B, C, and D) hA3C and hA3F were transfected into 293T cells together with B-NL4-3 Vif or with F-1 Vif or F-1 Vif mutant. A3 and Vif were detected by using anti-HA and anti-Vif antibodies. Tubulin served as a loading control. (E) The structures of wild-type F-1 Vif and F-1 Vif mutants were modeled by SWISS modeling. The internal interaction between residues 171 and 167 was analyzed by PyMOL and is shown as a red dashed line.
FIG 9.

Structural differences between B-NL4-3 Vif and F-1 Vif. (A and C) hA3C and hA3F or hA3G expression plasmids were cotransfected together with B-NL4-3 Vif and its mutants. A3 and Vif were detected by using anti-HA and anti-Vif antibodies. Tubulin served as a loading control. (B) The structures of B-NL4-3, F-1 Vif, and their variants were modeled by SWISS modeling. The internal interaction between residues 171, 167, and 101 were analyzed by PyMOL, and the distances between interacting side chains are between 3.4 and 3.8 Å. The internal interaction is shown as a red dashed line. (D) Structure analysis of HIV-1 Vif (PDB entry 4N9F) for hA3C/hA3F interaction sites. Three discontinuous HIV-1 Vif motifs (F1 box, F2 box, and F3 box) that interact with hA3C/F are shown in red. 167R that contacted the F3 box is shown in magenta.
Statistical analysis.
Data are represented as the means with standard deviations (SD) in all bar diagrams. Statistically significant differences between two groups were analyzed using the unpaired Student's t test with GraphPad Prism, version 5 (GraphPad Software, San Diego, CA, USA). A minimum P value of 0.05 was considered statistically significant. A P value of <0.001 was extremely significant (***), 0.001 to 0.01 very significant (**), 0.01 to 0.05 significant (*), and >0.05 not significant (ns).
Accession number(s).
Sequences for human A3C (NM_014508.2), chimpanzee A3C (NM_001251910.1), gorilla A3C (AH013821.1), African green monkey A3C (EU381232.1), sooty mangabey monkey A3C (KX405162), rhesus monkey A3C (JF714486.1), SIVpts1 (Tan1) (EF394356.1), and SIVpts2 (Tan2) (EF394357.1) were deposited in NCBI.
RESULTS
The sensitivity of primate A3C to lentiviral Vif.
A previous study has identified several residues in the hydrophobic V-shaped groove formed by the α2 and α3 helices of A3C involved in HIV-1 Vif interaction (63). This interaction interface is conserved in primate A3C proteins, although rhA3C has one substitution (E to K) at position 106 (Fig. 1A). To test the sensitivities of these A3C proteins to HIV-1 Vif, cotransfections of expression plasmids for A3Cs and HIV-1 Vif were performed. A3C steady-state levels were assessed by immunoblotting protein lysates of coexpressing cells. Previous studies demonstrated that both SIVagm and HIV-1 Vif were able to induce the degradation of human A3C by the proteasome (83). Based on this finding, we assume that the dominant mechanism of Vif against A3C in our study is Vif-triggered degradation of A3 as well. Results show that HIV-1 Vif reduced the stability of hA3C and cpzA3C, while smmA3C, agmA3C, and rhA3C were resistant (Fig. 1B). smmA3C was cloned from a white-crowned mangabey (Cercocebus torquatus lunulatus), which is one of the three subspecies of sooty mangabey monkeys. The resistance of smmA3C to HIV-1 Vif was unexpected, because the previously identified HIV-1 Vif interaction sites were identical between smmA3C and hA3C (Fig. 1A). HIV-1 Vif's block to deplete rhA3C may be caused by an E-to-K substitution at position 106. To prove this, we made a K106-to-E mutation in rhA3C and tested its sensitivity to HIV-1 Vif. However, the results showed that rhA3C.K106E still could not be degraded by HIV-1 Vif (Fig. 1B). These observations indicate that sites in addition to the already characterized residues in A3C are involved in the HIV-1 Vif interaction. We also observed that HIV-2, SIVmac, and SIVagm Vif depleted all A3C proteins, including rhA3C.K106E (Fig. 1B). These data suggest that Vif proteins from diverse HIV/SIV lineages have distinct binding sites in A3C which mediate its degradation.
FIG 1.

Sensitivity of primate A3C to lentiviral Vifs. (A) Sequence alignment of primate A3C proteins. Black stars represent important residues for HIV-1 Vif interaction. Green stars indicate the dimerization of A3C protein and anti-SIV activity. The red star is the A3C RNA binding site and indicates its viral incorporation. (B) 293T cells were cotransfected with A3C and HIV-1, HIV-2, SIVagm, or SIVmac Vif expression plasmids. Expression of A3Cs, Vifs, and tubulin was detected by immunoblotting using anti-HA, anti-V5, and anti-tubulin antibodies, respectively. h, cpz, gor, agm, smm, and rh represent human, chimpanzee, gorilla, African green monkey, sooty mangabey monkey, and rhesus monkey, respectively.
We next tested the anti-SIVmac activities of these primate A3C proteins in the presence of SIVmac or HIV-1 Vif proteins. Luciferase reporter viruses of SIVmac, SIVmacΔVif, and SIVmacΔVif plus Vif of HIV-1 were produced with hA3G, hA3C, cpzA3C, rhA3C, agmA3C, or smmA3C. The Vif-proficient virus SIVmac-Luc expresses Vif in its natural expression context; however, Vif lacks an epitope tag for detection. Viral particles were normalized by RT activity, and the luciferase activity of infected cells was quantified 2 days postinfection. All A3C proteins strongly inhibited the infectivity of Vif-deficient SIVmac, which was fully counteracted by SIVmac Vif, as was hA3G (Fig. 2A). The immunoblots of virus-producing cells indicated that SIVmac Vif reduced the stability of hA3G and all primate A3C proteins, which resulted in viral particles with much reduced A3 content (Fig. 2B). However, HIV-1 Vif counteracted the anti-SIVmac activity of hA3G, hA3C, and cpzA3C, and rhA3C, agmA3C, and smmA3C displayed resistance to HIV-1 Vif (Fig. 2C). The immunoblots showed that HIV-1 Vif decreased the protein level of hA3G, hA3C, and cpzA3C in cell lysates and viral particles but not that of rhA3C, agmA3C, and smmA3C (Fig. 2D). Since the HIV-1 Vif we used here contained a C-terminal V5 tag, we wondered whether the V5 tag in Vif would contribute to the Vif resistance of rhA3C, agmA3C, and smmA3C. We then tested the sensitivity of A3Cs to untagged HIV-1 Vif. We found that hA3C and cpzA3C were sensitive to antagonization by untagged HIV-1 Vif, whereas rhA3C, agmA3C, and smmA3C still were not degraded (Fig. 2E). Thus, the V5 tag in Vif did not contribute to the HIV-1 Vif resistance of rhA3C, agmA3C, and smmA3C. It is important to point out that cpzA3C was less efficiently depleted from cells by HIV-1 Vif (untagged and V5 tagged) than hA3C (Fig. 2D and E). The immunoblotting results showed that SIVcpz (SIVpts1 and SIVpts2) Vifs displayed stronger activity against hA3C and cpzA3C than HIV-1 Vif (Fig. 2F).
FIG 2.

SIVmac Vif, but not HIV-1 Vif, counteracts primate A3Cs. (A and C) 293T cells were transfected with expression plasmids for SIVmacΔVif-Luc or SIVmac-Luc (A) or SIVmacΔVif-Luc or SIVmacΔVif-Luc plus HIV-1 Vif (C), together with expression plasmids for hA3G and primate A3Cs. pcDNA3.1(+) was used as a control (vector). After normalizing for reverse transcriptase activity, viral infectivity was determined by quantification of luciferase activity in 293T cells. (B and D) Lysates of SIVmac producer cells were used to detect the expression of A3s, SIVmac capsid (p27), or HIV-1 Vif by anti-HA, anti-p27, or anti-V5 antibody, respectively. Tubulin served as a loading control. Encapsidation of A3s into SIVmac was detected by anti-HA antibody. (E) Primate A3Cs and HIV-1 Vif (without tag) expression plasmids were cotransfected into 293T cells. The expression of A3C and HIV-1 Vif was detected by anti-HA and anti-Vif antibodies. (F) Expression plasmids for hA3C and cpzA3C were cotransfected with HIV-1 Vif or SIVcpz Vif into 293T cells. The expression of A3C and Vif was detected by anti-HA and anti-V5 antibodies. Pts, Pan troglodytes schweinfurthii. cps, counts per second. VLP, virus-like particle. Asterisks represent statistically significant differences: ***, P < 0.001; ns, no significance (Dunnett's t test).
Identification of specific rh/smmA3C residues involved in resistance to HIV-1 Vif.
The results from Fig. 1B suggest that additional sites in hA3C are involved in the interaction with HIV-1 Vif. To identify the additional determinants, several rh/hA3C chimeras were constructed. Structures of A3 chimeras are shown in Fig. 3A, and their sensitivities to HIV-1 Vif were determined by immunoblotting of cell lysates expressing Vif and individual A3 constructs. The results showed that hA3C chimera 9 and chimera 11 were sensitive to HIV-1 Vif-mediated depletion, while rhA3C chimeras 13, 15, 17, and 31 were resistant and not degraded (Fig. 3B). The composition of the chimeras indicated that the C terminus (from amino acids 120 to 190) of rhA3C determined the resistance to HIV-1 Vif. smmA3C was also resistant to HIV-1 Vif-induced degradation (Fig. 1A). To narrow the scope of residues in A3C that are involved in the HIV-1 Vif interaction, 10 chimeras of human A3C and smmA3C were produced. The structures of smm/hA3C chimeras are displayed in Fig. 3C. We found that hA3C chimeras 2, 4, 6, 7, and 9 could be depleted by HIV-1 Vif, while smmA3C and other chimeras evidently were not degraded (Fig. 3D). Based on these results, amino acids 120 to 150 of smmA3C were identified as an essential domain that confers resistance to HIV-1 Vif.
FIG 3.

Sensitivity of rh/hA3C and h/smmA3C chimeras to HIV-1 Vif-mediated depletion. (A and C) Schematic structure of rh/hA3C and h/smmA3C chimeras. Amino acid positions 120, 150, and 190 in hA3C are indicated. (B and D) Immunoblots of protein lysates of cotransfected cells displaying the A3's sensitivity to HIV-1 Vif-triggered degradation. The expression of A3s and HIV-1 Vif were analyzed by using anti-HA and anti-V5 antibodies. Tubulin served as a loading control. +, with HIV-1 Vif; −, without HIV-1 Vif.
To identify the residues in rhA3C and smmA3C that are important for resistance to HIV-1 Vif-mediated counteraction, we compared the amino acids from 120 to 150 in primate A3C proteins, and four residues (130, 133, 140, and 141) were selected for further characterization. In hA3C and cpzA3C these residues are C130, E133, and 140QE141, while in agmA3C, rhA3C, and smmA3C these four positions are N/H130, Q133, and 140EK141, respectively (Fig. 1A). Thus, we mutated N130, Q133, and 140EK141 in rhA3C to the residues found in hA3C, termed rhA3C.CE+QE. Based on this construct, we performed the single-amino-acid substitution K106E, named rhA3C.K106E-CE+QE (Fig. 4A), because E106 in hA3C indicates it is binding to HIV-1 Vif (65). For smmA3C, four amino acid substitutions were produced, and the construct was termed smmA3C.CE+QE (Fig. 4A). The sensitivities of these constructs together with wild-type hA3C, rhA3C, and smmA3C to HIV-1 Vif-triggered depletion were tested by immunoblotting. The results showed that rhA3C.CE+QE was still resistant to HIV-1 Vif, but rhA3C.K106E-CE+QE was sensitive to hA3C (Fig. 4B). This result confirmed the importance of E106 in hA3C involvement in the HIV-1 Vif interaction. The smmA3C.CE+QE protein was depleted by HIV-1 Vif, which suggests that C130, E133, and 140QE141 are also important for HIV-1 Vif-mediated degradation. In addition, the equivalent residues (C130, E133, and 140QE141) in hA3C were replaced by N130, Q133, and 140EK141, respectively, producing hA3C.CE-NQ, hA3C.QE-EK, and hA3C.NQ+QE (Fig. 4C). hA3C.E106K, which could not be antagonized by HIV-1 Vif, was used as a control. The results showed that hA3C.E106K was partially resistant to HIV-1 Vif but that hA3C.QE-EK was depleted (Fig. 4D), showing consistency with previous studies (63). Moreover, we found that hA3C.CE-NQ and hA3C.NQ+EK were resistant to HIV-1 Vif-mediated degradation (Fig. 4D). Taken together, these results demonstrate that residues 106, 130, and 133 of primate A3C proteins determine their sensitivities to HIV-1 Vif, but residues 140QE141 were unimportant for HIV-1 Vif interaction.
FIG 4.
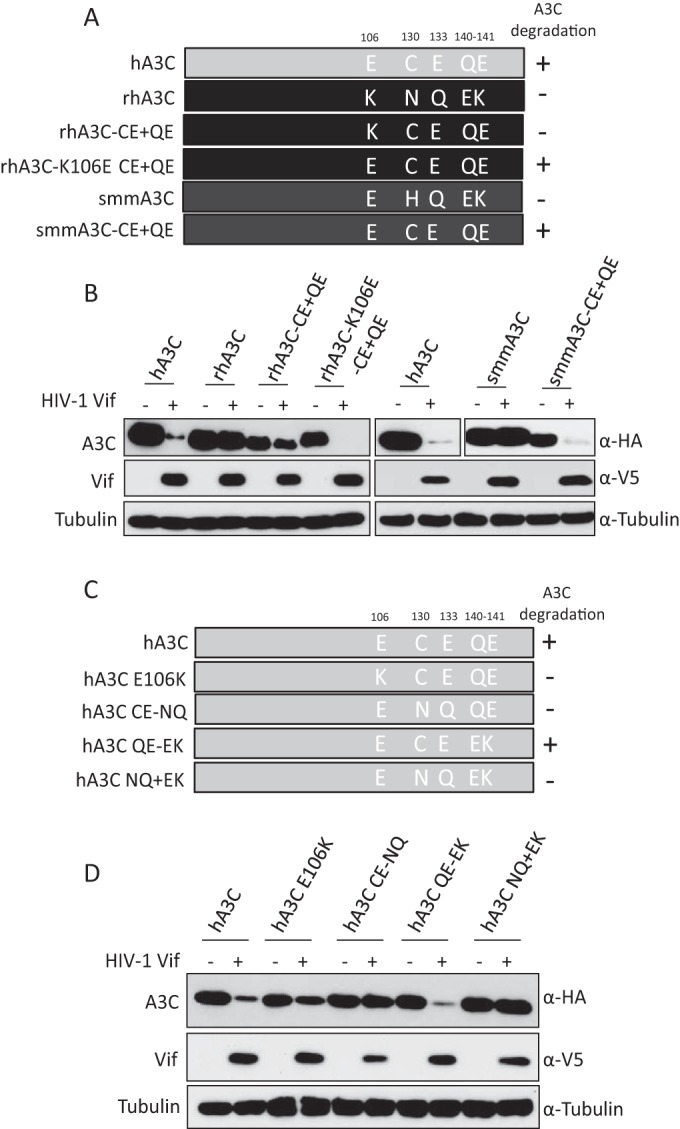
Identification of determinants in rhA3C and smmA3C that confer resistance to HIV-1 Vif. (A) The schematic structure of rhA3C and smmA3C mutants. The numbers represent amino acid positions in A3C. +, sensitive to HIV-1 Vif-induced degradation; −, resistant to HIV-1 Vif-induced degradation. (B and D) hA3C, rhA3C, smmA3C, or A3C mutants were cotransfected with HIV-1 Vif into 293T cells. A3, HIV-1 Vif, and tubulin were detected by using anti-HA, anti-V5, and anti-tubulin antibodies, respectively. (C) Schematic structure of hA3C mutants.
A3F and A3C have distinct Vif interaction interfaces.
After identification of the residues C130 and E133 in hA3C as being important for interaction with HIV-1 Vif, we expanded our experiments to hA3F. The equivalent residues (D313 and E316) in hA3F were replaced by H313 and Q316, which were present in rhA3F. In addition, we altered amino acids Q323E and E324K in hA3F (Fig. 5A). We analyzed the anti-HIV-1 activity of these hA3F mutants in the presence of HIV-1 Vif. HIV-1 Vif restored the infectivity in the presence of hA3F and hA3F.DE-HQ, while hA3F.QE-EK displayed antiviral activity in the presence and absence of HIV-1 Vif (Fig. 5B). The corresponding immunoblots of virus-producing cells showed that HIV-1 Vif reduced the steady-state level of hA3F and hA3F.DE-HQ but not of hA3F.QE-EK (Fig. 5C). Q323E and E324K alterations in hA3F impaired HIV-1 Vif-mediated depletion, consistent with previous studies (63, 67). However, we and other groups also observed that the equivalent residues (Q140 and E141) in hA3C were unimportant for HIV-1 Vif interaction (63) (Fig. 4D). We then compared the structures of hA3C and the C-terminal domain of hA3F and highlighted the HIV-1 Vif interaction surface investigated in our study. As shown in Fig. 5D, residue E106 in hA3C and the corresponding residue E289 in hA3F, which are located in the α3 helices, were involved in HIV-1 Vif interaction. In the α4 helices, the HIV-1 Vif interaction surface differs in hA3C and hA3F, shifting from the top to the bottom of α4 helices (Fig. 5D). Taken together, these results indicate that the Vif-A3 interaction interfaces in hA3C and hA3F are different.
FIG 5.

hA3F residues and HIV-1 Vif-induced depletion. (A) The schematic structure of hA3F mutants. +, sensitive to HIV-1 Vif induced degradation; −, resistant to HIV-1 Vif-induced degradation. (B) HIV-1ΔVif luciferase or HIV-1ΔVif luciferase plus HIV-1 Vif were produced in 293T cells in the presence of A3F or mutants. After normalizing for reverse transcriptase activity, viral infectivity was determined by quantification of luciferase activity in 293T cells. (C) A3s in HIV-1 producer cells and HIV-1 viral particles were detected by using anti-HA antibody. HIV-1 Vif and capsid (p24) were detected by anti-Vif and anti-p24 antibodies, respectively. Tubulin served as a loading control. (D) Superimposition of A3C crystal structure (PDB entry 3VOW) (green) and A3F-CTD crystal structure (PDB entry 4J4J) (light blue). Key residues E106, C130, and E133 are shown as orange surfaces in A3C with labels, and the essential residues (E289, Q323, and E324) in A3F are highlighted with gray surfaces and labels. cps, counts per second. Asterisks represent statistically significant differences: ***, P < 0.001; ns, no significance (Dunnett's t test).
To clarify whether the identified residues of A3C and A3F were directly involved in Vif binding, coimmunoprecipitation (co-IP) assays of Vif.SLQ/AAA, which binds A3s without inducing their degradation, were performed with HA-tagged A3C and A3F. Because the expression levels of primate A3C and several mutants were distinct, ImageJ software was used to evaluate the band density of A3C and Vif from immunoprecipitation. The ratio of Vif to A3 was calculated as A3-Vif binding activity in which we set wild-type A3C-Vif or wild-type A3F-Vif binding activity as 100%. As shown in Fig. 6, rhA3C and its mutant, rhA3C-K106E-CE+QE, which was sensitive to HIV-1 Vif, immunoprecipitated with HIV-1 Vif (Fig. 6A); however, binding was much reduced compared to Vif binding to hA3C. The smmA3C and smmA3C.CE+QE protein levels were much lower in the cell lysates, so they precipitated less than hA3C in co-IP complexes, whereas the amount of detected HIV-1 Vif was similar to that of the hA3C-Vif complex (Fig. 6A), indicating that smmA3C has high binding activity to HIV-1 Vif (Fig. 6B). The hA3C.E106K and hA3C.QE-EK mutants showed very low HIV-1 Vif binding, which was consistent with a previous study (63). However, hA3C.CE-NQ, which was not degraded by HIV-1 Vif, pulled down similar amounts of Vif compared to the wild-type hA3C-Vif complex (Fig. 6A). The HIV-1 Vif binding capacities of hA3F and hA3F.DE-HQ were similar, corresponding to similar sensitivities to HIV-1 Vif (Fig. 5B and 6B). Significantly reduced amounts of Vif were pulled down by hA3F.QE-EK, suggesting hA3F residues 323QE324 were involved in Vif binding. RhA3C and smmA3C were totally resistant to HIV-1 Vif-induced degradation (Fig. 4B and D); however, both of these proteins showed robust binding to HIV-1 Vif, suggesting that the Vif binding detected by co-IPs is unrelated to Vif-mediated degradation.
FIG 6.
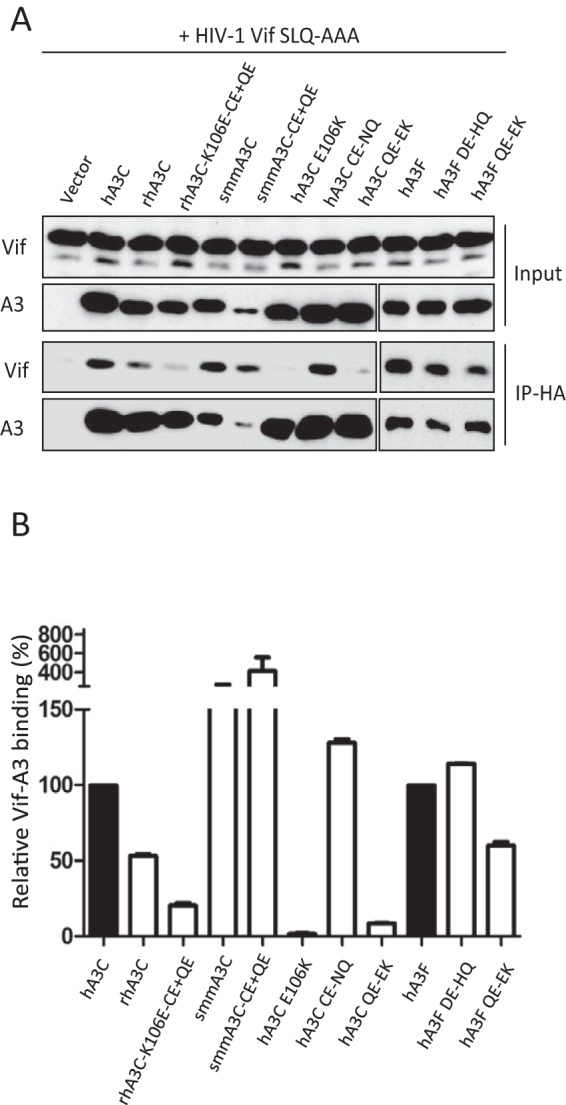
HIV-1 Vif binding to A3C and A3F mutants. (A) Coimmunoprecipitation of HIV-1 Vif SLQ/AAA with A3C and A3F mutants. Protein cell lysates (Input) and immunoprecipitated complexes (IP) were analyzed by immunoblotting with anti-Vif for HIV-1 Vif or anti-HA for A3. (B) ImageJ was used to evaluate the band density of A3C and Vif from immunoprecipitation. The ratio of Vif to A3 was calculated as A3-Vif binding activity. WT A3C-Vif and WT A3F-Vif binding activity was set as 100%.
Internal salt bridge of Vif protein influences degradation of hA3C/F.
Previous studies have identified three motifs of HIV-1 Vif specifically involved in the interaction with hA3F: the F1 box (14DRMR17), F2 box (74TGEDW79), and F3 box (171EDRW174) (66, 84). To identify additional determinants in HIV-1 Vif relevant for interaction with hA3C/F, 21 HIV-1 Vif alleles (71) were used for detecting their hA3C counteractivity, including five Vifs isolated from HIV-1 N and O subtypes. All Vif alleles were detectable by anti-Vif polyclonal antibody (Fig. 7A). The Vif variants (C1, C2, and C3) displayed lower-level signals in the immunoblots than the other Vifs, which might be caused by weaker antibody recognition (Fig. 7A). The 21 Vif alleles depleted hA3C to various degrees. To evaluate the degree of this counteraction, ImageJ was used to calculate the density of hA3C bands in which hA3C cotransfected with empty vector plasmid was set as the control (100% expression). The results showed that 10 Vif variants (A2, B-NL4-3, B-LAI, C3, D2, F-3, F-4, O-119, O-127, and N-116) strongly depleted hA3C (Fig. 7A and B). All other Vif variants, except Vif from F-1, moderately reduced the protein level of hA3C. The F-1 Vif had an expression level similar to those of B-NL4-3 and B-LAI Vif, but almost 80% of hA3C could be detected in the presence of F-1 Vif (Fig. 7A and B). A previous study demonstrated that F-1 Vif, but not Vif from F-2 and B-NL4-3, could mediate the degradation of hA3H hapII (71). To understand whether the resistance of hA3C to F-1 Vif is specific, three Vif variants (B-NL4-3, F-1, and F-2) were tested for counteraction of hA3H hapII, hA3G, hA3F, and hA3C. The results showed that the protein levels of both hA3C and hA3F could be reduced by B-NL4-3 and F-2 Vif but displayed resistance to F-1 Vif. hA3G was strongly depleted by these three Vif variants, while hA3H hapII was resistant to B-NL4-3 and F-2 Vif but sensitive to F-1 Vif (Fig. 7C), consistent with previous data (71). Taken together, these results indicated that the differences between F-1, F-2, and B-NL4-3 Vifs determine specific degradation of hA3C/F.
FIG 7.

hA3C depletion activities of HIV-1 Vif alleles. (A) The 21 HIV-1 Vif alleles induced depletion of hA3C to various degrees. hA3C and Vif alleles were cotransfected into 293T cells. Protein extracts of transfected cells were used for detecting the expression of hA3C, Vif, and tubulin by anti-HA, anti-Vif, and anti-tubulin antibodies, respectively. Vifs from N and O subtypes were detected by anti-myc antibody. This A3C experiment was repeated at least three times with similar results. (B) ImageJ was used to evaluate the band density of A3C and tubulin. The ratio of A3C to tubulin was calculated as A3C expression efficiency. A3C plasmid cotransfected with empty vector plasmid was set as 100% expression. (C) hA3C, hA3F, hA3G, and hA3H hapII expression plasmids were transfected together with B-NL4-3 expression plasmids and F-1 and F-2 Vif variants. A3 and Vif were detected by using anti-HA and anti-Vif antibodies. Tubulin served as a loading control.
After analyzing the sequences of B-NL4-3, F-1, and F-2 Vif, six residues outside the CUL5 and BC boxes were identified (positions 39, 48, 61, 62, 167, and 182) where all three Vifs differed from each other. Residues F39 and H48 in Vif determine the interaction with hA3H hapII (71). Based on the structure of Vif, residues 61 and 62 are on the other side of the F box that was identified for the interaction with hA3F. Thus, we focused on residues 167 and 182 in F-1 Vif. K167 and D182 in F-1 Vif were mutated to R and G, which are the corresponding residues in B-NL4-3 Vif both separately and in combination (Fig. 8A). Wild-type Vif and Vif mutants were tested in coexpression experiments for their effect on the steady-state levels of hA3C and hA3F. The immunoblot showed that replacing K167 with arginine recovered the F-1 Vif depletion activity against hA3C, while the introduction of glycine at position 182 did not help much to enhance A3C reduction (Fig. 8B). However, both K167R and D182G exchanges activated F-1 Vif counteraction of hA3F, which suggests that G182 is important for degradation of hA3F but not hA3C (Fig. 8B). We next replaced K167 in F-1 Vif by a negatively charged amino acid, D/E, as well as a nonpolar alanine, and analyzed their hA3C/hA3F inhibitory activities. We found that mutants K167A, K167D, and K167E depleted hA3C/hA3F as efficiently as B-NL4-3 Vif (Fig. 8C). The F3 box (171EDRW174) that interacts with hA3C/F is conserved in B-NL4-3 and F-1 Vif (Fig. 8A). Thus, we wondered whether the F3 box in F-1 Vif is still essential for inhibition of hA3C/hA3F. To address this question, 171ED172 to 171AA172 mutations were generated in both F-1 wild-type Vif and F-1 Vif.K167R constructs. The immunoblot revealed that replacing K167 with R recovered the F-1 Vif activity against hA3C/hA3F, but mutations in the F3 box together with K167R impaired degradation, suggesting that the F3 box in F-1 Vif is still key to the function of interaction with hA3C/hA3F (Fig. 8D). The Vifs from B-NL4-3 and F-1 share 90.3% identical residues. Thus, we modeled the structures of F-1 Vif and its mutants using the structure of B-NL4-3 Vif (PDB entry 4N9F) as a template and analyzed the interactions of residues 171 and 167 within these model variants. We found that E171 and K167 displayed a strong interaction in the F-1 Vif, while the models of K167A, K167D, and K167E displayed no interaction (Fig. 8E). Taken together, these results suggest that the side-chain interaction of E171 and K167 affect hA3C/hA3F-Vif interaction induced by E171, resulting in an F-1 Vif being inactive in the induction of the degradation of hA3C/hA3F.
Additionally, we exchanged R167 and G182 in B-NL4-3 Vif for K and D, which were found in F-1 Vif. As controls we included F2 and F3 box mutants (B-NL4-3.RR-AA and B-NL4-3.ED-AA). Wild-type Vif and Vif mutants were tested for affecting the protein stability of hA3C, hA3F, and hA3G in Vif/A3 coexpression experiments. The results showed that mutations in the F2 box and the F3 box indeed impaired Vif activity against hA3C and hA3F but not against hA3G (Fig. 9A). Surprisingly, substitution R167K or G182D or the combination of both mutations did not impair depletion of hA3C, hA3F, and hA3G by B-NL4-3 Vif (Fig. 9A). To analyze the internal structural changes of the R167K Vif mutant, we modeled the structures of B-NL4-3 Vif.R167K and analyzed the E171-K167 interaction. We found that residue K167 in B-NL4-3 Vif.R167K preferred to interact with D101 but not with E171 (Fig. 9B). In F-1 Vif, a glycine was present at position 101 which had no chance to form an internal interaction with K167 (Fig. 9B). Thus, replacing D101 in B-NL4-3 Vif.R167K with glycine could rebuild the connection of E171-K167 (Fig. 9B). To test this hypothesis, we analyzed the counteractivity of B-NL4-3 Vif.D101G and B-NL4-3 Vif.D101G+R107K against different amounts of hA3C- or hA3F-transfected (150 ng, 250 ng, or 350 ng) expression plasmid. We found that B-NL4-3 Vif.D101G+R107K reduced the capacity to induce depletion of hA3F and hA3C compared with B-NL4-3 wild-type Vif (Fig. 9C). As shown in Fig. 9D, three discontinuous motifs (shown in red) in Vif (B-NL4-3) formed the hA3F and hA3C interaction surface, while the residues R167 and D101, which determined internal interaction with the F3 box, are shown in magenta (Fig. 9D). Taken together, these results suggest that internal interactions in the Vif protein can also influence the Vif-A3 interaction.
DISCUSSION
The antiviral activity of A3C against HIV-1ΔVif is not as potent as that of A3G and A3F. While several studies found no antiviral activity of hA3C, others reported that the infectivity of HIV-1ΔVif can be reduced by 50% by hA3C and restored by its Vif protein (11, 19–23, 26–28). We do not know whether ΔVif mutants of the various HIV-1 subtypes differ in their sensitivity to hA3C. However, of the 21 Vif isolates we tested here from HIV-1 group M, N, and O subtypes, only four Vifs (A-1, B-1, D-1, and F-1) showed reduced counteractivity to hA3C. Thus, the capacity of HIV-1 Vifs to counteract hA3C is highly conserved. Our data indicate that HIV-1 Vif uses a different binding surface to interact with A3C and the related A3F and that impairing the anti-A3C/A3F activity does not prevent the counteraction of A3G.
A previous study identified several hydrophobic or negatively charged residues between the α2 and α3 helix of hA3C that bind to HIV-1 Vif (63), and it was also reported that C130A, C130L, and E133A substitutions in hA3C did not impair HIV-1 Vif-induced degradation. However, in our study, replacement of C130 and E133 in hA3C with residues N130 and Q133, which are found in rhA3C (hA3C CE-NQ mutant in Fig. 4C), resulted in resistance to HIV-1 Vif (Fig. 4D). Both cysteine and asparagine residues belong to polar amino acids, but the E133Q substitution alters the residue from negatively polar acidic to uncharged (whereas alanine is nonpolar). These observations suggest that the charge of residue 133 in hA3C is important for the interaction with HIV-1 Vif. In addition, we demonstrated the E324 of hA3F is essential for HIV-1 Vif interaction, while the equivalent residue E141 in hA3C is not. These results are consistent with Kitamura et al. (63). In the Vif protein, a recent study described that mutations in a basic Vif interface patch (R17, E171, and R173) had distinct influence on degradation of hA3C and hA3F (66). In our experiments, we found that the additional residue G182 of HIV-1 Vif had different effects on the counteraction of hA3C and hA3F (Fig. 8B and 9C). These findings strongly support that HIV-1 Vif interaction interfaces in hA3C and hA3F are not identical.
A recent study reported that amino acids P16, M19, E20, P44, H45, and V47 of HIV-2 Vif are important for interaction with A3F, whereas the 140AGARV144 motif in the N-terminal half of A3F is critical for interaction with HIV-2 Vif (74). Here, we found that all tested A3C proteins were sensitive to HIV-2, SIVagm, and SIVmac Vif. hA3C and the C terminus of hA3F are homologous (5). A3C proteins are strong inhibitors of SIVΔVif, while SIVagm and SIVmac Vifs reduced the protein levels of hA3C, cpzA3C, smmA3C, agmA3C, and rhA3C (Fig. 1B and 2). This counteraction might promote SIV cross-species transmissions between primates. Compared to HIV-1 Vif, SIVcpz Vif is more active against hA3C and cpzA3C (Fig. 2F). The role of the human A3C protein during SIVcpz transmission to the human population is currently unclear. Letko et al. reported that gorilla A3G resists degradation of HIV-1 and most SIV Vifs, serving as a barrier to SIV transmission (51). Compared to the sequence of cpzA3C or hA3C, the region of amino acids 128 to 133 of gorA3C is quite different (128YPCYQE133 presents in cpzA3C and hA3C, 128DTDYQE133 in gorA3C). Whether the replacement of 128YPC130 with 128DTD130 affects gorA3C degradation by HIV-1 or SIV Vifs needs further investigation.
Here, we found that rhA3C and smmA3C were resistant to HIV-1 Vif (Fig. 1B). However, coimmunoprecipitation assays showed that rhA3C, smmA3C, and the hA3C.CE-NQ mutant, which was insensitive to HIV-1 Vif, still bound to HIV-1 Vif. These results indicate that binding detected by co-IPs is not a proper reporter for Vif-mediated counteraction affecting the protein levels. Indeed, several previous studies reported that binding of Vif and A3 is not the only determinant for complete Vif-mediated degradation (53, 57, 83–85). Kitamura et al. identified several residues between α2 and α3 helices of A3C binding to HIV-1 Vif (63). These residues are conserved in smmA3C (Fig. 1A), which might be the reason that binding of smmA3C to HIV-1 Vif was observed in our study. Recently a wobble model of the evolution of the Vif-A3 interaction was presented (64), implicating that among the Vif-A3 interactions, some binding sites are essential while others provide additional stabilizing contacts. Based on this idea, where only Vif forms a sufficient network of interactions with A3s, a functional interaction is formed. Suboptimal, destabilized interactions could be restored by the evolution of compensatory changes in the Vif-A3 interface. Thus, it is possible that the Vif interaction sites in hA3C identified by Kitamura et al. are the main interactions (63), while C130 and E133 in hA3C represent one of the relevant additional interacting points for hA3C-Vif complex formation.
Previous studies have identified three discontinuous motifs of HIV-1 Vif specifically involved in interaction with hA3F: F1-(14DRMR17), F2-(74TGEDW79), and F3-(171EDRW174) (66, 84). In addition, conserved tryptophans in Vif outside the F1, F2, and F3 box have been reported to be important for hA3F degradation (86). Here, we identified HIV-1 Vif subtype F-1, which did not degrade hA3C and hA3F, as was determined by residue K167. Our experimental results suggest that the A3-Vif interaction surface differs in Vif proteins of B-NL4-3 and F-1. Structure modeling suggested that E171, K167, and D101 in HIV-1 Vif can form internal noncovalent interactions, while the mechanism of how this internal interaction influences the Vif-A3 interaction needs further investigation, especially due to its intrinsic disordered nature (87). Vif proteins of different F-1 isolates show variability at 101 and 167 sites (G101D or K167Q/R), but E171 is conserved (data not shown). The hA3F/hA3C degradation activities of these F-1 Vif variants require further investigation. Interestingly, Vif from the F-1 subtype did not degrade hA3C and hA3F, whereas it counteracted hA3G and hA3H hapII (Fig. 7C) (71). It is unknown how the F-1 subtype virus, an isolate from Brazil, escapes the restriction of hA3F. Recently, single-nucleotide polymorphism (SNP) haplotypes of hA3F were reported, and an I231V variant with an allele (V) frequency of 48% in European Americans was associated with significantly lower set-point viral load and lower rate of progression to AIDS (88). Thus, it is possible that the hA3F of the host of our F-1 subtype had a unique haplotype that can be counteracted by F-1 Vif.
In summary, we found that rhA3C and smmA3C were resistant, but hA3C was sensitive, to HIV-1 Vif-triggered depletion, which was regulated by residues 130 and 133. Moreover, we identified that the Vif of HIV-1 F-1 subtype could not degrade hA3C as well as hA3F but was counteractive against hA3G and hA3H hapII. Residues 167 and 182 of F-1 Vif were critical for its inactivity against hA3C and hA3F, which was due to the intramolecular interaction with the F3 box. These findings provide an important addition to the model of the HIV-1 Vif and hA3C/F interaction and also advance our understanding of host-virus interactions during cross-species transmission and viral evolution.
ACKNOWLEDGMENTS
We thank Wioletta Hörschken for excellent technical assistance. We thank Neeltje Kootstra, Nathanial R. Landau, and Viviana Simon for reagents. The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 HXB2 Vif antiserum (number 2221) from Dana Gabuzda, monoclonal antibody to HIV-1 p24 (AG3.0) from Jonathan Allan, and SIVCPZTAN1.910 (number 11496) and SIVCPZTAN2.69 (number 11497) from Jun Takehisa, Matthias H. Kraus, and Beatrice H. Hahn.
REFERENCES
- 1.Sharp PM, Hahn BH. 2011. Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med 1:a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris RS, Dudley JP. 2015. APOBECs and virus restriction. Virology 479-480:131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simon V, Bloch N, Landau NR. 2015. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol 16:546–553. doi: 10.1038/ni.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LaRue RS, Andresdottir V, Blanchard Y, Conticello SG, Derse D, Emerman M, Greene WC, Jonsson SR, Landau NR, Löchelt M, Malik HS, Malim MH, Münk C, O'Brien SJ, Pathak VK, Strebel K, Wain-Hobson S, Yu XF, Yuhki N, Harris RS. 2009. Guidelines for naming nonprimate APOBEC3 genes and proteins. J Virol 83:494–497. doi: 10.1128/JVI.01976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Münk C, Willemsen A, Bravo IG. 2012. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol Biol 12:71. doi: 10.1186/1471-2148-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ooms M, Brayton B, Letko M, Maio SM, Pilcher CD, Hecht FM, Barbour JD, Simon V. 2013. HIV-1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 14:411–421. doi: 10.1016/j.chom.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Refsland EW, Hultquist JF, Harris RS. 2012. Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog 8:e1002800. doi: 10.1371/journal.ppat.1002800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 10.Mariani R, Chen D, Schröfelbauer B, Navarro F, König R, Bollman B, Münk C, Nymark-McMahon H, Landau NR. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21–31. doi: 10.1016/S0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- 11.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. 2011. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol 85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J 23:2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG. 2007. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res 35:7096–7108. doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mbisa JL, Bu W, Pathak VK. 2010. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J Virol 84:5250–5259. doi: 10.1128/JVI.02358-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Ao Z, Chen L, Kobinger G, Peng J, Yao X. 2012. The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication. J Virol 86:3777–3786. doi: 10.1128/JVI.06594-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmes RK, Koning FA, Bishop KN, Malim MH. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem 282:2587–2595. [DOI] [PubMed] [Google Scholar]
- 17.Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, Engelman A, Pathak VK. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol 81:7099–7110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillick K, Pollpeter D, Phalora P, Kim EY, Wolinsky SM, Malim MH. 2013. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J Virol 87:1508–1517. doi: 10.1128/JVI.02587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Q, Chen D, Konig R, Mariani R, Unutmaz D, Landau NR. 2004. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem 279:53379–53386. doi: 10.1074/jbc.M408802200. [DOI] [PubMed] [Google Scholar]
- 20.Stauch B, Hofmann H, Perkovic M, Weisel M, Kopietz F, Cichutek K, Münk C, Schneider G. 2009. Model structure of APOBEC3C reveals a binding pocket modulating ribonucleic acid interaction required for encapsidation. Proc Natl Acad Sci U S A 106:12079–12084. doi: 10.1073/pnas.0900979106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perkovic M, Schmidt S, Marino D, Russell RA, Stauch B, Hofmann H, Kopietz F, Kloke BP, Zielonka J, Strover H, Hermle J, Lindemann D, Pathak VK, Schneider G, Löchelt M, Cichutek K, Münk C. 2009. Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J Biol Chem 284:5819–5826. doi: 10.1074/jbc.M808853200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaipan C, Smith JL, Hu WS, Pathak VK. 2013. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J Virol 87:444–453. doi: 10.1128/JVI.00676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dang Y, Wang X, Esselman WJ, Zheng YH. 2006. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol 80:10522–10533. doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warren CJ, Xu T, Guo K, Griffin LM, Westrich JA, Lee D, Lambert PF, Santiago ML, Pyeon D. 2015. APOBEC3A functions as a restriction factor of human papillomavirus. J Virol 89:688–702. doi: 10.1128/JVI.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahasan MM, Wakae K, Wang Z, Kitamura K, Liu G, Koura M, Imayasu M, Sakamoto N, Hanaoka K, Nakamura M, Kyo S, Kondo S, Fujiwara H, Yoshizaki T, Mori S, Kukimoto I, Muramatsu M. 2015. APOBEC3A and 3C decrease human papillomavirus 16 pseudovirion infectivity. Biochem Biophys Res Commun 457:295–299. doi: 10.1016/j.bbrc.2014.12.103. [DOI] [PubMed] [Google Scholar]
- 26.Marin M, Golem S, Rose KM, Kozak SL, Kabat D. 2008. Human immunodeficiency virus type 1 Vif functionally interacts with diverse APOBEC3 cytidine deaminases and moves with them between cytoplasmic sites of mRNA metabolism. J Virol 82:987–998. doi: 10.1128/JVI.01078-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol 14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 28.Langlois MA, Beale RC, Conticello SG, Neuberger MS. 2005. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res 33:1913–1923. doi: 10.1093/nar/gki343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ooms M, Krikoni A, Kress AK, Simon V, Münk C. 2012. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J Virol 86:6097–6108. doi: 10.1128/JVI.06570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doehle BP, Schafer A, Cullen BR. 2005. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 31.Bogerd HP, Wiegand HL, Doehle BP, Cullen BR. 2007. The intrinsic antiretroviral factor APOBEC3B contains two enzymatically active cytidine deaminase domains. Virology 364:486–493. doi: 10.1016/j.virol.2007.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahieux R, Suspene R, Delebecque F, Henry M, Schwartz O, Wain-Hobson S, Vartanian JP. 2005. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J Gen Virol 86:2489–2494. [DOI] [PubMed] [Google Scholar]
- 33.Burns MB, Temiz NA, Harris RS. 2013. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet 45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Land AM, Wang J, Law EK, Aberle R, Kirmaier A, Krupp A, Johnson WE, Harris RS. 2015. Degradation of the cancer genomic DNA deaminase APOBEC3B by SIV Vif. Oncotarget 6:39969–39979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, Getz G, Gordenin DA. 2013. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van't Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain I, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. 2013. Signatures of mutational processes in human cancer. Nature 500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nik-Zainal S, Wedge DC, Alexandrov LB, Petljak M, Butler AP, Bolli N, Davies HR, Knappskog S, Martin S, Papaemmanuil E, Ramakrishna M, Shlien A, Simonic I, Xue Y, Tyler-Smith C, Campbell PJ, Stratton MR. 2014. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat Genet 46:487–491. doi: 10.1038/ng.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS. 2013. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife 2:e00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu R, Zhang X, Zhang W, Fang Y, Zheng S, Yu XF. 2007. Association of human APOBEC3 cytidine deaminases with the generation of hepatitis virus B x antigen mutants and hepatocellular carcinoma. Hepatology 46:1810–1820. doi: 10.1002/hep.21893. [DOI] [PubMed] [Google Scholar]
- 40.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 41.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. 2012. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Du J, Evans SL, Yu Y, Yu XF. 2012. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481:376–379. [DOI] [PubMed] [Google Scholar]
- 43.Derse D, Hill SA, Princler G, Lloyd P, Heidecker G. 2007. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc Natl Acad Sci U S A 104:2915–2920. doi: 10.1073/pnas.0609444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Löchelt M, Romen F, Bastone P, Muckenfuss H, Kirchner N, Kim YB, Truyen U, Rosler U, Battenberg M, Saib A, Flory E, Cichutek K, Münk C. 2005. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc Natl Acad Sci U S A 102:7982–7987. doi: 10.1073/pnas.0501445102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stavrou S, Nitta T, Kotla S, Ha D, Nagashima K, Rein AR, Fan H, Ross SR. 2013. Murine leukemia virus glycosylated Gag blocks apolipoprotein B editing complex 3 and cytosolic sensor access to the reverse transcription complex. Proc Natl Acad Sci U S A 110:9078–9083. doi: 10.1073/pnas.1217399110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosales Gerpe MC, Renner TM, Belanger K, Lam C, Aydin H, Langlois MA. 2015. N-linked glycosylation protects gammaretroviruses against deamination by APOBEC3 proteins. J Virol 89:2342–2357. doi: 10.1128/JVI.03330-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jaguva Vasudevan AA, Perkovic M, Bulliard Y, Cichutek K, Trono D, Häussinger D, Münk C. 2013. Prototype foamy virus Bet impairs the dimerization and cytosolic solubility of human APOBEC3G. J Virol 87:9030–9040. doi: 10.1128/JVI.03385-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bogerd HP, Doehle BP, Wiegand HL, Cullen BR. 2004. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc Natl Acad Sci U S A 101:3770–3774. doi: 10.1073/pnas.0307713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schröfelbauer B, Chen D, Landau NR. 2004. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc Natl Acad Sci U S A 101:3927–3932. doi: 10.1073/pnas.0307132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K, Pathak VK. 2004. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc Natl Acad Sci U S A 101:5652–5657. doi: 10.1073/pnas.0400830101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Letko M, Silvestri G, Hahn BH, Bibollet-Ruche F, Gokcumen O, Simon V, Ooms M. 2013. Vif proteins from diverse primate lentiviral lineages use the same binding site in APOBEC3G. J Virol 87:11861–11871. doi: 10.1128/JVI.01944-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshikawa R, Takeuchi JS, Yamada E, Nakano Y, Ren F, Tanaka H, Münk C, Harris RS, Miyazawa T, Koyanagi Y, Sato K. 2015. Vif determines the requirement for CBF-beta in APOBEC3 degradation. J Gen Virol 96:887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stern MA, Hu C, Saenz DT, Fadel HJ, Sims O, Peretz M, Poeschla EM. 2010. Productive replication of Vif-chimeric HIV-1 in feline cells. J Virol 84:7378–7395. doi: 10.1128/JVI.00584-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dang Y, Wang X, York IA, Zheng YH. 2010. Identification of a critical T(Q/D/E)x5ADx2(I/L) motif from primate lentivirus Vif proteins that regulate APOBEC3G and APOBEC3F neutralizing activity. J Virol 84:8561–8570. doi: 10.1128/JVI.00960-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaur R, Strebel K. 2012. Insights into the dual activity of SIVmac239 Vif against human and African green monkey APOBEC3G. PLoS One 7:e48850. doi: 10.1371/journal.pone.0048850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hatziioannou T, Princiotta M, Piatak M Jr, Yuan F, Zhang F, Lifson JD, Bieniasz PD. 2006. Generation of simian-tropic HIV-1 by restriction factor evasion. Science 314:95. doi: 10.1126/science.1130994. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Z, Gu Q, Jaguva Vasudevan AA, Hain A, Kloke BP, Hasheminasab S, Mulnaes D, Sato K, Cichutek K, Häussinger D, Bravo IG, Smits SH, Gohlke H, Münk C. 2016. Determinants of FIV and HIV Vif sensitivity of feline APOBEC3 restriction factors. Retrovirology 13:46. doi: 10.1186/s12977-016-0274-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larue RS, Lengyel J, Jonsson SR, Andresdottir V, Harris RS. 2010. Lentiviral Vif degrades the APOBEC3Z3/APOBEC3H protein of its mammalian host and is capable of cross-species activity. J Virol 84:8193–8201. doi: 10.1128/JVI.00685-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huthoff H, Malim MH. 2007. Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and Virion encapsidation. J Virol 81:3807–3815. doi: 10.1128/JVI.02795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Letko M, Booiman T, Kootstra N, Simon V, Ooms M. 2015. Identification of the HIV-1 Vif and human APOBEC3G protein interface. Cell Rep 13:1789–1799. doi: 10.1016/j.celrep.2015.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhen A, Wang T, Zhao K, Xiong Y, Yu XF. 2010. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J Virol 84:1902–1911. doi: 10.1128/JVI.01509-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ooms M, Letko M, Binka M, Simon V. 2013. The resistance of human APOBEC3H to HIV-1 NL4-3 molecular clone is determined by a single amino acid in Vif. PLoS One 8:e57744. doi: 10.1371/journal.pone.0057744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitamura S, Ode H, Nakashima M, Imahashi M, Naganawa Y, Kurosawa T, Yokomaku Y, Yamane T, Watanabe N, Suzuki A, Sugiura W, Iwatani Y. 2012. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat Struct Mol Biol 19:1005–1010. doi: 10.1038/nsmb.2378. [DOI] [PubMed] [Google Scholar]
- 64.Richards C, Albin JS, Demir O, Shaban NM, Luengas EM, Land AM, Anderson BD, Holten JR, Anderson JS, Harki DA, Amaro RE, Harris RS. 2015. The binding interface between human APOBEC3F and HIV-1 Vif elucidated by genetic and computational approaches. Cell Rep 13:1781–1788. doi: 10.1016/j.celrep.2015.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith JL, Pathak VK. 2010. Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3DE and African green monkey APOBEC3F that interact with HIV-1 Vif. J Virol 84:12599–12608. doi: 10.1128/JVI.01437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakashima M, Ode H, Kawamura T, Kitamura S, Naganawa Y, Awazu H, Tsuzuki S, Matsuoka K, Nemoto M, Hachiya A, Sugiura W, Yokomaku Y, Watanabe N, Iwatani Y. 2016. Structural insights into HIV-1 Vif-APOBEC3F interaction. J Virol 90:1034–1047. doi: 10.1128/JVI.02369-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Albin JS, LaRue RS, Weaver JA, Brown WL, Shindo K, Harjes E, Matsuo H, Harris RS. 2010. A single amino acid in human APOBEC3F alters susceptibility to HIV-1 Vif. J Biol Chem 285:40785–40792. doi: 10.1074/jbc.M110.173161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Land AM, Shaban NM, Evans L, Hultquist JF, Albin JS, Harris RS. 2014. APOBEC3F determinants of HIV-1 Vif sensitivity. J Virol 88:12923–12927. doi: 10.1128/JVI.02362-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zennou V, Bieniasz PD. 2006. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology 349:31–40. doi: 10.1016/j.virol.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 70.Russell RA, Pathak VK. 2007. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J Virol 81:8201–8210. doi: 10.1128/JVI.00395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Binka M, Ooms M, Steward M, Simon V. 2012. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J Virol 86:49–59. doi: 10.1128/JVI.06082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salter JD, Morales GA, Smith HC. 2014. Structural insights for HIV-1 therapeutic strategies targeting Vif. Trends Biochem Sci 39:373–380. doi: 10.1016/j.tibs.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dang Y, Davis RW, York IA, Zheng YH. 2010. Identification of 81LGxGxxIxW89 and 171EDRW174 domains from human immunodeficiency virus type 1 Vif that regulate APOBEC3G and APOBEC3F neutralizing activity. J Virol 84:5741–5750. doi: 10.1128/JVI.00079-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smith JL, Izumi T, Borbet TC, Hagedorn AN, Pathak VK. 2014. HIV-1 and HIV-2 Vif interact with human APOBEC3 proteins using completely different determinants. J Virol 88:9893–9908. doi: 10.1128/JVI.01318-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zielonka J, Marino D, Hofmann H, Yuhki N, Löchelt M, Münk C. 2010. Vif of feline immunodeficiency virus from domestic cats protects against APOBEC3 restriction factors from many felids. J Virol 84:7312–7324. doi: 10.1128/JVI.00209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takehisa J, Kraus MH, Decker JM, Li Y, Keele BF, Bibollet-Ruche F, Zammit KP, Weng Z, Santiago ML, Kamenya S, Wilson ML, Pusey AE, Bailes E, Sharp PM, Shaw GM, Hahn BH. 2007. Generation of infectious molecular clones of simian immunodeficiency virus from fecal consensus sequences of wild chimpanzees. J Virol 81:7463–7475. doi: 10.1128/JVI.00551-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, Flory E, Schumann GG, Münk C. 2006. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem 281:22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- 78.Zennou V, Perez-Caballero D, Gottlinger H, Bieniasz PD. 2004. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J Virol 78:12058–12061. doi: 10.1128/JVI.78.21.12058-12061.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bähr A, Singer A, Hain A, Vasudevan AA, Schilling M, Reh J, Riess M, Panitz S, Serrano V, Schweizer M, Konig R, Chanda S, Häussinger D, Kochs G, Lindemann D, Münk C. 2016. Interferon but not MxB inhibits foamy retroviruses. Virology 488:51–60. doi: 10.1016/j.virol.2015.10.034. [DOI] [PubMed] [Google Scholar]
- 80.Goncalves J, Jallepalli P, Gabuzda DH. 1994. Subcellular localization of the Vif protein of human immunodeficiency virus type 1. J Virol 68:704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simm M, Shahabuddin M, Chao W, Allan JS, Volsky DJ. 1995. Aberrant Gag protein composition of a human immunodeficiency virus type 1 vif mutant produced in primary lymphocytes. J Virol 69:4582–4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. 2009. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc 4:1–13. [DOI] [PubMed] [Google Scholar]
- 83.Zhang W, Huang M, Wang T, Tan L, Tian C, Yu X, Kong W, Yu XF. 2008. Conserved and non-conserved features of HIV-1 and SIVagm Vif mediated suppression of APOBEC3 cytidine deaminases. Cell Microbiol 10:1662–1675. doi: 10.1111/j.1462-5822.2008.01157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.He Z, Zhang W, Chen G, Xu R, Yu XF. 2008. Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J Mol Biol 381:1000–1011. doi: 10.1016/j.jmb.2008.06.061. [DOI] [PubMed] [Google Scholar]
- 85.Baig TT, Feng Y, Chelico L. 2014. Determinants of efficient degradation of APOBEC3 restriction factors by HIV-1 Vif. J Virol 88:14380–14395. doi: 10.1128/JVI.02484-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tian C, Yu X, Zhang W, Wang T, Xu R, Yu XF. 2006. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J Virol 80:3112–3115. doi: 10.1128/JVI.80.6.3112-3115.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Auclair JR, Green KM, Shandilya S, Evans JE, Somasundaran M, Schiffer CA. 2007. Mass spectrometry analysis of HIV-1 Vif reveals an increase in ordered structure upon oligomerization in regions necessary for viral infectivity. Proteins 69:270–284. doi: 10.1002/prot.21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.An P, Penugonda S, Thorball CW, Bartha I, Goedert JJ, Donfield S, Buchbinder S, Binns-Roemer E, Kirk GD, Zhang W, Fellay J, Yu XF, Winkler CA. 2016. Role of APOBEC3F gene variation in HIV-1 disease progression and pneumocystis pneumonia. PLoS Genet 12:e1005921. doi: 10.1371/journal.pgen.1005921. [DOI] [PMC free article] [PubMed] [Google Scholar]