

ABSTRACT
Herpes simplex viruses (HSVs) are unusual in that unlike most enveloped viruses, they require at least four entry glycoproteins, gB, gD, gH, and gL, for entry into target cells in addition to a cellular receptor for gD. The dissection of the herpes simplex virus 1 (HSV-1) entry mechanism is complicated by the presence of more than a dozen proteins on the viral envelope. To investigate HSV-1 entry requirements in a simplified system, we generated vesicular stomatitis virus (VSV) virions pseudotyped with HSV-1 essential entry glycoproteins gB, gD, gH, and gL but lacking the native VSV fusogen G. These virions, referred to here as VSVΔG-BHLD virions, infected a cell line expressing a gD receptor, demonstrating for the first time that the four essential entry glycoproteins of HSV-1 are not only required but also sufficient for cell entry. To our knowledge, this is the first time the VSV pseudotyping system has been successfully extended beyond two proteins. Entry of pseudotyped virions required a gD receptor and was inhibited by HSV-1 specific anti-gB or anti-gH/gL neutralizing antibodies, which suggests that membrane fusion during the entry of the pseudotyped virions shares common requirements with the membrane fusion involved in HSV-1 entry and HSV-1-mediated syncytium formation. The HSV pseudotyping system established in this study presents a novel tool for systematic exploration of the HSV entry and membrane fusion mechanisms.
IMPORTANCE Herpes simplex viruses (HSVs) are human pathogens that can cause cold sores, genital herpes, and blindness. No vaccines or preventatives are available. HSV entry into cells—a prerequisite for a successful infection—is a complex process that involves multiple viral and host proteins and occurs by different routes. Detailed mechanistic knowledge of the HSV entry is important for understanding its pathogenesis and would benefit antiviral and vaccine development, yet the presence of more than a dozen proteins on the viral envelope complicates the dissection of the HSV entry mechanisms. In this study, we generated heterologous virions displaying the four essential entry proteins of HSV-1 and showed that they are capable of cell entry and, like HSV-1, require all four entry glycoproteins along with a gD receptor. This HSV pseudotyping system pioneered in this work opens doors for future systematic exploration of the herpesvirus entry mechanisms.
INTRODUCTION
To enter living cells to replicate, viruses must overcome the barrier of the cellular membrane. Enveloped viruses accomplish this task by facilitating the merger of their envelope with a target cell membrane, during which capsids are delivered into the cytosol and infection ensues. Entry is initiated by binding of a virus to an appropriate receptor on the surface of the host cell and is catalyzed by a virus-encoded membrane fusogen. In most enveloped viruses, the receptor binding and the fusogenic functions are executed by a single protein (1).
Herpesviruses are double-stranded DNA, enveloped viruses with intricate envelopes that contain at least a dozen proteins (2). Herpesvirus entry is a complex process that requires three conserved proteins plus additional nonconserved glycoproteins specific to individual herpesviruses (3–5) and could be further modulated by viral and host proteins (6–9).
Herpes simplex virus 1 (HSV-1) is the prototype of the diverse herpesvirus family (10). HSV-1 envelopes contain at least 14 different proteins (2), but only four, gB, gD, gH, and gL, are required for entry, which was established by assaying the infectivity of HSV-1 mutants containing single gene deletions (11–14). gD is the receptor-binding protein (13) that engages one of its three cellular entry receptors: nectin-1, a cell adhesion molecule found at cell junctions; a herpesvirus entry mediator (HVEM) (15–17); or a non-protein receptor 3-O-modifed heparan sulfate (18). gB, gH, and gL are present in all herpesviruses and constitute their core fusion machinery (3–5). Of these three proteins, gB is the most highly conserved, and it is a class III viral fusion protein (19, 20), presumably, directly involved in bringing the viral and host cell membranes together to enable their fusion. gH/gL does not resemble a viral fusion protein and, instead, probably regulates the fusogenic activity of gB (21).
The four HSV-1 glycoproteins essential for entry are also sufficient for cell-cell fusion when coexpressed in transfected cells in the presence of a gD receptor (22, 23). But whether they are sufficient for viral entry is unknown. Construction of an HSV-1 mutant lacking all glycoproteins except for the essential four has not yet been attempted and may be technically challenging due to the need to delete at least 10 genes.
The requirements and the mechanisms of HSV-1-mediated membrane fusion have been primarily investigated using cell-cell fusion of cells transfected with the four essential glycoproteins. This robust system has provided important insights into the mechanisms of membrane fusion and contributions of individual glycoproteins. It is well appreciated that virus-cell and cell-cell fusion are distinct processes.
In this study, we developed a simplified system for investigating HSV-1 entry mechanism. We generated VSV virions pseudotyped with HSV-1 gB, gD, gH, and gL through transcomplementation but lacking the native vesicular stomatitis virus (VSV) fusogen G (VSV-G). These virions, which we termed VSVΔG-BHLD, were capable of infecting a cell line expressing gD receptors, demonstrating for the first time that gB, gD, gH, and gL are not only required but also sufficient for cell entry. Entry of pseudotyped virions was blocked by HSV-1-specific anti-gB or anti-gH/gL neutralizing antibodies, which suggests that membrane fusion during the entry of the pseudotyped virions is subject to requirements similar to those for membrane fusion involved in HSV-1 entry. The VSV pseudotyping system provides a potential platform for future dissection of the contributions of HSV-1 envelope proteins to entry.
MATERIALS AND METHODS
Cells, plasmids, and antibodies.
BHK (a gift from J. White [University of Virginia]), 293T (a gift from J. M. Coffin [Tufts University]), and B78H1 (a gift from R. J. Eisenberg and G. H. Cohen [University of Pennsylvania]) cells were grown in Dulbecco's modified Eagle medium containing high glucose and sodium pyruvate (DMEM; Gibco) supplemented with 5% fetal bovine serum (FBS; Gibco). C10 cells (24) (a gift from R. J. Eisenberg and G. H. Cohen), which are B78H1 derivatives that stably express nectin-1, were grown in DMEM supplemented with 5% FBS and 250 μg/ml of Geneticin (Gibco). V529 cells (25) (a gift from D. M. Knipe [Harvard Medical School]) were grown in DMEM supplemented with 10% FBS and 400 μg/ml of Geneticin. All cell lines were grown at 37°C with 5% CO2.
Plasmids pPEP98, pPEP99, pPEP100, and pPEP101 carry the full-length HSV-1 (strain KOS) gB, gD, gH, and gL genes, respectively, in a pCAGGS vector (26) and were gifts from P. G. Spear (Northwestern University). Plasmid pCMV-VSVG was provided by J. White.
Rabbit anti-HSV-1 gH/gL polyclonal antibody (PAb) R137, anti-HSV-1 gD PAb R7, anti-HSV-1 gB PAb R68, and mouse anti-HSV-1 gB monoclonal antibodies (MAbs) SS10 and SS55 were gifts of G. H. Cohen and R. J. Eisenberg. Mouse anti-HSV-1 gH/gL MAb LP11 was a gift of H. Browne (U. of Cambridge). Mouse anti-VSV-G MAb IE9F9, anti-VSV-M MAb 23H12, and neutralizing anti-VSV-G MAb 8G5F11 were purchased from KeraFast. Goat anti-rabbit and goat anti-mouse PAbs were purchased from Bio-Rad.
Pseudotyped virions.
Recombinant VSVΔG-G was generated previously by M. Whitt (27) and provided by J. White (University of Virginia). VSVΔG-gB, VSVΔG-G, or VSVΔG was generated by transfecting BHK cells with plasmids pPEP98 (gB), pCMV-VSVG, or pCAGGS and infecting with them with VSVΔG-G 24 h posttransfection at a multiplicity of infection (MOI) of 3 to 5 IU/cell for 1 h at 37°C in a 5% CO2 incubator using serum-free DMEM. VSVΔG-G encodes green fluorescent protein (GFP) instead of VSV-G, and infection is monitored by GFP expression beginning 12 to 24 h postinfection. The cells were washed 3 times using phosphate-buffered saline containing calcium and magnesium (PBS++) (Corning) and incubated in DMEM containing 5% FBS for 24 h at 37°C. Supernatants containing virions were harvested by two rounds of centrifugation at 1,500 × g for 10 min at 4°C. Virions were isolated from the supernatant by pelleting through a 20% sucrose cushion at 100,000 × g in an SW28 rotor and resuspended in 10% sucrose in 20 mM HEPES, pH 7.4, and 150 mM NaCl (HN buffer). Virions were further purified and concentrated by band purification using a 20 to 60% step gradient at 40,000 × g for 12 to 16 h using an SW55 rotor. The band was extracted from the 20 to 60% sucrose interface, diluted 2-fold in HN buffer, and pelleted through a 20% sucrose cushion at 100,000 × g.
VSVΔG-BHLD virions were prepared by following a modified protocol for pseudotyping hepatitis C Virus envelope proteins (28). 293T cells were cotransfected with pPEP98, pPEP99, pPEP100, and pPEP101 using polyethylenimine (PEI) and 24 h posttransfection were infected with VSVΔG-G at an MOI of 3 to 5 IU/cell for 2 h at 37°C in serum-free DMEM. The cells were washed 2 or 3 times using PBS++ and incubated for 48 to 72 h with DMEM containing 5% FBS at 30°C. Supernatants containing virions were cleared by two rounds of centrifugation at 1,500 × g for 10 min at 4°C and stored at −80°C until use or until further purification, as with other virions. Titers of VSVΔG-BHLD virion preparations were determined by serial dilution on C10 cells using fluorescence microscopy to detect GFP in the presence of anti-VSV-G MAb 8G5F11. Typical yields were 105 to 106 IU/ml.
The recombinant HSV-1 KOS8GFP virus expresses ICP8 with C-terminal GFP in the Kos1.1 background (25) and was kindly provided by D. M. Knipe. The HSV-1 KOS8GFP virus was propagated on V529 cells.
Western blotting.
Virion preparations were separated by SDS-PAGE using a 12% or 4 to 15% TGX gel (Bio-Rad) and visualized by staining with GelCode blue (ThermoFisher Scientific). Western blot analysis was used to confirm the presence of specific VSV and HSV-1 proteins in virions.
EM and immunogold labeling.
Purified VSVΔG-gB was diluted 10-fold in double-distilled water (ddH2O) and absorbed onto a glow-discharged carbon-coated 200-mesh copper grid at room temperature. Immunogold labeling was performed using 1% bovine serum albumin (BSA) for blocking, anti-gB MAb SS10 diluted 1:20, rabbit anti-mouse secondary PAb diluted 1:100, and protein A conjugated to 5-nm gold particles. Labeled samples were stained using 1% phosphotungstic acid (PTA) solution, pH 7.4. Purified VSVΔG-BHLD samples were diluted 10-fold in ddH2O, absorbed onto a glow-discharged Formvar/carbon-coated 200-mesh copper grid, and stained using 1% PTA. Samples were visualized using Tecnai G2 Spirit BioTWIN transmission electron microscope equipped with an AMT 2k charge-coupled device camera microscope at the Harvard Medical School electron microscopy (EM) core facility.
Cell entry and fluorescence microscopy.
C10 and B78H1 cells were seeded in a 24-well plate with glass coverslips at 1 × 105/well and reached confluence the following day. VSVΔG-BHLD, VSVΔG-G, and HSV-1 KOS8GFP were added equally to both cell types and visualized 16 to 24 h later by microscopy after fixation using 4% PFA-PBS (4% paraformaldehyde solution in phosphate-buffered saline [Boston BioProducts]). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The medium was replaced 2 h later and the cells were incubated overnight before visualization by fluorescence microscopy.
Antibody inhibition experiments.
C10 cells were seeded in a 24-well plate at 1 × 105/well and reached confluence the following day. Antibody blocking experiments were performed by incubating virions with appropriate antibody for 1 h at 4°C prior to addition to the cells. Anti-gH/gL LP11 IgG was used at 2.5 μg/ml, anti-VSV-G (8G5F11) was used at 0.5 μg/ml, and SS55 (anti-gB) in ascites fluid was used at a 1:100 dilution. GFP expression 16 to 24 h postinfection was quantified by fluorescence-activated cell sorting (FACS) and expressed as the percentage of GFP-positive cells versus that under control (no antibody) conditions. For immunofluorescence imaging, VSVΔG-BHLD and VSVΔG-G were preincubated with anti-VSV-G (8G5F11) at 1 μg/ml, SS55 (anti-gB) in ascites fluid at 1:50, or no antibody at 4°C prior to addition to the cells.
PEG-mediated fusion.
Stock solution of polyethylene glycol 6000 (PEG 6000; Fluka) in PBS was prepared by autoclaving 58% (wt/wt) solution of PEG 6000 in PBS at 121°C for 15 min, cooling it to room temperature, and diluting it to 44% prior to the experiment. Cells were seeded in a 24-well plate at 1 × 105 cells/well and reached confluence the following day. The medium was replaced with cold DMEM containing 30 mM HEPES, pH 7.4, and cells were chilled to 4°C. VSVΔG-BHLD, VSVΔG-B, VSVΔG-BD, or VSVΔG-HLD was added and incubated at 4°C for 2 h to allow attachment. Cells were washed twice in PBS, incubated with warm 44% PEG 6000 or PBS for 1 min at 37°C, and washed five times with PBS. Cells were then incubated overnight at 37°C in DMEM containing 5% FBS. The percentage of GFP-positive cells was quantified by FACS.
RESULTS
Generation and characterization of VSV pseudotyped with HSV-1 glycoprotein B.
We used a pseudotyping system based on recombinant VSV, in which the gene for the native fusogen G was replaced with GFP (VSVΔG) (27). VSVΔG virions can be pseudotyped with a glycoprotein of interest through transcomplementation whereby cells are transfected with an expression plasmid encoding the glycoprotein gene and then infected with the “helper” virus VSVΔG-G, which is pseudotyped with the VSV native fusogen G.
Initially, we generated VSVΔG virions pseudotyped with HSV-1 gB alone (VSVΔG-gB). “Bald” VSVΔG virions lacking any glycoproteins and VSVΔG-G virions pseudotyped with the native VSV-G served as controls. The average gB incorporation in VSVΔG-gB virions was comparable to that of native VSV-G in the control VSVΔG-G virions as judged by SDS-PAGE and GelCode blue staining (Fig. 1A). Robust incorporation was achieved without substituting the native 109-amino-acid cytoplasmic domain of gB for the 29-amino-acid cytoplasmic tail of VSV-G, a strategy that is often used to improve incorporation of challenging glycoproteins (29, 30). The presence of HSV-1 gB in VSVΔG-gB preparations was verified by Western blotting with anti-gB PAb R68 (Fig. 1B) and by immunogold labeling with anti-gB MAb SS10 (Fig. 1C). To ascertain that the VSVΔG-gB preparations were free from contamination with the VSVΔG-G helper virus, they were tested by Western blotting with an anti-VSV-G MAb (Fig. 1B). No VSV-G was detected in VSVΔG-gB preparations, even at a high exposure (data not shown).
FIG 1.
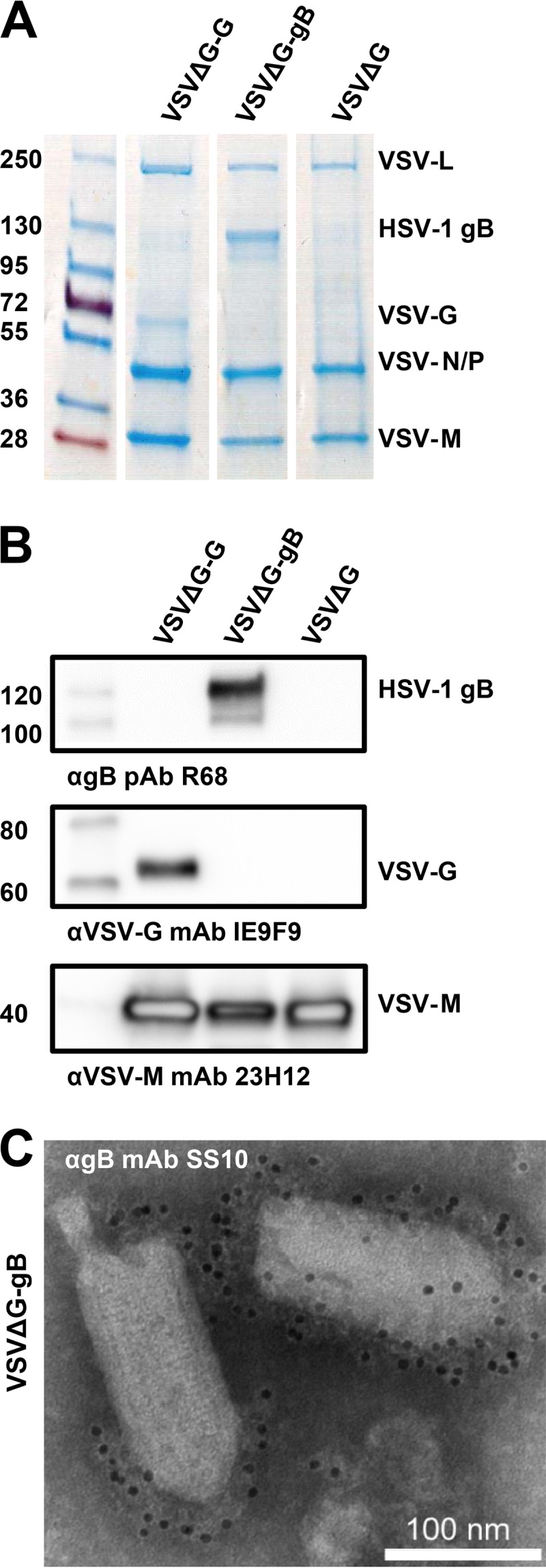
Characterization of VSVΔG virions pseudotyped with the HSV-1 glycoprotein gB. (A and B) Pseudotyped VSVΔG virions were separated by SDS-PAGE and analyzed by GelCode blue staining (A) or by Western blotting (B). Molecular mass standards (kDa) are labeled on the left, and the proteins are labeled on the right. (A) Virions pseudotyped with HSV-1 gB (VSVΔG-gB), VSV-G (VSVΔG-G), or no glycoproteins (VSVΔG) were purified from BHK cells and normalized for VSV-M content using densitometry. (B) Pseudotyped virions from panel A were analyzed by Western blotting using the indicated antibodies. (C) VSVΔG-B virions were visualized by electron microscopy using immunogold labeling with anti-gB MAb SS10 and negative staining with 1% PTA.
Generation and characterization of VSV pseudotyped with HSV-1 gB, gD, gH, and gL.
While required for viral entry and for membrane fusion (14, 23), HSV-1 gB alone is insufficient for cell-cell fusion (23), so it was not surprising that VSVΔG-gB virions were unable to enter cells (data not shown). In contrast, cells cotransfected with HSV-1 gB, gD, gH, and gL undergo robust fusion with cells bearing gD receptors (23, 26), which demonstrates that these four HSV-1 glycoproteins are sufficient for cell-cell fusion. To determine whether gB, gD, gH, and gL are also sufficient for viral entry, we generated VSVΔG virions pseudotyped with all four HSV-1 glycoproteins, gB, gH, gL, and gD (VSVΔG-BHLD), by transcomplementation. 293T cells were cotransfected with four plasmids and then infected with the VSVΔG-G helper virus.
The presence of HSV-1 glycoproteins in VSVΔG-BHLD virion preparations was determined by SDS-PAGE with GelCode Blue staining as well as Western blot analysis (Fig. 2A and B). The VSVΔG-BHLD virions contained all four glycoproteins, with gB incorporated in greater amounts than gD or gH, as judged by relative band intensity in GelCode blue-stained gels (Fig. 2A). gL binds Coomassie stain poorly even in purified gH/gL preparations (data not shown). This likely explains the apparent lack of a band corresponding to gL in Fig. 2A. The Western blot signal for gL was also low with anti-gH/gL PAb R137. Yet gL is expected to be present in equimolar amounts with gH because gH needs gL for proper folding (31, 32) and in its absence is not expressed on cell surface (33) or incorporated into virions (12). To verify the presence of gL in VSVΔG-BHLD virions, another, comparable VSVΔG-BHLD preparation was tested with an anti-gL MAb, showing that gL is present in purified VSVΔG-BHLD despite the low signal from the R137 blot (Fig. 2C).
FIG 2.

Characterization of VSVΔG virions pseudotyped with the HSV-1 glycoproteins gB, gD, gH, and gL. (A to C) Virions pseudotyped with HSV-1 gB, gD, gH, and gL (VSVΔG-BHLD) were separated by SDS-PAGE and analyzed by GelCode blue staining (A) or by Western blotting (B and C). Molecular mass standards (kDa) are labeled on the left, and the proteins are labeled on the right. (A) VSVΔG-BHLD virions were purified from 293T cells. (B) VSVΔG-BHLD virions from panel A were analyzed by Western blotting using the indicated antibodies. (C) VSVΔG-BHLD virions prepared as described for panels A and B were analyzed by Western blotting using anti-gL MAb L1. (D) VSVΔG-BHLD virions were visualized by electron microscopy using negative staining with 1% PTA.
Negative-staining electron microscopy images of VSVΔG-BHLD virions showed the presence of glycoprotein spikes on the viral surface (Fig. 2D), although the identity of glycoproteins cannot be assigned with certainty.
VSVΔG-BHLD virions require a gD receptor for cellular entry.
To determine whether gB, gD, gH, and gL are sufficient for viral entry, we tested their ability to infect C10 cells. C10 cells express the gD receptor nectin-1 and were generated previously from the cell line B78H1 (32). As a positive control, we used HSV-1 recombinant virus ICP8-GFP, which expresses ICP8 containing a C-terminal GFP tag (25). GFP expression was used as a readout of viral entry. VSVΔG-BHLD virions entered C10 cells efficiently as judged by GFP expression (Fig. 3). Entry was dependent on the presence of a gD receptor because the virions were unable to infect the parental B78H1 cells, which lack any known gD receptors (24, 34). As expected, HSV-1 KOS8GFP infected gD-receptor-bearing C10 cells but not B78H1 cells (Fig. 3). Thus, entry of VSVΔG-BHLD virions, like that of HSV-1, requires a gD receptor. In contrast, VSVΔG-G was able to infect the two cell types equally well, demonstrating that the B78H1 cells are permissive for VSV (Fig. 3). The ability of the VSVΔG-BHLD virions to infect cells demonstrates for the first time that the core entry glycoproteins gB, gD, gH, and gL are sufficient for cell entry.
FIG 3.

VSVΔG-BHLD virions require a gD receptor for cellular entry. Entry of VSVΔG-BHLD, VSVΔG-G, and HSV-1 KOS8GFP was visualized by confocal microscopy. VSVΔG-BHLD and VSVΔG-G express GFP in infected cells, and HSV-1 KOS8GFP expresses ICP8-GFP. B78H1 cells lack any known gD receptors, while C10 cells, derived from B78H1, stably express gD receptor nectin-1. Cells were seeded on glass coverslips, infected the following day, and fixed using 4% PFA-PBS. Nuclei were stained with DAPI.
Entry of VSVΔG-BHLD virions is blocked by neutralizing antibodies.
A number of antibodies against HSV-1 entry glycoproteins are neutralizing; that is, they inhibit HSV-1 entry. These antibodies also reduce fusion of cells transiently expressing the four entry glycoproteins and would be expected to block entry of VSVΔG-BHLD virions. To determine the effect of neutralizing antibodies on entry of VSVΔG-BHLD, virions were preincubated with the anti-gB MAb SS55 (35), the anti-gH/gL MAb LP11 (36), or anti-VSV-G MAb 8G5F11 as a negative control. Both SS55 and LP11 reduced entry of VSVΔG-BHLD virions (Fig. 4A and B) as well as HSV-1 KOS8GFP (Fig. 4A and D) but had no effect on the entry of VSVΔG-G virions (Fig. 4A and C). Only anti-gH/gL and anti-gB MAbs reduced entry of VSVΔG-BHLD and HSV-1 KOS8GFP, whereas anti-VSV-G MAb had no effect. HSV-1 KOS8GFP infection of the C10 cells was also reduced by either LP11 or SS55 but not by anti-VSV-G MAb (Fig. 4D). As expected, VSVΔG-G infection was blocked only by preincubation with anti-VSV-G MAb and not by the anti-HSV-1 MAbs (Fig. 4A and C). Thus, both gB and gH/gL are required for entry of VSVΔG-BHLD virions.
FIG 4.

VSVΔG-BHLD infection is blocked by anti-HSV-1 antibodies. (A) VSVΔG-BHLD or VSVΔG-G virions were incubated with anti-VSV-G, anti-gB MAb SS55, or no antibody prior to being added to C10 cells. Nuclei were stained with DAPI. (B to D) VSVΔG-BHLD, VSVΔG-G, or HSV-1 KOS8GFP virions were incubated with the neutralizing antibodies anti-gH/gL MAb LP11, anti-VSV-G MAb 8G5F11, or anti-gB MAb SS55 prior to being added to C10 cells. Cells were fixed in 4% PFA-PBS, and GFP expression was quantified using FACS. Each experiment was performed in triplicate and normalized to no-antibody controls, with the mean graphed. Error bars represent standard deviations.
The anti-gH/gL MAb LP11 efficiently neutralizes virus infectivity, inhibits cell-cell spread (36), and blocks cell-cell fusion of cells transiently expressing the four entry glycoproteins (21). It has been proposed to block an essential interaction between gH/gL and gB (21), which could be its mechanism of neutralization. The anti-gB MAb SS55 neutralizes virus infectivity (35, 37) and blocks cell-cell fusion of cells transiently expressing the four entry glycoproteins (38). SS55 blocks binding of soluble gB ectodomain to synthetic lipid vesicles (37), and its binding site (37) is located in the vicinity of the fusion loops, which interact with membranes (39–41). SS55 could thus inhibit entry and fusion by interfering with the gB fusion loop insertion into membrane, a critical step in membrane fusion (37). The ability of these antibodies to block entry of VSVΔG-BHLD and HSV-1 suggests that membrane fusion during the entry of the pseudotyped virions shares common mechanistic features with membrane fusion involved in HSV-1 entry and in HSV-1-mediated cell-cell fusion.
VSVΔG-BHLD virions require gB, gH/gL, and gD for entry into C10 cells.
To confirm that each of the four HSV-1 entry glycoproteins, gB, gH, gL, and gD, are required for entry into the C10 cells, we generated VSVΔG virions pseudotyped with incomplete entry glycoprotein sets: VSVΔG-B, VSVΔG-BD, VSVΔG-BHL, and VSVΔG-HLD (Fig. 5A to D). All proteins were incorporated into the virions (Fig. 5A to C). A band corresponding to gD was detected in VSVΔG-BHLD, VSVΔG-BD, and VSVΔG-HLD preparations by GelCode blue staining (Fig. 5A). Western blotting was used to confirm the presence of gB and gH because their respective bands migrate too closely together to be adequately distinguished by GelCode blue staining alone. The relative amounts of gH were lower than those of gB or gD, judging by GelCode blue staining (Fig. 5A). Also, less gH was present in the VSVΔG-HLD than in VSVΔG-BHL virions (Fig. 5C) despite comparable analyzed amounts of VSVΔG-BHL and VSVΔG-HLD virions, as judged by VSV-M content (Fig. 5A and D). The anti-gH/gL PAb R137 reacts poorly with gL, and while gL can be detected in BHLD preparations, it appears below the detection limit in HLD and BHL preparations. However, given that in the absence of gL, gH is not expressed on the cell surface (33) or incorporated into HSV-1 virions (12), we conclude that both BHL and HLD preparations contain gL.
FIG 5.

VSVΔG-BHLD virions require gB, gH/gL, and gD for entry into C10 cells. (A to D) Pseudotyped VSVΔG virions were separated by SDS-PAGE and analyzed by GelCode blue staining (A) or by Western blotting (B to D). Molecular mass standards (kDa) are labeled on the left, and the proteins are labeled on the right. (A) Virions pseudotyped with HSV-1 gB, gH/gL, and gD or a subset of these were purified from 293T cells and normalized for VSV-M content using densitometry. (B to D) Pseudotyped virions from panel A were analyzed by Western blotting using the indicated antibodies. (E) Equal volumes of VSVΔG-BHLD, VSVΔG-B, VSVΔG-BD, or VSVΔG-HLD preparations were incubated with C10 cells at 4°C to permit attachment and then treated with PBS or 44% PEG 4000. GFP expression was quantified using FACS. The experiment was performed in triplicate, with the means graphed. Error bars represent standard deviations.
Only VSVΔG-BHLD virions were capable of infecting C10 cells (Fig. 4E), demonstrating the requirement for all four essential entry glycoproteins in this minimal system. To rule out that the productive entry of VSVΔG virions containing incomplete entry glycoprotein sets was blocked at an attachment or a postfusion step, we forced fusion of VSVΔG-BHLD virions with the plasma membrane using polyethylene glycol (Fig. 5E), an approach that has been used with HSV-1 (42, 43). Approximately 10% of PEG-treated cells were productively infected (Fig. 5E). Thus, gB, gD, gH, and gL are necessary for entry of VSVΔG virions. The increase in the infectivity of VSVΔG-BHLD virions in the presence of PEG is probably due to PEG-forced entry of particles that are normally incapable of entry.
DISCUSSION
The recombinant VSVΔG system has been used successfully to study the entry of fusogens from many viruses, especially for viruses for which biosafety is a concern (44–50). In this study, we applied this technology to HSV-1, which contains over a dozen proteins in its envelope. We generated VSVΔG virions bearing the four essential HSV-1 entry glycoproteins, gB, gD, gH, and gL, which are also sufficient to mediate fusion of transiently transfected cells. To our knowledge, this is the first time the VSVΔG pseudotyping system has been extended beyond two proteins. By establishing a pseudotyping system for HSV-1, we showed that the four essential entry glycoproteins are sufficient for entry into gD receptor-bearing cells.
Prepared pseudotyped virions were free from contamination with residual VSV-G from helper virus as judged by SDS-PAGE, Western blotting, and the inability of the neutralizing anti-VSV-G antibody to block VSVΔG-BHLD infectivity. Successful incorporation of heterologous glycoproteins into VSV particles often requires that their cytoplasmic tail be replaced with that of VSV-G (29, 30). But the incorporation of gB into the pseudotyped virions was robust and comparable to that of native VSV-G despite the presence of a relatively large cytoplasmic domain. In contrast, the incorporation of gD and especially gH/gL was lower and could, potentially, be improved by including the cytoplasmic tail of VSV-G. However, efficient fusion requires the intact cytoplasmic tail of gH (51–53), and its replacement would likely reduce infectivity of VSVΔG-BHLD virions.
The VSVΔG-BHLD virions successfully infected a gD receptor-bearing cell line, demonstrating for the first time that HSV-1 gB, gD, gH, and gL are not only required but also sufficient for cell entry. It is well known that the four HSV-1 entry glycoproteins mediate fusion of cells transiently expressing these proteins. However, the process of viral entry into a cell differs from cell-cell fusion in several regards. During viral entry, a small viral particle attaches to and fuses its lipid envelope with the membrane of a much larger cell, whereas during cell-cell fusion, juxtaposed membranes of comparable size fuse. While studies of cell-cell fusion of transfected cells have greatly advanced our understanding of membrane fusion mediated by herpesviral glycoproteins, the transfected cell system may not fully recapitulate fusion during viral entry due to differences in membrane curvature, contact area, cytoskeleton, among other factors. Moreover, mutations in herpesvirus entry glycoproteins have been shown to have distinct effects on viral entry versus cell-cell fusion (54, 55). Our results demonstrate that the four essential HSV-1 entry glycoproteins function as the core fusion machinery both during viral entry and during cell fusion, highlighting their role in mediating membrane fusion, which is an essential step in both processes.
Entry of VSVΔG-BHLD virions required the presence of gD receptor on the target cells and was sensitive to inhibition by neutralizing antibodies against gB or gH/gL. Thus, entry of the pseudotyped virions is subject to requirements similar to those for entry of HSV-1. Whether it recapitulates all aspects of HSV-1 entry is yet unknown. For example, VSV pseudotypes have a different membrane composition than native HSV-1 virions due to different cellular budding locations, plasma membrane (56) versus cytosolic membranes derived from the trans-Golgi network or endosomes (57, 58). Nevertheless, the pseudotyping system described here provides the means to manipulate glycoprotein composition in the context of a viral particle, which could be advantageous for studying contributions of HSV glycoproteins to viral entry and membrane fusion.
HSV-1, along with other herpesviruses, infects multiple cell types. Moreover, HSV-1 has been reported to enter different cells by one of three cell-type specific routes: fusion at the plasma membrane, pH-dependent endocytosis, or pH-independent endocytosis (5, 59, 60). Entry by any of three routes requires the presence of gB, gD, gH/gL, and a gD receptor (6), and what dictates the choice of a route in a particular cell type is unclear. Future work will investigate the entry routes of VSVΔG-BHLD virions.
The four essential entry glycoproteins are sufficient for membrane fusion. Yet herpesviruses, including HSV-1, have many other glycoproteins. The roles of these so-called nonessential glycoproteins have not yet been fully elucidated, but some of these may play important roles during viral entry into specific cell types (8). It is possible that one of the nonessential HSV-1 proteins may specify entry routes or increase efficiency of entry into different cell types. The pseudotyping system pioneered in this work opens doors for future systematic exploration of the herpesvirus entry pathways and the role of the nonessential glycoproteins.
ACKNOWLEDGMENTS
We thank Stephen Kwok and Allen Parmelee at the Tufts Laser Cytometry Core facility for their help with FACS experiments; Marta Gaglia, James Schwob, and the members of the Schwob laboratory for help with the microscopy experiments; Maria Eriksson at the HMS EM Core Facility for help with sample preparation and data acquisition; Ellen White for performing negative-stain EM on VSVΔG-BHLD virions; Roselyn Eisenberg, Gary Cohen, Helena Browne, and David Knipe for the gifts of antibodies and cell lines; David Knipe for the gift of HSV-1 strain KOS8GFP; and Joan Mecsas for the gift of DAPI stain.
REFERENCES
- 1.Harrison SC. 2015. Viral membrane fusion. Virology 479-480C:498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci 65:1653–1668. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicola AV, Straus SE. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol 78:7508–7517. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komala Sari T, Pritchard SM, Cunha CW, Wudiri GA, Laws EI, Aguilar HC, Taus NS, Nicola AV. 2013. Contributions of herpes simplex virus 1 envelope proteins to entry by endocytosis. J Virol 87:13922–13926. doi: 10.1128/JVI.02500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jambunathan N, Charles AS, Subramanian R, Saied AA, Naderi M, Rider P, Brylinski M, Chouljenko VN, Kousoulas KG. 9 December 2015. Deletion of a predicted beta-sheet domain within the amino-terminus of herpes simplex virus glycoprotein K (gK) conserved among alphaherpesviruses prevents virus entry into neuronal axons. J Virol doi: 10.1128/JVI.02468-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roizman B, Pellett PE. 2001. The family Herpesviridae: a brief introduction, p 2381–2398. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 4th ed Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 11.Davis-Poynter N, Bell S, Minson T, Browne H. 1994. Analysis of the contribution of herpes simplex virus type 1 membrane proteins to the induction of cell-cell fusion. J Virol 68:7586–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roop C, Hutchinson L, Johnson DC. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J Virol 67:2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ligas MW, Johnson DC. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J Virol 62:1486–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai WH, Gu B, Person S. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J Virol 62:2596–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol 6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 16.Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. 2001. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell 8:169–179. doi: 10.1016/S1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- 17.Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog 7:e1002277. doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tiwari V, O'Donnell C, Copeland RJ, Scarlett T, Liu J, Shukla D. 2007. Soluble 3-O-sulfated heparan sulfate can trigger herpes simplex virus type 1 entry into resistant Chinese hamster ovary (CHO-K1) cells. J Gen Virol 88:1075–1079. doi: 10.1099/vir.0.82476-0. [DOI] [PubMed] [Google Scholar]
- 19.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 20.Backovic M, Jardetzky TS. 2009. Class III viral membrane fusion proteins. Curr Opin Struct Biol 19:189–196. doi: 10.1016/j.sbi.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muggeridge MI. 2000. Characterization of cell-cell fusion mediated by herpes simplex virus 2 glycoproteins gB, gD, gH and gL in transfected cells. J Gen Virol 81(Part 8):2017–2027. [DOI] [PubMed] [Google Scholar]
- 23.Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol 72:873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller CG, Krummenacher C, Eisenberg RJ, Cohen GH, Fraser NW. 2001. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol Ther 3:160–168. doi: 10.1006/mthe.2000.0240. [DOI] [PubMed] [Google Scholar]
- 25.Taylor TJ, McNamee EE, Day C, Knipe DM. 2003. Herpes simplex virus replication compartments can form by coalescence of smaller compartments. Virology 309:232–247. doi: 10.1016/S0042-6822(03)00107-7. [DOI] [PubMed] [Google Scholar]
- 26.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 27.Whitt MA. 2010. Generation of VSV pseudotypes using recombinant DeltaG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J Virol Methods 169:365–374. doi: 10.1016/j.jviromet.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tani H, Komoda Y, Matsuo E, Suzuki K, Hamamoto I, Yamashita T, Moriishi K, Fujiyama K, Kanto T, Hayashi N, Owsianka A, Patel AH, Whitt MA, Matsuura Y. 2007. Replication-competent recombinant vesicular stomatitis virus encoding hepatitis C virus envelope proteins. J Virol 81:8601–8612. doi: 10.1128/JVI.00608-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Owens RJ, Rose JK. 1993. Cytoplasmic domain requirement for incorporation of a foreign envelope protein into vesicular stomatitis virus. J Virol 67:360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson LR, Whelan SP. 2016. Infectious entry pathway mediated by the human endogenous retrovirus K envelope protein. J Virol 90:3640–3649. doi: 10.1128/JVI.03136-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J Virol 66:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng T, Ponce de Leon M, Novotny MJ, Jiang H, Lambris JD, Dubin G, Spear PG, Cohen GH, Eisenberg RJ. 1998. Structural and antigenic analysis of a truncated form of the herpes simplex virus glycoprotein gH-gL complex. J Virol 72:6092–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gompels UA, Minson AC. 1989. Antigenic properties and cellular localization of herpes simplex virus glycoprotein H synthesized in a mammalian cell expression system. J Virol 63:4744–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Randazzo BP, Kesari S, Gesser RM, Alsop D, Ford JC, Brown SM, Maclean A, Fraser NW. 1995. Treatment of experimental intracranial murine melanoma with a neuroattenuated herpes simplex virus 1 mutant. Virology 211:94–101. doi: 10.1006/viro.1995.1382. [DOI] [PubMed] [Google Scholar]
- 35.Bender FC, Samanta M, Heldwein EE, de Leon MP, Bilman E, Lou H, Whitbeck JC, Eisenberg RJ, Cohen GH. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J Virol 81:3827–3841. doi: 10.1128/JVI.02710-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gompels U, Minson A. 1986. The properties and sequence of glycoprotein H of herpes simplex virus type 1. Virology 153:230–247. doi: 10.1016/0042-6822(86)90026-7. [DOI] [PubMed] [Google Scholar]
- 37.Cairns TM, Fontana J, Huang ZY, Whitbeck JC, Atanasiu D, Rao S, Shelly SS, Lou H, Ponce de Leon M, Steven AC, Eisenberg RJ, Cohen GH. 2014. Mechanism of neutralization of herpes simplex virus by antibodies directed at the fusion domain of glycoprotein B. J Virol 88:2677–2689. doi: 10.1128/JVI.03200-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84:3825–3834. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J Virol 83:6825–6836. doi: 10.1128/JVI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hannah BP, Heldwein EE, Bender FC, Cohen GH, Eisenberg RJ. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J Virol 81:4858–4865. doi: 10.1128/JVI.02755-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maurer UE, Zeev-Ben-Mordehai T, Pandurangan AP, Cairns TM, Hannah BP, Whitbeck JC, Eisenberg RJ, Cohen GH, Topf M, Huiskonen JT, Grunewald K. 2013. The structure of herpesvirus fusion glycoprotein B-bilayer complex reveals the protein-membrane and lateral protein-protein interaction. Structure 21:1396–1405. doi: 10.1016/j.str.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker EB, Pritchard SM, Cunha CW, Aguilar HC, Nicola AV. 2015. Polyethylene glycol-mediated fusion of herpes simplex type 1 virions with the plasma membrane of cells that support endocytic entry. Virol J 12:190. doi: 10.1186/s12985-015-0423-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wille PT, Wisner TW, Ryckman B, Johnson DC. 2013. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 4:e00332-13. doi: 10.1128/mBio.00332-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe R, Matsuyama S, Shirato K, Maejima M, Fukushi S, Morikawa S, Taguchi F. 2008. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved S protein as revealed by pseudotype virus bearing cleaved S protein. J Virol 82:11985–11991. doi: 10.1128/JVI.01412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rojek JM, Perez M, Kunz S. 2008. Cellular entry of lymphocytic choriomeningitis virus. J Virol 82:1505–1517. doi: 10.1128/JVI.01331-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ray N, Whidby J, Stewart S, Hooper JW, Bertolotti-Ciarlet A. 2010. Study of Andes virus entry and neutralization using a pseudovirion system. J Virol Methods 163:416–423. doi: 10.1016/j.jviromet.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 47.Mulherkar N, Raaben M, de la Torre JC, Whelan SP, Chandran K. 2011. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 419:72–83. doi: 10.1016/j.virol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsuura Y, Tani H, Suzuki K, Kimura-Someya T, Suzuki R, Aizaki H, Ishii K, Moriishi K, Robison CS, Whitt MA, Miyamura T. 2001. Characterization of pseudotype VSV possessing HCV envelope proteins. Virology 286:263–275. doi: 10.1006/viro.2001.0971. [DOI] [PubMed] [Google Scholar]
- 49.Cheresiz SV, Kononova AA, Razumova YV, Dubich TS, Chepurnov AA, Kushch AA, Davey R, Pokrovsky AG. 2014. A vesicular stomatitis pseudovirus expressing the surface glycoproteins of influenza A virus. Arch Virol 159:2651–2658. doi: 10.1007/s00705-014-2127-y. [DOI] [PubMed] [Google Scholar]
- 50.Clemente R, de la Torre JC. 2009. Cell entry of Borna disease virus follows a clathrin-mediated endocytosis pathway that requires Rab5 and microtubules. J Virol 83:10406–10416. doi: 10.1128/JVI.00990-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rogalin HB, Heldwein EE. 23 September 2015. The interplay between the HSV-1 gB cytodomains and the gH cytotail during cell-cell fusion. J Virol doi: 10.1128/JVI.02391-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silverman JL, Heldwein EE. 2013. Mutations in the cytoplasmic tail of herpes simplex virus 1 gH reduce the fusogenicity of gB in transfected cells. J Virol 87:10139–10147. doi: 10.1128/JVI.01760-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jones NA, Geraghty RJ. 2004. Fusion activity of lipid-anchored envelope glycoproteins of herpes simplex virus type 1. Virology 324:213–228. doi: 10.1016/j.virol.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 54.Fan Z, Grantham ML, Smith MS, Anderson ES, Cardelli JA, Muggeridge MI. 2002. Truncation of herpes simplex virus type 2 glycoprotein B increases its cell surface expression and activity in cell-cell fusion, but these properties are unrelated. J Virol 76:9271–9283. doi: 10.1128/JVI.76.18.9271-9283.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harman A, Browne H, Minson T. 2002. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J Virol 76:10708–10716. doi: 10.1128/JVI.76.21.10708-10716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fuller S, von Bonsdorff CH, Simons K. 1984. Vesicular stomatitis virus infects and matures only through the basolateral surface of the polarized epithelial cell line, MDCK. Cell 38:65–77. doi: 10.1016/0092-8674(84)90527-0. [DOI] [PubMed] [Google Scholar]
- 57.Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, Elliott G. 2012. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J 31:4204–4220. doi: 10.1038/emboj.2012.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 59.Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol 79:6655–6663. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol 77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]