

ABSTRACT
Mycobacteriophage DS6A is unique among the more than 8,000 isolated mycobacteriophages due to its ability to form plaques exclusively on mycobacteria belonging to the Mycobacterium tuberculosis complex (MTBC). Speculation surrounding this specificity has led to unsupported assertions in published studies and patents that nontuberculous mycobacteria (NTM) are wholly resistant to DS6A infection. In this study, we identified two independent nonessential regions in the DS6A genome and replaced them with an mVenus-expressing plasmid to generate fluorescent reporter phages Φ2GFP12 and Φ2GFP13. We show that even though DS6A is able to form plaques only on MTBC bacteria, infection of various NTM results in mVenus expression in transduced cells. The efficiency of DS6A in delivering DNA varied between NTM species. Additionally, we saw a striking difference in the efficiency of DNA delivery between the closely related members of the Mycobacterium abscessus complex, M. abscessus and Mycobacterium massiliense. We also demonstrated that TM4 and DS6A, two phages that do not form plaques on M. massiliense, differ in their ability to deliver DNA, suggesting that there is a phage-specific restriction between mycobacterial species. Phylogenetic analysis reveals that the DS6A genome has a characteristically mosaic structure but provided few insights into the basis for the specificity for MTBC hosts. This study demonstrates that the inability of the MTBC-specific phage DS6A to form plaques on NTM is more complex than previously thought. Moreover, the DS6A-derived fluorophages provide important new tools for the study of mycobacterial biology.
IMPORTANCE The coevolution of bacteria and their infecting phages involves a constant arms race for bacteria to prevent phage infection and phage to overcome these preventions. Although a diverse array of phage defense systems is well characterized in bacteria, very few phage restriction systems are known in mycobacteria. The DS6A mycobacteriophage is unique in the mycobacterial world in that it forms plaques only on members of the Mycobacterium tuberculosis complex. However, the novel DS6A reporter phages developed in this work demonstrate that DS6A can infect nontuberculous mycobacteria at various efficiencies. By comparing the abilities of DS6A and another phage, TM4, to infect and form plaques on various mycobacterial species, we can begin to discern new phage restriction systems employed within the genus.
INTRODUCTION
The standard treatment for a drug-susceptible Mycobacterium tuberculosis infection is four drugs (rifampin, isoniazid, pyrazinamide, and ethambutol) for 2 months, followed by 4 months of two drugs (rifampin and isoniazid). One of the key aspects of successfully treating tuberculosis is ensuring that the infecting strain is susceptible to each of the antibiotics given; therefore, quickly diagnosing antibiotic resistance is important for successful patient outcome as well as for lowering the incidence of the generation of drug-resistant mutants from improper antibiotic administration (1). Unfortunately, even with the advent of modern genomic techniques that can rapidly identity M. tuberculosis resistance alleles, the gold standard of antibiotic susceptibility testing is still to examine M. tuberculosis growth in the presence of antibiotics (2, 3), and in many resource-limited countries, this remains the only available assay. However, the culture method is very time-consuming, preventing access to appropriate chemotherapy regimens by a patient for up to 2 months.
Reporter mycobacteriophages provide an alternative method for quickly diagnosing M. tuberculosis infections and profiling their antibiotic susceptibilities (4). These phages have been modified to contain a fluorophore, and infected cells will “light up,” providing a visual cue for their survival in the presence of antibiotics. The first generation of reporter mycobacteriophages was developed by Jacobs et al. (4); luciferase was inserted into mycobacteriophage D29, providing a way to rapidly determine the antibiotic susceptibility profile of infecting M. tuberculosis. While highly informative, these phages were limited to efficient function only in M. tuberculosis in laboratory settings, performing poorly with clinical samples and in populations of M. tuberculosis with mixed antibiotic susceptibilities (1). In second-generation reporter phages, this original concept was improved through the use of fluorescent reporters (5). During infection with these second-generation reporter phages, bacteria fluoresce, which can be readily detected by microscopy or flow cytometry. When assaying for surviving bacteria following antibiotic treatment, these reporter phages provide exquisite sensitivity and the ability to detect partially or fully resistant cultures by allowing any remaining subpopulation of bacteria to be easily monitored through fluorescence detection methods (5).
The third generation of fluorescent reporter phages (Φ2GFP10), through the use of an advanced fluorescent reporter (mVenus) and stronger phage promoter (L5-Pleft), emits a fluorescent signal 100 times stronger than the second-generation fluorescent reporter phages and is able to detect M. tuberculosis in patient sputum (6). Φ2GFP10 phage has been shown to be sensitive for detecting M. tuberculosis infections in numbers as few as 104 bacteria per sputum sample when infected with 108 phage, has been evaluated for use in both patient diagnostics and antibiotic resistance screening, and is currently being utilized in clinical studies in KwaZulu-Natal, South Africa (7).
In mycobacteria, shuttle phasmids (8) have been critical to the development of genetic manipulation systems for mycobacteria, including the development of transformation systems, transposon mutagenesis, recombination substitution mutagenesis, and transfer of point mutations (9–13), as reviewed extensively by Hatfull and Jacobs (14). Thus, TM4- and D29-based recombinant mycobacteriophages make attractive tools for mycobacterial research due to their broad host range, which includes many species of mycobacteria. Unfortunately, that same property limits the ability of TM4- and D29-derived fluorophages to differentiate between strains of M. tuberculosis and nontuberculous mycobacteria (NTM), some of which may cause symptoms similar to tuberculosis but require different treatments (15). Ideally, to improve the technology, a fluorophage would be created from a mycobacteriophage with a host range limited to M. tuberculosis.
Mycobacteriophage DS6A has demonstrated a permissive host range of only members of the Mycobacterium tuberculosis complex (MTBC) bacteria (http://phagesdb.org/phages/DS6A/), including the species M. tuberculosis, M. bovis, M. microti, M. canettii, and M. africanum, with this limited host range established by the inability of DS6A to form plaques on any NTM. This restricted host range led to the initial usage of DS6A as a therapeutic (16–18) and served as the basis for several patents for its implementation as a diagnostic tool (19–22). Since its discovery over 50 years ago (23), DS6A has garnered much speculation that its specificity to members of the MTBC is due to complete resistance by nonpermissive hosts (24). This speculation has driven the idea that reporter phages based on DS6A could be specific to mycobacteria of the MTBC and therefore could also be powerful tools for studying these bacteria, treating infections, and acting as a much-needed tool in diagnosing patients. For over 2 decades, the benefit of DS6A has remained speculative, as no DS6A-based fluorophage had been constructed.
In this study, we examined the restriction of the DS6A host range to the MTBC using two independent genetically engineered DS6A-based reporter phages. Furthermore, phylogenetic analysis of this phage has been performed to determine the evolutionary linkage of this narrow-host-range phage by comparison to over 1,500 sequenced phage genomes (25) (http://phagesdb.org/hosts/genera/1/). Our results show that the inability of DS6A to form plaques on NTM does not equate to a lack of infectibility by DS6A. Additionally, our work demonstrates the possible presence of novel restriction methods utilized by multiple species of mycobacteria.
MATERIALS AND METHODS
Reagents.
General laboratory chemicals were obtained from Sigma or Thermo Fisher. Enzymes used in cloning were obtained from New England BioLabs, unless otherwise noted.
Bacterial species/strains.
All strains and DNA constructs are listed in Table 1. Strain mc26230 (ΔpanCD ΔRD1) (6) was grown at 37°C in our standard growth medium (SGM) (7H9 [BD Difco] supplemented with 1% glycerol, 10% oleic acid-albumin-dextrose-catalase [OADC], 1% pantothenate [Acros Organics], and 0.1% Tween 80) (14). All other mycobacterial strains were grown in 7H9 medium containing 1% glycerol, 10% OADC, and 0.1% Tween 80 at 37°C, except for Mycobacterium marinum, which was grown at 30°C. Escherichia coli strain HB101 was grown in LB medium (BD Difco) supplemented with 0.2% maltose and 10 mM MgSO4. Transformed E. coli cells were grown in either liquid or solid LB medium with 100 μg/ml carbenicillin (Gold Bio).
TABLE 1.
Bacteria and mycobacteriophages used in this work
| Strain or phage construct | Species or parental phage | Genotypea | Reference(s) |
|---|---|---|---|
| Strains | |||
| mc26230 | M. tuberculosis | ΔRD1 ΔpanCD | 6 |
| mc2155 | M. smegmatis | eptC1 | 13, 33 |
| mc27231 | M. asiaticum | WT | 34 |
| mc27232 | M. fortuitum | WT | 34 |
| mc27233 | M. massiliense | WT | 35 |
| mc27234 | M. avium | WT | 34 |
| mc27235 | M. abscessus | WT | 35 |
| mc27236 | Mycobacterium avium-M. intracellulare 1403 | WT | 34 |
| mc27237 | Mycobacterium avium-M. intracellulare 193B | WT | 34 |
| mc27238 | M. marinum | WT | 34 |
| Phage constructs | |||
| Ph90 | DS6A | WT | 23 |
| phAE900 | DS6A | Δ(gp42-gp65)::PacI/pYUB328 PacI | This work |
| phAE901 | DS6A | Δ(gp66-gp72)::PacI/pYUB328 PacI | This work |
| Φ2GFP10 | TM4 | TM4ts::Δ(gp48-gp65)::PacI/pYUB1551 PacI | 6; see also Fig. S1 in the supplemental material |
| Φ2GFP12 | DS6A | Δ(gp42-gp65)::PacI/pYUB1552 PacI | This work |
| Φ2GFP13 | DS6A | Δ(gp66-gp72)::PacI/pYUB1552 PacI | This work |
WT, wild type; TM4ts, temperature-sensitive TM4.
Amplification of mycobacteriophages.
Mycobacteriophage DS6A and DS6A-based fluorophages were amplified by growing mc26230 cells to an optical density at 600 nm (OD600) of 0.8, concentrating the cells 10-fold through centrifugation and resuspension in fresh SGM medium without Tween, aliquoting 600 μl to a fresh tube, and mixing in 100 μl of diluted DS6A phage to yield ∼105 to 106 PFU per plate. Bacterium-phage mixtures were incubated for 20 min at 37°C to allow for phage adsorption. Next, 6 ml of 7H9 top agar (supplemented with 10% OADC and 1% pantothenate) was added to each mixture, which was then plated via soft-agar overlay onto SGM plates. Plates were incubated for 7 to 8 days, and then 4 ml of MP buffer (150 mM NaCl, 10 mM MgSO4, 2 mM CaCl2, 50 mM Tris [pH 7.5]) (14) was added; plates were gently shaken at room temperature for 30 min, and then all the soft agar and liquid were scraped off into a 50-ml conical tube. Tubes were centrifuged at 4,200 rpm for 30 min and then filtered through a 0.45-μm Steriflip filter (Millipore). The titers of the recovered phages were determined, and stocks were stored at 4°C. D29 and TM4 phages were amplified using standard protocols (26). Φ2GPF10 fluorophage was amplified using the same procedure as for D29 but with incubations at 30°C due to the temperature sensitivity of the phage.
Isolating mycobacteriophage DNA.
To extract phage DNA, 500 μl of phage lysate (titer, ≥1010 PFU/ml) was mixed with 10 μl of 10 mg/ml lysozyme (Sigma) in a 1.5-ml microcentrifuge tube and incubated at 50°C for 15 min. Next, 50 μl of 10% SDS solution was added and mixed in gently, and then 10 μl of 10-mg/ml proteinase K (Sigma) was added with gentle mixing. The tube was then incubated at 50°C for 30 min before mixing in 100 μl of 5 M NaCl, followed by 100 μl of prewarmed hexadecyltrimethylammonium bromide (CTAB), mixing by inversion, and incubating at 50°C for 30 min. Next, 770 μl of phenol-chloroform was added, and the mixture was shaken vigorously for 1 to 2 min prior to centrifugation for ≥5 min at 15,000 rpm. The upper fraction was transferred to a new microcentrifuge tube, to which 760 μl of chloroform-methanol alcohol solution (2:1 ratio) was then added, followed by vigorous shaking for 1 to 2 min and then centrifugation for ≥5 min at 15,000 rpm. The upper fraction was transferred to a new microcentrifuge tube, isopropanol (0.7 times the sample volume) was added, and gentle inversion was done to precipitate the DNA. The DNA was pelleted by pulse centrifugation at 15,000 rpm; the supernatant was removed, and the pellet was washed in 1 ml 70% ethanol and recentrifuged in the same manner. The ethanol was removed, and the DNA pellet was resuspended in 50 μl of ultrapure water and stored at 4°C.
Generation of DS6A shuttle phasmid.
Shuttle phasmids were constructed as described by Jacobs et al. (8). High-quality intact phage DNA served as the genetic starting material for shuttle phasmid construction. T4 DNA ligase was used to ligate intact phage DNA into concatemers at room temperature overnight. Concatemers were partially digested using Sau3AI in five 2-fold serial dilutions of enzyme, beginning at 2.5 units/reaction, for exactly 15 min at 37°C before being placed on ice. The extent of digestion was evaluated by gel electrophoresis, with analyses indicating that 0.6 and 0.3 unit of Sau3AI were appropriate for shuttle phasmid construction.
Plasmid fragments ligated to partially digested DS6A DNA were generated by first digesting pYUB328 (27) with XbaI for 2 h at 37°C. The digested DNA was then dephosphorylated with SuperSAP (Affymetrix) for 28 min at 37°C before denaturing at 80°C for 20 min. Finally, linear plasmid DNA was digested with BamHI at 37°C overnight, resulting in two asymmetrical BamHI cohesive ends (two DNA arms). The final product was verified two ways: (i) DNA arms were run on an agarose gel, with the resulting bands of 1,165 bp and 3,783 bp demonstrating the complete removal of intact pYUB328, and (ii) 2 μl of DNA fragments was ligated for 4 h, and the ligation was run on an agarose gel to confirm the BamHI cohesive ends after BamHI digestion and dephosphorylation efficiency after XbaI digestion.
Sau3AI partially digested phage DNA was mixed with pYUB328 plasmid fragments in a ratio of 1:4 and ligated overnight with T4 ligase at room temperature. The resulting ligation was λ-packaged (MaxPlax) and subsequently used to infect HB101 E. coli cells (standard protocol). Cells were spread on plates containing 100 μg/ml carbenicillin and incubated overnight at 37°C. The resulting colonies were harvested by scraping and grown in LB containing 100 μg/ml carbenicillin. DNA was isolated from the pooled colonies using a QIAprep Spin miniprep kit (Qiagen), and various DNA amounts were electroporated into 200 μl of M. tuberculosis strain mc26230 using a Bio-Rad electroporator with settings of 2.5 kV, 25 μF, and 1,000 Ω. The entire electroporation volume was plated via soft-agar overlay onto SGM plates. The plates were incubated at 37°C for 7 days and then examined for the presence of plaques. The resulting plaques were picked and amplified, and DNA was harvested via CTAB DNA prep, as described above. Sequencing primers CGGCCGCATAATACGACTCACTA and CCGCAATTAACCCTCACTAAAG were used to identify the plasmid insertion site in the DS6A DNA.
Generation of DS6A fluorescent reporter phages.
The two DS6A shuttle phasmids generated were digested with PacI and run on a 1% agarose gel. Of the two bands on the gel, the higher-molecular-weight band was excised and extracted using the QIAquick gel extraction kit (Qiagen). New fragments were constructed with pYUB1552, a pYUB328-derived vector expressing mVenus from the L5-Pleft promoter (see Fig. S1 in the supplemental material), as described above, except that PacI was substituted for BamHI in the second digestion. These fragments were ligated to the two gel-purified PacI-ended DS6A DNA fragments overnight at room temperature. DNA was λ-packaged and used to infect HB101 cells as described above. The resulting colonies were examined for green fluorescence, glowing colonies were picked and grown in LB containing 100 μg/ml carbenicillin, and DNA was extracted. DNA was electroporated into mc26230 as described above, and at 7 days, the resulting plaques were examined for green fluorescence. Glowing plaques were picked and amplified, and the resulting phages were stored at 4°C.
Phage infection of bacteria.
The efficiency of phage infections for all strains was determined via spot assays. Log-phase cells (600 μl) were mixed with 4 ml of top agar (SGM) and plated on SGM. Phages were diluted with MP buffer in 10-fold increments, and 4 μl of each dilution was spotted on each strain in duplicate and allowed to incubate for 7 days. Efficiency calculations were conducted from spots yielding from 2 to 15 plaques.
Phage infections for amplification of each phage were performed by growing wild-type M. smegmatis or M. tuberculosis to log phase, centrifuging the cells, and resuspending them at 5 times the original concentration with MP buffer. Next, 600 μl of cells was mixed with 100 μl of phage (diluted to yield ∼50,000 to 100,000 PFU/plate), allowed to adsorb for 25 min at 30°C or 37°C, mixed with 6 ml of top agar (SGM), and plated for 2 to 3 days for M. smegmatis or 7 to 8 days for M. tuberculosis. Phages were harvested by adding 4 ml of MP buffer to each plate and then gently shaking them at room temperature for 30 min. The soft agar and liquid were scraped off into 50-ml conical tubes, which were then centrifuged at 4,200 rpm for 30 min and filtered through a 0.45-μm Steriflip filter (Millipore). The titers of the recovered phages were determined, and stocks were stored at 4°C.
Infections for imaging.
To infect M. tuberculosis with fluorophages, 10 ml of cells was grown to an OD600 of 0.8, washed three times in MP buffer, resuspended in 1 ml of SGM, and then incubated at room temperature for 1 to 3 days. NTM bacteria were similarly washed but were resuspended in medium without pantothenate and incubated overnight at 4°C or at 37°C for 3 h prior to phage infection. To infect the cells, 2 μl of cells was aliquoted into a sterile 1.5-ml microcentrifuge tube, and 25 μl of 1010 PFU/ml fluorophage was mixed in. Mixtures were incubated at 37°C overnight prior to imaging.
Microscopy.
Infected cells were centrifuged (10,000 rpm, 2 min), and the supernatant was discarded. Pellets were resuspended in 5 μl of MP buffer before being transferred to a wet-mount slide. Imaging was performed using a Nikon-TI microscope at ×60 magnification (40×/numerical aperture [NA] of 0.75 at 1.5×), and image processing was performed using NIS Elements-AR software. Exposure was set for 40 ms for brightfield microscopy and 400 ms for detecting mVenus fluorescence.
Flow cytometry.
Cells were prepared as described above and then diluted after 18 h in 100 μl of MP buffer before being transferred to a flow cytometry tube. Samples were run on either a FACSCalibur flow cytometer in a biological safety hood or on a BD FACSCanto II cell analyzer, and 100,000 events were acquired. All flow cytometry data were analyzed using FlowJo (version 10), and gating was done on the negative sample of each strain. All bacterial infections were analyzed in duplicate.
Phylogenetic analysis and tree construction.
Genes of interest from the genome of DS6A were selected from the database at PhagesDB.org. BLAST analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) of the genes of interest from DS6A was performed against the full phage database using the BLAST 2.3.0+ function tblastn with an E value cutoff of 10−5. Hits were then aligned using Clustal Omega (28, 29). Phylogenetic relationships were computed using maximum parsimony analysis using the MEGA7 software (30). The maximum parsimony search method was subtree-pruning-regrafting with 10 initial trees.
RESULTS
Generation of DS6A shuttle phasmids.
The DS6A shuttle phasmid was generated using a multistep cloning method (Fig. 1) that resulted in the replacement of a nonessential region of the phage genome with a plasmid and that required high-quality intact phage genomic DNA. Standard amplification of mycobacteriophages is performed in M. smegmatis mc2155 (31). However, DS6A phage amplification requires MTBC strains of mycobacteria, but standard mycobacteriophage amplification protocols regularly yield low phage titers and fractured phage DNA (26). High-titer DS6A was obtained using a modified protocol in mc26230, a biosafety level 2 (BSL2)-laboratory-safe strain of M. tuberculosis (32), yielding high-quality phage DNA suitable for DS6A shuttle phasmid construction (see Fig. S2A in the supplemental material). Nonessential regions for phage propagation were determined by creating a random deletion library of DS6A in which the deletion region was replaced by the plasmid DNA. To delete 1 to 10 kb of DNA, harvested concatemerized DS6A DNA was partially digested by Sau3AI, a four-base “cutter” restriction enzyme with 287 randomly distributed recognition sites in DS6A, and the resulting DNA fragments were ligated with plasmid pYUB328 that had been previously digested with XbaI and BamHI, which generate termini compatible with Sau3AI-digested ends. This random library was amplified in E. coli and transformed into M. tuberculosis to select for partial phage genomes capable of producing phages, as confirmed by the ability to form plaques. Approximately 25 plaques were obtained on M. tuberculosis, and eight representative plaques were picked, amplified to 1010 PFU/ml, and digested with PacI to confirm the presence of plasmid DNA in these phages (see Fig. S2B). We observed two bands upon digestion, the larger band being the high-molecular-weight DS6A DNA and the smaller, at ∼3,900 bp, representing a single cos pYUB328 plasmid. It is important to note that these phages could infect M. tuberculosis but remained incapable of forming plaques on M. smegmatis or any other mycobacterial strain outside the MTBC. Thus, these genetically engineered DS6A phasmids retained the selectiveness of the parental DS6A.
FIG 1.

Schematic of the multistep construction of a DS6A shuttle phasmid. DS6A genomic DNA isolated from phage is linear and was ligated to concatemerize. Partial Sau3AI digestion was used to delete random regions of 1 to 10 kb. The cohesive ends generated by Sau3AI are compatible with the two BamHI fragments generated from pYUB328, and the ligation of the two pYUB328 fragments to Sau3AI-digested DS6A resulted in a linear DNA fragment flanked by λ-cos sites at each end. Incubation of this ligation mix with in vitro λ-packaging mix started the packaging reaction by cutting at the first cos site, followed by inserting the DNA from the first cos site to the second cos site, and terminating the packaging reaction by cutting the second cos site. Transduction of in vitro packaged λ to E. coli HB101 delivered the linear DNA from λ heads to E. coli. The two cohesive ends of the λ-cos site were ligated to circularize. This process generated a phasmid library where modified pYUB328 DNA (with one cos site) is inserted at random places in the DS6A genome. Transformation of this library into M. tuberculosis generates DS6A plaques in cases where the recombinant DNA has replaced a small nonessential region of DS6A DNA. The deletion sites in these phages were determined by sequencing. AmpR, ampicillin resistance; Amp100, 100 µg/ml ampicillin; Mtb, M. tuberculosis.
The deletion mutagenesis site mapping of these phages established that there are at least two nonessential regions in the DS6A phage genome, as evidenced by two unique DS6A shuttle phasmids, phAE900 {with a deletion of bp 32941 to 43686 [Δ(gp42-gp65)]} and phAE901 {with a deletion of bp 43912 to 47301 [Δ(gp66-gp72)]} (Fig. 2A). It was noted that while no genes previously thought to be essential for plaque formation were deleted in these phasmids, the deletion in phAE900 includes the integrase gene, whereas the phAE901 deletion includes mazG. While both these genes can play roles in phage life cycles, they appear to have no obvious detrimental effects on the DS6A phasmids, as the two mutants gave rise to similarly sized plaques and could be amplified to wild-type titers.
FIG 2.

Generation of DS6A fluorescent reporter phages. (A) The two nonessential regions in DS6A identified after deletion mutagenesis site mapping of all the recombinant DS6A phages are shown. The first deletion of ∼11 kb spanned genes gp42 to gp65, and the resulting phage was named phAE900. The second deletion mapped to an adjacent 3.4-kb region spanning gp66 to gp72, and the resulting phage was named phAE901. (B) The pYUB328 plasmid inserted in phAE900 and phAE901 was replaced by the pYUB1552 plasmid, resulting in Φ2GFP12 (i) and Φ2GFP13 (ii), respectively. Comparable expression of mVenus from the Pleft (L5) promoter resulted in plaque boundaries of similar fluorescence intensities after electroporation of Φ2GFP12 and Φ2GFP13 into M. tuberculosis. Genome maps were generated using Phamerator (53), with rightward- and leftward-transcribed genes shown as colored boxes above and below the genome, respectively. Genes are sorted into phamilies according to sequence similarity (37); the pham assignment number is shown above or below each gene with the number of phamily members in parentheses. Genes are colored according to their pham assignment, and white-colored genes are those that do not have homologues among other mycobacteriophages.
Generation of DS6A reporter mycobacteriophages.
A salient feature of these shuttle phasmids is the introduction of two PacI sites at each end of the deletion junction. The PacI site was introduced because its 8-bp recognition site is 100% AT rich and not found in the heavily GC-rich (∼67% GC content) genomes of mycobacteria. The DS6A genome shares both this GC nucleotide bias (68.4% GC content) and the lack of a PacI site, making a PacI site ideal for the easy insertion of recombinant DNA in phAE900 and phAE901. These shuttle phasmids were converted into reporter phages by replacing pYUB328 with pYUB1552, a plasmid that expresses mVenus under the highly constitutive L5-Pleft promoter, generating Φ2GFP12 and Φ2GFP13, respectively, from phAE900 and phAE901. All of the ∼20 plaques arising on M. tuberculosis lawns after electroporation with Φ2GFP12 and Φ2GFP13 had fluorescent green borders, indicating that the fluorescent reporter cassette had integrated successfully into phAE900 and phAE901 (Fig. 2B). The successful insertion of a single cos pYUB1552 in the DS6A genome was also confirmed by PacI digestion (data not shown). As with phAE900/901, these phasmids were unable to form plaques on any tested mycobacteria from outside the MTBC, including M. smegmatis, M. fortuitum, M. abscessus, and M. massiliense, demonstrating that the reporter version of this phage again retained its selectivity. Additionally, these phages could be amplified to titers similar to those of parental DS6A (1010 PFU/ml), demonstrating the production of mVenus in infected cells was not deleterious to phage production.
Comparison of infection efficiencies of DS6A/Φ2GFP12 and TM4/Φ2GFP10 in M. tuberculosis.
The percentage of fluorescent cells in the population and the fluorescence intensity of infected M. tuberculosis are dependent upon both the infection efficiency and multiplicity of infection (MOI) (6). Both the TM4-based Φ2GFP10 and the DS6A-based Φ2GFP12 express the mVenus gene from the same Pleft promoter of phage L5. We reasoned that if the two phages infect M. tuberculosis with similar efficiencies, comparable portions of the population should turn fluorescent, with similar mean fluorescence intensities, when infected at comparable MOIs.
Results show that Φ2GFP10 and Φ2GFP12 (Fig. 3A) induced infected M. tuberculosis cells to fluoresce at similar intensities, plateauing at 18 to 20 h postinfection. When overlaid with their respective brightfield images, the fluorescence microscopy images indicated that 90 to 95% of the cells were infected by these two phages in multiple microscopic fields. These data indicate that the DS6A reporter phage is highly efficacious at infecting and expressing the reporter gene in M. tuberculosis. Fluorescence microscopy provided a good representative “snapshot” of the value of Φ2GFP12 as a reporter phage; however, it is a qualitative observation. Flow cytometry analysis was performed to quantitate the infection efficiency of Φ2GFP12 compared to that of Φ2GFP10 (Fig. 3B). With Φ2GFP10 infection of M. tuberculosis, 87.7% of the cells were positive for mVenus, and Φ2GFP12 mirrored those results, with 86.1% of cells fluorescing, demonstrating that the newly generated fluorophage was efficient at both infecting and generating fluorescence in M. tuberculosis.
FIG 3.
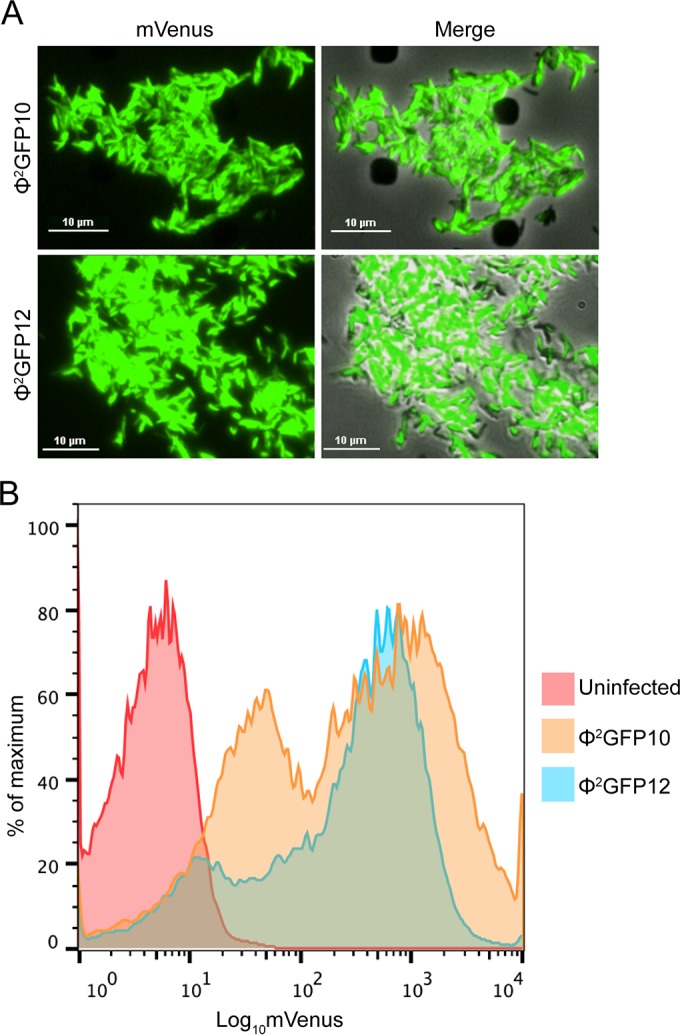
Comparison of DS6A- and TM4-derived fluorescent reporter phages. The fluorescence intensity of M. tuberculosis after infection with Φ2GFP10 and Φ2GFP12 was evaluated by fluorescence microscopy (A) and flow cytometry (B).
DS6A can inject its DNA but is unable to productively infect NTM.
Prior to the generation of DS6A reporter phage, it was possible to observe productive infection by DS6A phage only by scoring for the efficiency of plaque formation on the host. DS6A reporter phage, for the first time, provide a visual alternative to determine whether the initial steps, such as phage adsorption, receptor binding, and DNA injection, are occurring even when DS6A is unable to form plaques on the bacterial lawn. The presence of fluorescence after Φ2GFP12 infection would mean that DS6A is able to perform the first three steps of the phage life cycle. Four species of NTM were tested: the M. smegmatis laboratory strain mc2155 (33) and clinical strains Mycobacterium avium-M. intracellulare complex 1403 (34), M. abscessus (35), and M. fortuitum. To our surprise, all 5 strains fluoresced, with the fluorescence intensity of individual cells on par with that of M. tuberculosis cells infected by Φ2GFP12 (Fig. 4A); however, there were differences in efficiencies of infection. Visual estimates indicated M. smegmatis was infected the most efficiently, with 30 to 50% of cells fluorescing, whereas Mycobacterium avium-M. intracellulare complex 1403, M. abscessus, and M. fortuitum had significantly fewer fluorescing cells, in the range of 5 to 20%. Additionally, time-lapse imaging was performed, and cells were monitored for fluorescence initiation and termination (data not shown). In each strain, cell fluorescence ceased 2 to 4 h after initiation; however, differentiation between phage-induced lysis and phage-induced cell death was indeterminate due to the two events yielding the same cellular phenotype of a residual dark shell (data not shown).
FIG 4.

Comparison of DS6A- and TM4-derived fluorescent reporter phage infections of NTM. (A) The fluorescence intensity of M. tuberculosis, M. smegmatis, Mycobacterium avium-M. intracellulare 1403, M. abscessus, and M. fortuitum after infection with Φ2GFP12, shown by fluorescence microscopy and brightfield imaging. (B) Comparison of fluorescence intensities and percentages of the population infected using Φ2GFP12 and Φ2GFP10 at similar MOIs. Flow cytometry analysis of M. smegmatis, Mycobacterium avium-M. intracellulare 1403, M. abscessus, M. fortuitum, M. marinum, M. asiaticum, M. avium, M. massiliense, and Mycobacterium avium-M. intracellulare 193B is shown. In each case, uninfected cells were used to control for autofluorescence. (C) Plaque formation efficiencies of representative mycobacteriophages on MTBC and NTM.
To quantify the ability of DS6A to infect NTM, flow cytometry analysis was performed on nine NTM species/strains, i.e., M. smegmatis, M. abscessus, Mycobacterium avium-M. intracellulare 1403, Mycobacterium avium-M. intracellulare 193B, M. asiaticum (34), M. marinum (34), M. fortuitum, M. avium (34), and M. massiliense (35), which were infected with either Φ2GFP12 or Φ2GFP10 (Fig. 4B). Uninfected cells were used as controls for autofluorescence. Flow cytometry data revealed several trends in the ability of each phage to infect a mycobacterial host (Table 2). First, although all mycobacterial strains were infected by Φ2GFP10, the degree of infectivity varied. M. tuberculosis, M. massiliense, Mycobacterium avium-M. intracellulare 193B, and M. smegmatis were all very efficiently infected, with infection rates of ≥80%. Each of the remaining strains exhibited differing infectivity efficiencies, ranging from 70% in M. marinum down to 30% in Mycobacterium avium-M. intracellulare 1403. In all cases, there was a clear population shift from nonfluorescent to fluorescent, and in many cases, there were two distinct fluorescent populations (Fig. 3B and 4B). The basis for these distinct fluorescent populations is unknown, but we hypothesize it is caused by either phage infection of the more metabolically active cells or the cells that are about to undergo division within the population. Second, eight of the nine NTM were susceptible to Φ2GFP12 infection, albeit, with the exception of M. tuberculosis, at a lower efficiency than with Φ2GFP10. M. asiaticum, M. smegmatis, M. avium, and Mycobacterium avium-M. intracellulare 193B were the most susceptible to Φ2GFP12, with infection efficiencies of ≥30%, whereas the efficiencies for M. marinum and M. fortuitum were around 20%. Mycobacterium avium-M. intracellulare 1403 and M. abscessus had infection rates of only 6%, and M. massiliense appeared to be completely resistant to Φ2GFP12. Taken together, all NTM besides M. massiliense were infectible by both phages. However, sensitivity of the NTM to infection with Φ2GFP10 was 2 to 4 times higher than with Φ2GFP12.
TABLE 2.
Φ2GFP12 and Φ2GFP10 infection efficiencies of NTM
| Species | % mVenus-positive cells for: |
Ratio of Φ2GFP10 value to Φ2GFP12 value | |
|---|---|---|---|
| Φ2GFP12 | Φ2GFP10 | ||
| M. tuberculosis | 86.1 | 87.7 | 1.02 |
| M. smegmatis | 33.1 | 77.8 | 2.35 |
| M. abscessus | 6.2 | 34.9 | 5.64 |
| Mycobacterium avium-M. intracellulare 1403 | 6.1 | 28.8 | 4.72 |
| Mycobacterium avium-M. intracellulare 193B | 28.1 | 80.4 | 2.86 |
| M. asiaticum | 39.2 | 46.3 | 2.35 |
| M. marinum | 23.8 | 68.5 | 2.88 |
| M. fortuitum | 19.7 | 42.2 | 2.14 |
| M. avium | 28.5 | 58.5 | 2.05 |
| M. massiliense | 1.1 | 84.1 | 76.45 |
Comparison of plaque formation efficiencies of various phages.
Flow cytometry data indicated that most strains of mycobacteria tested are infectible by DS6A, and all are infectible by Φ2GFP10. These results indicate that phage infection and induced fluorescence do not equate to phage plaque formation on a host, but it is not known if this phenomenon is DS6A specific. Prior work by Jacobs-Sera et al. demonstrated that the cluster G phages Halo and BPs have different efficiencies of plaque formation when infecting M. smegmatis and M. tuberculosis (36). To determine if infection/fluorescence correlates with plaque formation with other phages, we compared the efficiencies of plaque formation with phages DS6A, Φ2GFP12, TM4, D29, and Φ2GFP10 on M. tuberculosis and the four NTM species M. smegmatis, M. abscessus, M. fortuitum, and M. massiliense (Fig. 4C). As expected, all five phages produced plaques on M. tuberculosis, and all except DS6A produced plaques on M. smegmatis. However, none of the five phages were able to form plaques on the other NTM, which is contrary to what was expected considering the flow cytometry data demonstrating infectibility.
When these results are taken with the flow cytometry data, an interesting picture emerges. M. massiliense is wholly resistant to plaque formation with both fluorophages and yet exhibited fluorescence with Φ2GFP10 only. M. fortuitum showed moderate fluorescence with both phages but cannot produce plaques with either, whereas M. abscessus had moderate fluorescence with Φ2GFP10 but only minimal fluorescence with Φ2GFP12. Previously, bacteria that could not produce plaques with a phage were considered resistant; however, these data indicate that the relationship between resistance and plaque formation is far more complex.
Phylogenetic analysis of DS6A.
A large collection of sequenced genomes of phages infecting actinobacterial hosts (http://phagesdb.org) reveals a continuum of genetic diversity but with uneven representation of different types. The genomes can thus be grouped into separate clusters and subclusters according to their genomic relationships, but with sometimes poorly delineated boundaries between the different groups (37). This finding is consistent with the pervasive genomic mosaicism of phage genomes, whereby different DNA segments with different evolutionary histories are reassorted by homologous and illegitimate recombination.
DS6A is currently classified as a “singleton” genome, as it has no close relatives among the more than 1,500 sequenced phages of actinobacterial hosts (38). Furthermore, fewer than 50% of the predicted proteins have identifiable homologues in these other sequenced phages (Fig. 5), a characteristic of singleton phage genomes (37). For the 46 genes with identifiable homologues, the best protein matches typically share less than 60% amino acid identity and only two gene products have matches with greater than 70% amino acid identity. Moreover, the top matches correspond to representatives of 24 different clusters, subclusters, or singleton genomes, reflecting the characteristically mosaic nature of the DS6A genome (37). Rather than examining the phylogeny of the DS6A genome in its entirety, we compared a selected set of DS6A genes based on their functional and spatial distribution in the DS6A genome. Sequences of the DS6A structural proteins gp11 (major capsid protein), gp16 (major tail protein), and gp19 (tape measure protein), DNA-interacting proteins gp34 (DNA polymerase [Pol] III β-subunit), gp56 (integrase), gp70 (MazG), and gp77 (primase), and the late lysis proteins gp30 (lysin A) and gp31 (lysin B) were queried against all sequenced mycobacteriophages at the protein level to determine their closest relative and whether the phylogenetic analysis would provide some answers regarding DS6A evolution (see Fig. S3i to ix in the supplemental material).
FIG 5.

Representation of phylogenetic analysis of various DS6A proteins against sequenced mycobacteriophages. Proteins selected for analysis are boxed. The closest cluster relative is written underneath the analyzed protein. The closest relatives to the capsid and tape measure proteins belong to singleton phages and are specifically named. The phylogenetic trees are shown in Fig. S3 in the supplemental material. The map was generated as described in the Fig. 2 legend.
The phylogenetic data are summarized in Fig. 5 and Table 3. The major capsid protein had the closest similarity to the singleton phage Sparky, a singleton phage that is closely related to cluster X phages (37). The tape measure protein showed strongest relation to the singleton phages Muddy, ArV2, Pine5, and Lucky10, as well as many other phages. The major tail protein was most closely associated with the two cluster X phages Gaia and Nebkiss. Additionally, of the two DNA processing proteins MazG and primase, MazG was more closely associated with cluster X phages, whereas the primase was closely associated with both cluster X phages and cluster J phages. Based on the sequence similarity among the genes, it is difficult to state whether the presence of genes similar to cluster X in DS6A is a result of one major rearrangement or of several independent recombination events over time. Uniquely, the DNA polymerase III β-subunit most closely mapped to cluster AR phages. The integrase maps to a wholly different cluster, cluster F, as well as to a variety of other clusters. Last, the lysin protein lysin A was found to be highly conserved and present in a multitude of differing clustered phages, whereas lysin B had the strongest similarity to cluster A and cluster L phages.
TABLE 3.
Amino acid identities and similarities of DS6A gene products to their closest phage relatives
| Gene name | Gene product | Protein length (amino acids) | Closest phage relative(s) | % identity (amino acids) | % similarity (amino acids) |
|---|---|---|---|---|---|
| gp11 | Major capsid | 348 | Sparky | 70, 30 | 82, 44 |
| gp16 | Major tail | 223 | Gaia, Nebkiss | 56, 56 | 74, 74 |
| gp19 | Tape measure | 1,208 | Varied (most similar to Pine5, Muddy, ArV2) | 31, 32, 29 | 47, 45, 44 |
| gp30 | Lysin A | 486 | Varied (most similar to Avani) | 55 | 64 |
| gp31 | Lysin B | 345 | Cluster A, cluster L | 46, 45 | 58, 57 |
| gp34 | DNA Pol III β-subunit | 383 | ArV1 | 30 | 44 |
| gp56 | Integrase | 582 | Varied (most similar to Zapner) | 49 | 62 |
| gp70 | MazG | 174 | Gaia, Nebkiss | 60, 60 | 70, 70 |
| gp77 | Primase | 265 | Cluster X, cluster J | 39, 39 | 53, 54 |
An unusual feature of the DS6A genome is the organization of the genes near the center of the genome that are anticipated to be involved in the establishment and maintenance of lysogeny (Fig. 5). First, the integrase gene (gp56) encodes a member of the tyrosine-integrase family of site-specific recombinases but has an unusual structure in which an excise-like domain is present within the gene, inserted approximately at codon 380; HHPred analysis shows significant similarity (E value, 10−9) of this domain to the gp36 excise domain of mycobacteriophage Pukovnik (39). This is a highly unusual organization, and we are not aware of any other examples of this. Second, a predicted attP gene was identified through a 49-bp sequence (the common core; DS6A coordinates 36358 to 36406) shared with a putative attB site that overlaps a tRNAlys gene present in both M. smegmatis and M. tuberculosis H37Rv (Msmeg_4746 and NT02MT2737, respectively). However, the attP site is not located in its anticipated location adjacent to the integrase gene open reading frame but is rather positioned upstream in the gp50-gp51 intergenic region. Last, a gene encoding a putative DS6A DNA binding protein (gp58) is a good candidate for providing repressor function, and the gene is divergently transcribed from another predicted DNA binding protein gene, gp59 (Fig. 5). Thus, although the lysogeny of DS6A in M. tuberculosis has not been demonstrated, we predict that lysogeny can be established and maintained.
DISCUSSION
Our work reveals aspects of the DS6A phage unknown before the construction of DS6A fluorescent reporter phages. Past speculation for the usage of DS6A as a diagnostic tool manifested itself through several patents based on the idea that DS6A was so specific to MTBC bacteria that only they would fluoresce when infected with DS6A fluorophages or express DS6A phage DNA (19–22). However, in light of our current work, DS6A applications as a specific diagnostic phage are more limited than was earlier hypothesized. Currently, there are two mycobacteriophage-based diagnostic tests to screen for M. tuberculosis infections. The first test uses Φ2GFP10 (6) and has demonstrated early success for diagnosing drug-susceptible M. tuberculosis infections in South Africa (7). It is unclear if the DS6A-based fluorophage is a better alternative to Φ2GFP10, considering that the performances of DS6A and Φ2GFP10 are comparable on laboratory-grown cultures of M. tuberculosis, and that DS6A, in most cases, is relatively less efficient at infecting NTM in comparison to Φ2GFP10. The second test uses the commercially available FASTPlaque tuberculosis (TB) assay (40) in which D29 phages are used to infect patient samples; extracellular phages are then neutralized, and D29 progeny phages are screened by visualizing plaque formation on lawns of fast-growing M. smegmatis. Although our DS6A fluorophage does not form plaques on lawns of M. smegmatis, these phages could be used to modify this assay and potentially reduce the time to diagnosis from days to hours. As with D29 phage, DS6A fluorophages would be used to infect M. tuberculosis, but instead of screening for plaques after extracellular neutralization, samples would be screened for fluorescence, potentially yielding results both more quickly and more sensitively.
DS6A plaques are not evidently turbid on lawns of M. tuberculosis, suggesting that it is virulent in nature or that it is temperate but forms lysogens at relatively low frequencies, as reported for some other mycobacteriophages (41). DS6A genomic features reflect components characteristic of temperate phages, including a putative repressor gene (gp58), an integrase gene (gp56), and a putative attP site (gp50-gp51 intergenic region). Furthermore, we predict that if integration occurs, it will be at the tRNAlys gene present in both M. smegmatis and M. tuberculosis. Phylogenetic analysis of the integrase protein adds another level of complexity to this hypothesis; we show that the DS6A integrase is closely related to the integrase in cluster F phages as well as in one cluster K phage. However, cluster F phages are not known to readily infect M. tuberculosis, suggesting these phages either diverged from a common ancestor or coinfected an alternative host bacterium, which allowed for the genetic recombination to occur and further contributed to the observed DS6A mosaicism. As our fluorescence work in this study demonstrated, it is also possible that although cluster F phages cannot form plaques, they can inject their DNA into M. tuberculosis, facilitating the potential interaction between DS6A and a cluster F phage. Based on this phylogenetic analysis, in theory, DS6A could still act as a temperate phage, and we hypothesized that efficient lysogeny, leading to superinfection immunity, could be the reason behind the inability of DS6A to form plaques on NTM. However, as Φ2GFP12 is missing the attP site, repressor, and integrase genes and should therefore not be capable of lysogeny, this suggests that efficient lysogeny is not the basis for the inability of Φ2GFP12 or its parental strain DS6A to form plaques on NTM.
Host metabolism is known to influence the outcome of phage infection. The species of the MTBC that DS6A is known to infect (M. tuberculosis, M. bovis, M. canettii, M. microti, and M. africanum) grow slowly, with doubling times of at least 16 h. The presence of MazG, a nucleoside triphosphate pyrophosphohydrolase known to upregulate the host metabolism (42), was also thought to be required by DS6A for efficient propagation and to serve the need to propagate faster than the division time of M. tuberculosis. Surprisingly, the presence and absence of mazG in Φ2GFP12 and Φ2GFP13, respectively, did not affect fitness, and both phages generated titers comparable to that of wild-type DS6A. It is possible, however, that DS6A was able to utilize M. tuberculosis MazG to compensate for the loss of its own gene. The functional complementation of an important gene in the mycobacteriophage life cycle by the host gene is not unprecedented. Mycobacteriophages Omega and Corndog have been shown to exploit mycobacterial bacterial nonhomologous end-joining (NHEJ) DNA ligase (LigD) to circularize their short 4-bp cos ends (43). Creating ΔmazG mutants of M. tuberculosis and evaluating the ability of phAE901/Φ2GFP13 to form plaques on them could reveal another host system that mycobacteriophage can commandeer.
Bacteria utilize a wide variety of systems to prevent phage infection, including restriction-modification (R-M) systems, abortive infection systems (Abis), and clustered regularly interspaced short palindromic repeat (CRISPR) systems (44–46), but few such systems have been characterized in mycobacteria. Our novel DS6A fluorophages allow us to start evaluating if these systems operate in NTM. Differences in the efficiencies of plaque formation with phages on differing bacterial hosts have been demonstrated (36, 47–49), and although not yet well explored, mycobacterial restriction systems are a potential explanation. M. abscessus and M. massiliense are very closely related bacteria, and DS6A can infect M. abscessus at low levels, whereas M. massiliense appears to be completely resistant; possible mechanisms underlying these observations range from the lack of a specific phage receptor in M. massiliense to a defense system in M. abscessus that functions early in the phage life cycle, for example, an R-M system that interferes with genome replication, a necessary step in achieving a bright fluorescent signal. In contrast, 40% of M. asiaticum cells appeared infected, indicating a later inhibition in the phage life cycle, such as through an Abi, is the cause of the inability for DS6A to form plaques.
The trend of DS6A to infect without forming plaques was mirrored in our flow cytometry measurements and plaque formation results with Φ2GFP10. This phage was very efficient at generating fluorescence in M. massiliense and M. fortuitum, but it was unable to form plaques even at titers of 1010 PFU/ml. In the presequencing era, plaque formation efficiencies were an accurate method to differentiate various acid-fast mycobacterial species. Presently, with modern tools and technologies, these mycobacterial reporter phages are still useful for developing genetic tools to deliver recombinant DNA or for genetic manipulation. For example, although TM4 and its derivatives do not yield plaques in M. abscessus or M. chelonae (50), these phages can deliver recombinant DNA, such as fluorescent reporters, transposons, and allelic exchange substrates for genetic replacement (51), in M. abscessus. Similar tools will now be generated using DS6A shuttle phasmids to provide an alternative to the TM4-derived mycobacteriophages in cases where TM4 and its derivatives cannot be used.
Even when mycobacteriophage classification was in its infancy, DS6A was recognized as having distinct features related to its restricted host range. Realistically, there are likely many more phages like DS6A, those with unique or specialized host ranges; however, the usual selection method of finding new mycobacteriophages through growth on M. smegmatis, due to the safety concerns of working with pathogenic Mycobacterium species, is likely a source of limitation to finding other phages with unique host ranges. By expanding the number and types of mycobacterial strains utilized in phage enrichment and isolation methods, such as by using nonpathogenic M. tuberculosis derivatives certified for BSL2 usage (6, 10, 52) as well as NTM, the diversity of phages with unique characteristics will likely rise, thus helping to discover novel differences between slow-growing MTBC mycobacteria and their fast-growing NTM counterparts. The creation of these novel DS6A fluorophages has shed light on the meaning of resistance to plaque formation by DS6A, and these phages can be versatile tools in a fluorophage-based diagnostic system of M. tuberculosis. The specificity of DS6A plaque formation on MTBC bacteria remains a very interesting mystery, one that these novel fluorophages can now begin to help answer.
Supplementary Material
ACKNOWLEDGMENTS
This article is dedicated to Morris Kirschen (Papa), a firm believer in always looking toward what is next. We thank Paul Riska for providing strains Mycobacterium avium-M. intracellulare 1403, Mycobacterium avium-M. intracellulare 193B, M. asiaticum, and M. fortuitum for this work.
We declare no conflicts of interest.
Funding Statement
The funders have had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
This article is dedicated to the memory of Stoyan Bardarov.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00592-16.
REFERENCES
- 1.Jain P, Thaler DS, Maiga M, Timmins GS, Bishai WR, Hatfull GF, Larsen MH, Jacobs WR. 2011. Reporter phage and breath tests: emerging phenotypic assays for diagnosing active tuberculosis, antibiotic resistance, and treatment efficacy. J Infect Dis 204(Suppl 4):S1142–S1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Achkar JM, Lawn SD, Moosa MY, Wright CA, Kasprowicz VO. 2011. Adjunctive tests for diagnosis of tuberculosis: serology, ELISPOT for site-specific lymphocytes, urinary lipoarabinomannan, string test, and fine needle aspiration. J Infect Dis 204(Suppl 4):S1130–S1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearson RE, Jurgensen S, Sarkis GJ, Hatfull GF, Jacobs WR Jr. 1996. Construction of D29 shuttle phasmids and luciferase reporter phages for detection of mycobacteria. Gene 183:129–136. doi: 10.1016/S0378-1119(96)00530-6. [DOI] [PubMed] [Google Scholar]
- 4.Jacobs WR Jr, Barletta RG, Udani R, Chan J, Kalkut G, Sosne G, Kieser T, Sarkis GJ, Hatfull GF, Bloom BR. 1993. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science 260:819–822. doi: 10.1126/science.8484123. [DOI] [PubMed] [Google Scholar]
- 5.Piuri M, Jacobs WR Jr, Hatfull GF. 2009. Fluoromycobacteriophages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PLoS One 4:e4870. doi: 10.1371/journal.pone.0004870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jain P, Hartman TE, Eisenberg N, O'Donnell MR, Kriakov J, Govender K, Makume M, Thaler DS, Hatfull GF, Sturm AW, Larsen MH, Moodley P, Jacobs WR Jr. 2012. Φ2GFP10, a high-intensity fluorophage, enables detection and rapid drug susceptibility testing of Mycobacterium tuberculosis directly from sputum samples. J Clin Microbiol 50:1362–1369. doi: 10.1128/JCM.06192-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Donnell MR, Pym A, Jain P, Munsamy V, Wolf A, Karim F, Jacobs WR Jr, Larsen MH. 2015. A novel reporter phage to detect tuberculosis and rifampin resistance in a high-HIV-burden population. J Clin Microbiol 53:2188–2194. doi: 10.1128/JCM.03530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobs WR Jr, Tuckman M, Bloom BR. 1987. Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature 327:532–535. doi: 10.1038/327532a0. [DOI] [PubMed] [Google Scholar]
- 9.Bardarov S, Kriakov J, Carriere C, Yu S, Vaamonde C, McAdam RA, Bloom BR, Hatfull GF, Jacobs WR Jr. 1997. Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 94:10961–10966. doi: 10.1073/pnas.94.20.10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain P, Hsu T, Arai M, Biermann K, Thaler DS, Nguyen A, Gonzalez PA, Tufariello JM, Kriakov J, Chen B, Larsen MH, Jacobs WR Jr. 2014. Specialized transduction designed for precise high-throughput unmarked deletions in Mycobacterium tuberculosis. mBio 5(3):e01245-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vilcheze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, Weisbrod TR, Alland D, Sacchettini JC, Jacobs WR Jr. 2006. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat Med 12:1027–1029. doi: 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- 12.Bardarov S, Bardarov S Jr, Pavelka MS Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR Jr. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 13.Snapper SB, Lugosi L, Jekkel A, Melton RE, Kieser T, Bloom BR, Jacobs WR Jr. 1988. Lysogeny and transformation in mycobacteria: stable expression of foreign genes. Proc Natl Acad Sci U S A 85:6987–6991. doi: 10.1073/pnas.85.18.6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatfull G, Jacobs WR Jr. 2000. Molecular genetics of mycobacteria. ASM Press, Washington, DC. [Google Scholar]
- 15.Kendall BA, Varley CD, Choi D, Cassidy PM, Hedberg K, Ware MA, Winthrop KL. 2011. Distinguishing tuberculosis from nontuberculous mycobacteria lung disease, Oregon, USA. Emerg Infect Dis 17:506–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sula L, Sulova J, Stolcpartova M. 1981. Therapy of experimental tuberculosis in guinea pigs with mycobacterial phages DS-6A, GR-21 T, My-327. Czech Med 4:209–214. [PubMed] [Google Scholar]
- 17.Zemskova ZS, Dorozhkova IR. 1991. Pathomorphological assessment of the therapeutic effect of mycobacteriophages in tuberculosis. Probl Tuberk (11):63–66. (In Russian.) [PubMed] [Google Scholar]
- 18.Sula L, Sulova J, Stolepartova M. 1981. Phagotherapy with homologous and heterologous mycobacterial phages in experimental tuberculosis of rabbits infected with M. microti “MP” strain. Studia Pneumol Phtiseol Cechoslov. [Google Scholar]
- 19.Pearson RE, Dickson JA, Hamilton PT, Little MC, Beyer WF Jr. December 1995. Mycobacteriophage DSGA specific for the Mycobacterium tuberculosis complex. US patent 5,476,768.
- 20.Pearson RE, Dickson JA, Hamilton PT, Little MC, Beyer WF Jr. March 1997. Mycobacteriophage specific for the Mycobacterium Tuberculosis complex. US patent 5,612,182.
- 21.Pearson RE, Dickson JA, Hamilton PT, Little MC, Beyer WF Jr. December 1996. Mycobacteriophage specific for the Mycobacterium tuberculosis complex. US patent 5,582,969.
- 22.Pearson RE, Dickson JA, Hamilton PT, Little MC, Beyer WF Jr. May 1997. DNA polymerase III β-subunit from mycobacteriophage DS6A. US patent 5,633,159.
- 23.Redmond WB, Cater JC. 1960. A bacteriophage specific for Mycobacterium tuberculosis, varieties hominis and bovis. Am Rev Respir Dis 82:781–786. [DOI] [PubMed] [Google Scholar]
- 24.Petrova ZO, Broussard GW, Hatfull GF. 2015. Mycobacteriophage-repressor-mediated immunity as a selectable genetic marker: Adephagia and BPs repressor selection. Microbiology 161:1539–1551. doi: 10.1099/mic.0.000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowman BU. 1969. Properties of mycobacteriophage DS6A I. Immunogenicity in rabbits. Proc Soc Exp Biol Med 131:196–200. doi: 10.3181/00379727-131-33838. [DOI] [PubMed] [Google Scholar]
- 26.Hatfull G. Manufacturing high titer lysates. The Actinobacteriophage Database at PhagesDB.org. http://phagesdb.org/media/workflow/protocols/pdfs/Manufacturing_a_High_Titer_Lysate_6.2013_PDF_1.pdf.
- 27.Balasubramanian V, Pavelka MS Jr, Bardarov SS, Martin J, Weisbrod TR, McAdam RA, Bloom BR, Jacobs WR Jr. 1996. Allelic exchange in Mycobacterium tuberculosis with long linear recombination substrates. J Bacteriol 178:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. 2010. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res 38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatfull G. 2014. Molecular genetics of mycobacteriophages. Microbiol Spectr 2:1–36. [PMC free article] [PubMed] [Google Scholar]
- 32.Sambandamurthy VK, Derrick SC, Hsu T, Chen B, Larsen MH, Jalapathy KV, Chen M, Kim J, Porcelli SA, Chan J, Morris SL, Jacobs WR Jr. 2006. Mycobacterium tuberculosis DeltaRD1 DeltapanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine 24:6309–6320. doi: 10.1016/j.vaccine.2006.05.097. [DOI] [PubMed] [Google Scholar]
- 33.Panas MW, Jain P, Yang H, Mitra S, Biswas D, Wattam AR, Letvin NL, Jacobs WR Jr. 2014. Noncanonical SMC protein in Mycobacterium smegmatis restricts maintenance of Mycobacterium fortuitum plasmids. Proc Natl Acad Sci U S A 111:13264–13271. doi: 10.1073/pnas.1414207111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riska PF, Jacobs WR Jr, Bloom BR, McKitrick J, Chan J. 1997. Specific identification of Mycobacterium tuberculosis with the luciferase reporter mycobacteriophage: use of p-nitro-alpha-acetylamino-beta-hydroxy propiophenone. J Clin Microbiol 35:3225–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huth RG, Douglass E, Mondy K, Vasireddy S, Wallace RJ Jr. 2015. Treatment of Mycobacterium abscessus subsp. massiliense tricuspid valve endocarditis. Emerg Infect Dis 21:535–537. doi: 10.3201/eid2103.140577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, Petrova ZO, Dedrick RM, Pope WH, Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science Sea-Phages Program, Modlin RL, Hendrix RW, Hatfull GF. 2012. On the nature of mycobacteriophage diversity and host preference. Virology 434:187–201. doi: 10.1016/j.virol.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pope WH, Bowman CA, Russell DA, Jacobs-Sera D, Asai DJ, Cresawn SG, Jacobs WR, Hendrix RW, Lawrence JG, Hatfull GF, Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science, Phage Hunters Integrating Research and Education, Mycobacterial Genetics Course. 2015. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. eLife 4:e06416. doi: 10.7554/eLife.06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatfull GF. 2014. Mycobacteriophages: windows into tuberculosis. PLoS Pathog 10:e1003953. doi: 10.1371/journal.ppat.1003953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh S, Plaks JG, Homa NJ, Amrich CG, Heroux A, Hatfull GF, VanDemark AP. 2014. The structure of Xis reveals the basis for filament formation and insight into DNA bending within a mycobacteriophage intasome. J Mol Biol 426:412–422. doi: 10.1016/j.jmb.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marei AM, El-Behedy EM, Mohtady HA, Afify AF. 2003. Evaluation of a rapid bacteriophage-based method for the detection of Mycobacterium tuberculosis in clinical samples. J Med Microbiol 52:331–335. doi: 10.1099/jmm.0.05091-0. [DOI] [PubMed] [Google Scholar]
- 41.Broussard GW, Hatfull GF. 2013. Evolution of genetic switch complexity. Bacteriophage 3:e24186. doi: 10.4161/bact.24186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu LD, Sun Q, Fan XY, Zhong Y, Yao YF, Zhao GP. 2010. Mycobacterial MazG is a novel NTP pyrophosphohydrolase involved in oxidative stress response. J Biol Chem 285:28076–28085. doi: 10.1074/jbc.M109.088872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pitcher RS, Tonkin LM, Daley JM, Palmbos PL, Green AJ, Velting TL, Brzostek A, Korycka-Machala M, Cresawn S, Dziadek J, Hatfull GF, Wilson TE, Doherty AJ. 2006. Mycobacteriophage exploit NHEJ to facilitate genome circularization. Mol Cell 23:743–748. doi: 10.1016/j.molcel.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 44.Wilson GG, Murray NE. 1991. Restriction and modification systems. Annu Rev Genet 25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
- 45.Chopin MC, Chopin A, Bidnenko E. 2005. Phage abortive infection in lactococci: variations on a theme. Curr Opin Microbiol 8:473–479. doi: 10.1016/j.mib.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 46.Szczepankowska A. 2012. Role of CRISPR/Cas system in the development of bacteriophage resistance. Adv Virus Res 82:289–338. doi: 10.1016/B978-0-12-394621-8.00011-X. [DOI] [PubMed] [Google Scholar]
- 47.Jones WD Jr, Greenberg J. 1977. Host modification and restriction with a mycobacteriophage isolated from a pseudolysogenic Mycobacterium chelonei. J Gen Microbiol 99:389–395. doi: 10.1099/00221287-99-2-389. [DOI] [PubMed] [Google Scholar]
- 48.Jones WD., Jr 1975. Differentiation of known strains of BCG from isolates of Mycobacterium bovis and Mycobacterium tuberculosis by using mycobacteriophage 33D. J Clin Microbiol 1:391–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones WD., Jr 1979. Further studies of mycobacteriophage 33D (Warsaw) for differentiation of BCG from M. bovis and M. tuberculosis. Tubercle 60:55–58. doi: 10.1016/0041-3879(79)90057-6. [DOI] [PubMed] [Google Scholar]
- 50.Calado Nogueira de Moura V, Gibbs S, Jackson M. 2014. Gene replacement in Mycobacterium chelonae: application to the construction of porin knock-out mutants. PLoS One 9:e94951. doi: 10.1371/journal.pone.0094951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medjahed H, Reyrat JM. 2009. Construction of Mycobacterium abscessus defined glycopeptidolipid mutants: comparison of genetic tools. Appl Environ Microbiol 75:1331–1338. doi: 10.1128/AEM.01914-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sambandamurthy VK, Wang X, Chen B, Russell RG, Derrick S, Collins FM, Morris SL, Jacobs WR Jr. 2002. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat Med 8:1171–1174. doi: 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- 53.Cresawn SG, Bogel M, Day N, Jacobs-Sera D, Hendrix RW, Hatfull GF. 2011. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics 12:395. doi: 10.1186/1471-2105-12-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.